

Abstract
Understanding protein stability is central to combatting protein aggregation diseases and developing new protein therapeutics. At the high concentrations often present in biological systems, purified proteins can exhibit undesirable high solution viscosities and poor solubility mediated by short-range electrostatic and hydrophobic protein-protein interactions. The interplay between protein amino acid sequence, protein structure and solvent conditions to minimize protein-protein interactions is key to design of well-behaved pharmaceutical proteins. However, theoretical approaches have yet to yield a general framework to address these problems. Here, we analyzed the high concentration behavior of super-folder GFP (sfGFP) and two super-charged sfGFP variants engineered to have formal charges of −18 or +15. Under low cosolute conditions, sfGFP and the −18 variant formed a gel or phase separated at ~10 mg/ml. Under conditions that screen surface charges, including formulations with high histidine or high NaCl concentrations, all three variants attained concentrations up to 250 mg/ml with moderate viscosities. Moreover, all three variants exhibited very similar viscosity-concentration profiles over this range. This effect was not mimicked by high sugar concentrations that exert excluded volume effects without shielding charge. Collectively, these data demonstrate that charge shielding not only neutralizes long-range electrostatic interactions but also, surprisingly, short-range electrostatic effects due to surface charge anisotropy. This work shows that supercharged sfGFP behavior under high ionic strength is largely determined by particle geometry, a conclusion that is supported by colloid models and may be applicable to pharmaceutically-relevant proteins.
Keywords: Protein aggregation, protein anisotropy, hard sphere, viscosity, electrostatic interactions
Graphical abstract
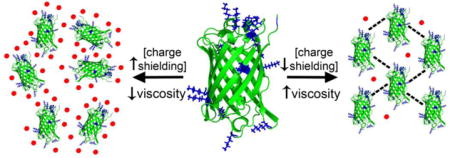
Introduction
Proteins have generated broad interest for their ubiquitous roles in cellular function as well as for therapeutic applications. However, many proteins have characteristics that predispose them to aggregation, which impedes protein activity. Aggregation of misfolded proteins underlies a growing number of diseases, including Alzheimer’s and Parkinson’s diseases.1–2 Similarly, proteins such as therapeutic monoclonal antibodies can reversibly associate into higher molecular weight species or irreversibly aggregate during manufacturing and storage.3 In this state, proteins have reduced biological activity, accelerated pharmacokinetic clearance, and increased risks of adverse immune reactions and precipitation. Intermolecular forces between proteins, namely short-range attractive and long-range repulsive forces, contribute to protein stability in vitro and in vivo.4
Understanding these weak protein-protein interactions is thus key to maintaining protein solubility. In dilute protein solutions, long-range electrostatic forces dominate protein-protein interactions.4–5 As protein concentration increases, the contributions from short-range interactions, including hydrogen bonds, van der Waals forces, hydrophobic and excluded volume interactions also increase and can drive protein-protein association.6 The overall impact of these interactions and the resulting propensity for association can be quantified by experimentally determined parameters including the osmotic second virial coefficient (B22),7 the Kirkwood-Buff integral (G22),8 and the dynamic light scattering interaction parameter (kD).9 Favorable interaction parameters can guide selection of a protein variant for therapeutic development as well as buffer formulation to stabilize a given protein. Conversely, unfavorable interaction parameters can explain why one protein allele is more prone to aggregation and severe disease than another.10 However, as proteins are complex molecules able to engage in a variety of protein-protein and protein-solvent interactions, models attempting to predict protein solution behavior at high concentrations have met with mixed results.9, 11–14
The contributions of electrostatics to protein behavior have been studied extensively by computational15–17 and experimental approaches.4, 18 Electrostatic interactions are typically modeled as long-range repulsive interactions originating from a protein’s net surface charge. These forces can be tuned by changing the protein amino acid sequence and thus the net charge or by changing the type and concentration of solute molecules present to shield protein surfaces. For instance, modulating the protein surface charge has been observed to improve protein thermostability,19–20 while adding charged residues adjacent to antibody binding sites can reduce aggregation.21 Similarly, optimization of the formulation buffer and salt concentrations can prevent protein self-association and decrease solution viscosity.14
A major outstanding question is how proteins retain solubility and activity at high protein concentrations. Proteins intended for pharmaceutical use require storage as a homogeneous preparation at concentrations up to ~250 mg/ml. This is often difficult to achieve due to protein association and the resulting high viscosity at these concentrations. When protein concentrations reach 100 mg/ml, the average protein-protein separation distance for an average size protein is less than 10 nm.22 At these concentrations, short-range attractive forces acting over nanometer and tens-of-nanometer length scales dominate over surface-charge-mediated, repulsive electrostatic interactions.23 These net attractive forces, due to hydrogen bonds, hydrophobic interactions, depletion-attraction, and electrostatic attractive interactions, combined with the close proximity of protein species in solution, promote aggregation and high viscosity. In contrast, the cellular cytoplasm comprises >300 mg/ml total protein with comparatively little protein aggregation.24
Because of the complex physics and chemistry of protein interactions at high concentrations, no general theories have been developed to predict electrostatic effects and guide design of proteins and formulations able to achieve high concentrations. For the monoclonal antibodies that can remain soluble at high concentrations, this behavior has been attributed to unknown features of the protein amino acid sequence.25 Protein surface charge appears to play a major role, as a net negative charge correlates with reduced aggregation while a net positive charge correlates with increased aggregation in at least some cases.26–27 The role of protein surface charge is supported by the observation that a common strategy to stabilize proteins is simply adjusting the solution pH away from the protein isoelectric point, which confers a net charge to the protein.28 These observations have led to considerable interest in development of rational strategies and predictive models to guide protein engineering and formulation design efforts.
Here, we investigated the impact of electrostatic anisotropy on protein solubility and viscosity at high protein concentrations. We chose a related set of proteins, the wild-type superfolder GFP (sfGFP) with a formal charge of −6 and two highly charged sfGFP variants engineered to have surface charges of −18 and +15.29–30 Under low cosolute concentrations typical of pharmaceutical formulations, the −18 and sfGFP variants aggregated extensively at 10 mg/ml protein. However, under high cosolute concentrations, including high concentrations of histidine, trehalose or salt, all three variants remained soluble at protein concentrations exceeding 250 mg/ml. Remarkably, the protein surface charge had minimal effect on the overall solution viscosity under conditions that screened surface charges and the viscosity profiles of the supercharged variants collapsed onto that of the sfGFP. These experimental results provide an experimentally tractable model system to bridge protein structure-based and colloid based models of protein behavior.
Experimental section
All chemicals were obtained from Fisher Scientific unless otherwise noted.
Protein expression, purification and characterization
The wild-type superfolder GFP (sfGFP), supercharged RscG-18 GFP (−18), and RscG+15 GFP (+15) variants were engineered and cloned into pET21a vectors for cytoplasmic bacterial expression as described.30 Proteins were expressed in E. coli BL21 DE3 cells in TB media at 25°C to OD600~2.0. Cells were induced with isopropyl β-D-1-thiogalactopyranoside (IPTG) for 18 hours at 18°C, then harvested and resuspended in cell lysis buffer (25 mM HEPES, 500 mM NaCl, 40 mM imidazole, pH 7.4) and lysed via french press (Thermo-Scientific) at 1500 psi. The cell debris was pelleted by centrifugation at 20,000 RPM for 20 minutes at 4°C (Beckman-Coulter) and the supernatant was dialyzed overnight into 500 mM NaCl, 10 mM Tris, pH 8.0. The dialysate was incubated with charged IMAC Sepharose 6 Fast Flow beads (GE Healthcare) for at least 3 hours and eluted with 1.5 M NaCl, 100 mM EDTA, 20 mM Tris, pH 8.0. The eluate was applied to a HiLoad 16/60 Superdex 200 prep grade size exclusion chromatography (SEC) column with phosphate buffered saline (PBS; 137 mM NaCl, 10 mM phosphate, 2.7 mM KCl, pH 7.4) as the running buffer.
Protein size and purity was monitored by sodium-dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) on 12% gels and analytical SEC with a Superdex 75 10/300 GL column (GE Healthcare) on a GE ÅKTA FPLC with PBS running buffer for the sfGFP and −18 variants, with calculated iso-electric points of 6.4 and 5.1, respectively. To prevent anionic interactions between the +15 variant and the column matrix which has a calculated isoelectric point of 10.5, 50 mM sodium phosphate buffer with 1 M NaCl, pH 7.2 was used as running buffer. The specific activity of the sfGFP variants was measured as the ratio of the absorbance at 488 nm normalized by the absorbance at 280 nm, which is directly proportional to the protein concentration.30 Absorbances were measured with a Cary spectrophotometer, with replicate technical measurements of replicate samples.
To measure the thermal melting temperatures via loss of intrinsic green fluorescence, purified sfGFP proteins were adjusted to 800 μg/ml in PBS and 0.2 mm filtered. Protein (12.5 μl) was combined with 5 μl Protein Thermal Shift Buffer (Thermo Pierce) and PBS for a 20 μl final volume in a MicroAmp Fast 96-well reaction plate. The plate was subjected to a melt curve on an Applied Biosystems Viia7 system using two heating steps between 25°C and 99°C, with ramp rates of 1.6°C/s and 0.05°C/s respectively. The melting temperature was determined as the temperature at which the derivative fluorescence of with respect to temperature is zero.
For CD analysis, purified sfGFP proteins were buffer exchanged into 5 mM sodium phosphate pH 4.5 (sfGFP and +15) or 5 mM Tris pH 8.4 (−18) with the pH adjusted using sulfuric acid, adjusted to ~0.3 mg/ml and filter sterilized. Samples were analyzed from 190–260 nm on a J-815 CD spectrometer using a 1 mm cuvette and the appropriate buffer for blanks.
Protein formulation
Proteins were concentrated by centrifugation as previously described.31 Purified sfGFP proteins were first buffer exchanged into the desired buffer by overnight dialysis (see Table 1 for the different compositions). The proteins were then concentrated using 10 kDa NMWL Amicon Ultra-15 regenerated cellulose centrifugal filters (EMD Millipore) at 3000g. The reduced protein volume was transferred to the smaller Amicon Ultra-4 regenerated cellulose centrifugal filters (EMD Millipore) for concentration at 3000g, and finally concentrated in 10 kDa NMWL Amicon Ultra-0.5 mL centrifugal filters at 10,000g to accommodate the reduced volumes. Proteins were concentrated until the protein concentration reached ~250 mg/ml or high viscosity prevented further concentration. The pH of all formulations was measured with a calibrated pH-meter with no added protein and at the highest protein concentration achieved and found to be within 0.1 unit of the initial pH for all formulations except the low sucrose formulation.
Table 1.
Formulation compositions and observations
| Formulation | Composition | Result |
|---|---|---|
| Low sucrose-low histidine | 10 mM histidine, 146 mM sucrose, pH 5.8 | Gelation/phase separation at ~10 mg/ml protein for sfGFP and −18 variant; +15 variant reached ~250 mg/ml |
| Low trehalose-low histidine | 10 mM histidine, 146 trehalose, pH 5.8 | Gelation/phase separation ~10 mg/ml for −18 sfGFP variant |
| High trehalose-high histidine, lyophilize | 50 mM phosphate, 320 mM histidine, 660 mM trehalose, pH 5.8, concentrated by lyophilization | All three sfGFP variants soluble up to ~250 mg/ml; exponential viscosity-protein profile |
| High trehalose-high histidine | 50 mM phosphate, 320 mM histidine, 660 mM trehalose, pH 5.8 | Exponential viscosity-protein profile for −18 sfGFP variant up to ~250 mg/ml |
| High trehalose-low histidine | 10 mM histidine, 660 mM trehalose, pH 5.8 | Viscosity increased rapidly relative to high trehalose-high histidine for −18 sfGFP variant |
| High histidine | 50 mM phosphate, 320 mM histidine, pH 5.8 | Relative viscosity was similar as compared to high trehalose-high histidine, but absolute viscosity was lower for −18 sfGFP variant |
| High trehalose-high salt | 10 mM histidine, 250 mg/ml trehalose, 210 mM NaCl, pH 5.8 | Similar viscosity profile as high trehalose-high histidine for −18 sfGFP variant |
| High salt-low histidine | 10 mM histidine, 210 mM NaCl, pH 5.8 | Absolute viscosity decreased as compared to high trehalose-high histidine, but relative viscosity was similar for −18 sfGFP variant |
| Low sucrose-low tris, pH 8.0 | 10 mM Tris buffer, 146 mM sucrose, pH 8.0 | −18 sfGFP variant soluble up to ~250 mg/ml; pH drift to 6.8 at high protein concentration |
All formulations were concentrated by centrifugation unless noted as lyophilization; all formulations used the the −18 sfGFP variant unless noted.
The high trehalose-high histidine formulation was initially generated using lyophilization. In this case, the proteins were first dialyzed overnight into 5 mM histidine buffer, pH 5.5. Proteins were next concentrated to ~20 mg/ml via Amicon Ultra-15 centrifugal filters with 10 kDa NMWL regenerated cellulose filters (EMD Millipore) as measured by microBCA (Thermo Fisher Pierce). The lyoprotectant α, α trehalose dihydrate was then added in a 1:1 mass ratio. Lyophilization was performed with a vacuum at 150 mTorr and −25°C for three days before increasing to 25°C over five hours. Lyophilized protein powder was weighed and reconstituted with 50 mM sodium phosphate buffer pH 7.2 and histidine stock solution such that the final conditions were ~250 mg/ml protein, ~660 mM trehalose, and ~320 mM histidine at pH 5.8.
Viscosity measurements
After equilibrating the sample temperature to 25°C, viscosities were measured in triplicate using a 25 gauge needle attached to a 1 mL syringe, as previously described.22 The time to draw the solution was correlated to a calibration curve following the Hagen-Pouiselle Equation fit using S200, N100, S60, N44, N35, N10 viscosity standards (Cannon Instrument Company, State College, PA) and distilled/deionized water (Figure S1). The solutions were serially diluted in the same solvent and viscosity measurements repeated. Solvent viscosities were measured using a Cannon-Fenske routine viscometer. All viscosity model fits were performed in MATLAB using the nlinfit non-linear regression function to the Ross-Minton viscosity equation (Equation 1).
Protein concentration measurements
Protein preparations were diluted such that the absorbance at 280 nm yielded values between 0.1 and 1.0. Protein concentrations were calculated using the A280 value and experimentally determined extinction coefficients for each protein (Figure S2). Absorbance measurements were collected with a Cary 3E UV-vis spectrophotometer (Agilent Technologies) using a quartz cuvette with a 1 cm path length (Hellma). The concentration of protein in all final preparations was measured with this method.
Zeta potential measurements
The theoretical charge of each sfGFP was calculated from the primary sequence using all ionizable amino acid side chains plus the C and N termini. The charge value of each of these groups was calculated from the Henderson-Hasselbalch equation , using known pKa values for each ionizable group. Changes in residue pKa values due to the surrounding environment were ignored and therefore these calculations are only a first approximation of the charge. To experimentally measure the zeta potential, two solutions of each sfGFP variant at low sucrose and high trehalose-high histidine conditions were prepared at a protein concentration of 1 mg/ml. The zeta potential was measured with a Malvern DTS 1070 folded capillary cell and Malvern Zetasizer Nano ZS. The zeta potential (ξ) was determined from the electrophoretic mobility measured at 25°C from 70 runs for each of the 12 individual measurements for each variant.
Electrostatic surface potential maps
All models are based on the structure of sfGFP (PDB 2B3P).29 Homology models of the −18 and +15 variants were generated using the Rosetta supercharge protocol: 11 to Asp or Glu for −18 or 16 to Arg or Lys for +15, such that each variant carried the specified charge at pH 7.30 The resulting PDB files were analyzed with PDB2PQR30 to adjust the pH to 5.8. The resulting PQR files were analyzed with DelPhi using the AMBER forcefield to fit charges and adjust the solution ionic strength (salt concentration) to the ionic strength of the solutions: ~6 mM for low cosolute conditions and ~270 mM for high cosolute conditions. Default values were used for all remaining variables to generate CUBE files.32–33 The PQR and CUBE files were loaded into PyMOL and the electrostatic potential maps were visualized with APBS.
Results
Here, we aimed to evaluate the role of patchy surface electrostatics in mediating protein solubility at high concentration. Since protein surface charge has been reported to influence protein-protein interactions and viscosity, we selected three sfGFP variants with different net surface charges (−18, −6 and +15 formal charges) as a model system. Attractive features of this system include sfGFP’s single domain structure, high stability and the previous description of a series of super-charged sfGFP variants.30 We selected the −18 and +15 variants, designed by an automated physics-based Rosetta process to include 11 and 15 residue changes, respectively (Figure S3), as representative positively and negatively super-charged variants. These variants were selected because they are highly expressed, soluble and thermostable. More highly charged variants (for example, −32 and +58) were not considered due to poor expression and/or altered folding characteristics.19, 30 After expression in E. coli and purification, the proteins were concentrated in the presence of different buffers, cosolute molecules and pHs with the resulting protein solubilities and viscosities monitored.
Low cosolute conditions prevent aggregation of only the negatively charged sfGFP variant
We initially selected a formulation with low levels of sucrose and histidine, similar to that used to formulate several commercial protein therapeutics (low sucrose-low histidine formulation: 10 mM histidine, 146 mM sucrose, pH 5.8; Table 1).34–35 This formulation is popular because it includes histidine as a pH buffer, sucrose to stabilize the protein and employs a pH that is below the isoelectric point (pI) of the typical antibody.28 Purified sfGFP proteins were dialyzed into this buffer at ~1 mg/ml and concentrated by centrifugation over a molecular weight filter.
Notably, the −18 and sfGFP proteins formed a gel or phase separated at concentrations near 10 mg/ml, respectively, suggesting the presence of strong inter-protein interactions. A similar low trehalose-low histidine formulation of the −18 sfGFP using 146 mM trehalose instead of sucrose also gelled upon concentration, indicating that the specific sugar molecule did not independently enhance solubility (Table 1). In contrast, the +15 variant was readily concentrated to ~250 mg/ml. Serial dilution of the highly concentrated +15 sfGFP from 250 mg/ml down to 75 mg/ml revealed an exponential relationship between protein concentration and solution viscosity η (Figure 1A). Since high concentrations of sugars increase the solvent viscosity ηo even in the absence of protein, these data are plotted as the relative viscosity, η/ηo, to clearly visualize the effects of protein concentration.36
Figure 1. Solution viscosity increases exponentially with sfGFP protein concentration under low cosolute and high cosolute conditions.
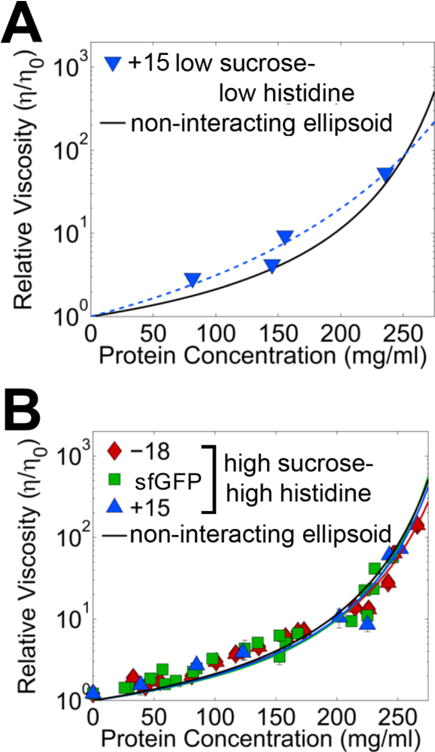
The solution viscosity was measured for each of three sfGFP variants at different protein concentrations in the two formulations. A, Under low cosolute conditions in low sucrose buffer (10 mM histidine, 146 mM sucrose, pH 5.8), only the +15 variant remained soluble, with moderate viscosities at concentrations >200 mg/ml. B, Under high cosolute conditions in histidine-high trehalose buffer (~320 mM histidine, ~660 mM trehalose and pH 5.8), all three sfGFP variants remained soluble, with moderate viscosities up to 250 mg/ml. Data for each protein variant was collected from at least two separate experiments with at least two different protein preparations. Error bars represent the range of replicate measurements; data points shown for each variant come from at least two independent formulations with two different protein preparations. The experimental data were fit to Ross and Minton’s modified Mooney viscosity model with the model fits shown as solid lines of the same color as the corresponding icons. The solid black line represents the theoretical Ross-Minton viscosity profile for a hard particle of the same aspect ratio sfGFP (see text for details).
High trehalose-high histidine formulation retains sfGFP variant solubility
We wondered if we might be able to solubilize all three proteins in the same formulation, using a lyophilization-based approach which previously supported formulation of several different proteins at high concentration with modest viscosities.22 The purified sfGFP variants were first dialyzed into 5 mM histidine buffer, pH 5.5, then trehalose was then added at a 1:1 mass ratio as a lyoprotectant and the resulting solution lyophilized. The powder was resuspended with 50 mM sodium phosphate buffer to produce a high trehalose-high histidine formulation with ~320 mM histidine and ~660 mM trehalose, at pH 5.8 and ~250 mg/ml protein. While this formulation is hyper-tonic relative to human serum and thus not suitable for injectable therapeutics, the approach resulted in clear solutions for all three sfGFP variants (Figure S4).
These protein solutions were serially diluted from ~250 mg/ml in high trehalose-high histidine buffer and the resulting relative solution viscosities plotted against protein concentration (Figure 1B). The viscosity again showed an exponential dependence on protein concentration, approaching the measured solvent viscosity (2.58 ± 0.03 cP) at low protein concentrations and retaining syringeable viscosities (<20 cP) at concentrations >200 mg/ml. Remarkably, these curves showed very close correspondence for all three sfGFP variants in this formulation, despite small differences in thermal stability due to supercharging.19 To determine whether the process used to reach high concentration, centrifugation or lyophilization, influenced the results, we also formulated the −18 variant in the high trehalose-high histidine formulation by centrifugation. Comparison of the viscosity-concentration data shows a close correspondence regardless of which process was used (Figure S5), as has been seen before.31
The purified protein for the sfGFP variants all exhibit a characteristic beta sheet structure as measured by circular dichroism and similar intrinsic stabilities, as measured by thermal melting temperature (Figure S6, Table 2). These results are similar to previous results for sfGFP and supercharged variants.19, 30 After formulation at ~250 mg/ml in high trehalose-high histidine buffer, the diluted proteins exhibited no indication of misfolding or irreversible aggregation. Analytical size exclusion chromatography traces showed that each of the sfGFP variants eluted in a single major peak (>99% all eluted protein), at a volume corresponding to monomeric protein (Figure 2A), while SDS-PAGE showed >95% pure protein with no observable aggregation (Figure 2B). Finally, GFP fluorescence is a sensitive probe of the folded state of the protein, as loss of tertiary structure surrounding the chromophore results in loss of fluorescence.29–30 The specific sfGFP activity, measured as the ratio of the absorbance at 488 nm to absorbance at 280 nm at 2 μM, was relatively constant, indicating no apparent change in protein folding (Figure 2C).
Table 2. Biophysical data for GFP solutions at low sucrose or high trehalose-high histidine conditions.
Estimates of the intrinsic viscosity [η] and effective crowding factor (k/ν) for high cosolute were extracted from the Ross-Minton equation fits of data in Figure 1. The error in the intrinsic viscosity and effective crowding factor k/ν is the error in the nonlinear fit to Equation 1, while the error in the zeta potential represents the standard deviation from twelve measurements with 70 runs determining the zeta potential for each measurement for two individual samples for each variant at low sucrose or high trehalose-high histidine conditions. For comparison, the theoretical intrinsic viscosity and effective crowding factor of a hydrated hard particle with the same aspect ratio as sfGFP is shown.
| sfGFP variant | Solvent viscosity (cP) | Intrinsic viscosity (ml/g) | k/ν | Zeta potential in low sucrose-low histidine conditions, 1 mg/ml protein (mV) | Zeta potential in high trehalose-high histidine conditions, 1 mg/ml protein (mV) | Calculated isoelectric point | Measured melting temperature (°C) |
|---|---|---|---|---|---|---|---|
| −18 | 2.58 ± 0.03 | 5.3 ± 0.5 | 0.5 ± 0.1 | −7.7 ± 0.9 | 0.1 ± 1.2 | 5.1 | 93.0 |
| sfGFP | 2.58 ± 0.03 | 4.9 ± 0.6 | 0.6 ± 0.1 | −1.2 ± 0.1 | −0.8 ± 2.1 | 6.4 | 92.4 |
| +15 | 2.58 ± 0.03 | 5.1 ± 1.4 | 0.6 ± 0.2 | 7 ± 2 | 1.4 ± 1.6 | 10.5 | 89.2 |
| Hard particle (L/D =1.8) | 2.58 | 5.5 | 0.5 | n/a | n/a | n/a | n/a |
Figure 2. Supercharged sfGFP variants do not irreversibly aggregate at high protein concentration in high trehalose-high histidine buffer.
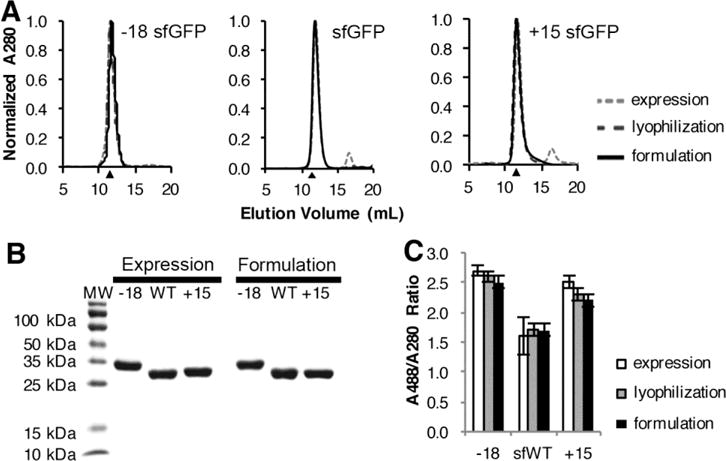
A, Analytical SEC was used to monitor the presence of soluble aggregates at different processing stages using a Superdex 75 column and an ÅKTA FPLC. Chromatograms were collected for a 100 μl injection of 1 mg/ml each sfGFP variant after initial purification, lyophilization, and dilution from high trehalose-high histidine conditions. Protein molecular weight standards indicated that the primary peaks eluted at volumes consistent with the 27–30 kDa sfGFP size; black triangles indicate the expected elution volume of a 27.5 kDa protein based on the calibration curve of protein molecular weight standards (Figure S5). B, SDS-PAGE analysis was used to monitor protein purity and the presence of covalent aggregates. Each sfGFP variant was analyzed after initial purification and after dilution from high concentration. The shift exhibited by the −18 variant is due to the altered electrophoretic mobility of the negatively charge protein. C, The specific activity for each sfGFP variant was measured as the ratio of absorbance at 488 nm divided by the absorbance at 280 nm, which proportional to total protein concentration. Error bars represent the standard deviation of replicate measurements; each experiment was repeated with at least two independent samples.
Comparison to colloid theory
The similarities among the viscosity curves for all three sfGFP charge variants in the high trehalose-high histidine formulation were particularly notable as they suggest the three proteins are experiencing nearly identical protein-protein interactions despite their very different surface charges. To explore this behavior more quantitatively, we fit the viscosity-concentration data to Ross and Minton’s modified Mooney equation (Equation 1). This equation was first used to describe non-idealities in hemoglobin solutions37 and is now commonly used to model viscosity versus concentration data for protein solutions.38 Unlike other models, this semi-empirical model accounts for protein shape and the excluded volume contribution to solution viscosity, without making assumptions about protein hydration states in the volume fraction.39
| (Equation 1) |
Here, η is the solution viscosity, η0 is the solvent viscosity, [η] is the intrinsic viscosity of the specific protein, c is the protein concentration, ν is the Simha shape parameter,40 and k is the crowding factor that is equivalent to the reciprocal of the maximum packing fraction of the particle. Typically, the crowding factor and Simha shape parameter are combined into a single effective crowding constant (k/ν), which gives an approximation of the effect of self-crowding. The solvent viscosity of each formulation was measured using a Cannon-Fenske viscometer, the protein concentration by absorbance at 280 nm and the solution viscosity with a syringe viscometer for each sample. The intrinsic viscosity [η] and crowding constant (k/ν) were determined from Equation 1, by determining which [η] and k/ν values provide the best fit between the experimentally obtained data and the model.
The modified Mooney model fits for the experimental data are shown as solid colored lines in Figure 1. Notably, the fitted intrinsic viscosity and effective packing fraction values were within error for all three sfGFP variants (Table 2), supporting the idea that these three protein variants behave identically under high trehalose-high histidine conditions despite the dramatically different surface charges and poor solubility under low sucrose-low histidine conditions for the −18 and sfGFP proteins.
Since the surface charges of the three sfGFP variants did not appear to affect their concentration-viscosity curves, we wondered about the strength of protein-protein interactions in these systems. Colloid theory predicts the behavior of non-interacting hard particles based only on steric constraints. Since this model does not account for specific protein-protein interactions due to electrostatics, this represents the lowest attainable viscosity for a system of homogeneous particles.
To apply hard particle theory to sfGFP, the following parameters were used. The protein was modeled as a prolate ellipsoid with an aspect ratio of 1.8, as measured from the sfGFP structure (PDB 2B3P). The crowding factor k was estimated as 1.43 from the maximum packing of a prolate ellipsoid,41 and ν was estimated as 2.82 from a table of shape parameters as a function of ellipsoid aspect ratios,40 resulting in a shape factor k/ν of 0.5. The dimensionless intrinsic viscosity of a prolate ellipsoid with 1.8 aspect ratio was estimated as 3.17, 40, 42 The approximate protein density was obtained from the sfGFP Matthew’s coefficient of 2.92 Å3/Da (PDB 2B3P),29 which is equivalent to 0.569 g/ml after units conversion.43 Together, these provide an estimated intrinsic viscosity of 5.5 ml/g for sfGFP.
The modified Mooney fits of the viscosity–concentration data of the supercharged variants are shown as solid black curves in Figure 1 and are strikingly similar to that of the sfGFP variants under high cosolute conditions. Moreover, the estimated intrinsic viscosity value of 5.5 ml/g is within error of the fitted parameter for each of three sfGFP variants (Table 2). A Debye length calculation shows a ~6.5-fold increase in the length scale of electrostatic interactions under low sucrose conditions versus the high trehalose-high histidine conditions. Thus, the high trehalose-high histidine conditions appear to screen electrostatic interactions leading to overall reduced protein-protein interactions between closely adjacent sfGFP molecules.
The similarities between these curves led us to speculate that the −18 and +15 variants were subject to the same attractive and repulsive forces as the wild-type sfGFP under high trehalose-high histidine conditions. Since the primary difference between these three proteins is the presence of charged, solvent exposed residues, shielding these charges would be expected to neutralize long-range electrostatics effects. However, charge shielding is expected to have less effect on short-ranged electrostatics due to the presence of charged patches on these proteins that can mediate formation of percolating protein networks that increase solution viscosity.44 These three sfGFP variants vary considerably in the type and spatial distribution of their surface charges yet this had no effect of protein rheology under charge shielding conditions. Moreover, charge shielding did not drive protein-protein association via hydrophobic interactions. Taken together, these data suggest that neutralization of the surface charges leads the sfGFP variant to approximate hard sphere behavior. If true, it should not matter how we neutralize the charge, whether by changing the pH, the solvent ionic strength or other ways.
Histidine is responsible for the low viscosity of the high trehalose-high histidine formulation
To determine which physico-chemical features of the high trehalose-high histidine formulation are responsible for low viscosity, we tested a series of related formulations designed to isolate crowding, hydrophobic and electrostatic shielding effects. For these experiments, we selected the −18 sfGFP variant since it exhibited poor solubility under low sucrose-low histidine conditions. We increased the protein concentration by centrifugation since this approach allowed us to adjust the trehalose concentration below that required for lyoprotection.
We first aimed to uncouple the contributions of histidine and trehalose in the high trehalose-high histidine formulation. The high concentrations of these molecules could support sfGFP solubility at high protein concentrations via a variety of chemical and physical mechanisms. The aromatic imidazole ring in histidine can interact with proteins via electrostatics and hydrogen bond interactions, as well as hydrophobic interactions via π-π stacking and polar cation-π interactions. In this way, histidine may not only act as a crowding molecule, but may also interact with water as well as shield solvent-exposed charged, polar, and hydrophobic side chains.45–48 Conversely, trehalose is unlikely to interact directly with a protein but instead interacts with the solvent and thereby entropically drives proteins to assume a more compact structure.49–50
To understand the roles of histidine and trehalose in the high co-solute formulation, we individually reduced the concentration of each excipient and analyzed the resulting viscosity-concentration data. We formulated −18 sfGFP in a high trehalose-low histidine buffer (660 mM trehalose and sufficient histidine to maintain the pH at 5.8, 10 mM) and concentrated the protein by centrifugation. This formulation showed a rapid increase in viscosity with increasing protein concentration that did not follow an exponential curve, suggesting the proteins experienced stronger attractive interactions (Figure 3A).
Figure 3. Histidine is responsible for low viscosity behavior of −18 sfGFP in the high trehalose-high histidine formulation.
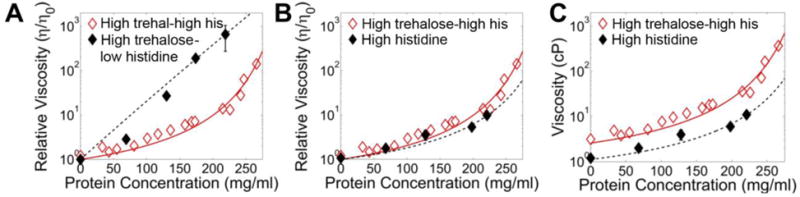
A, The relative viscosity in high trehalose buffer (10 mM Histidine and 660 mM trehalose, pH 5.8); B, the relative viscosity in high histidine buffer (320 mM histidine, pH 5.8) and C, the absolute viscosity in high histidine buffer are shown as solid black icons with black dashed lines to guide the eye. The high trehalose-high histidine data with the modified Mooney fit from Figure 1B is shown as hollow red icons for reference.
We next formulated −18 sfGFP in high histidine buffer (320 mM histidine, pH 5.8 with no trehalose). Interestingly, while the viscosity-concentration curve followed a similar exponential shape as compared to the initial high trehalose-high histidine formulation, the absolute viscosity was lower at all points (Figure 3B, C). The formulation pH of 5.8 is below the 6.0 pKa of histidine’s imidazole side chain yet above the 5.1 pI of the −18 sfGFP variant, suggesting that charged histidine molecules and the HCl counter-ions may be neutralizing protein surface charges. Taken together, these data suggest that histidine minimized the protein-protein interactions, resulting in enhanced protein solubility and reduced viscosity while trehalose independently increased the solvent viscosity. Thus, the main contribution of trehalose appears to be its well-known role as a protectant during lyophilization.51–52
Charge shielding explains the low viscosity of the high trehalose-high histidine formulation
Next, we wanted to determine whether histidine lowered the viscosity of the protein solution via excluded volume effects, an ionic strength effect or interactions between the imidazole ring and surface-exposed hydrophobic residues. To assess the effect of ionic strength, we used sodium chloride to replace the histidine in the high trehalose-high histidine formulation, resulting in a new high trehalose-high salt condition with 10 mM histidine pH 5.8, 660 mM trehalose and 210 mM NaCl. This formulation was chosen such that the final solution had the same buffer, pH, trehalose concentration and ionic strength as the initial high cosolute solution, but most of the 270 mM total ionic strength is supplied by NaCl. The resulting solution was visually clear at protein concentrations of ~250 mg/ml and the viscosity-concentration data were similar to those collected in high trehalose-high histidine buffer (Figure 4A). We next made a high salt-low histidine formulation, comprised of just 10 mM histidine pH 5.8 and 210 mM NaCl. This also yielded a clear solution at protein concentrations up to 250 mg/ml and similar viscosity-concentration profile (Figure 4B), albeit with lower absolute viscosities than trehalose containing formulations (Figure 4C). The sfGFP was also highly soluble in the high salt buffer, with no evidence of aggregation as measured by dynamic light scattering (DLS) at concentrations up to 137 mg/ml (Supplementary Figure S8). Together, these data suggest that histidine acts by shielding charges in this system.
Figure 4. Histidine lowers −18 sfGFP viscosity by charge shielding.
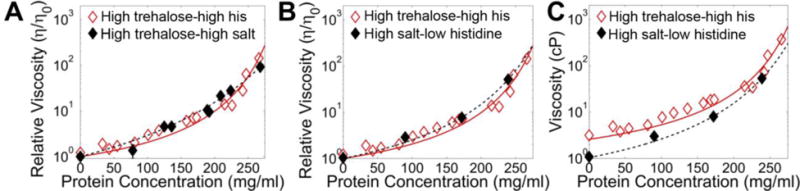
A, The relative viscosity in high trehalose-high salt buffer (10 mM histidine pH 5.8, 660 mM trehalose and 210 mM NaCl); B, the relative viscosity in high salt (10 mM histidine pH 5.8 and 210 mM NaCl); and C, the absolute viscosity in high salt buffer is shown as solid black icons with black dashed lines to guide the eye. High cosolute data with the modified Mooney fit from Figure 1B is shown for reference in hollow red icons.
Protein surface charge is altered by low and high cosolute conditions
To determine whether the other formulations tested can shield protein surface charges, we considered the sfGFP surface charge under the low and high co-solute conditions tested. Under these conditions, cosolutes and counter ions surrounding the protein can temper the observed surface charge. For instance, the surface charges for these proteins are expected to decrease with increasing pH, with the −18 variant having the lowest charge and the +15 variant the highest charge at all pH values (Figure 5A). To provide experimental data about the relative charge on the sfGFP variants in different cosolute conditions, the zeta potential was measured at a 1 mg/ml protein concentration (Figure 5B, Table 2). Under low sucrose-low histidine conditions, the sfGFP variants exhibited the expected trends in surface charge with −18 having the lowest and +15 having the highest zeta potential. However, under high trehalose-high histidine conditions, the measured charges were much smaller, supporting the idea that this high ionic strength shields the sfGFP surface charges. These experimental data are supported by computational electrostatic surface potentials under the same low or high cosolute conditions (Figure 5C). This charge shielding is expected to translate into greatly reduced long-range electrostatics forces and may explain why the different surface charges of the sfGFP variants had no observable effect on viscoelastic solution properties under high trehalose-high histidine conditions.
Figure 5. sfGFP variants have different surface charges.
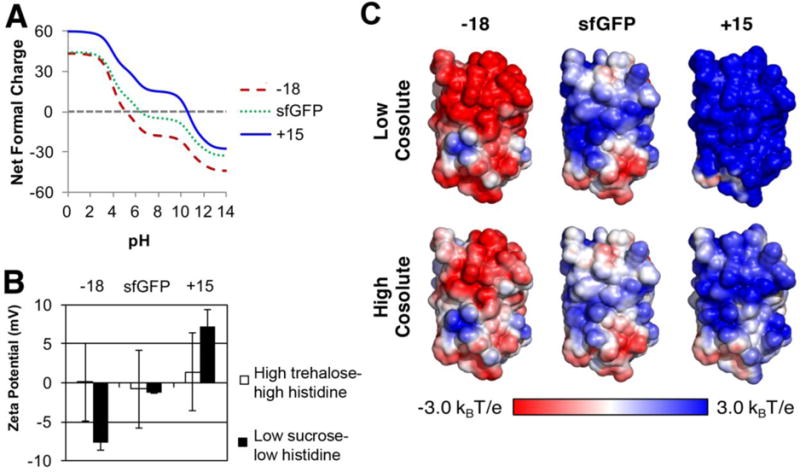
A, The theoretical net surface charge was calculated from the primary amino acid sequence of each sfGFP variant as a function of solvent pH using the Henderson-Hasselbach calculation. This approach predicts iso-electric points for the −18 variant of 5.1, for sfGFP of 6.4 and for the +15 variant of 10.5. B, The surface charge was experimentally measured by zeta potential for all three sfGFP variants at 1 mg/ml under low sucrose and high trehalose-high histidine conditions. C, Electrostatic surface potential of the sfGFP and the super-charged variants. Models of the supercharged sfGFP variants were constructed by introducing the amino acids changes into the wild-type sfGFP structure (PDB 2B3P) in PyMOL. Electrostatic maps were calculated with DelPhi under low (6 mM ionic strength, pH 5.8) and high cosolute conditions (270 mM total ionic strength, pH 5.8) and plotted between ±3 kBT/e. Note the overall charge and the anisotropy of the charge distribution.
Changing pH improves sfGFP solubility under low sucrose conditions
Since the isoelectric point is the primary difference between these preparations, we considered whether this could explain why only the +15 variant remained could attain high protein concentration in our initial low sucrose-low histidine conditions. Proteins are generally more soluble at pH values 1–2 units away from their isoelectric point, conditions under which the increased net surface charge is expected to increase long-range electrostatic repulsive forces and prevent protein-protein association. The calculated pIs of the −18 variant and sfGFP were calculated from the amino acid sequence to be 5.1 and 6.4, respectively, while that of the +15 variant was calculated at 10.5 (Figure 5A). At pH 5.8, the −18 and sfGFP variants are expected to have minimal net charge and a correspondingly low repulsive electrostatic force to off-set the attractive forces promoting protein association as protein concentration increases, as observed in our initial experiments (Figure S4). In contrast, the +15 variant would be highly protonated at pH 5.8 and better able to remain soluble, consistent with our initial data in low sucrose-low histidine buffer (Figure 1A).
To test this, we evaluated the behavior of the −18 variant in a low sucrose-low tris formulation at pH 8.0 instead of 5.8. Since histidine does not buffer in this regime, we replaced it with a 10 mM Tris buffer, pH 8.0 while maintaining sucrose at 146 mM. The protein was now readily concentrated to >200 mg/ml (Figure 6), which coincided with pH drift to 6.8, still well above the protein pI. Since the isoelectric point of this variant is 5.1, the protein is expected to be highly charged in this formulation. While the different buffer molecules could affect aggregation through buffer-protein interactions, the low buffer concentration renders this unlikely. Thus, even for highly charged, anisotropic proteins, charge shielding can substantially reduce the electrostatic interactions that lead to protein network formation and high viscosity.
Figure 6. High pH prevents −18 sfGFP aggregation under low sucrose conditions.
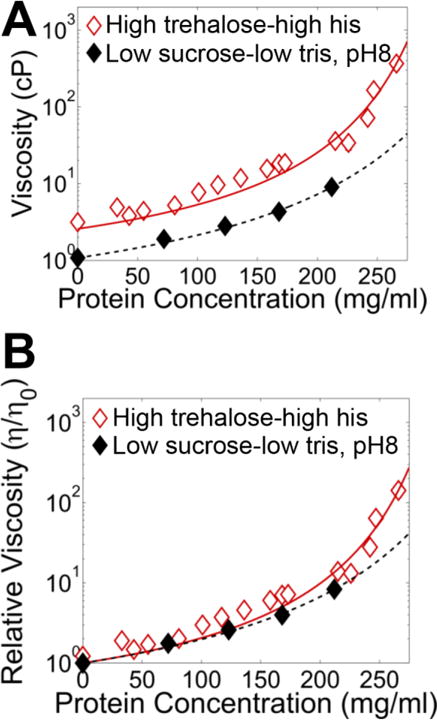
Viscosity-concentration curves for −18 sfGFP (pI = 5.1) in low sucrose conditions with an elevated pH. The protein was dialyzed against 10 mM tris, 146 mM sucrose, pH 8.0 buffer and concentrated by centrifugation to ~216 mg/ml. A, The absolute and B, relative viscosity-concentration dilution curves are shown as solid black icons with black dashed lines to guide the eye. High sucrose-high trehalose data with the modified Mooney fit from Figure 1B is shown for reference in hollow red icons.
Discussion
Despite several decades of research, there is still no clear model to predict the impact of protein-protein interactions on the relationship between viscosity and protein concentration. The ability to predict viscosity from low concentration data or from protein sequence/structure data would be of great interest to biophysicists and pharmaceutical chemists, most notably to aid in selection and development of protein therapeutics. Colloid theory, which models proteins as rigid particles of fixed volume with uniform surface charge distribution, appears to predict the behavior of some proteins, including bovine serum albumin (BSA), hemoglobin, alpha-crystallin and even mixtures of BSA and ovalbumin, with reasonable accuracy at concentrations up to 300–400 mg/ml.37–38
However, under conditions in which these proteins are destabilized, and for proteins with more complex shapes and characteristics, the colloid model has been less successful. At high concentration, these proteins can form networks mediated by weak, nonspecific protein-protein interactions that increase viscosity.44 The inability of colloid theories to reliably predict this behavior has been attributed to the changes in protein shape, excluded volume and higher order associations between multiple protein molecules due to attractive and repulsive interactions that occur at high concentrations.53–54 In the absence of a robust model, these theoretical approaches have been complemented by empirical efforts to identify biochemical and biophysical characteristics predictive of low viscosity. For example, the net charge on an antibody appears to correlate with low viscosity while antibody surface hydrophobicity and the presence of charged dipoles with increased viscosity.55–57
Remarkably, the three variants of sfGFP studied here all exhibited similar concentration-viscosity curves under conditions that screened surface electrostatics, whether this was achieved by changing pH, increasing histidine or increasing salt concentrations. We analyzed this by fitting experimental data relating protein concentration to the solution viscosity in the presence of different solutes to the modified Mooney equation with two adjustable parameters, the intrinsic viscosity and the shape parameter (Figures 1, 3, 4 and 5). Using this approach, the fitted intrinsic viscosity and packing factors were identical, within error, for all three sfGFP variants. The modified Mooney equation accounts for protein shape and specific volume but is based upon the assumption that individual proteins do not appreciably self-associate and does not account for long-ranged intermolecular interactions. As a possible explanation for these results, the experimental data compared closely to the viscosity predicted for a hard particle of the same shape and specific volume as sfGFP, suggesting that the proteins experience minimal protein-protein interactions under high cosolute conditions.
A key element of this analysis is the choice of specific molar volume for the sfGFP-like effective hard particle. This value reflects both steric and electrostatic repulsion and thus varies with pH and ionic strength.38 We chose to use the specific molar volume observed in sfGFP crystals as this value represents a hydrated protein in an experimentally observed, tightly packed system with 58% solvent. Protein-protein interactions in the context of a crystal are similar to those in high concentration pharmaceutical formulations.58 Recent reports59–61 have supported using hydrated protein volumes as more appropriate choices than the generic value of 1.35 g/ml, which represents the specific molar volume for average amino acid residues, without accounting for hydration or packing in the context of a folded protein. Calculation of protein density using the sfGFP molecular weight (27.5 kDa) and hydrodynamic radius (RH = 2.6–2.8 nm) measured in low concentration dynamic light scattering experiments, as reported in Godfrin et al,59 predicts a density of 0.646 – 0.497 g/ml, which is similar the value calculated from the Matthews coefficient and supports our approach.
This is surprising because the sfGFP proteins are highly charged, with the charges distributed in discrete anisotropic patches (Figure 5C) and the hard particle model does not account for surface charges. Previous experimental results showed that network formation at high protein concentrations is often dominated by electrostatic interactions. In general, when the protein carries a net charge, such as when the pH is less than the protein iso-electric point, long-range electrostatic repulsion prevents reversible protein-protein association and network formation. However, at high protein concentrations, charge anisotropy on the protein surface can allow for network formation via alignment of patches with complementary charge (aka dipole-dipole attraction) even when the protein carries a net charge.56, 62 As a test of this hypothesis, neutralization of a charged patch via substitution of a solvent-exposed glutamic acid with a tyrosine (E59Y) increased protein solubility and reduced viscosity for one antibody.63 For the sfGFP proteins studies here, anisotropy did not appear to play a role in enhancing viscosity. When the pH was different than the protein pI, the super-charged −18 and +15 sfGFP variants remained soluble at 250 mg/ml with low viscosity even under low sucrose conditions (Figures 1A, 5).
Electrostatic effects on protein-protein interactions can be tuned by increasing the salt concentration at the risk of decreasing repulsive protein-protein interactions responsible for increased solubility and decreased solution viscosities as well as magnifying the effects of hydrophobic protein-protein interactions. When repulsive electrostatics are sufficiently screened, short-range attractive hydrophobic interactions can mediate network formation at high protein concentrations, thus salt can reduce, increase or have no effect on the viscosity of a specific protein solution. One antibody, mAb G, was soluble at high protein concentrations and moderate ionic strength but formed a gel at high ionic strength. The removal of two hydrophobic CDR residues resulted in a variant that retained solubility even under high ionic strength64. For another antibody, substitution of solvent-exposed aromatic residues with charged residues similarly decreased protein self-association and solution viscosity.65 Classic DLVO theory indicates that charges are fully screened by ionic strengths of >100 mM. In our sfGFP experiments, the high salt formulation (with 210 mM) did not increase protein-viscosity relative to the low sucrose formulation when the pH was different than the protein pI (Figure 4).
A critical limitation to development of robust theoretical models to support this effort is access to sufficient quantities of protein and the ability to systematically alter parameters affecting protein-protein interactions through site-directed mutagenesis. As recombinant proteins, antibody variants can be readily generated, but the amounts required for the experiments shown here are prohibitive for many labs, requiring stable cell lines and large-scale expression efforts. This limits the number of antibody variants that can be analyzed; moreover, their complex, multi-domain structure is more challenging to model than a single domain protein. Common model proteins, such as BSA and ovalbumin, are readily available in large quantities but since they are purified from animals (cow serum and chicken eggs, respectively), cannot be readily altered at the protein amino acid sequence level. Moreover, they may be too simple and well-behaved to provide an accurate model for more complex proteins such as antibodies.
The sfGFP model system used here represents a compromise position between these previously used systems. GFP is a small, 27 kDa protein which is highly expressed, soluble and stable. As a single domain, 11-stranded beta-barrel protein, its shape approximates an ellipsoid that can pack tightly together and exhibits minimal conformational breathing that can lead to non-native aggregation. As a recombinant protein, the biophysical and biochemical properties can be readily tuned by amino acid changes and its high level bacterial expression renders it amenable to the high protein production required for the experiments shown here. The super-folder variant includes an A206V substitution that eliminates homo-dimerization even at high concentrations66 and an S30R substitution which mediates formation of an extended electrostatic charge network on the protein surface that increases global stability and confers a more compact structure as compared to other GFP variants.29 Moreover, sfGFP contains few solvent exposed hydrophobic residues (11 of the 118 residues with >20% relative accessible surface area, Table S1), some of which have been replaced by polar or charged residues in the super-charged variants to minimize the potential for hydrophobic effects on flow behavior.
Conclusions
While the factors affecting protein rheology at high concentration have been studied for decades, there are still no theories that accurately predict this behavior for real proteins. Here, we showed that sfGFP and its supercharged variants exhibit identical rheology at high protein concentrations under conditions that shield surface charges and that this behavior can be accurately modeled by simple colloid theory. The sfGFP proteins used here represent a special case of simple, well-behaved proteins, but other GFP variants have been described that exhibit more complex behavior, such as weak dimerization and reduced conformational stability. Analyzing the rheology of a wider range of GFP variants could provide an experimentally tractable system to guide modifications of colloid theory such that it can better predict the protein rheology. Finally, increasing protein solubility by charge screening takes advantage of basic physical features common to many proteins and may be applicable to antibodies and other pharmaceutically relevant proteins.
Supplementary Material
Acknowledgments
This work was supported by the Norman Hackerman Advanced Research Program under Grant No. 003658-0019-2011 and NIH grant R21 AI103763 to JAM and Welch grants F-1767 to JAM, F-1391 to KPJ and F-1696 to TMT. The authors thank Drs. Ameya Borwankar and Anna Simon for helpful discussions.
Abbreviations
- CD
circular dichroism
- Dc
collective diffusion coefficients
- Do
diffusion coefficient at infinite dilution
- DLS
dynamic light scattering
- sfGFP
superfolder GFP
- RH
hydrodynamic radius
- η
solution viscosity of the formulation including a specific protein concntration
- η0
solvent viscosity of the formulation without added protein
- [η]
intrinsic viscosity of the specific protein
- pI
isoelectric point
Footnotes
Author Contributions
JRL expressed and purified all proteins, while JRL, MLM, DEJ and AMD characterized the proteins after purification. JDG assisted with sfGFP expression. JRL and BJD made all protein formulations, while BJD collected and analyzed all viscosity data. JRL performed analytical characterization. KPJ, TMT, ADE and JAM assisted with project guidance and data interpretation. JRL and JAM wrote the manuscript and all authors contributed to the final version.
Supporting information available Figure S1 Syringe viscometer calibration curve; Figure S2 Experimental determination of sfGFP extinction coefficients from A280/BCA calibration; Figure S3 Amino acid sequence alignment of the sfGFP and −18 and +15 supercharged sfGFP variants; Figure S4 High concentration behavior of sfGFP variants; Figure S5 High cosolute conditions confer identical viscosity-concentration profiles when produced by centrifugation or lyophilization; Figure S6. Similar thermal stabilities and secondary structure of purified sfGFP variants; Figure S7 Analytical gel filtration column calibration curve; Figure S8 sfGFP does not form aggregates at high concentration in high salt formulation; Table S1. Relative solvent accessible surface area of each residue in the sfGFP variant sequences.
References
- 1.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–66. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 2.Selkoe DJ. Cell biology of protein misfolding: the examples of Alzheimer’s and Parkinson’s diseases. Nat Cell Biol. 2004;6(11):1054–61. doi: 10.1038/ncb1104-1054. [DOI] [PubMed] [Google Scholar]
- 3.Perchiacca JM, Tessier PM. Engineering aggregation-resistant antibodies. Annual review of chemical and biomolecular engineering. 2012;3:263–86. doi: 10.1146/annurev-chembioeng-062011-081052. [DOI] [PubMed] [Google Scholar]
- 4.Curtis RA, Prausnitz JM, Blanch HW. Protein-Protein and Protein-Salt Interactions in Aqueous Protein Solutions Containing Concentrated Electrolytes. Biotechnology and Bioengineering. 1998;57(1):11–21. doi: 10.1002/(sici)1097-0290(19980105)57:1<11::aid-bit2>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 5.Elcock AH, McCammon JA. Calculation of Weak Protein-Protein Interactions: the pH Dependence of the Second Virial Coefficient. Biophys J. 2001;80:613–625. doi: 10.1016/S0006-3495(01)76042-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saluja A, Kalonia DS. Nature and consequences of protein-protein interactions in high protein concentration solutions. Int J Pharm. 2008;358:1–15. doi: 10.1016/j.ijpharm.2008.03.041. [DOI] [PubMed] [Google Scholar]
- 7.Valente JJ, Payne RW, Manning MC, Wilson WW, Henry CS. Colloidal Behavior of Proteins: Effects of the Second Virial Coefficient on Solubility, Crystallization and Aggregation of Proteins in Aqueous Solution. Curr Pharm Biotechnol. 2005;6:427–439. doi: 10.2174/138920105775159313. [DOI] [PubMed] [Google Scholar]
- 8.Ghosh R, Calero-Rubio C, Saluja A, Roberts CJ. Relating Protein-Protein Interactions and Aggregation Rates From Low to High Concentrations. J Pharm Sci. 2016;105(3):1086–96. doi: 10.1016/j.xphs.2016.01.004. [DOI] [PubMed] [Google Scholar]
- 9.Connolly BD, Petry C, Yadav S, Demeule B, Ciaccio N, Moore JM, Shire SJ, Gokarn YR. Weak interactions govern the viscosity of concentrated antibody solutions: high-throughput analysis using the diffusion interaction parameter. Biophys J. 2012;103(1):69–78. doi: 10.1016/j.bpj.2012.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burns JN, Turnage KC, Walker CA, Lieberman RL. The stability of myocilin olfactomedin domain variants provides new insight into glaucoma as a protein misfolding disorder. Biochemistry. 2011;50(26):5824–33. doi: 10.1021/bi200231x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roberts CJ, Blanco MA. Role of anisotropic interactions for proteins and patchy nanoparticles. J Phys Chem B. 2014;118(44):12599–611. doi: 10.1021/jp507886r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bostrom M, Williams DR, Ninham BW. Specific ion effects: why DLVO theory fails for biology and colloid systems. Phys Rev Lett. 2001;87(16):168103. doi: 10.1103/PhysRevLett.87.168103. [DOI] [PubMed] [Google Scholar]
- 13.Kumar V, Dixit N, Zhou LL, Fraunhofer W. Impact of short range hydrophobic interactions and long range electrostatic forces on the aggregation kinetics of a monoclonal antibody and a dual-variable domain immunoglobulin at low and high concentrations. Int J Pharm. 2011;421(1):82–93. doi: 10.1016/j.ijpharm.2011.09.017. [DOI] [PubMed] [Google Scholar]
- 14.Liu J, Nguyen MD, Andya JD, Shire SJ. Reversible self-association increases the viscosity of a concentrated monoclonal antibody in aqueous solution. J Pharm Sci. 2005;94(9):1928–40. doi: 10.1002/jps.20347. [DOI] [PubMed] [Google Scholar]
- 15.Strickler SS, Gribenko AV, Gribenko AV, Keiffer TR, Tomlinson J, Reihle T, Loladze VV, Makhatadze GI. Protein stability and surface electrostatics: a charged relationship. Biochemistry. 2006;45(9):2761–6. doi: 10.1021/bi0600143. [DOI] [PubMed] [Google Scholar]
- 16.Tzul FO, Schweiker KL, Makhatadze GI. Modulation of folding energy landscape by charge-charge interactions: linking experiments with computational modeling. Proc Natl Acad Sci U S A. 2015;112(3):E259–66. doi: 10.1073/pnas.1410424112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Buell AK, Tartaglia GG, Birkett NR, Waudby CA, Vendruscolo M, Salvatella X, Welland ME, Dobson CM, Knowles TP. Position-dependent electrostatic protection against protein aggregation. Chembiochem. 2009;10(8):1309–12. doi: 10.1002/cbic.200900144. [DOI] [PubMed] [Google Scholar]
- 18.Chari R, Jerath K, Badkar AV, Kalonia DS. Long- and Short-Range Electrostatic Interactions Affect the Rheology of Highly Concentrated Antibody Solutions. Pharm Res. 2009;26(12):2607–2618. doi: 10.1007/s11095-009-9975-2. [DOI] [PubMed] [Google Scholar]
- 19.Lawrence MS, Phillips KJ, Liu DR. Supercharging proteins can impart unusual resilience. J Am Chem Soc. 2007;129(33):10110–2. doi: 10.1021/ja071641y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miklos AE, Kluwe C, Der BS, Pai S, Sircar A, Hughes RA, Berrondo M, Xu J, Codrea V, Buckley PE, Calm AM, Welsh HS, Warner CR, Zacharko MA, Carney JP, Gray JJ, Georgiou G, Kuhlman B, Ellington AD. Structure-based design of supercharged, highly thermoresistant antibodies. Chem Biol. 2012;19(4):449–55. doi: 10.1016/j.chembiol.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perchiacca JM, Ladiwala AR, Bhattacharya M, Tessier PM. Aggregation-resistant domain antibodies engineered with charged mutations near the edges of the complementarity-determining regions. Protein Eng Des Sel. 2012;25(10):591–601. doi: 10.1093/protein/gzs042. [DOI] [PubMed] [Google Scholar]
- 22.Johnston KP, Maynard JA, Truskett TM, Borwankar AU, Miller MA, Wilson BK, Dinin AK, Khan TA, Kaczorowski KJ. Concentrated dispersions of equilibrium protein nanoclusters that reversibly dissociate into active monomers. ACS Nano. 2012;6(2):1357–69. doi: 10.1021/nn204166z. [DOI] [PubMed] [Google Scholar]
- 23.Sear RP. Interactions in protein solutions. Current Opinion in Colloid & Interface Science. 2006;11:35–39. [Google Scholar]
- 24.Ellis RJ. Macromolecular crowding: obvious but underappreciated. Trends Biochem Sci. 2001;26(10):597–604. doi: 10.1016/s0968-0004(01)01938-7. [DOI] [PubMed] [Google Scholar]
- 25.Yadav S, Laue TM, Kalonia DS, Singh SN, Shire SJ. The influence of charge distribution on self-association and viscosity behavior of monoclonal antibody solutions. Molecular pharmaceutics. 2012;9(4):791–802. doi: 10.1021/mp200566k. [DOI] [PubMed] [Google Scholar]
- 26.Liu J, Nguyen MDH, Andya JD, Shire SJ. Reversible Self-Association Increases the Viscosity of a Concentrated Monoclonal Antibody in Aqueous Solution. J Pharm Sci. 2005;94(9):1928–1940. doi: 10.1002/jps.20347. [DOI] [PubMed] [Google Scholar]
- 27.Kanai S, Liu J, Patapoff TW, Shire SJ. Reversible Self-Association of a Concentrated Monoclonal Antibody Solution Mediated by Fab-Fab Interaction That Impacts Solution Viscosity. J Pharm Sci. 2008;97(10):4219–4227. doi: 10.1002/jps.21322. [DOI] [PubMed] [Google Scholar]
- 28.Cleland JL, Powell MF, Shire SJ. The development of stable protein formulations: a close look at protein aggregation, deamidation, and oxidation. Crit Rev Ther Drug Carrier Syst. 1993;10(4):307–77. [PubMed] [Google Scholar]
- 29.Pedelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol. 2006;24(1):79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- 30.Der BS, Kluwe C, Miklos AE, Jacak R, Lyskov S, Gray JJ, Georgiou G, Ellington AD, Kuhlman B. Alternative computational protocols for supercharging protein surfaces for reversible unfolding and retention of stability. PLoS One. 2013;8(5):e64363. doi: 10.1371/journal.pone.0064363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borwankar A, Dinen A, Laber J, Twu A, Wilson B, Maynard J, Truskett T, Johnston K. Tunable equilibrium nanocluster dispersions at high protein concentrations. Soft Matter. 2013;9:1776–1771. [Google Scholar]
- 32.Smith N, Witham S, Sarkar S, Zhang J, Li L, Li C, Alexov E. DelPhi web server v2: incorporating atomic-style geometrical figures into the computational protocol. Bioinformatics. 2012;28(12):1655–7. doi: 10.1093/bioinformatics/bts200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sarkar S, Witham S, Zhang J, Zhenirovskyy M, Rocchia W, Alexov E. DelPhi Web Server: A comprehensive online suite for electrostatic calculations of biological macromolecules and their complexes. Commun Comput Phys. 2013;13(1):269–284. doi: 10.4208/cicp.300611.201011s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.insert], S. p. Johnson and Johnson. 2009.
- 35.insert], I. p. Novartis. 2009.
- 36.He F, Woods CE, Litowski JR, Roschen LA, Gadgil HS, Razinkov VI, Kerwin BA. Effect of sugar molecules on the viscosity of high concentration monoclonal antibody solutions. Pharm Res. 2011;28(7):1552–60. doi: 10.1007/s11095-011-0388-7. [DOI] [PubMed] [Google Scholar]
- 37.Ross PD, Minton AP. Hard Quasispherical Model for the Viscosity of Hemoglobin Solutions. Biochem Biophys Res Commun. 1977;76(4):971–976. doi: 10.1016/0006-291x(77)90950-0. [DOI] [PubMed] [Google Scholar]
- 38.Minton AP. The Effective Hard Particle Model Provides a Simple, Robust, and Broadly Applicable Description of Nonideal Behavior in Concentrated Solutions of Bovine Serum Albumin and Other Nonassociating Proteins. J Pharm Sci. 2007;96(12):3466–3469. doi: 10.1002/jps.20964. [DOI] [PubMed] [Google Scholar]
- 39.Yadav S, Shire SJ, Kalonia DS. Factors affecting the viscosity in high concentration solutions of different monoclonal antibodies. J Pharm Sci. 2010;99(12):4812–29. doi: 10.1002/jps.22190. [DOI] [PubMed] [Google Scholar]
- 40.Mehl JW, Oncley JL, Simha R. Viscosity and the Shape of Protein Molecules. Science. 1940;92(2380):132–3. doi: 10.1126/science.92.2380.132. [DOI] [PubMed] [Google Scholar]
- 41.Li S, Zhao J, Lu P, Xie Y. Maximum packing densities of basic 3D objects. Chinese Science Bulletin. 2010;55:114–119. [Google Scholar]
- 42.Sun Y, Li X, Duzgunes N, Takaoka Y, Ohi S, Hirota S. The shape parameter of liposomes and DNA-lipid complexes determined by viscometry utilizing small sample volumes. Biophys J. 2003;85(2):1223–32. doi: 10.1016/s0006-3495(03)74558-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matthews BW. Solvent content of protein crystals. J Mol Biol. 1968;33(2):491–7. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- 44.Buck PM, Chaudhri A, Kumar S, Singh SK. Highly viscous antibody solutions are a consequence of network formation caused by domain-domain electrostatic complementarities: insights from coarse-grained simulations. Molecular pharmaceutics. 2015;12(1):127–39. doi: 10.1021/mp500485w. [DOI] [PubMed] [Google Scholar]
- 45.Loewenthal R, Sancho J, Fersht AR. Histidine-aromatic interactions in barnase. Elevation of histidine pKa and contribution to protein stability. J Mol Biol. 1992;224(3):759–70. doi: 10.1016/0022-2836(92)90560-7. [DOI] [PubMed] [Google Scholar]
- 46.Liao SM, Du QS, Meng JZ, Pang ZW, Huang RB. The multiple roles of histidine in protein interactions. Chem Cent J. 2013;7(1):44. doi: 10.1186/1752-153X-7-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Du QS, Meng JZ, Liao SM, Huang RB. Energies and physicochemical properties of cation-pi interactions in biological structures. J Mol Graph Model. 2012;34:38–45. doi: 10.1016/j.jmgm.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 48.Dougherty DA. Cation-pi interactions in chemistry and biology: a new view of benzene, Phe, Tyr, and Trp. Science. 1996;271(5246):163–8. doi: 10.1126/science.271.5246.163. [DOI] [PubMed] [Google Scholar]
- 49.Lee JC, Timasheff SN. The stabilization of proteins by sucrose. Journal of Biological Chemistry. 1981;256(14):7193–7201. [PubMed] [Google Scholar]
- 50.Lins RD, Pereira CS, Hunenberger PH. Trehalose-Protein Interaction in Aqueous Solution. Proteins: Structure, Function, and Bioinformatics. 2004;55:177–186. doi: 10.1002/prot.10632. [DOI] [PubMed] [Google Scholar]
- 51.Liao YH, Brown MB, Quader A, Martin GP. Protective mechanism of stabilizing excipients against dehydration in the freeze-drying of proteins. Pharm Res. 2002;19(12):1854–61. doi: 10.1023/a:1021497625645. [DOI] [PubMed] [Google Scholar]
- 52.Cleland JL, Lam X, Kendrick B, Yang J, Yang TH, Overcashier D, Brooks D, Hsu C, Carpenter JF. A specific molar ratio of stabilizer to protein is required for storage stability of a lyophilized monoclonal antibody. J Pharm Sci. 2001;90(3):310–21. doi: 10.1002/1520-6017(200103)90:3<310::aid-jps6>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 53.Sarangapani PS, Hudson SD, Migler KB, Pathak JA. The limitations of an exclusively colloidal view of protein solution hydrodynamics and rheology. Biophys J. 2013;105(10):2418–26. doi: 10.1016/j.bpj.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sarangapani PS, Hudson SD, Jones RL, Douglas JF, Pathak JA. Critical examination of the colloidal particle model of globular proteins. Biophys J. 2015;108(3):724–37. doi: 10.1016/j.bpj.2014.11.3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sharma VK, Patapoff TW, Kabakoff B, Pai S, Hilario E, Zhang B, Li C, Borisov O, Kelley RF, Chorny I, Zhou JZ, Dill KA, Swartz TE. In silico selection of therapeutic antibodies for development: viscosity, clearance, and chemical stability. Proc Natl Acad Sci U S A. 2014;111(52):18601–6. doi: 10.1073/pnas.1421779112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li L, Kumar S, Buck PM, Burns C, Lavoie J, Singh SK, Warne NW, Nichols P, Luksha N, Boardman D. Concentration dependent viscosity of monoclonal antibody solutions: explaining experimental behavior in terms of molecular properties. Pharm Res. 2014;31(11):3161–78. doi: 10.1007/s11095-014-1409-0. [DOI] [PubMed] [Google Scholar]
- 57.Agrawal NJ, Helk B, Kumar S, Mody N, Sathish HA, Samra HS, Buck PM, Li L, Trout BL. Computational tool for the early screening of monoclonal antibodies for their viscosities. MAbs. 2016;8(1):43–8. doi: 10.1080/19420862.2015.1099773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brader ML, Baker EN, Dunn MF, Laue TM, Carpenter JF. Using X-Ray Crystallography to Simplify and Accelerate Biologics Drug Development. J Pharm Sci. 2017;106(2):477–494. doi: 10.1016/j.xphs.2016.10.017. [DOI] [PubMed] [Google Scholar]
- 59.Godfrin PD, Zarraga IE, Zarzar J, Porcar L, Falus P, Wagner NJ, Liu Y. Effect of Hierarchical Cluster Formation on the Viscosity of Concentrated Monoclonal Antibody Formulations Studied by Neutron Scattering. J Phys Chem B. 2016;120(2):278–91. doi: 10.1021/acs.jpcb.5b07260. [DOI] [PubMed] [Google Scholar]
- 60.Roosen-Runge F, Hennig M, Zhang F, Jacobs RM, Sztucki M, Schober H, Seydel T, Schreiber F. Protein self-diffusion in crowded solutions. Proc Natl Acad Sci U S A. 2011;108(29):11815–20. doi: 10.1073/pnas.1107287108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roos M, Ott M, Hofmann M, Link S, Rossler E, Balbach J, Krushelnitsky A, Saalwachter K. Coupling and Decoupling of Rotational and Translational Diffusion of Proteins under Crowding Conditions. J Am Chem Soc. 2016;138(32):10365–72. doi: 10.1021/jacs.6b06615. [DOI] [PubMed] [Google Scholar]
- 62.Arora J, Hu Y, Esfandiary R, Sathish HA, Bishop SM, Joshi SB, Middaugh CR, Volkin DB, Weis DD. Charge-mediated Fab-Fc interactions in an IgG1 antibody induce reversible self-association, cluster formation, and elevated viscosity. MAbs. 2016;8(8):1561–1574. doi: 10.1080/19420862.2016.1222342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nichols P, Li L, Kumar S, Buck PM, Singh SK, Goswami S, Balthazor B, Conley TR, Sek D, Allen MJ. Rational design of viscosity reducing mutants of a monoclonal antibody: hydrophobic versus electrostatic inter-molecular interactions. MAbs. 2015;7(1):212–30. doi: 10.4161/19420862.2014.985504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pindrus M, Shire SJ, Kelley RF, Demeule B, Wong R, Xu Y, Yadav S. Solubility Challenges in High Concentration Monoclonal Antibody Formulations: Relationship with Amino Acid Sequence and Intermolecular Interactions. Molecular pharmaceutics. 2015;12(11):3896–907. doi: 10.1021/acs.molpharmaceut.5b00336. [DOI] [PubMed] [Google Scholar]
- 65.Geoghegan JC, Fleming R, Damschroder M, Bishop SM, Sathish HA, Esfandiary R. Mitigation of reversible self-association and viscosity in a human IgG1 monoclonal antibody by rational, structure-guided Fv engineering. MAbs. 2016;8(5):941–50. doi: 10.1080/19420862.2016.1171444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zacharias DA, Violin JD, Newton AC, Tsien RY. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296(5569):913–6. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.