

Abstract
The eponymous DNA-binding domain of ETS (E26 transformation-specific) transcription factors binds a single sequence-specific site as a monomer over a single helical turn. Following our previous observation by titration calorimetry that the ETS member PU.1 dimerizes sequentially at a single sequence-specific DNA-binding site to form a 2:1 complex, we have carried out an extensive spectroscopic and biochemical characterization of site-specific PU.1 ETS complexes. Whereas 10 bp of DNA was sufficient to support PU.1 binding as a monomer, additional flanking bases were required to invoke sequential dimerization of the bound protein. NMR spectroscopy revealed a marked loss of signal intensity in the 2:1 complex, and mutational analysis implicated the distal surface away from the bound DNA as the dimerization interface. Hydroxyl radical DNA footprinting indicated that the site-specifically bound PU.1 dimers occupied an extended DNA interface downstream from the 5′-GGAA-3′ core consensus relative to its 1:1 counterpart, thus explaining the apparent site size requirement for sequential dimerization. The site-specifically bound PU.1 dimer resisted competition from nonspecific DNA and showed affinities similar to other functionally significant PU.1 interactions. As sequential dimerization did not occur with the ETS domain of Ets-1, a close structural homolog of PU.1, 2:1 complex formation may represent an alternative autoinhibitory mechanism in the ETS family at the protein-DNA level.
Keywords: DNA-binding protein, DNA-protein interaction, ETS transcription factor family, nuclear magnetic resonance (NMR), protein-DNA interaction, Negative cooperativity, PU.1
Introduction
The differentiation of distinct lineages of blood cells from a single progenitor species occurs in a multistep process, termed hematopoiesis, that is intricately controlled at the transcriptional level. The ETS family transcription factor PU.1 ranks among the most essential hematopoietic regulators in ensuring the continued self-renewal of this progenitor, the hematopoietic stem cell (1). PU.1 is also essential for directing correct differentiation of the hematopoietic stem cell to various cell lineages. Crucially, PU.1 governs cell fate specification and functions in a transient, dosage– and cell stage–dependent manner (2). In mature cells, graded PU.1 activity is also required for key cellular processes and the specialization of mature cells into function-specific subtypes. Aberrant PU.1 activity is linked to a spectrum of diseases, including rheumatism (3), hematologic cancers (4–6), and Alzheimer's disease (7). Clearly, knowledge of the regulatory mechanisms of PU.1 is essential to an understanding of its role in normal biology and in disease.
Biological modulation of PU.1 activity is generally attributed to up- or down-regulation at the level of expression. With a metabolic half-life of ∼50 h, a period that spans the entire cell cycle (8), the cellular persistence of PU.1 means that down-regulation of its own expression cannot alone provide a complete description of PU.1 regulation, as additional dampening mechanisms are required to prevent open-ended escalation of PU.1 activity during its lifetime in the cell.
Outside of down-regulated expression, few inhibitory mechanisms have been described for PU.1. The best understood is the mutual antagonism between PU.1 and the zinc finger transcription factor GATA-1, wherein each protein inhibits DNA binding by the other during myelopoiesis (9). In addition, PU.1 is one of only a few ETS family members that lack so-called autoinhibition, a regulatory mechanism in which helices adjacent to the ETS DNA-binding domain allosterically reduce DNA-binding affinity (Fig. 1A) (10). In the case of Ets-1, the archetypal autoinhibited ETS paralog, interactions with partner proteins, such as Pax5 (11) and AML1/RUNX1/CBFα2 (12), relieve autoinhibition and restore high-affinity binding. Thus, in the absence of lineage-specific inhibitory partners such as GATA-1 or some intrinsic regulatory alternative to autoinhibition, PU.1 would be continuously poised in a functionally uncontrolled, transcriptionally permissive state.
Figure 1.
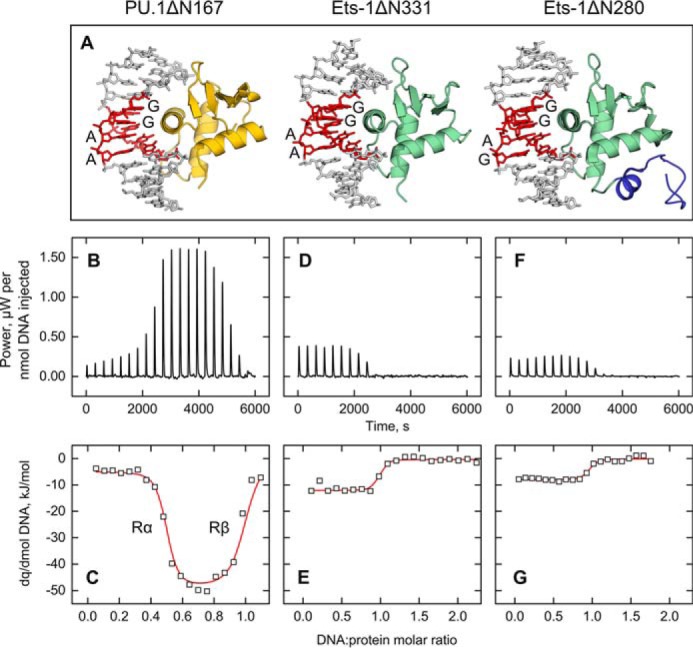
Dimerization at a single cognate binding site is intrinsic to the ETS domain of PU.1, but not its structural homolog Ets-1. A, the co-crystal structures of PU.1 (gold; PDB code 1PUE) and Ets-1 (green), the latter with (1MDM) or without (1K79) part of its autoinhibitory domain (blue). All three structures show the canonical 1:1 binding stoichiometry with oligomeric DNA harboring a core 5′-GGAX-3′ consensus (red), as labeled. Note that the cognate DNA sequences in the co-crystal structures are not sequence-identical to the experimental sequences in this study. B, D, and F, representative ITC thermograms at 25 °C of DNA-into-protein titrations for the ETS domains of PU.1 (B) and Ets-1 (minimal = ΔN331 (D); autoinhibited = ΔN280 (F)). The ordinate is baseline-subtracted and normalized to the amount of DNA delivered per injection to aid comparison; exothermic response is upward. C, E, and G, the titration data for PU.1ΔN167 was empirically fitted as a negatively cooperative interaction. The two phases in the PU.1 titration (marked Rα and Rβ in C) represent the successive formation of a protein/DNA = 2:1 complex (protein in excess) followed by the 1:1 complex. For Ets-1ΔN331 (E) and Ets-1ΔN280 (G), a 1:1 model was empirically fitted to the data. The stronger and more complex apparent heats associated with the PU.1ΔN167 titrations included the dimerization and binding of PU.1ΔN167 as a 2:1 complex, which dissociates to two 1:1 complexes as DNA reached molar equivalence, in addition to more enthalpically driven 1:1 binding than Ets-1. The details of the thermodynamic deconvolution are provided under “Materials and methods.”
In previous work, we have observed in calorimetric titrations the potential for the eponymous DNA-binding domain of PU.1 to dimerize at a single cognate DNA-binding site (13). Whereas dimerization of ETS domains of other ETS family homologs bound to two sites (i.e. 2:2 complexes) has been reported (14–18), self-association at a single site is poorly understood. We have carried out an extensive series of spectroscopic and biochemical experiments to directly characterize the variable binding modes of PU.1 as a function of DNA sequence and site size. The results show a site-specific 2:1 complex in exchange between free PU.1 on the one hand and the 1:1 site-specifically bound state on the other, while contacting the DNA over an extended interface beyond the single helical turn observed in the 1:1 co-crystal structure. Sequential dimerization imposes the dual requirements of specific DNA as well as a site size longer than 10 bp. Nonspecific DNA forms oligomeric but not 1:1 complexes at equilibrium. Sequential dimerization of site-specifically bound PU.1, which sequesters excess circulating PU.1 from accessible DNA target sites, suggests itself as a potential mechanism of negative feedback in the absence of inhibitory binding partners.
Results
A 1:1 protein/DNA site stoichiometry is universally observed in co-crystal structures of ETS family transcription factors, including PU.1 (19, 20) (Fig. 1A). Nevertheless, calorimetric measurements of DNA binding by the PU.1 ETS domain (encoded by the C-terminal fragment, PU.1ΔN167) showed that PU.1 bound site-specific targets with non-1:1 stoichiometry (13). When site-specific DNA was titrated into PU.1ΔN167, the protein bound the DNA in a negatively cooperative manner (Fig. 1, B and C). Dimerization was strictly noncovalent, as PU.1ΔN167 harbored no cysteine residue. To broaden our observations and determine whether 2:1 binding was a class property of ETS domains, we measured high-affinity site-specific binding by the ETS domain of Ets-1 (encoded by the C-terminal fragment Ets-1ΔN331), which contained two free cysteines. Although PU.1ΔN167 and Ets-1ΔN331 represent sequence-divergent ETS members, their backbones are superimposable in their DNA-bound states (21). At comparable concentrations as PU.1ΔN167 (∼40 μm) and under reducing but otherwise identical conditions, Ets-1ΔN331 bound site-specific DNA at strictly 1:1 stoichiometry (Fig. 1, D and E). Moreover, the inclusion of flanking elements known to autoinhibit Ets-1 (Ets-1ΔN280) did not affect the binding stoichiometry (Fig. 1, F and G). Thus, dimerization at a single site was not shared by Ets-1 but was particular to PU.1 and possibly other sequence-similar ETS homologs.
Although the ITC7 titrations could be fitted empirically with model-dependent profiles, the high concentrations (>10−5 m) required for the experiments precluded an accurate quantitative determination of binding affinities due to the strong dissociation constant of the 1:1 complex (10−9 m). We therefore titrated a 20-bp Cy3-labeled duplex oligonucleotide harboring the same high-affinity site as used in the ITC experiments and measured binding from changes in fluorescence polarization of the DNA probe (Fig. 2A). The binding data yielded a biphasic profile to which a sequential binding model (22) was fitted with dissociation constants of 7.0 ± 1.3 nm and (1.2 ± 0.8) × 103 nm, or a (negative) cooperativity parameter of ∼170. Constraining the model to 1:1 binding significantly compromised the fit to the data (green curve in Fig. 2A). To determine whether a single helical turn of contact interface, as observed in the co-crystal PU.1/DNA structure (23), was sufficient to support sequential binding of PU.1, we repeated the titration with a DNA construct in which only the core 10 bp of the cognate site (5′-AGCGGAAGTG-3′) was duplex. Binding to the 10-bp duplex exhibited monophasic binding that was well-described by a 1:1 model with a ∼2-fold reduction in dissociation constant (12 ± 2 nm). To further determine whether the excess binding to the 23-bp construct represented nonspecific interactions, we measured PU.1ΔN167 binding to an isomer of the 20-bp DNA in which the core 5′-GGAA-3′ consensus was mutated to 5′-GAGA-3′. In contrast with the specific site, binding to the nonspecific site was >100-fold weaker (2.1 ± 0.2 μm) than either site-specific DNA and yielded a Hill coefficient of well above unity, indicative of concerted binding of two or more equivalents of PU.1ΔN167 (Fig. 2B). The titrations therefore showed that sequential dimerization of the PU.1 ETS domain, wherein excess protein self-titrated the canonical 1:1 PU.1/DNA complex to form a 2:1 complex, was exclusive to site-specific DNA longer than 10 bp. Although nonspecific DNA bound PU.1 in multiple equivalents, it did not form a 1:1 complex at equilibrium (Fig. 2B).
Figure 2.
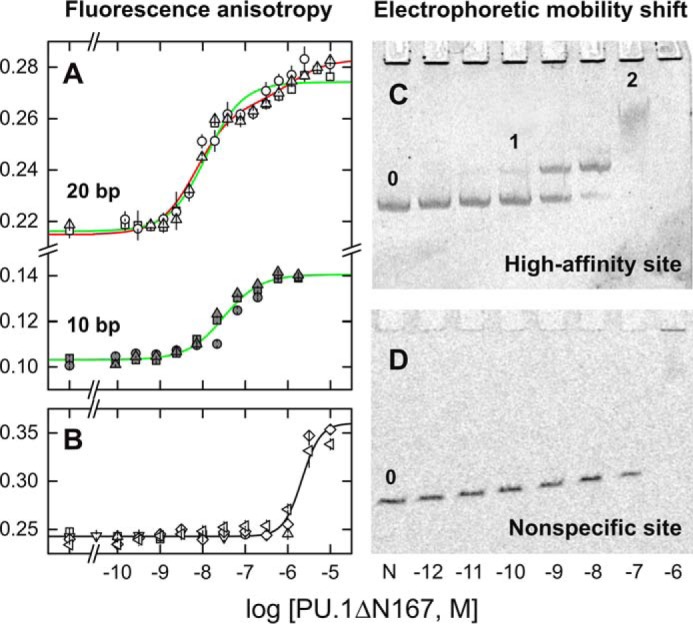
Sequence and site size requirements for sequential dimerization of the specific DNA-bound ETS domain of PU.1. A and B, fluorescence polarization titrations of Cy3-labeled 20-bp DNA probes (22) harboring the high-affinity site (A) 5′-AGCGGAAGTG-3′ or its isomeric nonspecific variant (B) 5′-AGCGAGAGTG-3′ with PU.1ΔN167. The 10-bp duplex in A was constructed by annealing the labeled strand with a 10-bp complement encoding only the core ETS-binding site. Symbols represent data from independent replicate experiments. Curves represent a least-square fit of the data from triplicate experiments to a sequential 2:1 binding model (red) or constrained to a 1:1 model (green). The latter afforded a significantly poorer fit of the data (p < 1 × 10−4, Fisher's F-test on sums of squares). The nonspecific data were fitted with the Hill equation (black). Error bars, S.E. C, electrophoretic mobility shift titration of a 209-bp DNA fragment (1 nm, marked 0) harboring a single copy of the high-affinity site with PU.1ΔN167. Following formation of the 1:1 complex (marked 1), a discrete, low-mobility species was present at 0.1 μm protein (labeled 2). At 1 μm protein (103-fold excess), a nonspecific complex finally formed, which did not enter the gel, as confirmed with a fragment harboring the nonspecific sequence (D). The shadows lining the wells in C represent an irregular imaging artifact of the stained gel, not protein-bound DNA, as it was observed even in the negative-control lane containing no PU.1 (marked 0).
To evaluate site-specific PU.1 dimerization in the presence of excess nonspecific DNA, as would be encountered under genomic conditions, we titrated a 209-bp fragment harboring a single high-affinity PU.1-binding site with PU.1ΔN167. Resolution of the DNA by native gel electrophoresis (Fig. 2C) showed a PU.1-bound band that transitioned to a less mobile band at ∼100-fold excess protein with respect to the specific site (1 nm), in agreement with the sequential site-specific dimer observed in the fluorescence polarization titrations. The low mobility and broadness of the dimer peak suggested that this complex was interconverting between free and bound states at rates comparable with electrophoretic separation of the two complexes (24). The dimeric peak occurred in advance of a final nonspecific complex that was not detected in the gel. The latter state was verified with an isomeric nonspecific DNA fragment that failed to yield PU.1ΔN167 at any defined stoichiometry (Fig. 2D). Thus, the electrophoretic data showed that the sequential dimerization of PU.1 at a single embedded cognate site effectively resisted competition from excess nonspecific DNA.
Hydrodynamic characterization of PU.1/DNA complexes by NMR spectroscopy
To characterize the solution behavior of the PU.1/DNA complex directly, we interrogated PU.1ΔN167 with site-specific and nonspecific DNA oligonucleotides hydrodynamically by diffusion-ordered NMR spectroscopy (see supplemental Methods). At 250 μm protein, we measured a self-diffusion coefficient of (9.1 ± 0.1) × 10−11 m2/s for unbound PU.1ΔN167 (13.0 kDa) in D2O at 25 °C. Comparison with a computed value (25) under equivalent conditions for a PU.1ΔN167 monomer derived from the co-crystal structure of the 1:1 PU.1 ETS/DNA complex (see “Materials and methods”) found good agreement (8.8 × 10−11 m2/s). An unbound PU.1 dimer modeled as a pair of rigid spheres would exhibit a diffusion coefficient at 75% of the monomer (26), or ∼7 × 10−11 m2/s. Thus, although unbound PU.1 was known to dimerize at very high concentrations (13, 27) in vitro, PU.1ΔN167 was monomeric under the conditions of the DOSY experiments.
We tracked the self-diffusion coefficients of PU.1ΔN167 at graded stoichiometric ratios of DNA (supplemental Figs. S2 and S3; parametric values in supplemental Table S1). Titration with 16-bp high- or low-affinity site-specific DNA lowered the apparent diffusion coefficient to a minimum of (5.9 ± 0.1) × 10−11 m2/s at, within experimental uncertainty, a DNA/PU.1 ratio of 0.5 (i.e. PU.1/DNA = 2:1). The subsequent addition of site-specific DNA past this point increased the diffusion coefficient to a stable value of (7.5 ± 0.2) × 10−11 m2/s at 1:1 equivalence and beyond (supplemental Figs. S3A). This biphasic behavior was consistent with the other titration data by ITC, fluorescence polarization, and gel mobility shift. This change in DOSY intensity was not due to simple contributions from added DNA because we had carefully avoided peaks that overlapped with DNA (supplemental Fig. S2). The measured diffusion coefficient upon reaching molar equivalence also agreed with a computed value (25) of 7.3 × 10−11 m2/s based on the 1:1 PU.1/DNA co-crystal structure. Finally, the sequential transitions in diffusion coefficients at half and unit molar equivalence were incompatible with a 2:2 complex. Thus, the DOSY titrations indicated that PU.1 formed exclusively a 2:1 complex with site-specific DNA at PU.1/DNA up to 2:1, followed by a 1:1 complex at molar equivalence and above.
In contrast with site-specific DNA, nonspecific binding by PU.1ΔN167 showed qualitatively different behavior (supplemental Fig. S3C). Specifically, titration of PU.1ΔN167 with 16-bp nonspecific DNA yielded only a single inflection point at a DNA/PU.1 ratio of 0.5 and a stable diffusion coefficient of (6.6 ± 0.2) × 10−11 m2/s, between the site-specific 1:1 and 2:1 complexes. Thus, the DOSY titration data pointed to the exclusive formation of a defined dimeric nonspecific 16-bp complex. An alternative scenario in which PU.1ΔN167 formed a mixture of 2:1 and 1:1 complexes was not likely, as the apparent diffusion coefficient would be composition-dependent and change upon continued titration of DNA.
As the fluorescence polarization titration showed that the 10 bp of site-specific DNA bound PU.1ΔN167 in a 1:1 complex but was insufficient to invoke sequential dimerization, we repeated the DOSY titrations with a 10-bp duplexes. In stark contrast with their 16-bp parents, all of the 10-bp complexes regardless of sequence gave indistinguishable hydrodynamic profiles showing single inflections (supplemental Fig. S3, D–F). If the site-specific 10-bp 1:1 complexes maintained the structure observed in the co-crystal structure, their computed (25) diffusion coefficient would be 7.9 × 10−11 m2/s under the present experimental conditions. Thus, the measured diffusion coefficients of the 10-bp complexes (∼6.5 × 10−11 m2/s), which were significantly lower even than their 16-bp 1:1 counterpart, were unexpected for a 10-bp 1:1 complex. We confirmed the 1:1 stoichiometry of the 10-bp PU.1/DNA complexes by examining the 1H spectra of the 10-bp high-affinity DNA in the imino region at graded PU.1ΔN167 concentration. Resonances corresponding to free DNA were exhausted by unit molar protein/DNA ratio (supplemental Fig. S4). Thus, PU.1 bound 10-bp DNA exclusively as monomers even at excess concentrations, and sequential dimerization of PU.1 was limited to site-specific DNA longer than 10 bp. Moreover, the data implied that the protein underwent significant conformational changes to hydrodynamically larger structures than the same protein bound to 16-bp site-specific DNA.
Structural properties of the site-specific PU.1 ETS dimer
We recorded 1H-15N HSQC spectra of uniformly 15N-labeled PU.1ΔN167, which showed a structured protein with well-dispersed cross-peaks in the absence of DNA (Fig. 3A). Upon the addition of 16-bp high-affinity DNA (Fig. 3, B–D) to a DNA/PU.1 ratio of 1:2, we observed the immediate disappearance of ∼80% of the NH resonances and a marked loss of chemical shift dispersion, but no sign of precipitation even after prolonged incubation (>24 h). The addition of a second half-equivalent of DNA to DNA/PU.1 = 1:1 promptly restored the NH resonances, with significant chemical shift perturbations compared with the free protein. The further addition of excess DNA produced no further changes to the HSQC spectrum.
Figure 3.
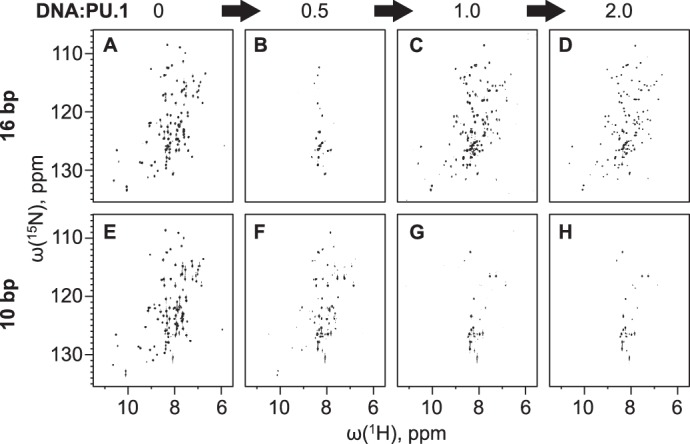
1H-15N HSQC NMR spectroscopy of PU.1/DNA complexes. Uniformly 15N-labeled PU.1ΔN167 was titrated with a 16-bp (A–D) or 10-bp (E–H) unlabeled high-affinity DNA (5′-GCAAGCGGAAGTGAGC-3′) at the indicated molar ratios. Each series of spectra was acquired with the same sample and intensity adjusted to the same noise level.
When we repeated the HSQC titration using the same high-affinity 10-bp construct as in the DOSY experiments, we observed a progressive disappearance of resonances past the half-equivalence point and no further change past the 1:1 equivalence point (Fig. 3, E–H). The monotonic transition for the 10-bp DNA tracked the changes in diffusion coefficient (Fig. 3, E–H), in contrast with the sharply biphasic behavior of the 16-bp site.
To better understand the effect of binding site size on the bound protein's conformation, we probed DNA-bound PU.1ΔN167 with 8-anilino-1-naphthalenesulfonate (ANS), an indicator dye of solvent-exposed hydrophobic moieties. Unbound PU.1 at 50 μm induced strong blue-shifted ANS fluorescence associated with a significant number of untitrated basic residues, which paired with the anionic dye (28), in the absence of DNA (Fig. 4A). DNA alone induced a negligible effect on ANS fluorescence. After subtraction of a dye-only control, ANS fluorescence of PU.1 was reduced about 3-fold upon binding a half-equivalent of the 16-bp site-specific DNA (2:1 complex), and another 8-fold at unit equivalence (1:1 complex) (Fig. 4B). The higher sensitivity to ANS, together with the NMR DOSY data, suggest that the DNA-bound PU.1 dimer may be less structured than in the 1:1 complex.
Figure 4.
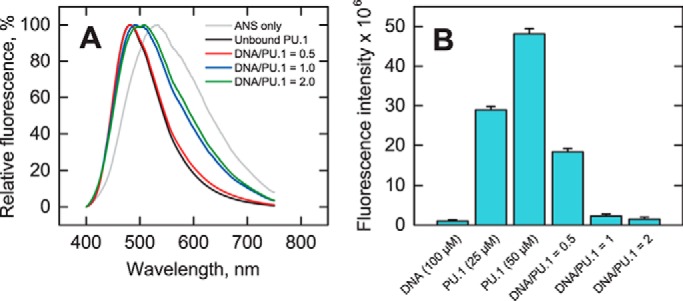
Biochemical characterization of PU.1/DNA complex conformation. A, fluorescence spectra of 50 μm PU.1ΔN167 alone or with 0.5, 1.0, or 2.0 molar eq of 16-bp site-specific DNA after mixing with 200 μm ANS. Samples were excited at 375 nm. B, fluorescence intensity at 530 nm of PU.1ΔN167 with or without 16-bp DNA after subtraction of an ANS-only control, shown as average ± S.D. (error bars) of triplicate experiments.
Topology of the site-specifically bound PU.1 dimer
The attenuation in NMR signal from the 2:1 complex suggests intermediate exchange between these two states. As a result, although the disappearance of 80% of cross-peaks in the DNA-bound PU.1 dimer (cf. Fig. 3B) precluded a direct identification of the residues involved in 2:1 complex formation, the remaining resonances still provided valuable clues to the location of the dimerization interface. We overlaid the HSQC spectra for free and bound PU.1 to 16-bp DNA and identified resonances that showed strong overlap throughout the titration (Fig. 5, A–E). Using a reported 1H-15N HSQC assignment of the PU.1 residues 167–260 (27), which applied well to PU.1ΔN167 (supplemental Fig. S5), we identified well-resolved, well-overlapped resonances for Arg173, Ala210, Lys224, Gly238, Gly241, Lys247, and Ser255. Resolvable resonances that overlapped only in the free and 1:1-bound states (i.e. no detectable signal in the 2:1 state), including Ser184, Trp192, Trp193, Asp197, and Thr200, mapped to solvent-exposed surfaces away from the DNA (Fig. 5F). Thus, the HSQC data implicated the distal surface of PU.1ΔN167 opposite the DNA-binding site as a major part of the dimerization interface of the site-specific 2:1 complex.
Figure 5.

Mapping the dimerization interface of the site-specific 2:1 complex. A, overlay of 1H-15N HSQC spectra in the absence (green) or presence of 16-bp site-specific DNA at 0.5 (red) and 1.0 (blue) molar ratios. Peaks labeled in orange that showed strong overlap among all three states (blue/red/green) were taken to represent residues not involved in site-specific dimerization. Peaks labeled in purple that overlapped only in the unbound and 1:1-bound states (blue/green) were taken to represent residues involved in dimerization. Assigned resonances were as reported for residues 167–260 by Jia et al. (27). Boxes indicate regions that are magnified in B–E. F, mapping of the (purple) residues implicated in PU.1 dimerization to the 1:1 co-crystal structure (PDB code 1PUE). G, continuum electrostatic surface potential of PU.1 in the co-crystal structure. The residues 195DKDK198 are shown as spheres. H, DNA-binding profiles of a 195NINI198 mutant of PU.1ΔN167 by fluorescence polarization (20 bp) and gel mobility shift (209 bp) under the same experimental conditions as in Fig. 2. Symbols represent replicate experiments; the curve represents a 1:1 fit to the data. Error bars, S.E.
To further understand how the distal surface of the PU.1 ETS domain was involved in dimerization, we examined the amino acids that mapped to that surface and noticed a sequence of four alternately charged residues, 195DKDK198, that comprise part of a β-pleated sheet. These residues include (Asp197) or are proximal to residues (Trp192, Trp193, and Thr200) whose resonances became reversibly undetectable in the 2:1 complex (Fig. 5, A and D). The 195DKDK198 sequence gave rise to a charge distribution that suggested an electrostatically complementary interface for dimerization (Fig. 5G). This hypothesis was further motivated by the low level of sequence conservation in Ets-1 (357TGDG360) and within the ETS family in general (29). We therefore cloned a PU.1ΔN167 mutant harboring 195NINI198, which abrogated the charges but maintained similar side-chain structures and secondary structure propensities (30). In fluorescence polarization and gel mobility shift experiments, the mutant gave titration profiles that showed a single binding mode at up to 10 μm, a concentration at which DNA-bound wild-type PU.1 had undergone two binding transitions (Fig. 5H; cf. Fig. 2). The anisotropy and electrophoretic mobility of the bound mutant corresponded to the 1:1 complex formed by wild-type protein. Thus, the 195NINI198 mutant confirmed that the distal surface was involved in PU.1 dimerization. In addition, the mutant bound DNA ∼10-fold more weakly (88 ± 11 nm) than wild-type PU.1ΔN167, suggesting coupling between DNA binding and dimerization of the bound state.
The 2:1 site-specific PU.1/DNA complex occupies an expanded DNA-binding site
To define the contact interface of the 2:1 PU.1/DNA complex, we carried out hydroxyl radical (•OH) footprinting titration of a 130-bp radiolabeled DNA fragment harboring the same high-affinity binding site used in the other experiments. Previous footprinting studies of the 1:1 site-specific PU.1/DNA complex by our group (13, 31) and others (32, 33) have established that two spaced clusters of minor-groove contacts flanking the 5′-GGAA-3′ core consensus generate a highly characteristic •OH footprint on the 5′-TTCC-3′ strand. We therefore used this biochemical signature to track changes in the DNA-binding site at graded concentration of wild-type PU.1ΔN167 (Fig. 6A). Upon reaching PU.1ΔN167 concentrations of ∼10−7 m that saturated 1:1 complex (marked P1 and P2 in Fig. 6B), additional PU.1ΔN167 gave rise to protected positions near P2 (marked P*). In total, the footprint of the 2:1 complex spanned ∼20 bp of DNA. Quantitation of the protection from •OH at the protected bases as a function of PU.1 concentration clearly recapitulated the sequential formation of the 1:1 followed by the 2:1 complex observed in the other experiments (Fig. 6C). In addition, the hypersensitive positions between P1 and P2, which is also diagnostic of site-specific 1:1 binding, was preserved in the 2:1 complex and tracked the titration profiles produced by fluorescence anisotropy (cf. Fig. 2A).
Figure 6.

Expansion of the DNA contact interface in the 2:1 PU.1/DNA complex. A singly end-radiolabeled DNA fragment was titrated at equilibrium with PU.1ΔN167 and digested with •OH under single-hit conditions. A C + T reaction was included to index the digested DNA following denaturing electrophoresis. A, image of the sequencing gel. N and U denote DNA digested without protein and undigested DNA, respectively. A second footprint was observed at a cryptic binding site (5′-ATGGGAATTC-3′) encoded by pUC19 vector further downstream from the cloned high-affinity site. The lower affinity of this site (48) meant that it did not generate the sequential 2:1 complex beyond the 1:1 footprint at the maximum PU.1 concentration used. B, traces of the indicated lanes. Brackets and red dots denote protected and hypersensitive positions at the indicated and higher protein concentrations, respectively, relative to a distal control peak marked with a hollow dot (○). C, titration of the summed integrated intensities of the protected bases marked P1 and P2 (white squares) associated with the 1:1 complex and P* (black squares) produced by the 2:1 complex in B, normalized to the control peak intensity and scaled to (0, 1). Curves represent empirical fits to the Hill equation. D, titration of the summed integrated intensities of the hypersensitive peaks (red circles), scaled to (0, 1) but normalized to the intensity at the highest PU.1ΔN167 concentration tested. The curve represents a fit by a sequential 2:1 binding model.
Thus, the •OH footprints showed, at single-nucleotide resolution, that the site-specific PU.1ΔN167 dimer made extended contacts with the DNA minor groove although the dimerization interface was distal from the DNA. The extended footprint exerted by the 2:1 complex also explained the 10-bp specific site's apparent incompatibility with PU.1 dimerization, which required several more flanking bases downstream of the 5′-GGAA-3′ core consensus, although its affinity for the 1:1 complex was only modestly compromised relative to longer DNA (cf. Fig. 2A).
Discussion
When bound to sufficiently long site-specific DNA, the ETS domain of PU.1 self-associates sequentially to a defined dimer, a behavior that is heretofore unknown for ETS transcription factors (19). The reversibility of dimerization is demonstrated by its independence on directionality. In gel mobility shift, fluorescence anisotropy, or DNA footprinting titrations in which excess protein was titrated into site-specific DNA, the 2:1 complex was produced subsequently to the canonical 1:1 complex. When the titration was reversed (DNA into protein), as was the case in the NMR and ITC studies, limiting concentrations of DNA directly yielded the 2:1 complex. Impressively, the NMR titrations showed that even after prolonged co-incubation, the further addition of site-specific DNA converted the 2:1 complex rapidly and quantitatively into its 1:1 counterpart. Structurally, the identification of the solvent-exposed surface distal from the DNA as the dimerization interface on the one hand, and the expansion of the DNA footprint of the 2:1 complex on the other, suggested an allosteric coupling between the PU.1 dimers and their bound DNA.
Although many DNA-binding domains are known to self-associate when they bind to site-specific DNA (34), this behavior is associated with systems in which the protein protomers bind multiple DNA subsites independently, such as the Trp repressor (35), or with positive cooperativity, such as the p53 core domain (36). Ets-1 and several other non-PU.1 ETS members can also bind as homodimers, but only to two tandem DNA sites (14–18). To our knowledge, negatively cooperative binding to a single DNA site, as the PU.1 ETS domain is able to execute, has not been reported previously.
Flanking sequence length as a specificity determinant of PU.1/DNA binding
We tested a range of DNA lengths to define the site size requirements for PU.1 dimerization in the bound state and to probe the relevance of dimerization in the presence of excess nonspecific DNA. We observed that the binding modes accessible to the ETS domain of PU.1 depended on a threshold length of bases flanking the core 10-bp binding site. Within the range of DNA lengths tested in the various experiments, 16-bp and longer DNA invoked sequential dimerization of bound PU.1. In stark contrast, in the absence of flanking bases, the 10-bp DNA bound PU.1ΔN167 exclusively with 1:1 stoichiometry (Figs. 2B and 3). Thus, 10 bp of site-specific DNA was insufficient to elicit the full site-specific behavior of the PU.1 ETS domain. Available evidence indicates that flanking sequence identity is not a determinant because we had observed two other site-specific DNA sequences yielding the same ITC profiles for PU.1ΔN167 (13).
The 10-bp complex represented a distinct binding mode as the bound PU.1 monomer was structurally different from its 16-bp counterpart as judged by their HSQC spectra (Fig. 3). This observation was unexpected, given the single turn of contacted double helical DNA in the co-crystal structure of the high-affinity PU.1/DNA complex (23) and ETS/DNA structures more generally. Of relevance is the report that DNA with staggered ends was absolutely required for diffraction-quality crystals of the PU.1/DNA complex (37). The overhangs, which paired end-to-end between asymmetric units, would result in essentially continuous DNA in the crystal. Additional interactions with flanking bases that are not part of the core sequence therefore stabilize the bound protein, and without this stabilization, dimerization becomes prohibitive. In summary, flanking sequence length represents an essential additional determinant to fully specify cognate binding by PU.1 in solution.
Functional relevance of self-titration as a potential negative feedback mechanism for PU.1 transactivation
PU.1 is a highly inducible protein, ranging from <10 to >200 copies of mRNA per cell in murine bone marrow progenitors, depending on the stage of hematopoietic development (38). Under physiologic induction, PU.1 mRNA levels matching and even exceeding that of glyceraldehyde 3-phosphate dehydrogenase, an abundant glycolytic housekeeping enzyme, have been measured in cultured (39) and primary (40) human cells. This inducible expression profile suggests that interactions spanning a large range in affinity are likely to be biologically relevant. For instance, NMR characterization of the functionally essential PU.1/GATA-1 interaction estimated its dissociation constant to be no stronger than 10−4 m in vitro (41) and did not appear to involve (as judged by chemical shift changes) the dimerization interface of PU.1.
Many ETS family transcription factors, such as Ets-1, ERG, and members of the ETV subfamily, are regulated at the protein/DNA level by inhibitory helices that pack against their DNA-binding domain in the unbound state (Fig. 1A). Perturbing these helices imposes an energetic penalty on DNA-binding that maintains, by default, an autoinhibited state. Binding partners that disrupt the autoinhibitory interactions thus induce a transcriptionally permissive state (19). ETS paralogs, such as PU.1, that lack this mechanism would therefore be locked in a permissive state in the absence of some mechanism for negative regulation. Whereas functionally antagonistic binding partners, such as GATA-1, would serve such an inhibitory role, their expression profiles only partially overlap with that of PU.1 (the common myeloid progenitor in the case of GATA-1 (42)). An intrinsic negative feedback mechanism is hitherto unknown in PU.1. Our observation that PU.1 forms a reversible, negatively cooperative 2:1 complex with site-specific DNA suggests “self-titration” as a potential mechanism of negative feedback: even if the 2:1 complex retains the functional activity of the 1:1 complex, removal of circulating PU.1 alone would attenuate transactivation of target genes. Consistent with this notion, we observed self-titration only with site-specific DNA and not nonspecific DNA. Moreover, we did not observe dimer formation with the structural homolog Ets-1, with or without its autoinhibition helices, when its cysteines were maintained in a reduced state (Fig. 1, D–G). Interestingly, a 2:1 Ets-1/DNA complex was reported under non-reducing conditions (43), reflecting the strong propensity for its two cysteine residues (which are not present in the PU.1 ETS domain) to form non-native disulfide linkages.
The dissociation constant for binding to oligomeric nonspecific sites (∼10−6 m) (31), such as that used in our NMR experiments, is only ∼10-fold higher than the sequential affinity of the second equivalent of PU.1ΔN167. It might therefore appear that the abundance of nonspecific DNA relative to specific sites would overwhelm self-titration of specific complexes. Our gel mobility data on binding to polymeric DNA (cf. Fig. 2C) provide a useful insight into this question. Compared with titration of oligomeric site-specific DNA, formation of the 2:1 complex at an embedded binding site flanked by substantial nonspecific DNA (∼100 bp on each side) occurred at ∼10-fold lower concentration (∼10−7 m) and clearly preceded any nonspecific binding. The footprinting data showed the same behavior at a shorter (∼130-bp) DNA fragment. This difference reflected the favorable contribution to reaching an embedded site from linear diffusion that was absent for an isolated counterpart. Thus, a complete description of the effect of excess nonspecific flanking DNA (as would be expected under genomic conditions) includes a competitive effect that is more than offset by favorable contributions from linear diffusion.
Nonspecifically bound PU.1 is oligomeric
The 16-bp nonspecific site, involving only the isomeric reversal of two adjacent positions in the core consensus (5′-GGAA-3′ to 5′-GAGA-3′), forced the exclusive formation of a dimeric complex. No 1:1 complex was detectable at equilibrium. In the context of self-titration as a potential negative regulatory mechanism, this behavior suggests that the role of site-specific DNA (i.e. sequences harboring the core consensus) is not only to provide a much higher-affinity binding site for PU.1 but, perhaps more importantly, to “unlock” the transcriptionally active 1:1 conformation. It may therefore be more appropriate to consider dimeric PU.1, as the default autoinhibited state, which becomes activated, by a coupled dissociation/order transition, upon encountering a specific DNA site at permissively low protein concentrations.
Conclusion
We report, for the first time, a 2:1 complex formed by PU.1 with a single cognate binding site. This complex forms negatively cooperatively with respect to the canonical, transcriptionally active 1:1 complex and resists competition from nonspecific DNA. It is kinetically stable (on the order of many hours) and interconverts efficiently with the 1:1 complex (within minutes) upon the addition of DNA. These biophysical properties of self-titration of PU.1 at site-specific DNA are biologically compatible and, indeed, physiologically appropriate given the significant accumulation of PU.1 under induction (>10−6 m), when negative feedback would be most required to dampen its transcriptional response. Self-titration therefore represents a potential buffering mechanism for self-regulation in ETS paralogs, such as PU.1, that lack autoinhibitory elements in their structures.
Materials and methods
Proteins
Recombinant constructs representing the ETS domain of murine PU.1 (residues 167–272, designated PU.1ΔN167) and Ets-1 (residues 331–440, designated Ets-1ΔN331) were cloned with a thrombin-cleavable C-terminal His6 tag as described (44). A similarly tagged construct for autoinhibited Ets-1 (residues 280–440, Ets-1ΔN280) was a gift from Dr. Lawrence P. McIntosh (University of British Columbia). Unlabeled constructs were overexpressed in Escherichia coli in LB medium. Uniformly 15N-labeled PU.1ΔN167 was expressed from 5-ml starter cultures in LB broth grown at 37 °C for ∼8 h. All of the culture was inoculated into 250 ml of LB broth, grown at 37 °C for ∼16 h, and harvested. The cell pellet was resuspended in standard M9 medium containing 15NH4Cl, MgSO4, CaCl2, trace metals, minimal essential medium vitamins, and glucose. Protein expression was induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside overnight at 25 °C. Both unlabeled and isotopically labeled constructs were purified as described (22). In brief, cleared lysate was first purified by immobilized metal affinity chromatography, cleaved with thrombin, dialyzed against 10 mm NaH2PO4/Na2HPO4 (pH 7.4) containing 0.5 m NaCl, and polished on Sepharose SP (GE Healthcare). Buffers used with Ets-1 constructs, which harbored reduced cysteines, additionally contained 0.5 mm tris(2-carboxyethyl)phosphine-HCl. Protein concentrations were determined by UV absorption at 280 nm using the following extinction coefficients (in m−1 cm−1): 22,460 (PU.1ΔN167), 32,430 (Ets-1ΔN331), and 39,880 (Ets-1ΔN280). The labeling efficiency of 15N-labeled constructs was >98%, as judged by mass spectrometry (supplemental Fig. S1).
Nucleic acids
Synthetic DNA oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA) and annealed to form duplex binding sites harboring the high-affinity 5′-AGCGGAAGTG-3′, low-affinity 5′-AAAGGAATGG-3′, or nonspecific 5′-AGCGAGAGTG-3′ DNA sequence (ETS-specific core consensus in boldface type). Fluorescent DNA probes were constructed by annealing a Cy3-labeled oligonucleotide with excess unlabeled complementary strand as described (22).
Fluorescence polarization titrations
ETS protein binding to fluorescently labeled DNA sites was measured using a Molecular Devices Paradigm plate reader as described (29). In brief, DNA probe (0.5 nm) was incubated to equilibrium with graded concentrations of purified PU.1ΔN167 in a total volume of 30 μl of 10 mm Tris-HCl (pH 7.4) containing 150 mm total Na+ and 0.1 mg/ml bovine serum albumin. Steady-state fluorescence parallel and perpendicular to the incident polarized light was acquired at 595/35 nm upon excitation at 535/25 nm. Each data point represents the mean ± S.E. of five consecutive measurements as an indication of instrumental noise. Anisotropy data were fitted with a 1:1 or sequential binding model (22) to directly estimate the dissociation constants of the PU.1/DNA 1:1 and 2:1 complexes.
2D 1H-15N HSQC NMR
Purified [15N]PU.1ΔN167 (∼0.5 mm) was extensively dialyzed together with various duplex DNA constructs (∼2 mm) against 11 mm NaH2PO4/Na2HPO4, pH 7.6, 167 mm NaCl, and 0.1% NaN3 and adjusted to 10% D2O. DNA was titrated into protein to achieve the desired DNA/protein ratios. 1H-15N correlated measurements were made using a phase-sensitive, double inept transfer with a GARP decoupling sequence, and solvent suppression (hsqcf3gpph19). Spectra were acquired with 1024 × 144 data points and zero-filled to 4096 × 4096.
ANS fluorescence
ANS (ammonium salt, Alfa Aesar) was prepared at 2 mm in 10 mm NaH2PO4/Na2HPO4 (pH 7.4) buffer containing 150 mm NaCl and stored in the dark at 4 °C. Triplicate samples of PU.1 with or without 16-bp high-affinity DNA plus various controls were prepared in the same buffer before the addition of ANS to 200 μm. Final concentrations of PU.1 and DNA were as indicated under “Results.” After incubation for 30 min, the fluorescence intensity of each sample was read at 370/530 nm or scanned from 400 to 750 nm with a Paradigm plate reader.
Hydroxyl radical DNA footprinting
A 130-bp DNA fragment harboring a copy of the high-affinity PU.1-binding site 5′-AGCGGAAGTG-3′ was generated by PCR using two primers, of which the one encoding the 5′-CACTTCCGCT-3′ strand had been 5′-end–labeled with [32P]ATP. After purification by agarose gel electrophoresis, the radiolabeled fragment (<1 nm) was incubated to equilibrium with graded concentrations of PU.1ΔN167. Each sample was digested with hydroxyl radical, purified, resolved by denaturing polyacrylamide gel electrophoresis, and digitized by phosphorimagery as described (31). Lane traces were constructed, and bands were indexed using a C + T chemical sequencing reaction. Peaks were fitted as a superposition of Gaussian distributions, numerically integrated, and normalized to a band outside of the binding site to quantify fractional protection relative to the unbound sample.
Structure-based calculations
Self-diffusion constants for unbound and 1:1 DNA-bound PU.1ΔN167 were computed using the software HydroPRO (25). DNA-bound and unbound PU.1 structures were templated from the co-crystal structure with DNA (PDB code 1PUE) (23), appended with additional residues present in PU.1ΔN167, and relaxed by all-atom molecular dynamics simulation for 200 ns following our established protocol (29). Computations were carried out using volumetric values for D2O at 25 °C, namely a density of 1.107 g/ml, viscosity of 1.25 centipoises, and partial specific volume of 0.70 ml/g.
Continuum electrostatics of PU.1 in the co-crystal structure were computed using APBS (45). Calculations were performed for an aqueous solution containing 0.15 m NaCl at 25 °C and rendered on the solvent-accessible surface from −1 to +1 kT/e.
ITC
Purified PU.1 or Ets-1 was dialyzed extensively together with 23-bp DNA harboring the protein's respective optimal target (5′-AGCGGAAGTG-3′ for PU.1; 5′-GCCGGAAGTG-3′ for Ets-1) in separate compartments, against 50 mm NaH2PO4/Na2HPO4, pH 7.4, 150 mm total Na+, 0.1 mm EDTA, and 1 mm dithiothreitol. Titrations were performed by injecting DNA (initial concentration ∼500 μm) into protein (∼50 μm) in a Nano ITC instrument (TA Instruments). Data fitting to empirical 1:1 and cooperative models was performed as described (46) only to demonstrate the models qualitatively, not for quantitative estimation of the binding affinities, due to the very strong dissociation constant of the 1:1 complex (10−9 m).
To compare the calorimetric enthalpies for DNA binding by PU.1 with those for Ets-1, which exhibited strictly 1:1 behavior, the calorimetric enthalpies for PU.1/DNA binding were decomposed as follows to extract the enthalpy changes for the 1:1 complex. Whereas PU.1ΔN167 dimerizes in both unligated and DNA-bound states, the former occurs at considerably higher concentrations (near 10−3 m) (13, 27) than those used in the “reverse” DNA-into-protein titrations shown in Fig. 1 (A and B). Under these conditions, the biphasic profile arises from the 1:1 complex being strongly favored and yielding a 2:1 complex only in excess protein at the initial phase (Rα) of the titration as shown in Scheme 1, where P and D represent PU.1ΔN167 and site-specific DNA in their various free and bound states. The two phases Rα and Rβ are marked in the reverse titration shown in Fig. 1B. Because the transition from the 1:1 to 2:1 PU.1/DNA complex occurs sequentially, both phases are well-defined and extracted according to the technique of “total association at partial saturation” (47). To compare the enthalpy changes meaningfully with the manifestly 1:1 binding for Ets-1 in Fig. 1 (C–F), the complex heats in the reverse titrations are dissected to account for the thermodynamics of coupled dimerization and dissociation of PU.1 as shown in Scheme 2. The calorimetric enthalpy marked Fβ has been measured previously for PU.1ΔN167 under the same solutions (13). Based on ΔHRβ = −44.2 ± 1.4 kJ/mol (cf. Fig. 1B) and ΔHFβ = 17.1 ± 0.7 kJ/mol (13) at 25 °C, the enthalpy change for the formation of the canonical 1:1 complex was −27.1 ± 1.6 kJ/mol. Thus, the enthalpy change for formation of the 1:1 complex from unbound constituents was larger in magnitude than that for minimal (ΔH1:1 = −12.0 ± 0.4 kJ/mol) and autoinhibited Ets-1 (ΔH1:1 = −8.1 ± 0.4 kJ/mol) at 25 °C.
Scheme 1.

Scheme 2.
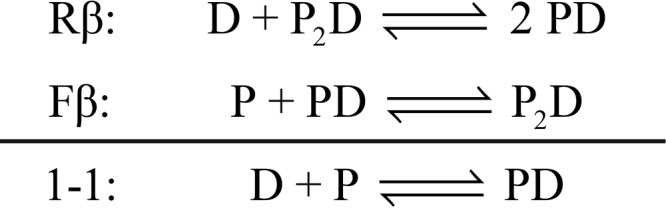
Author contributions
S. E. conducted the dye binding and NMR experiments, the latter in collaboration with M. G. E. N. E. performed the fluorescence polarization titrations. G. M. K. P. carried out the ITC, gel mobility shift, and DNA footprinting experiments. All authors analyzed the results. G. M. K. P. and M. W. G. conceived the idea for the project and wrote the paper with the other co-authors. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. W. David Wilson for insightful discussion and Kenneth Huang for molecular dynamics simulation of PU.1ΔN167.
This work was supported by National Science Foundation Grant MCB 15451600 and National Institutes of Health Grant R21 HL129063 (to G. M. K. P.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Methods, Table S1, and Figs. S1–S5.
- ITC
- isothermal titration calorimetry
- DOSY
- diffusion-ordered spectroscopy
- HSQC
- heteronuclear single quantum coherence
- ANS
- 8-anilinonaphthalene-1-sulfonate
- PDB
- Protein Data Bank.
References
- 1. Iwasaki H., Somoza C., Shigematsu H., Duprez E. A., Iwasaki-Arai J., Mizuno S., Arinobu Y., Geary K., Zhang P., Dayaram T., Fenyus M. L., Elf S., Chan S., Kastner P., Huettner C. S., et al. (2005) Distinctive and indispensable roles of PU.1 in maintenance of hematopoietic stem cells and their differentiation. Blood 106, 1590–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mak K. S., Funnell A. P., Pearson R. C., and Crossley M. (2011) PU.1 and haematopoietic cell fate: dosage matters. Int. J. Cell Biol. 2011, 808524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dozmorov M. G., Wren J. D., and Alarcón-Riquelme M. E. (2014) Epigenomic elements enriched in the promoters of autoimmunity susceptibility genes. Epigenetics 9, 276–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rosenbauer F., Wagner K., Kutok J. L., Iwasaki H., Le Beau M. M., Okuno Y., Akashi K., Fiering S., and Tenen D. G. (2004) Acute myeloid leukemia induced by graded reduction of a lineage-specific transcription factor, PU.1. Nat. Genet. 36, 624–630 [DOI] [PubMed] [Google Scholar]
- 5. Tatetsu H., Ueno S., Hata H., Yamada Y., Takeya M., Mitsuya H., Tenen D. G., and Okuno Y. (2007) Down-regulation of PU.1 by methylation of distal regulatory elements and the promoter is required for myeloma cell growth. Cancer Res. 67, 5328–5336 [DOI] [PubMed] [Google Scholar]
- 6. Jundt F., Kley K., Anagnostopoulos I., Schulze Pröbsting K., Greiner A., Mathas S., Scheidereit C., Wirth T., Stein H., and Dörken B. (2002) Loss of PU.1 expression is associated with defective immunoglobulin transcription in Hodgkin and Reed-Sternberg cells of classical Hodgkin disease. Blood 99, 3060–3062 [DOI] [PubMed] [Google Scholar]
- 7. Gjoneska E., Pfenning A. R., Mathys H., Quon G., Kundaje A., Tsai L.-H., and Kellis M. (2015) Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer's disease. Nature 518, 365–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kueh H. Y., Champhekhar A., Nutt S. L., Elowitz M. B., and Rothenberg E. V. (2013) Positive feedback between PU.1 and the cell cycle controls myeloid differentiation. Science 341, 670–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nerlov C., Querfurth E., Kulessa H., and Graf T. (2000) GATA-1 interacts with the myeloid PU.1 transcription factor and represses PU.1-dependent transcription. Blood 95, 2543–2551 [PubMed] [Google Scholar]
- 10. Pufall M. A., and Graves B. J. (2002) Autoinhibitory domains: modular effectors of cellular regulation. Annu. Rev. Cell Dev. Biol. 18, 421–462 [DOI] [PubMed] [Google Scholar]
- 11. Fitzsimmons D., Lukin K., Lutz R., Garvie C. W., Wolberger C., and Hagman J. (2009) Highly cooperative recruitment of Ets-1 and release of autoinhibition by Pax5. J. Mol. Biol. 392, 452–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goetz T. L., Gu T. L., Speck N. A., and Graves B. J. (2000) Auto-inhibition of Ets-1 is counteracted by DNA binding cooperativity with core-binding factor α2. Mol. Cell. Biol. 20, 81–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Poon G. M. (2012) DNA binding regulates the self-association of the ETS domain of PU.1 in a sequence-dependent manner. Biochemistry 51, 4096–4107 [DOI] [PubMed] [Google Scholar]
- 14. Baillat D., Bègue A., Stéhelin D., and Aumercier M. (2002) ETS-1 transcription factor binds cooperatively to the palindromic head to head ETS-binding sites of the stromelysin-1 promoter by counteracting autoinhibition. J. Biol. Chem. 277, 29386–29398 [DOI] [PubMed] [Google Scholar]
- 15. Lamber E. P., Vanhille L., Textor L. C., Kachalova G. S., Sieweke M. H., and Wilmanns M. (2008) Regulation of the transcription factor Ets-1 by DNA-mediated homo-dimerization. EMBO J. 27, 2006–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hou C., and Tsodikov O. V. (2015) Structural basis for dimerization and DNA binding of transcription factor FLI1. Biochemistry 54, 7365–7374 [DOI] [PubMed] [Google Scholar]
- 17. Mimeault M. (2000) Structure-function studies of ETS transcription factors. Crit. Rev. Oncog. 11, 227–253 [PubMed] [Google Scholar]
- 18. Jolma A., Yan J., Whitington T., Toivonen J., Nitta K. R., Rastas P., Morgunova E., Enge M., Taipale M., Wei G., Palin K., Vaquerizas J. M., Vincentelli R., Luscombe N. M., Hughes T. R., et al. (2013) DNA-binding specificities of human transcription factors. Cell 152, 327–339 [DOI] [PubMed] [Google Scholar]
- 19. Hollenhorst P. C., McIntosh L. P., and Graves B. J. (2011) Genomic and biochemical insights into the specificity of ETS transcription factors. Annu. Rev. Biochem. 80, 437–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Poon G. M. K., and Kim H. M. (2017) Signatures of DNA target selectivity by ETS transcription factors. Transcription 8, 193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. He G., Tolic A., Bashkin J. K., and Poon G. M. (2015) Heterogeneous dynamics in DNA site discrimination by the structurally homologous DNA-binding domains of ETS-family transcription factors. Nucleic Acids Res. 43, 4322–4331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stephens D. C., Kim H. M., Kumar A., Farahat A. A., Boykin D. W., and Poon G. M. K. (2016) Pharmacologic efficacy of PU.1 inhibition by heterocyclic dications: a mechanistic analysis. Nucleic Acids Res. 44, 4005–4013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kodandapani R., Pio F., Ni C. Z., Piccialli G., Klemsz M., McKercher S., Maki R. A., and Ely K. R. (1996) A new pattern for helix-turn-helix recognition revealed by the PU.1 ETS-domain-DNA complex. Nature 380, 456–460 [DOI] [PubMed] [Google Scholar]
- 24. Cann J. R. (1996) Theory and practice of gel electrophoresis of interacting macromolecules. Anal. Biochem. 237, 1–16 [DOI] [PubMed] [Google Scholar]
- 25. Ortega A., Amorós D., and García de la Torre J. (2011) Prediction of hydrodynamic and other solution properties of rigid proteins from atomic- and residue-level models. Biophys. J. 101, 892–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Altieri A. S., Hinton D. P., and Byrd R. A. (1995) Association of biomolecular systems via pulsed-field gradient NMR self-diffusion measurements. J. Am. Chem. Soc. 117, 7566–7567 [Google Scholar]
- 27. Jia X., Lee L. K., Light J., Palmer A. G. 3rd, Assa-Munt N. (1999) Backbone dynamics of a short PU.1 ETS domain. J. Mol. Biol. 292, 1083–1093 [DOI] [PubMed] [Google Scholar]
- 28. Matulis D., and Lovrien R. (1998) 1-Anilino-8-naphthalene sulfonate anion-protein binding depends primarily on ion pair formation. Biophys. J. 74, 422–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xhani S., Esaki S., Huang K., Erlitzki N., and Poon G. M. (2017) Distinct roles for interfacial hydration in site-specific DNA recognition by ETS-family transcription factors. J. Phys. Chem. B 121, 2748–2758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Costantini S., Colonna G., and Facchiano A. M. (2006) Amino acid propensities for secondary structures are influenced by the protein structural class. Biochem. Biophys. Res. Commun. 342, 441–451 [DOI] [PubMed] [Google Scholar]
- 31. Poon G. M. (2012) Sequence discrimination by DNA-binding domain of ETS family transcription factor PU.1 is linked to specific hydration of protein-DNA interface. J. Biol. Chem. 287, 18297–18307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gross P., Arrowsmith C. H., and Macgregor R. B. Jr. (1998) Hydroxyl radical footprinting of DNA complexes of the ets domain of PU.1 and its comparison to the crystal structure. Biochemistry 37, 5129–5135 [DOI] [PubMed] [Google Scholar]
- 33. Gross P., Yee A. A., Arrowsmith C. H., and Macgregor R. B. Jr. (1998) Quantitative hydroxyl radical footprinting reveals cooperative interactions between DNA-binding subdomains of PU.1 and IRF4. Biochemistry 37, 9802–9811 [DOI] [PubMed] [Google Scholar]
- 34. Siggers T., and Gordân R. (2014) Protein-DNA binding: complexities and multi-protein codes. Nucleic Acids Res. 42, 2099–2111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jin L., Yang J., and Carey J. (1993) Thermodynamics of ligand binding to trp repressor. Biochemistry 32, 7302–7309 [DOI] [PubMed] [Google Scholar]
- 36. Rippin T. M., Freund S. M., Veprintsev D. B., and Fersht A. R. (2002) Recognition of DNA by p53 core domain and location of intermolecular contacts of cooperative binding. J. Mol. Biol. 319, 351–358 [DOI] [PubMed] [Google Scholar]
- 37. Pio F., Ni C. Z., Mitchell R. S., Knight J., McKercher S., Klemsz M., Lombardo A., Maki R. A., and Ely K. R. (1995) Co-crystallization of an ETS domain (PU.1) in complex with DNA: engineering the length of both protein and oligonucleotide. J. Biol. Chem. 270, 24258–24263 [DOI] [PubMed] [Google Scholar]
- 38. Warren L., Bryder D., Weissman I. L., and Quake S. R. (2006) Transcription factor profiling in individual hematopoietic progenitors by digital RT-PCR. Proc. Natl. Acad. Sci. U.S.A. 103, 17807–17812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang G., Zhou B., Li S., Yue J., Yang H., Wen Y., Zhan S., Wang W., Liao M., Zhang M., Zeng G., Feng C. G., Sassetti C. M., and Chen X. (2014) Allele-specific induction of IL-1β expression by C/EBPbeta and PU.1 contributes to increased tuberculosis susceptibility. PLoS Pathog. 10, e1004426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Imoto A., Okada M., Okazaki T., Kitasato H., Harigae H., and Takahashi S. (2010) Metallothionein-1 isoforms and vimentin are direct PU.1 downstream target genes in leukemia cells. J. Biol. Chem. 285, 10300–10309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liew C. W., Rand K. D., Simpson R. J. Y., Yung W. W., Mansfield R. E., Crossley M., Proetorius-Ibba M., Nerlov C., Poulsen F. M., and Mackay J. P. (2006) Molecular analysis of the interaction between the hematopoietic master transcription factors GATA-1 and PU.1. J. Biol. Chem. 281, 28296–28306 [DOI] [PubMed] [Google Scholar]
- 42. Arinobu Y., Mizuno S., Chong Y., Shigematsu H., Iino T., Iwasaki H., Graf T., Mayfield R., Chan S., Kastner P., and Akashi K. (2007) Reciprocal activation of GATA-1 and PU.1 marks initial specification of hematopoietic stem cells into myeloerythroid and myelolymphoid lineages. Cell Stem Cell 1, 416–427 [DOI] [PubMed] [Google Scholar]
- 43. Samorodnitsky D., Szyjka C., and Koudelka G. B. (2015) A role for autoinhibition in preventing dimerization of the transcription factor ETS1. J. Biol. Chem. 290, 22101–22110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang S., Linde M. H., Munde M., Carvalho V. D., Wilson W. D., and Poon G. M. (2014) Mechanistic heterogeneity in site recognition by the structurally homologous DNA-binding domains of the ETS family transcription factors Ets-1 and PU.1. J. Biol. Chem. 289, 21605–21616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dolinsky T. J., Nielsen J. E., McCammon J. A., and Baker N. A. (2004) PDB2PQR: an automated pipeline for the setup of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Poon G. M. (2010) Explicit formulation of titration models for isothermal titration calorimetry. Anal. Biochem. 400, 229–236 [DOI] [PubMed] [Google Scholar]
- 47. Bains G., and Freire E. (1991) Calorimetric determination of cooperative interactions in high affinity binding processes. Anal. Biochem. 192, 203–206 [DOI] [PubMed] [Google Scholar]
- 48. Poon G. M., and Macgregor R. B. Jr. (2003) Base coupling in sequence-specific site recognition by the ETS domain of murine PU.1. J. Mol. Biol. 328, 805–819 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.