

Abstract
Tumor cell invasion involves targeted localization of proteins required for interactions with the extracellular matrix and for proteolysis. The localization of many proteins during these cell–extracellular matrix interactions relies on membrane trafficking mediated in part by SNAREs. The SNARE protein syntaxin4 (Stx4) is involved in the formation of invasive structures called invadopodia; however, it is unclear how Stx4 function is regulated during tumor cell invasion. Munc18c is known to regulate Stx4 activity, and here we show that Munc18c is required for Stx4-mediated invadopodium formation and cell invasion. Biochemical and microscopic analyses revealed a physical association between Munc18c and Stx4, which was enhanced during invadopodium formation, and that a reduction in Munc18c expression decreases invadopodium formation. We also found that an N-terminal Stx4-derived peptide associates with Munc18c and inhibits endogenous interactions of Stx4 with synaptosome-associated protein 23 (SNAP23) and vesicle-associated membrane protein 2 (VAMP2). Furthermore, expression of the Stx4 N-terminal peptide decreased invadopodium formation and cell invasion in vitro. Of note, cells expressing the Stx4 N-terminal peptide exhibited impaired trafficking of membrane type 1 matrix metalloproteinase (MT1-MMP) and EGF receptor (EGFR) to the cell surface during invadopodium formation. Our findings implicate Munc18c as a regulator of Stx4-mediated trafficking of MT1-MMP and EGFR, advancing our understanding of the role of SNARE function in the localization of proteins that drive tumor cell invasion.
Keywords: cell invasion, epidermal growth factor receptor (EGFR), invadopodia, matrix metalloproteinase (MMP), trafficking, Munc18c, syntaxin4
Introduction
The process of metastasis is dependent on tumor cell invasion through the extracellular matrix (ECM),2 a supportive scaffold that acts to compartmentalize tissues (1). Tumor cell invasion through the ECM barrier can be viewed as a multistep process comprising cell adhesion, ECM proteolysis, and cell migration (2). To move through the ECM, tumor cells that have acquired an invasive phenotype utilize integrin receptors to engage with ECM components and also secrete matrix metalloproteinases (MMPs) to facilitate degradation of the ECM, ultimately promoting migration. Recent models suggest that tumor cell invasion can be mediated by subcellular structures called invadopodia. Invadopodia are F-actin–driven cell membrane protrusions, related to podosomes, that facilitate matrix metalloproteinase–mediated degradation of the ECM (3). Invadopodia have been studied in cancer cells in a variety of microenvironments in vitro (4, 5), and evidence from in vivo studies supports their role in the dissemination of tumor cell populations (6, 7).
Membrane trafficking of proteins to invadopodia is required for their formation and function in support of tumor cell invasion (8). Intracellular trafficking of cellular cargo is dependent on SNAREs, a family of membrane proteins that form complexes bridging apposed membranes and allowing membrane fusion (9). SNAREs are divided into two subfamilies: R- and Q-SNAREs, based on conserved arginine or glutamine residues, respectively. R-SNAREs are generally found on vesicles, whereas Q-SNAREs reside on target membranes. Fusion of membranes requires the formation of a trans-SNARE complex in which the SNARE domains of Q-SNAREs and R-SNAREs interact (9). Current models suggest that cellular invasion and invadopodium formation are dependent on the SNARE-mediated trafficking of proteins that facilitate invasion through the ECM (10–13) as well as cell migration (14–18). Syntaxin4 (Stx4) is a SNARE protein implicated in the trafficking of membrane type 1 matrix metalloproteinase (MT1-MMP) to the plasma membrane (19), including sites of invadopodium formation in MDA-MB-231 cells (11). To examine the regulation of Stx4 during tumor cell invasion, we analyzed the function of Munc18c, a known regulator of Stx4 activity belonging to the Sec1/Munc18 (SM) family of proteins (20). SM proteins are ∼65-kDa proteins found both in the cytosol and associated with membranes via interaction with cognate SNAREs (21, 22). SM proteins are viewed as regulators of SNARE function as well as mediators of SNARE complex formation that facilitate membrane fusion (23). Munc18c has broad tissue distribution (24) and has been implicated in several Stx4-mediated pathways, including insulin-stimulated glucose uptake in adipocytes (24–27), insulin exocytosis (26), neutrophil secretion (28), and pancreatic exocrine secretion (29). Insulin also stimulates the assembly of Stx4-containing complexes that involves the phosphorylation of Munc18c on Tyr-521 (27, 30).
To study the role of Munc18c during tumor cell invasion, we examined the effects of Munc18c knockdown and inhibition of endogenous Munc18c–Stx4 interactions in MDA-MB-231 and HT-1080 cells. We report that Munc18c and Stx4 association is enhanced under conditions in which cells are forming invadopodia, and knockdown of Munc18c impaired invadopodium formation. Expression of a peptide comprising the N-terminal 29 amino acids of Stx4 interfered with the interaction of endogenous Munc18c and Stx4, decreased trafficking of MT1-MMP and epidermal growth factor receptor (EGFR) to the cell surface, impaired invadopodium formation, and reduced cell invasion. These findings suggest that Munc18c is required to regulate Stx4 function during tumor cell interactions with the ECM.
Results
Stx4 and Munc18c association is enhanced during invadopodium formation
Previous studies have shown that Stx4 and Munc18c interact and that Munc18c plays an important role during Stx4-mediated exocytosis through this interaction (20, 29). To test whether Munc18c and Stx4 associate in MDA-MB-231 cells, co-immunoprecipitation experiments were performed. Cells were lysed in situ, and analysis of Munc18c immunoprecipitates revealed co-immunoprecipitation of Stx4 (Fig. 1A). Immunoprecipitates of Munc18c were also probed for syntaxin3 (Stx3), but co-immunoprecipitation of Stx3 was not observed (Fig. 1B). Consistent with the co-immunoprecipitation of Stx4 and Munc18c, confocal immunofluorescence microscopy revealed that Stx4 and Munc18c have a partly overlapping distribution near the ventral membrane of cells, whereas Stx3 and Munc18c do not (Fig. 1C).
Figure 1.
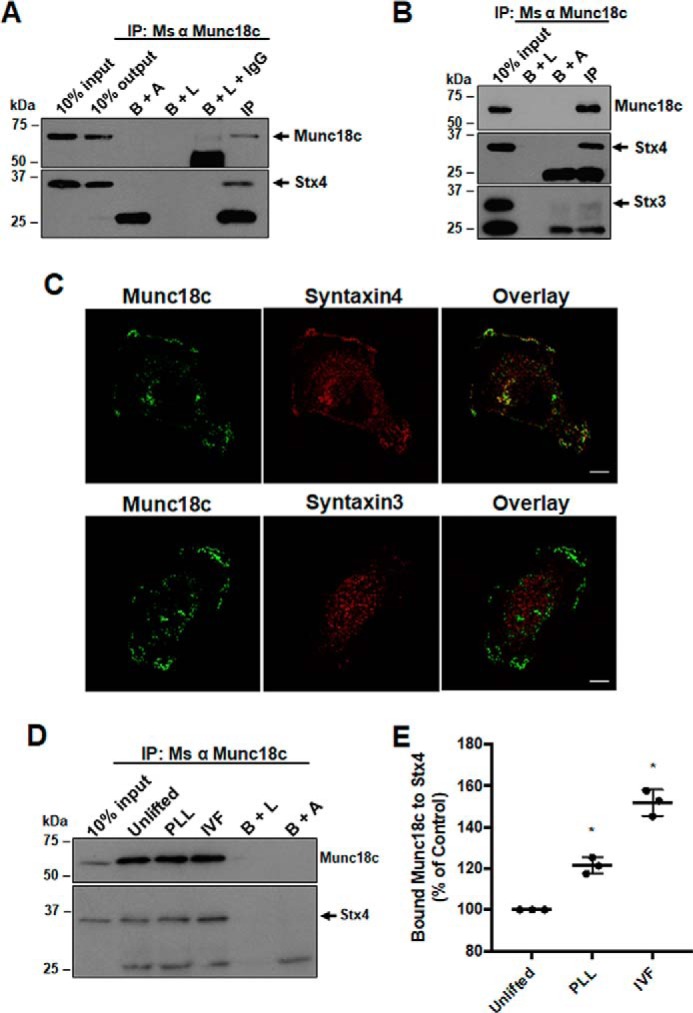
Analysis of Stx4 and Munc18c association in MDA-MB-231 cells. A, Munc18c immunoprecipitates were probed for Munc18c (arrow) and Stx4 (arrow). B+A, beads plus antibody; B+L, beads plus lysate; B+L+IgG, beads plus lysate plus unrelated IgG; IP, immunoprecipitation; Ms, mouse. B, Munc18c immunoprecipitates were probed for Munc18c, Stx4 (arrow), and Stx3 (arrow). C, cells were grown for 4 h on glass coverslips, fixed, permeabilized, and stained for either Munc18c and Stx4 or Munc18c and Stx3. A representative confocal section from the ventral region of a cell is shown. Scale bars = 10 μm. D, analysis of Stx4 and Munc18c association during invadopodium formation (IVF). Cells were seeded onto coverslips coated with PLL or gelatin, incubated for 4 h, lysed, and analyzed by immunoprecipitation/Western blotting. Munc18c immunoprecipitates were probed for Munc18c and Stx4 (arrow). E, quantification of Stx4 co-immunoprecipitated with Munc18c. Percent of controls are from three independent experiments ± S.D. Asterisks denote values significantly different from control unlifted cells (*, p < 0.05). All data represent three or more biological replicates with at least three technical replicates.
To assess the association of Stx4 and Munc18c during invadopodium formation, cells were seeded onto gelatin (an ECM analogue) or a non-ECM substrate (poly-l-lysine, PLL) as a control. Cells were lysed in situ, and analysis of Munc18c immunoprecipitates determined that the amount of Stx4 associated with Munc18c was increased in samples plated on gelatin when normalized to the amount of Munc18c immunoprecipitated in each sample (Fig. 1D). An increase of 21.4% ± 2.3% in the amount of Stx4 co-immunoprecipitated with Munc18c was observed when cells were seeded onto PLL relative to unlifted cells, and an increase of 52.0% ± 3.7% was observed during invadopodium formation on gelatin (Fig. 1E). These results suggest that Munc18c and Stx4 associate in a manner that increased significantly during invadopodium formation.
Knockdown of Munc18c impairs invadopodium formation
Involvement of Munc18c at invadopodia was supported by microscopic analysis of the localization of Munc18c during invadopodium formation. Munc18c was observed, along with Stx4, at invadopodia marked by β1 integrin and focal points of degradation in labeled gelatin (Fig. 2A). To test the requirement for Munc18c function in invadopodium formation, cells were transfected with a pool of siRNA targeting human Munc18c or nonspecific control siRNA. Knockdown of Munc18c was seen after 48 h, when Munc18c protein levels were reduced by 47.5% ± 2.8%. Protein levels of β1 integrin and MT1-MMP were unaffected (0.05% ± 0.08% and 0.08% ± 0.04%, respectively) (Fig. 2, B and C). Invadopodium formation was subsequently analyzed, and the number of cells forming invadopodia was reduced by 56.2% ± 4.0% in samples with decreased levels of Munc18c expression relative to controls (Fig. 2, D and E).
Figure 2.

RNAi-mediated knockdown of Munc18c impairs invadopodium formation. A, distribution of Munc18c during the formation of invadopodia. Cells were analyzed by confocal microscopy after being plated on Alexa Fluor 594–labeled gelatin for 4 h, fixed, permeabilized, and stained as indicated: β1 integrin and Stx4 (cyan, white arrows), Munc18c (green, yellow arrows). Dark spots in the gelatin field indicate sites of gelatin degradation corresponding to invadopodia. B, cells were transfected with siRNA targeting Munc18c or nonspecific control siRNA, lysed, and analyzed for Munc18c by SDS-PAGE/Western blotting. C, quantification of Munc18c knockdown normalized to actin. D, cells with F-actin puncta overlying dark spots of gelatin degradation were counted as cells forming invadopodia. Shown are percentages of cells forming invadopodia, normalized to control (GFP-transfected) cells. E, invadopodium-based degradation of Alexa Fluor 594 gelatin by cells co-transfected with GFP and control siRNA or Munc18c siRNA. Cells were transfected for 44 h, seeded onto gelatin for 4 h, and then fixed, permeabilized, stained for F-actin, and analyzed by confocal microscopy. Percent of controls are from three independent experiments ± S.D. Asterisks denotes values significantly different from control cells (*, p < 0.05). Scale bars = 10 μm. All data represent three or more biological replicates with at least three technical replicates.
An Stx4 N-terminal peptide associates with Munc18c and inhibits cognate SNARE binding with endogenous Stx4
Previous work has shown that the N-terminal 29 amino acids of Stx4 are required for binding to Munc18c in vitro and in vivo (31). In vitro pulldown experiments showed that the presence of this polypeptide reduced the degree of association between Munc18c and Stx4, suggesting that this N-terminal domain can act as a competitive inhibitor of Munc18c and Stx4 interactions (31). We hypothesized that an exogenously expressed peptide corresponding to the N-terminal 29 amino acids of Stx4 would bind to endogenous Munc18c and therefore impair normal Munc18c-dependent SNARE complex formation involving Stx4. A GFP-tagged construct encoding the N-terminal 29 amino acids of Stx4 (GFP–Stx4–N-term) was used to derive a stable cell line from MDA-MB-231 cells. Stable cell lines expressing GFP or GFP-Stx4-FL were also generated. Co-immunoprecipitations were done using GFP cells and GFP–Stx4–N-term cells lysed in situ. GFP immunoprecipitates from GFP–Stx4–N-term cells were found to contain a GFP-tagged protein with a molecular mass of ∼31 kDa, as predicted for the GFP–Stx4–N-term construct. These immunoprecipitates also contained Munc18c, consistent with the association of Munc18c and the N-terminal peptide construct (Fig. 3A). To assess whether the GFP–Stx4–N-term construct was able to compete with full-length Stx4 for binding to Munc18c, parental MDA-MB-231 cells and GFP–Stx4–N-term stable cells were transiently transfected with Stx4-FL-3xFLAG. Cells were lysed 24 h after transfection, and FLAG was immunoprecipitated (Fig. 3B). In GFP–Stx4–N-term stable cells, a 62.45% ± 6.65% decrease in the amount of Munc18c co-immunoprecipitated compared with parental cells was observed (Fig. 3C).
Figure 3.

Association of Stx4 N-terminal peptide reduces endogenous Munc18c binding to Stx4-FL 3xFLAG. A, parental MDA-MB-231 cells, GFP-expressing stable cells, and GFP–Stx4–N-term–expressing stable cells were lysed, and GFP was immunoprecipitated. Immunoprecipitates (IP) were probed for Munc18c (arrow). B+A, beads plus antibody; FLAG IgG, beads plus mouse anti-FLAG antibody plus GFP–Stx4–N-term peptide lysate; Rabbit IgG, rabbit IgG plus Stx4 N-terminal peptide lysate. B, parental and stable cells expressing GFP–Stx4–N-term were transiently transfected with Stx4-FL 3xFLAG for 24 h and lysed, and FLAG was immunoprecipitated. Immunoprecipitates were probed for Munc18c and FLAG (arrow). B+L, beads plus lysate from parental cells. C, densitometry of the amount of Munc18c co-immunoprecipitated relative to Stx4-FL 3xFLAG as shown in B. All data are presented as percent of control ± S.D. The asterisk denotes values significantly different from control (parental cells) (*, p < 0.05). All data represent three or more biological replicates with at least three technical replicates.
To determine whether expression of GFP–Stx4–N-term inhibits endogenous Stx4 from forming cognate SNARE complexes, we immunoprecipitated SNAP23 from cells stably expressing GFP–Stx4–N-term and observed nearly undetectable amounts of Stx4 associated with SNAP23 compared with cells expressing GFP-Stx4-FL or non-transfected MDA-MB-231 control cells (Fig. 4, A and B). VAMP2 was also immunoprecipitated from GFP-Stx4-FL and GFP–Stx4–N-term stable cells, and co-immunoprecipitation of endogenous Stx4 was assessed (Fig. 4C). GFP–Stx4–N-term stable cells showed a decrease of 93.25% ± 24.11% in the amount of Stx4 that was co-immunoprecipitated with VAMP2 compared with GFP-Stx4-FL cells (Fig. 4D).
Figure 4.

Association of Stx4 N-terminal peptide with Munc18c inhibits Stx4–SNAP23 and Stx4–VAMP2 interaction. A, parental MDA-MB-231 cells, and stable cells expressing either GFP-Stx4-FL or GFP–Stx4–N-term were lysed, and SNAP23 was immunoprecipitated. Immunoprecipitates (IP) were probed for Stx4 (arrow) and SNAP23. B+A, beads plus antibody; B+L, beads plus lysate from parental cells. B, densitometry of the amount of Stx4 co-immunoprecipitated relative to SNAP23 as shown in A. C, parental MDA-MB-231 cells and stable cells expressing either GFP-Stx4-FL or GFP–Stx4–N-term were lysed, and VAMP2 was immunoprecipitated. Immunoprecipitates were probed for Stx4 and VAMP2. D, densitometry of the amount of Stx4 co-immunoprecipitated relative to VAMP2 as shown in C. All data are presented as percent of control ± S.D. Asterisks denote values significantly different from control (parental cells) (*, p < 0.05). All data represent three or more biological replicates with at least three technical replicates.
Expression of Stx4 N-terminal peptide impairs invadopodium formation and gelatin degradation
Having observed inhibition of Stx4-SNAP23 complex formation caused by expression of GFP–Stx4–N-term, the effect of transient expression of this construct on invadopodium formation was examined. Overexpression of GFP–Stx4–N-term reduced the number of cells forming invadopodia by 65.1% ± 1.3% (Fig. 5, A and B). Transfection of cells with the N terminus of Stx3 did not alter invadopodium formation (Fig. 5B). To determine whether expression of the Stx4 N-terminal peptide and Munc18c knockdown were affecting the same pathway during invadopodium formation, we co-transfected cells with GFP–Stx4–N-term and Munc18c siRNA. Co-transfection did not cause a significantly different change in invadopodium formation compared with transfections with either the GFP–Stx4–N-term or Munc18c siRNA alone (Fig. 5B). ECM degradation was examined by transfecting cells with GFP, GFP-Stx4-FL, or GFP–Stx4–N-term for 24 h and then plating onto fluorescent gelatin for 24 h. Relative to GFP-expressing cells, no significant change in gelatin degradation was seen for GFP-Stx4-FL–expressing cells. GFP–Stx4–N-term–expressing cells exhibited a 62.2% ± 2.0% decrease in gelatin degradation compared with GFP-Stx4-FL–expressing cells (Fig. 5C).
Figure 5.

Stx4 N-terminal peptide impairs invadopodium formation and gelatin degradation. A, invadopodium-based degradation of gelatin by cells transfected with GFP (Control), GFP-Stx4-FL, and GFP–Stx4–N-term. Cells were transfected for 20 h, seeded onto fluorescent gelatin for 4 h, and then fixed, permeabilized, stained for F-actin, and analyzed by confocal microscopy. Scale bars = 10 μm. B, quantification of invadopodium formation. Cells were transfected with the following: GFP alone, GFP-Stx4-FL, GFP–Stx4–N-term, GFP-Stx3 N-terminal peptide, GFP + control siRNA, GFP + Munc18c siRNA, GFP-Stx4-FL + siRNA Control, GFP-Stx4-FL + siRNA Munc18c, GFP–Stx4–N-term + siRNA control, and GFP–Stx4–N-term + Munc18c siRNA. Cells were transfected for 44 h and then seeded onto fluorescent gelatin and processed as in A. Cells with F-actin puncta overlying dark spots of gelatin degradation were counted as cells forming invadopodia. Percentages of cells forming invadopodia, normalized to GFP alone, were determined by counting 50 cells/sample. C, cells expressing either GFP, GFP-Stx4-FL, or GFP–Stx4–N-term were transfected for 24 h, seeded onto fluorescent gelatin for 24 h, and then fixed. Parental cells and cells expressing GFP were analyzed for dark areas of degradation and scored as described under “Experimental procedures.” All data are presented as percent of control ± S.D. Asterisks denote values significantly different from control (*, p < 0.05). All data represent three or more biological replicates with at least three technical replicates.
Stable cell lines were also used to assess invadopodium formation, and similar results were found. Relative to parental MDA-MB-231 cells, no significant change in invadopodium formation was observed for GFP or GFP-Stx4-FL cell lines. The GFP–Stx4–N-term cell line displayed a 50.7% ± 5.4% decrease in invadopodium formation (Fig. 6, A and B). In gelatin degradation assays, no significant difference was observed between GFP or GFP-Stx4-FL cells. GFP–Stx4–N-term cells displayed a 74.4% ± 0.8% decrease in gelatin degradation compared with GFP-Stx4-FL control cells (Fig. 6C).
Figure 6.

Stable cell lines expressing Stx4 N-terminal peptide have reduced invadopodium formation and gelatin degradation. A, parental MDA-MB-231 cells and stable cell lines expressing GFP, GFP-Stx4-FL, or GFP–Stx4–N-term were seeded onto Alexa Fluor 594–labeled gelatin and incubated for 4 h prior to fixation, staining for F-actin, and analysis by confocal microscopy. Scale bars = 10 μm. B, quantification of invadopodium formation. Cells with F-actin puncta overlying dark spots of gelatin degradation were counted as cells forming invadopodia. Percentages of cells forming invadopodia are shown from three independent experiments in which 100 cells/sample were counted and normalized to parental MDA-MB-231 cells. C, quantification of gelatin degradation. Parental, GFP, GFP-Stx4-FL, and GFP–Stx4–N-term stable cells were plated onto fluorescent gelatin for 24 h and then fixed. Cells were analyzed for dark areas of degradation and scored as described under “Experimental Procedures.” Percentages of cells degrading gelatin are shown from three independent experiments in which 50 cells/sample were analyzed. All data are presented as percent of control ± S.D. Asterisks denote values significantly different from control (*, p < 0.05). All data represent three or more biological replicates with at least three technical replicates.
Expression of the Stx4 N-terminal peptide inhibits cellular invasion and trafficking of MT1-MMP and EGFR to the cell surface
Stx4 and SNAP23 have been shown previously to contribute to trafficking of MT1-MMP in MDA-MB-231 cells (11). Given the observed reduction in Stx4–SNAP23 and Stx4–VAMP2 interactions in cells expressing GFP–Stx4–N-term (Fig. 4B), we analyzed the trafficking of key regulators required for invadopodium formation and ECM invasion. The amount of MT1-MMP, EGFR, and β1 integrin at the plasma membrane during invadopodium formation was assessed by biotinylating cell surface proteins (Fig. 7, A and B). It was found that MT1-MMP, EGFR, and β1 integrin cell surface levels on GFP-Stx4-FL stable cells were reduced by 41.2% ± 0.1% and 24.7% ± 0.1%, respectively, compared with parental cells (Fig. 7B). Cell-surface MT1-MMP was further reduced by 40.0% ± 0.2%, and cell surface EGFR was further reduced by 48.1% ± 0.1% in cells stably expressing GFP–Stx4–N-term compared with those expressing GFP-Stx4-FL. The levels of β1 integrin at the cell surface, although variable to some extent, were not found to be significantly different in any of the cell lines. Given that invadopodium formation, gelatin degradation, and trafficking of MT1-MMP and EGFR were decreased in cells stably expressing GFP–Stx4–N-term, the ability of these cells to invade was examined using Matrigel-based invasion assays. Cells expressing GFP–Stx4–N-term exhibited a 77.0% ± 2.9% reduction in invasion compared with cells expressing GFP-Stx4-FL (Fig. 7C). No significant difference was found between parental MDA-MB-231 cells, GFP cells, and GFP-Stx4-FL cells.
Figure 7.

Stx4 N-terminal peptide expression reduces cell surface levels of MT1-MMP and EGFR as well as cellular invasion. A, parental MDA-MB-231 cells and stable cell lines expressing GFP, GFP-Stx4-FL, or GFP–Stx4–N-term were plated onto gelatin for 4 h, exposed to biotin, and then lysed and analyzed by precipitation with streptavidin beads. Cell surface protein levels of β1 integrin, MT1-MMP, EGFR, and β-tubulin were assessed by Western blotting. Parental biotin −ve indicates cells exposed to buffer only. B, densitometric analysis of the amount of β1 integrin, EGFR, and MT1-MMP in streptavidin precipitations, as in A, relative to control. C, parental, GFP, GFP-Stx4-FL, and GFP–Stx4–N-term stable cells were serum-starved for 24 h, seeded onto transwell membranes coated with Matrigel or uncoated membranes (control), and allowed to invade for 20 h. Percentages of cells invading are shown from experiments in which at least 10 fields of view were counted per treatment. All data represent percent of control ± S.D. Asterisks denote values significantly different from control (*, p < 0.05). All data represent three or more biological replicates with at least three technical replicates.
Inhibition of expression and function of Munc18c shows a conserved phenotype between tumor cell lines
To determine whether similar phenotypes are seen in other invasive cell lines, the effect of siRNA knockdown and inhibition of Munc18c on invadopodium formation and gelatin degradation was performed in HT-1080 fibrosarcoma cells. Immunoprecipitation of Munc18c from HT-1080 cells revealed co-immunoprecipitation of Stx4 but not Stx3 (Fig. 8A). HT-1080 cells were then transfected with a pool of siRNA targeting human Munc18c or nonspecific control siRNA (Fig. 8B), and clear knockdown of Munc18c expression was observed after 48 h (Fig. 8, B and C). Invadopodium formation was then analyzed, and the number of cells forming invadopodia was reduced by 73.0% ± 10.5% in samples with reduced Munc18c expression (Fig. 8D). Consistent with this observation, expression of GFP–Stx4–N-term caused a decrease (44.79% ± 14.06%) in invadopodium formation compared with cells expressing GFP-Stx4-FL (Fig. 8E). Inhibiting Munc18c through expression of GFP–Stx4–N-term in HT-1080 cells also decreased gelatin degradation by 57.76% ± 5.00% compared with GFP-Stx4-FL control cells (Fig. 8F).
Figure 8.
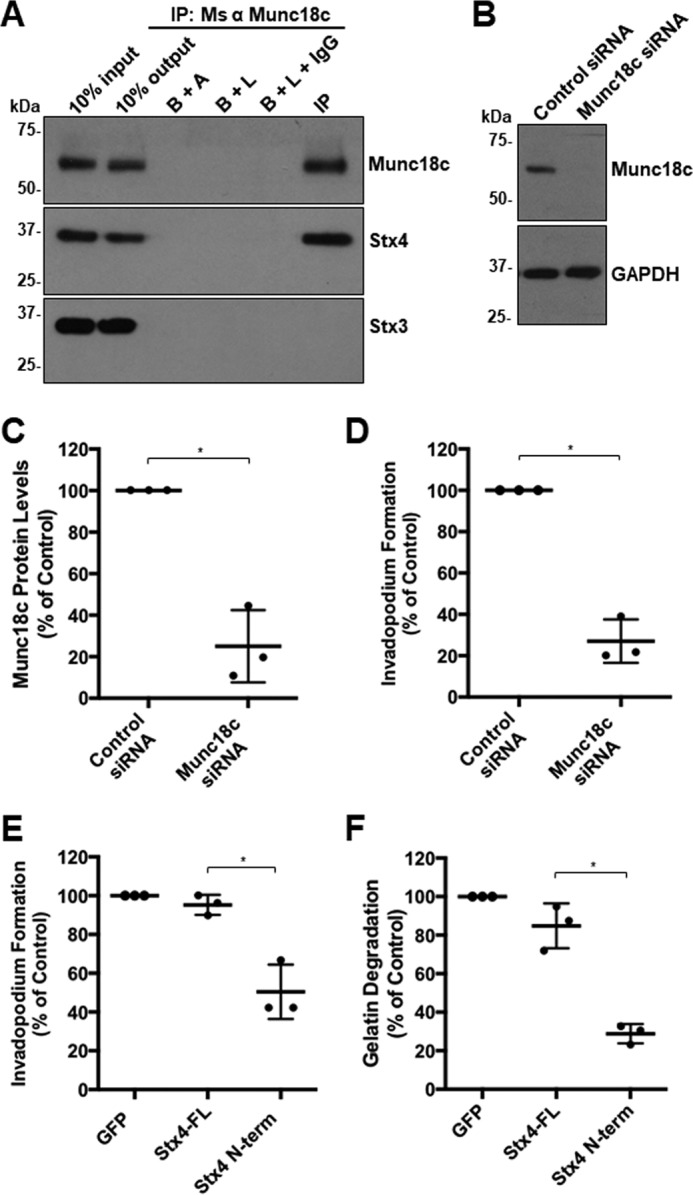
Inhibition of expression and function of Munc18c shows a conserved phenotype in HT-1080 cells. A, Munc18c immunoprecipitates (IP) were probed for Munc18c and Stx4. B+A, beads plus antibody; B+L, beads plus lysate; B+L+IgG, beads plus lysate plus unrelated IgG. B, HT-1080 cells were transfected with siRNA targeting Munc18c or nonspecific control siRNA, lysed, and analyzed for Munc18c by SDS-PAGE/Western blotting. C, quantification of Munc18c knockdown normalized to GAPDH. D, quantification of invadopodium formation. Cells were transfected with siRNA targeting Munc18c or nonspecific control siRNA for 44 h and then plated onto fluorescent gelatin for 4 h. Cells with F-actin puncta overlying dark spots of gelatin degradation were counted as cells forming invadopodia. Percentages of cells forming invadopodia are shown from three independent experiments in which 50 cells/sample were counted and normalized to parental MDA-MB-231 cells. E, invadopodium-based degradation of Alexa Fluor 594 gelatin by cells transfected with GFP (control), GFP-Stx4-FL, or GFP–Stx4–N-term. Cells were transfected for 20 h, seeded onto fluorescent gelatin for 4 h, and then fixed, permeabilized, stained for F-actin, and analyzed by confocal microscopy. Percentages of cells forming invadopodia are shown from three independent experiments in which 50 cells/sample were counted and normalized to GFP-expressing cells. F, cells were transfected with GFP (control), GFP-Stx4-FL, or GFP–Stx4–N-term for 24 h, plated onto fluorescent gelatin for 24 h, and then fixed. Cells were analyzed for dark areas of degradation and scored as described under “Gelatin degradation assay.” Percentages of cells degrading gelatin are presented as the mean ± S.D. from three independent experiments in which 50 cells/sample were analyzed. Asterisks denote values significantly different from control (*, p < 0.05). All treatments are a percentage of GFP alone, which was used as the control group. All data represent three or more biological replicates with at least three technical replicates.
Discussion
SNARE-mediated trafficking is important for several aspects of tumor cell invasion (10–13), including invadopodium formation (8, 9), cell migration (14–18), and MMP-directed ECM degradation (19). Although SNAREs have been shown to play important roles in these processes, there remains much to be understood about how SNARE function is regulated during tumor cell invasion. Previous research has described the SM protein Munc18c as both a negative (32, 33) and positive regulator of its binding partner Stx4 (25, 31, 34). Here we demonstrate that Munc18c contributes to invadopodium formation and cell invasion in MDA-MB-231 and HT-1080 cells by facilitating Stx4-mediated trafficking. Stx4-mediated membrane traffic is required for delivery of MT1-MMP and EGFR to the plasma membrane and can thus support modulation of ECM interactions during tumor cell invasion. SM proteins form interactions with their cognate syntaxins and play an important role in regulating membrane fusion (21). Munc18c and Stx4 have been shown to associate in some cell lines (e.g. 3T3-L1 adipocytes) (20, 25); however, their interaction has not been well studied in cancer cell lines. Our findings suggest that the interaction of Munc18c and Stx4 is a novel and important point of regulation in the control of MT1-MMP-mediated ECM degradation by invasive tumor cells.
The observed distribution of Stx4 in MDA-MB-231 cells is consistent with its function at the plasma membrane and possibly on membrane-bound compartments that contribute to the transport of material to and from the cell surface (Fig. 1B). Analyses of the distributions of Munc18c and Stx4 revealed that these two proteins partly co-localize in cells at rest or forming invadopodia. Co-immunoprecipitation of Munc18c and Stx4 was enhanced in cells forming invadopodia, compared with non-invading cells, without an obvious change in the co-localization of the proteins. This suggests that Munc18c and Stx4 might interact in a manner that increases in affinity during invadopodium formation. Munc18c and Stx4 were found to co-immunoprecipitate; however, in some cases we did observe a small increase in the molecular weight of the Munc18c bound to Stx4. It is possible that this represents a posttranslationally modified form of Munc18c, which could influence its affinity for Stx4, and future studies are planned to explore this.
Previous studies have shown that inhibition or RNAi-mediated knockdown of Munc18c impairs Stx4-mediated exocytic events such as GLUT4 translocation (25, 27). Here, RNAi-mediated knockdown of Munc18c was observed to significantly inhibit invadopodium formation in MDA-MB-231 and HT-1080 cells, similar to that observed when Stx4 was knocked down by siRNA (11). Together, the observations support a model wherein Stx4-mediated trafficking to the plasma membrane facilitates the delivery of proteins required for invadopodium formation in a manner that is regulated, in part, by Munc18c. How Munc18c functions to regulate Stx4 function is not clearly understood. Studies have demonstrated that Munc18c binds to the N terminus of Stx4, which may place Stx4 into a more open conformation that facilitates SNARE complex assembly (31). Consistent with this, the binding of Munc18c to Stx4 has been found to involve Phe-119 of Munc18c and accelerate the formation of SNARE complexes (31). There is evidence that Munc18c can associate with other SNAREs but may only do so when interacting with the N-terminal domain of Stx4 (31, 35). Therefore, it is reasonable to predict that overexpression of the N-terminal 29 amino acids of Stx4 could compete for binding of Stx4 to Munc18c, and it has been shown that this polypeptide can reduce the interaction of recombinant Munc18c and Stx4 in pulldown experiments (31). Here we expressed a GFP-tagged version of an Stx4 N-terminal peptide in MDA-MB-231 cells and observed perturbations of the Stx4–SNAP23 interaction, Stx4–VAMP2 interaction, MT1-MMP and EGFR trafficking, invadopodium formation, and cell invasion. Our observations of reduced interaction between SNAP23 and Stx4, as well as Stx4 and VAMP2, resulting from expression of the Stx4 N-terminal peptide are consistent with previous findings that the Munc18c–Stx4 interaction promotes complex formation involving Stx4, VAMP2, and SNAP23 (31). Furthermore, experiments performed in HT-1080 cells revealed that regulation of Stx4 by Munc18c is conserved between two invasive cancer cell lines. Studies to examine the effect of Stx4 N-terminal peptide expression in primary cells (for example, using a Tat-tagged construct) can now be undertaken to further our understanding of how Stx4 contributes to cell invasion.
It has been demonstrated previously that SNAREs are involved in the trafficking of key invadopodial proteins to the cell surface, including EGFR (12) and MT1-MMP (11). Here we find that the regulation of Stx4 by Munc18c is required for MT1-MMP and EGFR trafficking to the cell surface and that disruption of this contributes to decreased invadopodium formation and cell invasion. This is consistent with a role for Stx4 in invadopodium formation and cell invasion, as determined previously by siRNA knockdown of Stx4 (11). It has also been shown that a significant decrease in EGFR levels at the cell surface results when SNAP23 and syntaxin13 function are impaired (12), which is also consistent with our findings. Decreases in MT1-MMP and EGFR at the cell surface were observed in cells stably expressing GFP-Stx4-FL, but these changes did not correlate with alteration of invadopodium formation or cell invasion.
Over the past decade it has become clear that trafficking of MMPs and adhesion molecules has an essential role in the remodeling of the ECM during tumor cell invasion, facilitating tumor cell movement through interstitial tissues. The model that has emerged involves formation of invadopodia and subsequent matrix degradation by invasive tumor cells through coordinated cytoskeletal rearrangements and membrane trafficking. Although membrane traffic mediated by SNAREs, exocyst complexes, and other components of the membrane fusion machinery is crucial to tumor cell invasion (8), the direct roles of regulators of SNARE function in cell invasion remain to be defined. To our knowledge, our findings represent the first direct evidence of an SM family member regulating SNARE function during tumor cell invasion. Previous studies have identified gelsolin as an interaction partner of Stx4 (36), and gelsolin has been shown separately to have a role during invadopodium formation (37). Whether the interaction between gelsolin and Stx4 influences invadopodium formation and cell invasion is not known and remains to be studied. Our findings implicate the interaction of Stx4 and Munc18c in trafficking of MT1-MMP and EGFR, and this advances our understanding of the SNARE-mediated mechanisms that coordinate the localization of proteins that drive tumor cell invasion.
Experimental procedures
Reagents and cDNA constructs
Reagents and chemicals were purchased from either Fisher Scientific Ltd. (Nepean, ON, Canada) or Sigma-Aldrich Co. (St. Louis, MO) unless otherwise indicated. Antibodies were purchased from the following suppliers: rabbit anti-VAMP2 (Affinity Bioreagents, PA1 766); rabbit anti-MMP14, mouse anti-MMP14, rabbit anti-GFP, and rabbit anti-Munc18c (Abcam, ab3644, ab78738, ab290, and ab175238, respectively); mouse anti-Munc18c, mouse anti-SNAP23, mouse anti-syntaxin3, and rabbit anti-EGFR (Santa Cruz Biotechnology, sc-373813, sc-166244, sc-393518, and sc-03, respectively); mouse anti-syntaxin4 (BD Biosciences, 610439); rabbit anti-syntaxin4 (Alomone Labs, ANR-004); mouse anti-actin (Pierce, MA5-15739); mouse anti-β1 integrin, mouse anti-β-tubulin, and mouse anti-GAPDH (Developmental Hybridoma Studies Bank, P4C10, E7-s, and DSHB-hGAPDH-2G7, respectively); and mouse anti-FLAG and mouse anti β-actin, (Sigma-Aldrich, F3165 and A2228, respectively). All fluorescently labeled secondary antibodies, Hoechst 33342, and Alexa Fluor 647–conjugated phalloidin were purchased from Life Technologies (Mississauga, ON, Canada). Munc18c and control siRNA were purchased from Santa Cruz Biotechnologies (SC-42312 and SC-37007, respectively).
GFP-Stx4-FL is a construct of wild-type Stx4 with GFP fused to the C terminus. The GFP moiety is lumenal or extracellular when at the plasma membrane. The Stx4–N-terminal peptide is soluble. The GFP-Stx4-FL construct was cloned from a syntaxin4-myc-myc-His plasmid purchased from Addgene (plasmid 12377) into pEGFP-N1 using XhoI and KpnI. The following oligonucleotides were used as primers: forward Stx4-FL (5′-TTCACCTCGAGATGCGGGACAGGACCCAC-3′) and reverse Stx4-FL (5′-TTCAGGTACCTCCAACCACTGTGACGCCAATG-3′). Stx4-FL 3xFLAG was PCR-amplified from pEGFP-N1-Stx4-FL (above) and cloned into pcDNA3.1 3xFLAG using HindIII and KpnI. The following oligonucleotides were used as primers: forward Stx4-FL 3xFLAG (5′-TTCACAAGCTTATGCGGGACAGGACCCAC-3′) and the same reverse primer as used for GFP-Stx4-FL (above). N-terminal peptide from Stx4 was PCR-amplified from pEGFP-N1-Stx4-FL (above) and cloned into pEGFP-N1 using XhoI and KpnI to create the Stx4 N-terminal peptide fused to GFP (GFP–Stx4–N-term). The following oligonucleotides were used as primers: forward Stx4 N-terminal (5′-TGACGGTAAATGGCCCGCCTGGCATTATG-3′) and reverse Stx4 N-terminal (5′-TTTATCATTGGTACCGGGTGCACCACCAGCGCG-3′). Expression of GFP-Stx4-FL and GFP–Stx4–N-term were compared with endogenous Stx4 by immunofluorescence microscopy (supplemental Fig. 1). Expression of Stx4-FL 3xFLAG in parental and GFP–Stx4–N-term stable cells was determined by immunofluorescence microscopy (supplemental Fig. 2).
The N-terminal peptide from Stx3 was cloned from the syntaxin3-myc-myc-His plasmid purchased from Addgene (plasmid 12372) and cloned into pEGFP-N1 using HindIII and KpnI to create the plasmid Stx3 N-terminal peptide fused to GFP. The following oligonucleotides were used as primers: forward Stx3 N-terminal (5′-TTCACAAGCTTATGAAGGACCGTCTGGAGCAGCTGAAGG-3′) and reverse Stx3 N-terminal (5′-TTCACGGTACCCGTGTTGTCGATAGCAATCTCAACCG-3′).
Cell culture and transfection
MDA-MB-231 and HT-1080 cells were obtained from the American Type Culture Collection (Manassas, VA) and cultured in DMEM supplemented with 10% BCS. Stable cell lines derived from MDA-MB-231 cells were cultured in selection medium comprising DMEM supplemented with 10% BCS and 200 μg/ml G418 (BioShop). Growth conditions were kept at 37 °C with humidity and a 5% CO2 atmosphere. Cells were lifted by using 5 mm EDTA/PBS (pH 7.4). For all experiments, cells were used between 5 and 20 passages. All cells were passaged at most 24 h before each experiment.
Cells were transfected using jetPRIME Polyplus (VWR International) according to the protocol of the manufacturer. All transiently transfected constructs were expressed for at least 24 h. Cells were transfected with 50 nm siRNA and underwent knockdown for 48 h. Co-transfections were performed using a 1:10 molar ratio of marker pEGFP-N1 plasmid to siRNA for a total of 48 h.
Creation of stable cell lines
MDA-MB-231 cells were transfected with either GFP-Stx4-FL, GFP–Stx4–N-term, or pEGFP-N1 alone (GFP control). After 24 h, cells were lifted in selection medium and split at a ratio of 1:4. When distinct colonies had formed and cells in the control non-transfected plate died, 18 separate colonies were lifted using a P200 pipette tip and reseeded. When confluent, each colony-derived population of cells was split, and Western blot analysis was used to confirm expression. The cell lines that indicated the highest level of expression were propagated. Cells were also viewed under epifluorescence to confirm that all cells were GFP-positive. Equal expression between cell lines was determined by Western blotting (supplemental Fig. 3A) and epifluorescence microscopy (supplemental Fig. 3B).
Invadopodium formation assay
Invadopodium formation was performed as described previously (11). Glass coverslips were coated with 50 μg/ml PLL/PBS, followed by cross-linking with 0.5% gluteraldehyde/PBS. Coverslips were then inverted onto 70 μl of Alexa Fluor 594–labeled gelatin. The coated coverslips were then incubated with 5 mg/ml NaBH3/PBS and subsequently washed with PBS. All images of cells plated on fluorescent gelatin for invadopodium formation (Figs. 2, A and E, 5A, and 6A) were incubated on gelatin for 8 h instead of 4 h to allow for degradation that could be easily captured by confocal microscopy. Tissue culture plates were coated similarly, the exception being that plates were coated with 0.2% unlabeled gelatin/PBS.
Co-immunoprecipitation
Antibodies were coupled to protein G Dynabeads (Invitrogen) or cyanogen bromide–activated Sepharose beads (Sigma) according to the instructions of the manufacturer. Cells were lysed in situ with cold lysis buffer comprising 1% Nonidet P-40, 10% glycerol, 0.5% sodium deoxycholate, 137 mm NaCl, 20 mm Tris-HCl (pH 8.0), 10 mm NaF, 10 mm Na2P4O7, 0.2 mm Na3VO4, and protease inhibitor mixture (Sigma). Lysate was incubated with antibody-bound protein G Dynabeads for 1 h at 4 °C, washed three times with cold PBS, and eluted with 2.5× Laemmli loading buffer heated to 70 °C. Alternatively, lysate was incubated with antibody-bound Sepharose beads overnight at 4 °C and washed three times with cold lysis buffer. Bound proteins were eluted using 2.5× Laemmli loading buffer heated to 100 °C. Proteins were separated using SDS-PAGE and analyzed by Western immunoblotting.
Confocal immunofluoresence microscopy
Cells were either grown on glass coverslips for 4 h or seeded onto 0.2% gelatin-coated coverslips (as described under “Invadopodium formation assay”). Cells were fixed in 4% paraformaldehyde/PBS and then washed in 150 mm glycine/PBS at room temperature or overnight at 4 °C with gentle agitation. Cells were permeabilized in 0.1% Triton X-100/PBS and blocked in 5% BSA/PBS prior to staining with primary and secondary antibody. Samples analyzed by confocal microscopy were imaged through a 63× (numerical aperture 1.4) oil immersion lens using a Leica DM-IRE2 inverted microscope with a TCS SP2 scanning head (Leica, Heidelberg, Germany). Images were captured using Leica confocal software. All images were processed and analyzed using ImageJ software (National Institutes of Health, Bethesda, MD). Alexa Fluor 594–labeled gelatin images were converted from red to white using ImageJ to improve the contrast of areas of degradation.
Cell surface protein labeling
Cells were plated onto 0.2% gelatin (as described under “Invadopodium formation assay”) for 4 h. Cells were then washed with cold PBS once and incubated with 0.5 mg/ml Sulfo-NHS-SS-Biotin (APExBIO) dissolved in 10 mm boric acid and 150 mm NaCl (pH 8.0) at 4 °C with occasional agitation. Plates where then washed with 15 mm glycine/PBS and lysed using 1% Nonidet P-40, 10% glycerol, 0.5% NaDOC, 137 mm NaCl, 20 mm Tris-HCl (pH 8.0), 10 mm NaF, 10 mm Na2P4O7, 0.2 mm Na3VO4,and protease inhibitor mixture. Lysate was incubated with streptavidin-agarose beads (Novex, Thermo Fisher Scientific, a kind gift from Dr. Nina Jones, University of Guelph) overnight at 4 °C. Beads were then washed three times with cold lysate buffer. Bound products were eluted using 2.5× Laemmli loading buffer heated to 100 °C.
Gelatin degradation assays
Gelatin degradation assays were performed as described previously (38). Briefly, coverslips were coated with Alexa Fluor 594–labeled gelatin as described under “Invadopodium formation assay.” Cells were seeded at 30% confluency and incubated for 24 h. Degradation areas made by transfected cells were counted and scored as the percentage of area degraded per cell (+1 for fully degraded, +0.5 for partially degraded, and 0 for no degradation).
Cell invasion assay
Cell culture inserts were prepared as described previously (12). Briefly, the bottoms of transwell inserts (8-μm pore diameter, Corning Inc.) were coated with 20 μg/ml fibronectin/PBS and the tops with 0.125 mg/ml Matrigel (BD Biosciences). Parental and stable cells were serum-starved for 24 h, lifted, seeded into chambers, and allowed to invade for 20 h. The cells that invaded toward the lower chamber (10% BCS/0.1% BSA in DMEM) were fixed with 4% paraformaldehyde, stained with Hoechst, and counted. Cells that did not invade were removed with a cotton swab prior to fixation of the sample. Ten fields of cells per membrane were counted per treatment. Data are presented as percent of control.
Statistical analysis
The percent of controls for three experimental replicates is shown, with error bars representing the standard deviation. The horizontal bar for each treatment indicates the mean, in all graphs, unless otherwise indicated. For all experiments, each experimental group was compared with its respective control, vehicle, or wild-type treatment by Student's t test, with a statistical significance threshold of p = 0.05. Treatments that differed significantly from the control (p < 0.05) are indicated by an asterisk in the figures. All statistical analysis was done using Microsoft Excel. Graphs were prepared using GraphPad Prism version 7.0 (GraphPad Software, La Jolla, CA).
Author contributions
M. I. B. and D. M. M. planned and conducted experiments, prepared figures, and wrote and revised the manuscript. O. R. G. conducted experiments, prepared figures, and revised the manuscript. A. H. and E. S. S. conducted experiments. J. U. planned experiments and revised the manuscript. M. G. C. planned experiments and wrote and revised the manuscript.
Supplementary Material
Acknowledgment
We thank Esther Matus for work on supplemental Fig. 2.
This work was supported by the Natural Sciences and Engineering Research Council of Canada. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. 1–3.
- ECM
- extracellular matrix
- MMP
- matrix metalloproteinase
- SM
- Sec1/Munc18
- EGFR
- EGF receptor
- PLL
- poly-l-lysine
- FL
- full-length
- BCS
- bovine calf serum.
References
- 1. Hanahan D., and Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 [DOI] [PubMed] [Google Scholar]
- 2. Bravo-Cordero J. J., Hodgson L., and Condeelis J. (2012) Directed cell invasion and migration during metastasis. Curr. Opin. Cell Biol. 24, 277–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Murphy D. A., and Courtneidge S. A. (2011) The “ins” and “outs” of podosomes and invadopodia: characteristics, formation and function. Nat. Rev. Mol. Cell Biol. 12, 413–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Artym V. V., Swatkoski S., Matsumoto K., Campbell C. B., Petrie R. J., Dimitriadis E. K., Li X., Mueller S. C., Bugge T. H., Gucek M., and Yamada K. M. (2015) Dense fibrillar collagen is a potent inducer of invadopodia via a specific signaling network. J. Cell Biol. 208, 331–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tolde O., Rösel D., Veselý P., Folk P., and Brábek J. (2010) The structure of invadopodia in a complex 3D environment. Eur. J. Cell Biol. 89, 674–680 [DOI] [PubMed] [Google Scholar]
- 6. Clark E. S., Brown B., Whigham A. S., Kochaishvili A., Yarbrough W. G., and Weaver A. M. (2009) Aggressiveness of HNSCC tumors depends on expression levels of cortactin, a gene in the 11q13 amplicon. Oncogene 28, 431–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lohmer L. L., Kelley L. C., Hagedorn E. J., and Sherwood D. R. (2014) Invadopodia and basement membrane invasion in vivo. Cell Adhes. Migr. 8, 246–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Poincloux R., Lizárraga F., and Chavrier P. (2009) Matrix invasion by tumour cells: a focus on MT1-MMP trafficking to invadopodia. J. Cell Sci. 122, 3015–3024 [DOI] [PubMed] [Google Scholar]
- 9. Chen Y. A., and Scheller R. H. (2001) SNARE-mediated membrane fusion. Nat. Rev. Mol. Cell Biol. 2, 98–106 [DOI] [PubMed] [Google Scholar]
- 10. Steffen A., Le Dez G., Poincloux R., Recchi C., Nassoy P., Rottner K., Galli T., and Chavrier P. (2008) MT1-MMP-dependent invasion is regulated by TI-VAMP/VAMP7. Curr. Biol. 18, 926–931 [DOI] [PubMed] [Google Scholar]
- 11. Williams K. C., McNeilly R. E., and Coppolino M. G. (2014) SNAP23, Syntaxin4, and vesicle-associated membrane protein 7 (VAMP7) mediate trafficking of membrane type 1-matrix metalloproteinase (MT1-MMP) during invadopodium formation and tumor cell invasion. Mol. Biol. Cell. 25, 2061–2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Williams K. C., and Coppolino M. G. (2014) SNARE-dependent interaction of Src, EGFR and β1 integrin regulates invadopodia formation and tumor cell invasion. J. Cell Sci. 127, 1712–1725 [DOI] [PubMed] [Google Scholar]
- 13. Kean M. J., Williams K. C., Skalski M., Myers D., Burtnik A., Foster D., and Coppolino M. G. (2009) VAMP3, syntaxin-13 and SNAP23 are involved in secretion of matrix metalloproteinases, degradation of the extracellular matrix and cell invasion. J. Cell Sci. 122, 4089–4098 [DOI] [PubMed] [Google Scholar]
- 14. Tayeb M. A., Skalski M., Cha M. C., Kean M. J., Scaife M., and Coppolino M. G. (2005) Inhibition of SNARE-mediated membrane traffic impairs cell migration. Exp. Cell Res. 305, 63–73 [DOI] [PubMed] [Google Scholar]
- 15. Riggs K. A., Hasan N., Humphrey D., Raleigh C., Nevitt C., Corbin D., and Hu C. (2012) Regulation of integrin endocytic recycling and chemotactic cell migration by syntaxin 6 and VAMP3 interaction. J. Cell Sci. 125, 3827–3839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Day P., Riggs K. A., Hasan N., Corbin D., Humphrey D., and Hu C. (2011) Syntaxins 3 and 4 mediate vesicular trafficking of α5β1 and α3β1 integrins and cancer cell migration. Int. J. Oncol. 39, 863–871 [DOI] [PubMed] [Google Scholar]
- 17. Veale K. J., Offenhäuser C., Lei N., Stanley A. C., Stow J. L., and Murray R. Z. (2011) VAMP3 regulates podosome organisation in macrophages and together with Stx4/SNAP23 mediates adhesion, cell spreading and persistent migration. Exp. Cell Res. 317, 1817–1829 [DOI] [PubMed] [Google Scholar]
- 18. Proux-Gillardeaux V., Gavard J., Irinopoulou T., Mège R. M., and Galli T. (2005) Tetanus neurotoxin-mediated cleavage of cellubrevin impairs epithelial cell migration and integrin-dependent cell adhesion. Proc. Natl. Acad. Sci. U.S.A. 102, 6362–6367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miyata T., Ohnishi H., Suzuki J., Yoshikumi Y., Ohno H., Mashima H., Yasuda H., Ishijima T., Osawa H., Satoh K., Sunada K., Kita H., Yamamoto H., and Sugano K. (2004) Involvement of syntaxin 4 in the transport of membrane-type 1 matrix metalloproteinase to the plasma membrane in human gastric epithelial cells. Biochem. Biophys. Res. Commun. 323, 118–124 [DOI] [PubMed] [Google Scholar]
- 20. Yu H., Rathore S. S., Lopez J. A., Davis E. M., James D. E., Martin J. L., and Shen J. (2013) Comparative studies of Munc18c and Munc18–1 reveal conserved and divergent mechanisms of Sec1/Munc18 proteins. Proc. Natl. Acad. Sci. U.S.A. 110, 3271–3280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Carr C. M., and Rizo J. (2010) At the junction of SNARE and SM protein function. Curr. Opin. Cell Biol. 22, 519–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Südhof T. C., and Rothman J. E. (2009) Membrane fusion: grappling with SNARE and SM proteins. Science 323, 474–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gurung P., Lukens J. R., and Kanneganti T. (2016) A direct role for the Sec1-Munc18c-family protein Vps33 as a template for SNARE assembly. Science 21, 193–201 [Google Scholar]
- 24. Tellam J. T., McIntosh S., and James D. E. (1995) Molecular identification of two novel Munc-18 isoforms expressed in non-neuronal tissues. J. Biol. Chem. 270, 5857–5863 [DOI] [PubMed] [Google Scholar]
- 25. Thurmond D. C., Kanzaki M., Khan A. H., and Pessin J. E. (2000) Munc18c function is required for insulin-stimulated plasma membrane fusion of GLUT4 and insulin-responsive amino peptidase storage vesicles. Mol. Cell. Biol. 20, 379–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhu D., Xie L., Karimian N., Liang T., Kang Y., Huang Y. C., and Gaisano H. Y. (2015) Munc18c mediates exocytosis of pre-docked and newcomer insulin granules underlying biphasic glucose stimulated insulin secretion in human pancreatic β-cells. Mol. Metab. 4, 418–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jewell J. L., Oh E., Ramalingam L., Kalwat M. A., Tagliabracci V. S., Tackett L., Elmendorf J. S., and Thurmond D. C. (2011) Munc18c phosphorylation by the insulin receptor links cell signaling directly to SNARE exocytosis. J. Cell Biol. 193, 185–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brochetta C., Vita F., Tiwari N., Scandiuzzi L., Soranzo M. R., Guérin-Marchand C., Zabucchi G., and Blank U. (2008) Involvement of Munc18 isoforms in the regulation of granule exocytosis in neutrophils. Biochim. Biophys. Acta 1783, 1781–1791 [DOI] [PubMed] [Google Scholar]
- 29. Jewell J., Oh E., and Thurmond D. (2010) Exocytosis mechanisms underlying insulin release and glucose uptake: conserved roles for Munc18c and syntaxin 4. Am. J. Physiol. 298, 517–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kioumourtzoglou D., Gould G. W., and Bryant N. (2014) Insulin stimulates Syntaxin4 SNARE complex assembly via a novel regulatory mechanism. Mol. Cell Biol. 34, 1271–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Latham C. F., Lopez J. A., Hu S. H., Gee C. L., Westbury E., Blair D. H., Armishaw C. J., Alewood P. F., Bryant N. J., James D. E., and Martin J. L. (2006) Molecular dissection of the Munc18c/syntaxin4 interaction: implications for regulation of membrane trafficking. Traffic 7, 1408–1419 [DOI] [PubMed] [Google Scholar]
- 32. Tamori Y., Kawanishi M., Niki T., Shinoda H., Araki S., Okazawa H., and Kasuga M. (1998) Inhibition of insulin-induced GLUT4 translocation by Munc18c through interaction with Syntaxin4 in 3T3-L1 adipocytes. J. Biol. Chem. 273, 19740–19746 [DOI] [PubMed] [Google Scholar]
- 33. Thurmond D. C., Ceresa B. P., Okada S., Elmendorf J. S., Coker K., and Pessin J. E. (1998) Regulation of insulin-stimulated GLUT4 translocation by Munc18c in 3T3L1 adipocytes. J. Biol. Chem. 273, 33876–33883 [DOI] [PubMed] [Google Scholar]
- 34. Oh E., Spurlin B. A., Pessin J. E., and Thurmond D. C. (2005) Munc18c heterozygous knockout mice display increased susceptibility for severe glucose intolerance. Diabetes 54, 638–647 [DOI] [PubMed] [Google Scholar]
- 35. Peng R., and Gallwitz D. (2004) Multiple SNARE interactions of an SM protein: Sed5p/Sly1p binding is dispensable for transport. EMBO J. 23, 3939–3949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kalwat M. A., Wiseman D. A., Luo W., Wang Z., and Thurmond D. C. (2012) Gelsolin associates with the N terminus of syntaxin 4 to regulate insulin granule exocytosis. Mol. Endocrinol. 26, 128–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Crowley J. L., Smith T. C., Fang Z., Takizawa N., and Luna E. J. (2009) Supervillin reorganizes the actin cytoskeleton and increases invadopodial efficiency. Mol. Biol. Cell. 20, 948–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hoover H., Muralidharan-Chari V., Tague S., and D'Souza-Schorey C. (2005) Investigating the role of ADP-ribosylation factor 6 in tumor cell invasion and extracellular signal-regulated kinase activation. Methods Enzymol. 404, 134–147 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.