

Abstract
Oxidative stress has been implicated in multiple human neurological and other disorders. Proteasomes are multi-subunit proteases critical for the removal of oxidatively damaged proteins. To understand stress-associated human pathologies, it is important to uncover the molecular events underlying the regulation of proteasomes upon oxidative stress. To this end, we investigated H2O2 stress–induced molecular changes of the human 26S proteasome and determined that stress-induced 26S proteasome disassembly is conserved from yeast to human. Moreover, we developed and employed a new proteomic approach, XAP (in vivo cross-linking–assisted affinity purification), coupled with stable isotope labeling with amino acids in cell culture (SILAC)–based quantitative MS, to capture and quantify several weakly bound proteasome-interacting proteins and examine their roles in stress-mediated proteasomal remodeling. Our results indicate that the adapter protein Ecm29 is the main proteasome-interacting protein responsible for stress-triggered remodeling of the 26S proteasome in human cells. Importantly, using a disuccinimidyl sulfoxide–based cross-linking MS platform, we mapped the interactions of Ecm29 within itself and with proteasome subunits and determined the architecture of the Ecm29–proteasome complex with integrative structure modeling. These results enabled us to propose a structural model in which Ecm29 intrudes on the interaction between the 20S core particle and the 19S regulatory particle in the 26S proteasome, disrupting the proteasome structure in response to oxidative stress.
Keywords: MS, oxidative stress, proteasome, protein cross-linking, protein purification, protein–protein interaction, structural model
Introduction
Oxidative stress has been associated with the aging process and implicated in many human diseases, particularly neurodegenerative disorders (1). Protein oxidation can lead to unwanted changes in protein structure and function, resulting in the accumulation of severely oxidized proteins, subsequent cytotoxicity, and, ultimately, cell death. Therefore, oxidatively damaged proteins must be repaired or removed in a timely fashion to maintain cell homeostasis. Most oxidized proteins undergo selective proteolysis, and abundant evidence has indicated that proteasomes play a critically important role in the removal of oxidized proteins to preserve cell viability in response to oxidative stress (2–4). In addition, proteasomes are highly regulated during cellular responses to oxidative stresses. However, the molecular details underlying such modulation remain largely unexplored, particularly in human cells.
The 26S proteasome is a macromolecular machine responsible for ubiquitin/ATP-dependent protein degradation and comprises two subcomplexes: a 20S core particle (CP)3 and a 19S regulatory particle (RP) (5, 6). The 20S CP harbors various catalytic activities, including chymotrypsin-like, trypsin-like, and caspase-like peptidase activities. It is composed of seven α and seven β subunits in eukaryotes that form a conserved cylindrical structure of four heptameric stacked rings assembled in the order of αββα. Activation of the 20S CP requires binding to proteasome activator proteins (5). The 19S RP is one of the major proteasome activators and consists of at least 19 distinct subunits that constitute the base and lid subcomplexes. The base is composed of six ATPases (Rpt1–6) and four non-ATPase subunits (Rpn1, 2, 10, and 13), whereas the remaining nine subunits (Rpn3, 5–9, 11, and 12 and Rpn15/Sem1) comprise the lid structure. The 19S RP carries multiple functions to facilitate substrate degradation, including substrate recognition, deubiquitination, protein unfolding, substrate translocation, and gating of the 20S CP. In contrast to the highly ordered and stable structure of the 20S CP, the 19S RP appears to be much more flexible and dynamic. Nevertheless, the overall architectures of the 19S RP and the 26S holocomplex are highly conserved from yeast to human (7–11).
During oxidative stress, the proteasome system is highly regulated to fulfill its function in maintaining cell homeostasis (3, 4, 12). To facilitate the removal of oxidatively damaged proteins, ubiquitin/ATP-independent degradation by the 20S CP is significantly enhanced because of the increased amount of free 20S CP in cells, which is not the result of transcriptional control but, rather, of oxidative stress–triggered disassembly of the 26S proteasome (13–15). In yeast, such proteasome dissociation is dependent on the proteasome-interacting protein Ecm29 (13). However, it remains unclear whether human Ecm29 possesses a similar function because of low sequence similarity to its yeast ortholog (∼20%) and the relative complexity of human systems. In addition, Ecm29-dependent regulation of the proteasome system can have a multifaceted effect on cell physiology (11, 13, 16–19) by inhibiting ubiquitin-dependent protein degradation (11, 19), stabilizing proteasomes (16, 17), and assisting membrane-associated localization of proteasomes (20, 21) and TLR3-dependent signaling (22). However, how Ecm29 regulates the activity of the 26S proteasome in human cells, particularly in response to oxidative stress, is largely unknown. Therefore, further studies are needed to fully describe the molecular details underlying stress-mediated regulation of the 26S proteasome.
Here we quantitatively examined oxidative stress–mediated changes in the human 26S proteasome by developing a new affinity purification-MS strategy. In addition, we investigated the recruitment of Ecm29 to the proteasome and its associated biological implications. Moreover, we employed cross-linking mass spectrometry to define interactions within the Ecm29–proteasome complex, which were used for integrative structure modeling. Together, the results allow us to propose a structural model in which Ecm29 intrudes on the interaction between the 20S CP and 19S RP, thus modulating the function of the proteasome in response to oxidative stress.
Results
H2O2-mediated molecular changes in the human 26S proteasome
To evaluate the compositional changes of the human 26S proteasome under oxidative stress, we applied a single-step affinity purification procedure using an HTBH-tagged proteasome subunit (i.e. Rpn11-HTBH) as bait (23). This method has proven to be fast and effective to obtain functional human proteasome complexes and to identify proteasome-interacting proteins (23, 24). However, one of the key proteasome regulators, Ecm29, was not co-purified with human proteasomes. This is not surprising, as dynamic or transient interactions are often lost during conventional affinity purification (24, 25). To circumvent this problem, we developed a new strategy named in vivo cross-linking–assisted affinity purification MS (XAP-MS) (Fig. 1A). XAP-MS integrates mild in vivo formaldehyde (FA) cross-linking (<0.1%) prior to cell lysis, which enables better preservation of 26S proteasome intactness and proteolytic activities during native lysis (26). To quantify H2O2-induced changes, we coupled XAP-MS with SILAC-based quantitation, in which one population of 293Rpn11-HTBH cells was grown in light (L) medium and treated with H2O2, whereas the other population of the same cells was grown in heavy (H) medium as a control without treatment. The relative abundances of proteasome subunits between treated and untreated cells are represented by average peptide SILAC ratios (i.e. L/H) of the two compared samples (Fig. 1B and supplemental Table S2). As shown, all identified 19S RP subunits have SILAC ratios close to 1, indicating that the abundances of these subunits in the purified samples were unaffected by H2O2 stress (Fig. 1B). In contrast, the SILAC ratios of all 20S CP subunits decreased substantially with SILAC ratios of <0.4, demonstrating that oxidative stress resulted in dissociation of the 20S CP from the 19S RP. These results were validated using quantitative immunoblot analysis (supplemental Fig. S1). Similarly, we carried out XAP-SILAC MS experiments using 293α7/Pre10-HTBH cells. As expected, 20S CP subunits remained unchanged, whereas co-purified 19S RP subunits decreased substantially upon H2O2 stress (supplemental Fig. S2). Taken together, we confirmed that H2O2 stress–induced disassembly of the 26S proteasome is conserved in mammalian cells.
Figure 1.

Determination of 26S proteasome disassembly and enrichment of Ecm29 upon oxidative stress in human cells. A, the general workflow of the SILAC-based quantitative XAP-MS strategy. B, relative abundance changes (i.e. SILAC (L/H) ratios) of the human 26S subunits purified from 293Rpn11-HTBH cells in the presence and absence of H2O2 treatment. L ([12C14N]Arg/Lys), treated cells; H ([13C15N]Arg/Lys), untreated control cells. C, MS spectrum of a representative Ecm29 tryptic peptide pair (m/z 763.352+ versus 767.352+) with the SILAC ratio (L/H) as 6.0 when comparing treated with untreated samples. ○, treated (L); •, untreated (H). D, immunoblot analysis of Ecm29 abundance in proteasomes purified from 293Rpn11-HTBH cells before and after H2O2 treatment. IP, immunoprecipitation. E, immunoblot analysis of protein complexes purified using FLAG–Ecm29 that was transiently transfected in 293Rpn11-HTBH cells. Specific antibodies against Rpt6 and α7/Pre10 were used to probe their abundance. Streptavidin–HRP was used to probe HTBH-tagged Rpn11. All of the treated cells were incubated with 2 mm H2O2 for 30 min; untreated cells served as controls. The numbers under the immunoblot bands represent quantitative measurements using a Fuji LAS4000 scanning system.
Ecm29-dependent regulation of the 26S proteasome upon oxidative stress
Although it is known that Ecm29 can be recruited to the 19S proteasome during oxidative stress to regulate the proteasome composition in yeast (13), it remains unclear whether Ecm29 is a general player in modulating proteasome structure upon oxidative stress in eukaryotic systems. To this end, we first determined H2O2-triggered enrichment of human Ecm29 in the 19S RP using quantitative XAP-MS and immunoblotting analyses (Fig. 1, C and D). This was further validated by reciprocal affinity purification using FLAG-Ecm29 and quantitative immunoblotting (Fig. 1E). As shown, increased amounts of the two selected 19S subunits (i.e. Rpn11 and Rpt6) were co-purified with Ecm29 upon H2O2 treatment, whereas the amount of a 20S subunit, α7/Pre10, did not change in the purified Ecm29 complex. This result suggests that, although there is a dramatic increase in Ecm29 binding to the 19S upon oxidative stress, there is no change in Ecm29 binding to the 26S, thus implying two populations of Ecm29 detected here.
To understand how human Ecm29 regulates the 26S proteasome, we generated two Ecm29 knockdown (KD) cells (i.e. 293Rpn11-TB_Ecm29KD#53 and 293Rpn11-TB_Ecm29KD#55) (Fig. 2A). Quantitative immunoblotting analysis of purified proteasomes revealed that the abundances of the selected proteasome subunits (i.e. Rpt6 and α7/Pre10) are similar in Ecm29 KD and control KD cells under unstressed conditions (Fig. 2B). In addition, the 26S proteasomal activities were comparable in the two knockdown cells (Fig. 2C). Together, these results show that human Ecm29 is not essential for the assembly and function of the 26S proteasome. However, when cells were treated with H2O2 to induce oxidative stress, the 26S proteasome was rapidly disassembled in control KD cells, but such dissociation was considerably reduced in 293Rpn11-TB_Ecm29KD#53 and 293Rpn11-TB_Ecm29KD#55 cells (Fig. 2B). As expected, H2O2 stress also significantly reduced 26S proteasomal activities in control KD cells but not in Ecm29 KD cells (Fig. 2C). The observed changes in proteasomal activities correlated well with the levels of ubiquitinated proteins detected in these cells in the presence and absence of H2O2 stress, respectively (Fig. 2D). Collectively, our data strongly suggests that Ecm29 regulates H2O2-induced 26S proteasome disassembly in human cells.
Figure 2.
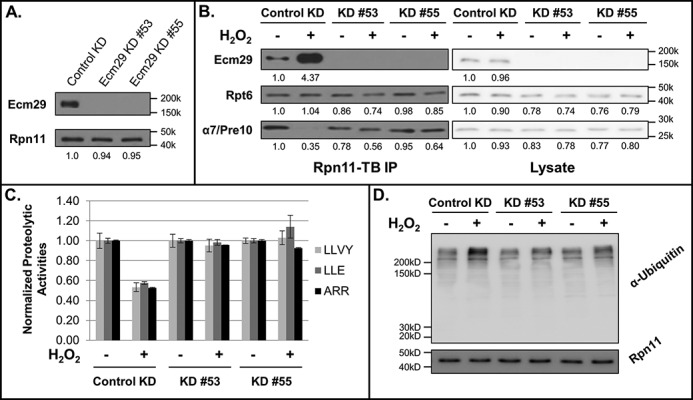
The effect of human Ecm29 on H2O2-induced 26S proteasome disassembly. A, evaluation of Ecm29 KD efficiency. B, the 26S proteasome was affinity-purified with Rpn11-TB from 293Rpn11-TB_controlKD (control KD), 293Rpn11-TB_ Ecm29KD#53 (KD #53), and 293Rpn11-TB_ Ecm29KD#55 (KD #55) cells and analyzed by quantitative Western blotting with antibodies against Rpt6 (19S) and α7/Pre10 (20S). C, effect of Ecm29 knockdown on the activity of the 26S proteasome. The proteasomal proteolytic activities in 293Rpn11-TB_controlKD (control KD), 293Rpn11-TB_ Ecm29KD#53 (KD #53), and 293Rpn11-TB_ Ecm29KD#55(KD #55) cells (treated or untreated) were determined by in-solution peptidase activity assays. Three fluorogenic peptide substrates were used: SUC-LLVY-AMC for chymotrypsin-like activity, SUC-LLE-AMC for peptide hydrolase activity, and SUC-ARR-AMC for trypsin-like activity. The activities were normalized to Rpn11 (a 19S subunit) in each sample. Data were obtained from three experiments. D, detection of total ubiquitin conjugates after H2O2-induced stress by Western blot analysis with an antibody against ubiquitin. Equivalent loading was determined by analysis of Western blots with an antibody against Rpn11 and by staining the membrane with Amido Black. The numbers under the immunoblot bands represent quantitative measurements using a Fuji LAS4000 scanning system.
To further evaluate the function of Ecm29, we overexpressed human FLAG-Ecm29 in 293Rpn11-HTBH cells. Quantitative immunoblotting analyses of the purified proteasomes revealed that overexpression of Ecm29 resulted in more proteasome-bound Ecm29 and, concurrently, less 19S RP-associated 20S CP under normal conditions (Fig. 3A and supplemental Fig. S3). This suggests that increased abundance of Ecm29 under non-stress conditions can disrupt normal 26S proteasome integrity, albeit to a lesser extent compared with the impact of oxidative stress. Interestingly, upon H2O2 stress, an increased amount of Ecm29 was also detected at the 19S RP even in the presence of overexpressed Ecm29, similar to wild-type cells, resulting in increased separation of the 20S CP from the 19S RP. These results were subsequently confirmed by the measurements of 26S proteasomal activities, as illustrated in Fig. 3B. Our data demonstrate that Ecm29 plays an evolutionarily conserved role in regulating the 26S proteasome, especially upon oxidative stress.
Figure 3.
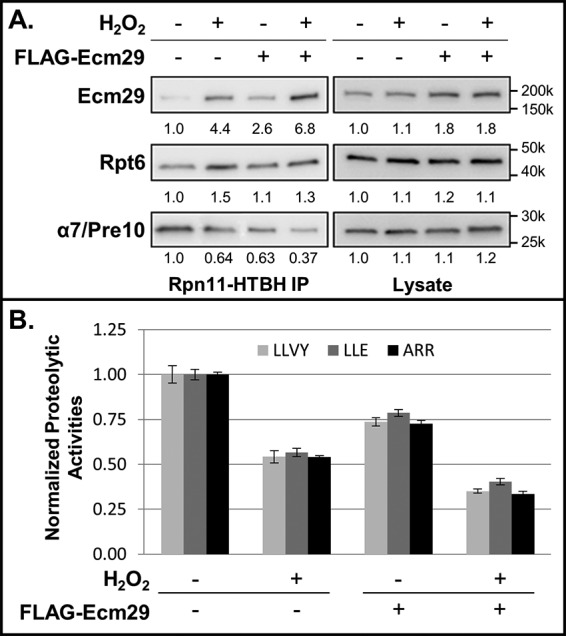
Modulation of the human 26S proteasome by Ecm29 overexpression. A, effect of Ecm29 overexpression on 26S proteasome integrity. Shown are quantitative immunoblot analyses of purified proteasomes (left) and cell lysates (right) in the presence or absence of overexpressed Ecm29. 293Rpn11-HTBH cells were transiently transfected with FLAG–Ecm29 either treated with H2O2 or untreated as a control. Quantitative Western blotting was performed with antibodies against Ecm29, Rpt6 (19S), and α7/Pre10 (20S). B, effect of Ecm29 overexpression on 26S proteasome activities. The proteasomal proteolytic activities were determined by in-solution peptidase activity assays with three fluorogenic peptide substrates: SUC-LLVY-AMC (LLVY); SUC-LLE-AMC (LLE), and SUC-ARR-AMC (ARR). The activities were normalized to Rpn11 (a 19S subunit) in each sample. Data were from three experiments. The numbers under the immunoblot bands represent quantitative measurements using a Fuji LAS4000 scanning system.
Ecm29 is the main PIP responsible for stress-induced proteasome disassembly
In addition to Ecm29, we identified 9 proteasome-interacting proteins (PIPs) that displayed H2O2-induced abundance changes alongside purified 19S RP (i.e. SILAC ratios ≥ 2) using quantitative XAP-MS analysis of 293Rpn11-HTBH cells (supplemental Table S2). Among them, five have known functions in the proteasome system: 19S assembly chaperones (p27/Nas2 and Rpn14/Gankyrin), deubiquitinase (Usp15), ubiquitin ligase (Ube3A), and Hsp70. Hsp70 has been shown to be important for proteasome reassembly after H2O2 stress (14). Three of the four remaining PIPs (i.e. Bag6, Ubl4A, and Trc35) are components of a ubiquitin ligase–associated multiprotein transmembrane recognition complex (TRC), which is unique to mammalian systems. With the exception of Bag6 (27), the other factors have not been linked to regulation of proteasome function. To confirm the MS results, respective immunoblotting analyses of purified Rpn11-containing proteasomes and FLAG–Ubl4A complexes were performed (supplemental Fig. S4). Our results demonstrate that the TRC complex interacts with the 26S proteasome via the 19S complex and that their interactions can be modulated by H2O2 stress. Given the similarity in H2O2-induced enrichment of the TRC complex to that of Ecm29 at the 19S RP, we initially hypothesized that the TRC complex may regulate the 26S proteasome complex in the same manner as human Ecm29 during oxidative stress. However, CRISPR-mediated knockout or siRNA-mediated silencing of Bag6 did not affect the interaction between 20S and 19S proteasomes in either normal or H2O2-treated cells (supplemental Fig. S5), suggesting that Bag6 is not required for H2O2-induced 26S proteasome disassembly. In addition, knockout of Bag6 did not interfere with H2O2-induced recruitment of Ecm29 to the 19S proteasome and vice versa. These results imply that Bag6 is not associated with Ecm29-dependent regulation of the 26S proteasome complex despite its increased association with the 19S RP upon H2O2 treatment. Because the TRC complex is thought to chaperone polypeptides en route to the proteasome to facilitate the degradation of folding-defective proteins, which include retrotranslocation products from the endoplasmic reticulum and mislocalized membrane proteins (28, 29), the increased association of Bag6 with the 19S complex may result from the loss of communication between the 19S and the 20S proteasome, which presumably traps ubiquitinated proteins together with their chaperones on the 19S complex. From these results, we concluded that, although oxidative stress-induced changes in the proteasome interactome are not limited to Ecm29, proteasome disassembly is specifically regulated by the 19S-associated Ecm29.
Physical interactions of Ecm29 with the proteasome
Because of the lack of structural details on Ecm29 alone and its complex with proteasomes, how Ecm29 is recruited to the 19S particle during oxidative stress is largely unknown. To understand how Ecm29 regulates the proteasome upon oxidative stress, we employed our previously developed XL-MS strategy to determine protein interaction contacts at specific residues (30). This XL-MS strategy enables simplified and accurate identification of cross-linked peptides by integrating an MS-cleavable homobifunctional amine-reactive NHS ester, disuccinimidyl sulfoxide (DSSO), with multistage tandem mass spectrometry (MSn) (30). To ensure the capture of a sufficient amount of Ecm29–proteasome complexes, we co-expressed HTBH-Ecm29 in 293HTBH-Rpt6 cells and treated the cells with H2O2 stress prior to cell lysis. Single-step affinity purification by binding to streptavidin beads was carried out to isolate Ecm29–proteasome complexes for in vitro DSSO cross-linking, similar as described previously (11). The resulting DSSO cross-linked peptides were analyzed by LC/MSn for identification (30). As an example, a representative MSn analysis of a DSSO interlinked peptide is illustrated in Fig. 4. As shown, the cleavage of either of the two symmetric MS-cleavable C–S bonds in the linker region of the DSSO interlinked peptide α-β (m/z 848.66974+) resulted in detection of two characteristic fragment pairs: αA/βT (m/z 762.412+ and 925.922+) and αT/βA (m/z 778.392+ and 909.942+) during MS2 analysis (Fig. 4A). MS3 sequencing of the αT and βA fragment ion pair yielded series of b and y ions that unambiguously identified them as 284HSVATAADLELKTSK287 of Ecm29 and 372KAMNVSPDVNYEELAR386 of Rpt5, respectively (Fig. 4B, left and right panels). Together, the MSn analysis determined a cross-link between Lys-285 of Ecm29 and Lys-372 of Rpt5 (Fig. 4). In total, LC/MSn analysis identified 69 unique Lys–Lys linkages involving Ecm29, seven of which were interprotein and 62 intraprotein interactions (supplemental Tables S3 and S4). Ecm29 was determined to interact with five 19S base subunits, Rpt1, Rpt4, Rpt5, Rpn1, and Rpn10, as illustrated in Fig. 5. Among them, Ecm29 has the most contacts with Rpt5, as their interactions are supported by multiple Lys–Lys linkages with the highest number of redundant counts. Based on the cross-link map (Fig. 5), we concluded that Ecm29 interacts with the 19S base subunits through its multiple HEAT repeat domains. Although the N termini of Rpt4 and Rpt5 are both proximal to the N terminus of Ecm29, the C termini of Rpt5, Rpt1, and Rpn10 also have close contacts with Ecm29. In addition, Lys-397 in Rpn1 near its T2 site was cross-linked to Lys-694 of Ecm29. Our results suggest that Ecm29 most likely interacts with the proteasome through multiple contact sites, with Rpt5 as the major docking point.
Figure 4.

Representative MSn analysis of a selected DSSO cross-link between Ecm29 and Rpt5. A, collision-induced dissociation (CID) cleavage of the DSSO cross-linked peptide α-β (m/z 848.66974+) in MS2 resulted in the formation of two predicted fragment ion pairs: αA2+/βT2+ and αT2+/βA2+. B, respective MS3 spectra of the two selected MS2 fragment ions αT2+ (m/z 778.392+) and βA2+ (909.942+). A series of b and y ions unambiguously identified the peptides as 284HSVATAADLELKTSK287 of Ecm29 and 372KAMNVSPDVNYEELAR386 of Rpt5, respectively. C, along with peptide mass matching at the MS1 level, three lines of evidence (including MS2 cross-linker fragmentation and individual MS3 peptide sequencing) confirm the identification of a DSSO cross-link between Lys-285 of Ecm29 and Lys-372 of Rpt5.
Figure 5.

Cross-link map of Ecm29-proteasome interactions. Diagram of unique Lys–Lys linkages between Ecm29 and the 19S base subunits Rpn1, Rpn10, Rpt1, Rpt4, and Rpt5. The remaining members of the AAA-ATPase ring are also included for spatial context. Cross-linked Ecm29 residues are labeled in green, whereas the residues of the respective 19S RP subunits are shown in blue. Various functional domains of individual proteins are labeled according to SMART: coiled-coil domain, orange; AAA-ATPase, blue; von Willebrand factor (VWA), green; ubiquitin-interacting motif (UIM), magenta; HEAT repeats, brown. The T1 and T2 domains of Rpn1 are shown in yellow and orange, respectively.
To evaluate the cross-links identified between Ecm29 and 19S RP subunits, we first performed phylogenetic alignment analysis of Ecm29 derived from five selected organisms:Saccharomyces cerevisiae, Caenorhabditis elegans, Drosophila, mouse, and human. Despite a relatively low degree of sequence homology, six of the seven lysine residues of Ecm29 that cross-linked with 19S base subunits were located proximally to conserved sequence regions (i.e. within five amino acid residues); only Lys-852 of Ecm29 was found to be relatively far from the nearest conserved site, ∼18 residues away (supplemental Table S5). When a similar phylogenetic alignment was performed for Rpt1, Rpt4, Rpt5, Rpn1, and Rpn10, the lysine residues on each respective subunit found to be cross-linked to Ecm29 were mostly located within or directly adjacent to highly conserved regions as well, with the exception of Lys-16 of Rpt5 (supplemental Table S5). Therefore, almost all of the residues cross-linked between Ecm29 and 19S base subunits correspond to evolutionarily conserved regions, suggesting that these cross-links more likely represent the functional protein interaction interfaces between Ecm29 and the proteasome complex.
Integrative modeling of the Ecm29–proteasome complex
To understand how Ecm29 docks onto the proteasome, the architecture of the Ecm29–proteasome complex was determined using the integrative structure modeling approach described previously (31–35). All available structural information on the Ecm29–proteasome complex was used for computational analyses (supplemental Fig. S6 and Methods). The proteasome was represented by the high-resolution structure of the human 26S proteasome (PDB code 5GJR) (10), whereas Ecm29 was represented by two comparative models built with MODELLER 9.17 (36) based on known template structures detected by HHPred (37) (supplemental Methods). Regions with unknown structures were modeled as flexible strings of beads. Finally, the proximity between specific residue pairs was determined by DSSO XL-MS experiments, which identified a total of 69 unique Lys–Lys linkages (supplemental Tables S3 and S4) describing seven Ecm29-containing interprotein interactions and 62 Ecm29 intraprotein interactions. The maximum Cα–Cα distance between any two lysine residues cross-linked by DSSO was estimated to be ∼30 Å, based on the spacer length of DSSO (10.1 Å), flexibility of lysine side chains, and backbone dynamics. Next, 3,750,000 Ecm29–proteasome models were computed by optimizing spatial proximities, as informed by cross-linking data, excluded volume, and sequence connectivity from 500 random initial models. This process yielded 109,951 good-scoring models (i.e. the ensemble) that satisfy the cross-linking data, the excluded volume, and sequence connectivity restraints used in computing the models. The clustering of the good-scoring models identified two distinct clusters (Fig. 6A), including 60% (89% of intersubunit cross-links satisfied; Fig. 6, B, left panel, and C) and 31% (100% of intersubunit cross-links satisfied; Fig. 6, B, right panel, and C) of the models; the precision of both clusters is 60 Å root mean square deviation for all Ecm29 Cαs (supplemental Fig. S7). In general, an ensemble of good-scoring models can be visualized as a localization probability density map. The map specifies the probability of any volume element being occupied by a given bead in superposed good-scoring models. The probability localization density for the structured regions of Ecm29 is sufficiently precise to define the position, but not the orientation, of Ecm29 relative to the proteasome for each of the two clusters (Fig. 6A). The binding sites on the proteasome are different between the two clusters, indicating that Ecm29 may interact with the proteasome in two different states (although it is also conceivable that we simply do not have enough data to define a single state). In the first state (cluster 1), Ecm29 interacts with the 19S within 10 Å of Rpn1, Rpt2, Rpt4, Rpt5, and Rpn10 (Fig. 6A, left panel). In this binding mode, Ecm29 does not overlap with the 20S particle and is proximal to the α1 subunit. In the second state (cluster 2), Ecm29 is similarly vicinal to the same partners in the 19S RP (Fig. 6A, right panel), although its relative position is different and inconsistent with the presence of 20S (Fig. 6A).
Figure 6.

Integrative structure modeling of the Ecm29–proteasome complex. A, localization probability density of the structured part of Ecm29 from cluster 1 (left panel) and 2 (right panel). The 19S structure is shown in dark gray, and the interacting partners of 19S with Ecm29 are colored. The 20S α ring is shown in light gray for reference. B, Euclidean Cα–Cα distance distributions of all measured cross-links in the ensemble of solutions for each cluster. The y axis provides the normalized number of cross-links that were mapped onto the model. The dashed red line denotes the expected maximum reach of a cross-link. C, Euclidean Cα–Cα distance statistics for each cross-link in both clusters (cluster 1 in green empty squares and cluster 2 in orange disks). The cross-links are sorted by average distance (ordinate axis); intra- and interprotein cross-links are separated (left and right, respectively). The error bars represent the standard deviation of the distance across all models in the clusters.
Discussion
Here we developed and employed the XAP-MS strategy to dissect the H2O2-dependent compositional dynamics of the human 26S proteasome complex. The results confirmed that the 26S proteasome is highly regulated during oxidative stress and that its disassembly yields more free 20S CP for the removal of oxidatively damaged proteins, corroborating previous reports (13, 14). In addition, with the XAP-MS strategy, we were able to co-purify human Ecm29 with proteasomes reliably with and without H2O2 treatment for the first time. This is significant, as the interaction of human Ecm29 with proteasomes appears to be much weaker and/or more transient than yeast Ecm29–proteasome interaction, thus preventing its capture using conventional affinity purification–MS approaches. The reliability of co-purification of human Ecm29 with proteasomes enabled us to confirm H2O2-induced enrichment of Ecm29 onto the 19S RP in human cells, similar to its yeast ortholog (13). Our results further indicate that the mild in vivo FA cross-linking implemented in the XAP-MS strategy is indeed beneficial for preserving weak, transient, and/or dynamic interactors of protein complexes under native conditions, as described previously (26, 38). Thus, it helps to maintain the integrity of protein complexes as well as to prevent reorganization and loss of protein–protein interactions. Therefore, the XAP-MS method can be applied to study the dynamic interactors of other protein complexes and identify their regulators through protein–protein interactions,
Ecm29 has been shown to be critical in modulating proteasome structure and function (11, 13, 16–19). Interestingly, knockdown of human Ecm29 did not seem to have much impact on the structure and function of proteasomes, as the 26S holocomplex remained intact, and its proteolytic activities were not impaired. These observations are consistent with those in yeast ECM29Δ cells (13) and Ecm29 (i.e. KIAA0368)-deficient mice (39). Collectively, our results suggest that Ecm29 is non-essential for the assembly, integrity, and function of proteasomes under unstressed conditions. However, the impact of Ecm29 on proteasomes becomes noticeably more apparent upon H2O2-induced stress. Here we demonstrated that Ecm29 has a conserved function in regulating oxidative stress-triggered 26S proteasome disassembly in eukaryotes, which has been shown to be important for cell survival, particularly for recovery from oxidative stress (13). In addition, we have shown that increased interaction between Ecm29 and the 19S RP is directly associated with remodeling of the 26S proteasome and that Ecm29–proteasome interaction is regulated by oxidative stress. These results indicate that the modes of Ecm29 function are diverse and depend on cellular conditions. Moreover, we found that overexpression of Ecm29 alone could not induce the same level of effects on the 26S proteasome as oxidative stress, suggesting that additional signal(s) would be needed for the recruitment of Ecm29 to dissociate the 20S CP from the 19S RP. It has been shown that oxidation of cysteine residues within proteasome subunits can activate 20S proteolytic activities by inducing gate opening (40) or modulate proteasome disassembly in yeast upon mitochondrial stress (15). Future studies are needed to determine whether protein oxidation and Ecm29 work hand in hand to reshape the structure of the 26S proteasome during oxidative stress.
We combined XL-MS studies and integrative structural modeling to explore the action mechanisms of Ecm29. Our XL-MS studies provide the first physical evidence at peptide resolution to model the positions of Ecm29 on the proteasome structure. It is not surprising that Ecm29 can have multiple contact sites on the proteasome, as it contains 1845 amino acids in the form of HEAT-like repeats throughout its sequence (41). In addition, EM imaging at low resolution has indicated that Ecm29 has an elongated and curved structure (16). Among the five 19S base subunits, Rpt5 seems to be the one that interacts most intimately with Ecm29, based on the number of detected Ecm29–Rpt5 cross-links. This is supported by previous studies showing that Ecm29 localizes in close proximity to Rpt5 in yeast (11, 25). We computed a structural model of the Ecm29–proteasome complex and show that either one of two similar states are consistent with the input cross-link data as well as atomic models of Ecm29 and the proteasome. The Ecm29–19S model shows an elongated structure for Ecm29, forming contacts with Rpt1, Rpt4, Rpt5, Rpn1, and Rpn10, with its C terminus reaching 20S, in agreement with an earlier EM study (16).
Apart from Rpt5, the 20S subunit α7 has been shown to interact with Ecm29 in yeast, depending on the phosphorylation of the α7 tail (15). Interestingly, α7 phosphorylation is constitutive and has been shown to be important for modulating the stability of the CP–RP interactions in a human system (42) but has not been associated with Ecm29 function. We found that phosphorylation of the α7 tail at Ser-250 did not change in response to oxidative stress (data not shown), suggesting that it might not be important for Ecm29 interaction during oxidative stress. In addition, integrative structure modeling has indicated that oxidative stress-mediated proteasome-bound Ecm29 is not in close proximity to α7. Indeed, either localization of Ecm29 on the 19S proteasome suggests that the closest distance between Ecm29 and the α7 subunit of 20S is more than 30 Å. However, this finding is not completely unexpected, as our XL-MS experiments were designed to localize Ecm29 on the 19S and not the 20S or 26S proteasomes.
XL-MS analysis of affinity-purified complexes often reveals multiple conformational states (11). The purified proteasome sample for DSSO cross-linking in this study is expected to contain two major populations of proteasomes: free 19S RP and the 26S proteasome. Each of the two good-scoring models of Ecm29–19S satisfies the input data equally well (Fig. 6), including six distinct interprotein cross-links (Fig. 6C, right panel). The major difference between the two clusters is that cluster 1 suggests a closer interaction between Ecm29 and Rpn1, whereas cluster 2 suggests a closer interaction with Rpt1. Although it is possible that we simply did not collected sufficient information to determine the Ecm29–19S structure precisely, it is also conceivable that Ecm29 interacts with the proteasome in multiple (at least two) conformations to fulfill its role in modulating the disassembly of the 26S proteasome upon oxidative stress. The two localizations of Ecm29 on the 19S RP suggest a possible mechanism for the dissociative role of Ecm29 on the proteasome under oxidative stress: recognition of the 19S (cluster 1, state 1) and its inhibition of the 20S–19S interaction (cluster 2, state 2) (Fig. 7). Thus, these binding modes imply two sequential events: the recruitment of Ecm29 to trigger the 26S proteasome disassembly and relocalization of Ecm29 on the 19S proteasome to block 26S proteasome reassembly. As shown, an increased amount of 19S-bound Ecm29 would lead to elevated competition between Ecm29 and 20S for the binding site on the 19S, thus keeping the 20S and the 19S separated after Ecm29-mediated dissociation upon oxidative stress.
Figure 7.

The proposed model of Ecm29-mediated disassembly of the 26S proteasome upon H2O2 stress. The amount of Ecm29 at the 19S proteasome increases with oxidative stress, which is illustrated with increased intensity in red.
In summary, we examined oxidative stress-triggered molecular changes in the human 26S proteasome using quantitative XAP-MS, biochemical methods, XL-MS, and integrative modeling. In addition, we were able to capture proteasome-bound Ecm29 and determined that Ecm29 binds to the 19S in response to H2O2 stress. Importantly, we demonstrated the biological role of human Ecm29 in modulating 26S proteasome disassembly and mapped specific residue–residue interactions between Ecm29 and multiple 19S RP subunits. The molecular architecture of the Ecm29–proteasome complex allows us to propose a model of Ecm29-dependent regulation of the 26S proteasome during oxidative stress. This model provides a basis for further exploring the diverse roles of Ecm29 in the proteasome system.
Experimental procedures
Materials
Regular DMEM, SILAC DMEM (deficient in lysine and arginine), ImmunoPure streptavidin, horseradish peroxidase–conjugated antibody, Super Signal West Pico chemiluminescent substrate, and TurboFect transfection reagent were obtained from Thermo Fisher Scientific. [13C615N4]arginine and [13C6 15N2]lysine were purchased from Cambridge Isotope Laboratories. [12C614N4]arginine, [12C6 14N2]lysine, anti-FLAG M2 affinity gel, and Ecm29 (KIAA0368) MISSION® shRNA bacterial glycerol stocks (catalog nos. TRCN0000263353 and TRCN0000263355) were obtained from Sigma. MISSION® pLKO.1-puro non-target shRNA bacterial glycerol stocks (catalog no. SHC016-1EA) were a kind gift from Dr. Anand Ganesan at the University of California, Irvine. Antibodies against human Rpt6 and Pre10 were obtained from Biomol International. Initially, human Ecm29 antibody was a kind gift from Dr. Carlos Gorbea (University of Utah, School of Medicine); later on it was purchased from Thermo Fisher Scientific. Ubiquitin antibody was from Santa Cruz Biotechnology. Endoproteinase Lys-C was from Wako Chemicals. Sequencing-grade trypsin was purchased from Promega. The proteasome substrates SUC-LLVY-AMC, SUC-LLE-AMC, and SUC-ARR-AMC were purchased from Boston Biochem. All other general chemicals for buffers and culture media were purchased from Thermo Fisher Scientific or VWR International.
Generation of Ecm29 knockdown cells and BAG6 knockdown cells
Lentiviruses were produced and knockdown cells were generated as described previously (43). Briefly, lentiviruses were generated by transfecting HEK293 cells with the pLKO.1-Ecm29shRNA vectors together with the packaging vectors pMDG and pCMVΔR8.91. Lentiviruses were collected 24 and 48 h post-transfection for target cell infection. 293Rpn11-TB(Hygro) (26) cells were transduced with recombinant lentivirus and selected with 2.5 μg/ml puromycin 48 h after viral infection to produce the stable cell line expressing Ecm29shRNA (293Rpn11-TB_Ecm29KD).
Bag6 knockout cells were generated by using CRISPR technology (44) from 293Rpn11-TB(Hygro) cells to get 293Rpn11-TB_Bag6KO cell lines.
Cloning of pQCXIP-HBTH-Ecm29
Ecm29 was PCR-amplified using FLAG-Ecm29 as the template with the following primers: forward, TTAATTAACGC-TGGAAAGGCCGGTGAAGGTG; reverse, GAATTCTCACATCCCTAACTCTCCTT-GAAAG. The CSN5 fragment in pQCXIP-HBTH-CSN5 (45) was removed, and Ecm29 PCR fragment was inserted.
Cell culture and purification of human 26S proteasomes
Nine cell lines (293Rpn11-HTBH, 293Rpn11-TB, 293HBTH-Rpt6, 293α7/Pre10-HTBH, 293HBTH-Ecm29, 293Rpn11-TB_controlKD, 293Rpn11-TB_Ecm29KD#53, 293Rpn11-TB_Ecm29KD#53, and 293Rpn11-TB_Bag6KO) were used in this work as listed in supplemental Table S1. Cells were grown to (90%) confluence in DMEM and either treated with 2 mm H2O2 for 30 min or left untreated as a control. Prior to harvesting, cells were incubated with 0.05% FA for 10 min at 37 °C. The human 26S proteasome was purified by binding to streptavidin–agarose resin (23), which was on-bead digested for MS analysis or eluted with SDS loading buffer for Western blotting. For SILAC experiments, stable cell lines were grown in SILAC DMEM as described previously (24). A Mix-After-Purification SILAC strategy was used to compare proteasome compositions before and after treatment (24).
Transient transfection and affinity purification of FLAG–Ecm29 and FLAG–Ubl4A
293Rpn11-HTBH cells were transiently transfected with FLAG–Ecm29 or FLAG–Ubl4A using TurboFect transfection reagent as described in the protocol of the manufacturer (Thermo Fisher Scientific). After 24 h, cells were treated with 2 mm H2O2 at 37 °C for 30 min or left untreated as a control, followed by 0.05% FA incubation for 10 min at 37 °C in PBS before harvesting. The respective Ecm29 and Ubl4A complexes were affinity-purified by anti-FLAG M2 affinity gel and eluted with 0.1 m glycine following the protocol of the manufacturer (Sigma).
Proteasome proteolytic activity assay
In-solution proteolytic activity assays for human proteasomes in cell lysates were performed with the fluorogenic peptide substrates SUC-LLVY-AMC, SUC-LLE-AMC, and SUC-ARR-AMC as described previously (23).
Quantitative immunoblot analysis
The purified proteasome complexes were analyzed by Western blotting as described previously (13). Primary antibodies against Rpt6, α6/MCP20, α7/Pre10, Ecm29, and Bag6 were utilized, followed by an HRP-conjugated mouse or rabbit secondary antibody against mouse IgG. Protein bands were detected and quantified using a Fuji LAS4000 scanning system (Fujifilm Life Sciences).
Protein identification and quantification by MS
Purified proteasome complexes were digested in-solution with Lys-C/trypsin and analyzed by LC/MS-MS using an Easy-nLC 1000 coupled with a linear ion trap (LTQ) Orbitrap XL mass spectrometer (Thermo Fisher, San Jose, CA) as described previously (13). The LC/MS-MS data were searched using Batch-Tag within a developmental version (v. 5.17.0) of Protein Prospector at the University of California, San Francisco against a decoy database consisting of a normal SwissProt database concatenated with its randomized version (SwissProt.2013.06.17.random.concat with a total of 455,294 protein entries) (13). Proteins were identified by at least two peptides with an FDR of ≤ 0.5%.
For SILAC experiments, the Search Compare program within Protein Prospector was used to calculate the relative abundance ratios of Arg/Lys-containing peptides based on ion intensities of monoisotopic peaks observed in the LC/MS spectra when the peptides were sequenced and subsequently identified during database searching as described previously (13, 24).
DSSO cross-linking of Ecm29–proteasome complexes
293HBTH-Rpt6 cells were transiently transfected with HBTH–Ecm29. After 24 h, the cells were treated with 5 mm H2O2at 37 °C for 30 min to maximize the interaction between Ecm29 and 19S RP, followed by 0.025% FA at 37 °C in PBS for 10 min. Single-step affinity purification of the human Ecm29–proteasome complexes was achieved by binding to streptavidin–agarose resin. The bound protein complexes were cross-linked on-bead in PBS buffer (pH 7.5) with 0.5 mm DSSO for 1 h at 37 °C and then quenched, reduced/alkylated, and digested as reported previously (11, 30). The resulting peptide mixture was extracted and desalted prior to LC/MSn analysis.
LC/MSn analysis of DSSO cross-linked peptides
LC MSn analysis was performed using a Thermo ScientificTM EASY-nLCTM 1200 ultrahigh pressure liquid chromatography (UPLC) system coupled with an Orbitrap Fusion LumosTM MS (46). Briefly, a 25 cm × 75 μm PepMap EASY-Spray column was used to separate peptides over acetonitrile gradients of 6% to 35% at a flow rate of 300 nl/min. Two different types of acquisition methods were utilized to maximize the identification of DSSO cross-linked peptides: top four data-dependent MS3 and targeted MS3 acquisition (46). Two biological replicates were analyzed, and each of them was analyzed with at least two technical replicates.
Data analysis to identify DSSO cross-linked peptides
MSn data were extracted, searched, and analyzed as described previously (11). Briefly, MS3 data were subjected to Batch-Tag against a decoy database consisting of a normal SwissProt database concatenated with its randomized version (SwissProt.2014.12.4.random.concat with a total of 20,194 protein entries). Peptides were identified from MS3 data with a FDR of 1.7%. MSn data and MS3 database search results were integrated in xl-Discoverer (an in-house script) to automatically generate, summarize, and validate identified cross-linked peptide pairs. The final FDR of identified interlinked peptides was determined to be 0.1%. The reduction in FDR for the identification of cross-linked peptides occurs as a result of MSn data integration, which improves identification accuracy.
Integrative modeling of the Ecm29–proteasome complex
Comparative and integrative modeling was carried out to elucidate the architecture of the human Ecm29–26S proteasome complex (11) (supplemental Methods).
Author contributions
L. H. conceived the study and directed the research. X. W., I. E. C., A. S., Y. Y., and L. H. designed the experiments. X. W. generated constructs and stable cell lines and carried out quantitative XAP-MS, biochemical, and XL-MS experiments and data analyses. C. Y. and R. V. performed the LC/MSn analysis. C. Y. analyzed the XL-MS data. I. E. C. performed integrative structure modeling. P. C. did the initial modeling analysis. X. W., C. Y., and I. E. C. prepared figures and tables. A. H. contributed to XL-MS data analysis. S. A. B. and S. D. R. synthesized the cross-linking reagent. Y. X. and Y. Y. provided biochemical reagents and generated CRISP-based Bag6 knockout cells. X. W., I. E. C., C. Y., Y. Y., A. S., and L. H. contributed to the writing of the manuscript.
Supplementary Material
Acknowledgments
We thank Prof. A. L. Burlingame and Dr. Robert Chalkley (University of California, San Francisco) for support of the development version of Protein Prospector.
This work was supported by National Institutes of Health Grants R01GM074830 (to L. H.), R01GM106003 (to L. H. and S. R.), and R01GM083960 and P41GM109824 (to A. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1–S7, Tables S1–S5, Methods, and References.
- CP
- core particle
- RP
- regulatory particle
- XAP-MS
- in vivo cross-linking–assisted affinity purification
- FA
- formaldehyde
- SILAC
- stable isotope labeling by amino acids in cell culture
- KD
- knockdown
- PIP
- proteasome-interacting protein
- TRC
- transmembrane recognition complex
- DSSO
- disuccinimidyl sulfoxide
- MSn
- multistage tandem mass spectrometry
- L
- light
- H
- heavy
- XL-MS
- cross-linking mass spectrometry
- HTBH tag
- Histidine-TEV-Biotin-Histidine tag
- HBTH tag
- Histidine-Biotin-TEV-Histidine tag
- TB tag
- TEV-Biotin tag
- XL-MS
- cross-linking mass spectrometry
- FDR
- false discovery rate.
References
- 1. Barnham K. J., Masters C. L., and Bush A. I. (2004) Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 3, 205–214 [DOI] [PubMed] [Google Scholar]
- 2. Byvaltsev V., Belykh E., Panasenkov S., Ivanov N., Tsyganov P., and Sorokovikov V. (2012) Nanostructural changes of intervertebral disc after diode laser ablation. World Neurosurgery 77, 6–7 [DOI] [PubMed] [Google Scholar]
- 3. Aiken C. T., Kaake R. M., Wang X., and Huang L. (2011) Oxidative stress-mediated regulation of proteasome complexes. Mol. Cell. Proteomics 10, R110.006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Goebl M. G., Yochem J., Jentsch S., McGrath J. P., Varshavsky A., and Byers B. (1988) The yeast cell cycle gene CDC34 encodes a ubiquitin-conjugating enzyme. Science 241, 1331–1335 [DOI] [PubMed] [Google Scholar]
- 5. Voges D., Zwickl P., and Baumeister W. (1999) The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu. Rev. Biochem. 68, 1015–1068 [DOI] [PubMed] [Google Scholar]
- 6. Finley D. (2009) Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 78, 477–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lander G. C., Estrin E., Matyskiela M. E., Bashore C., Nogales E., and Martin A. (2012) Complete subunit architecture of the proteasome regulatory particle. Nature 482, 186–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lasker K., Förster F., Bohn S., Walzthoeni T., Villa E., Unverdorben P., Beck F., Aebersold R., Sali A., and Baumeister W. (2012) Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc. Natl. Acad. Sci. U.S.A. 109, 1380–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schweitzer A., Aufderheide A., Rudack T., Beck F., Pfeifer G., Plitzko J. M., Sakata E., Schulten K., Förster F., and Baumeister W. (2016) Structure of the human 26S proteasome at a resolution of 3.9 A. Proc. Natl. Acad. Sci. U.S.A. 113, 7816–7821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huang X., Luan B., Wu J., and Shi Y. (2016) An atomic structure of the human 26S proteasome. Nat. Struct. Mol. Biol. 23, 778–785 [DOI] [PubMed] [Google Scholar]
- 11. Wang X., Cimermancic P., Yu C., Schweitzer A., Chopra N., Engel J. L., Greenberg C., Huszagh A. S., Beck F., Sakata E., Yang Y., Novitsky E. J., Leitner A., Nanni P., Kahraman A., et al. (2017) Molecular details underlying dynamic structures and regulation of the human 26S proteasome. Mol. Cell. Proteomics 16, 840–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ben-Nissan G., and Sharon M. (2014) Regulating the 20S proteasome ubiquitin-independent degradation pathway. Biomolecules 4, 862–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang X., Yen J., Kaiser P., and Huang L. (2010) Regulation of the 26S proteasome complex during oxidative stress. Sci. Signal. 3, ra88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Grune T., Catalgol B., Licht A., Ermak G., Pickering A. M., Ngo J. K., and Davies K. J. (2011) HSP70 mediates dissociation and reassociation of the 26S proteasome during adaptation to oxidative stress. Free Radic. Biol. Med. 51, 1355–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Livnat-Levanon N., Kevei É., Kleifeld O., Krutauz D., Segref A., Rinaldi T., Erpapazoglou Z., Cohen M., Reis N., Hoppe T., and Glickman M. H. (2014) Reversible 26S proteasome disassembly upon mitochondrial stress. Cell Rep. 7, 1371–1380 [DOI] [PubMed] [Google Scholar]
- 16. Leggett D. S., Hanna J., Borodovsky A., Crosas B., Schmidt M., Baker R. T., Walz T., Ploegh H., and Finley D. (2002) Multiple associated proteins regulate proteasome structure and function. Mol. Cell 10, 495–507 [DOI] [PubMed] [Google Scholar]
- 17. Kleijnen M. F., Roelofs J., Park S., Hathaway N. A., Glickman M., King R. W., and Finley D. (2007) Stability of the proteasome can be regulated allosterically through engagement of its proteolytic active sites. Nat. Struct. Mol. Biol. 14, 1180–1188 [DOI] [PubMed] [Google Scholar]
- 18. Park S., Kim W., Tian G., Gygi S. P., and Finley D. (2011) Structural defects in the regulatory particle-core particle interface of the proteasome induce a novel proteasome stress response. J. Biol. Chem. 286, 36652–36666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee S. Y., De la Mota-Peynado A., and Roelofs J. (2011) Loss of Rpt5 protein interactions with the core particle and Nas2 protein causes the formation of faulty proteasomes that are inhibited by Ecm29 protein. J. Biol. Chem. 286, 36641–36651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gorbea C., Goellner G. M., Teter K., Holmes R. K., and Rechsteiner M. (2004) Characterization of mammalian Ecm29, a 26 S proteasome-associated protein that localizes to the nucleus and membrane vesicles. J. Biol. Chem. 279, 54849–54861 [DOI] [PubMed] [Google Scholar]
- 21. Gorbea C., Pratt G., Ustrell V., Bell R., Sahasrabudhe S., Hughes R. E., and Rechsteiner M. (2010) A protein interaction network for Ecm29 links the 26S proteasome to molecular motors and endosomal components. J. Biol. Chem. 285, 31616–31633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gorbea C., Rechsteiner M., Vallejo J. G., and Bowles N. E. (2013) Depletion of the 26S proteasome adaptor Ecm29 increases Toll-like receptor 3 signaling. Sci. Signal. 6, ra86. [DOI] [PubMed] [Google Scholar]
- 23. Wang X., Chen C. F., Baker P. R., Chen P. L., Kaiser P., and Huang L. (2007) Mass spectrometric characterization of the affinity-purified human 26S proteasome complex. Biochemistry 46, 3553–3565 [DOI] [PubMed] [Google Scholar]
- 24. Wang X., and Huang L. (2008) Identifying dynamic interactors of protein complexes by quantitative mass spectrometry. Mol. Cell. Proteomics 7, 46–57 [DOI] [PubMed] [Google Scholar]
- 25. Guerrero C., Milenkovic T., Przulj N., Kaiser P., and Huang L. (2008) Characterization of the proteasome interaction network using a QTAX-based tag-team strategy and protein interaction network analysis. Proc. Natl. Acad. Sci. U.S.A. 105, 13333–13338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yu C., Yang Y., Wang X., Guan S., Fang L., Liu F., Walters K. J., Kaiser P., and Huang L. (2016) Characterization of dynamic UbR-proteasome subcomplexes by in vivo cross-linking (X) assisted bimolecular tandem affinity purification (XBAP) and label-free quantitation. Mol. Cell. Proteomics 15, 2279–2292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Akahane T., Sahara K., Yashiroda H., Tanaka K., and Murata S. (2013) Involvement of Bag6 and the TRC pathway in proteasome assembly. Nat. Commun. 4, 2234. [DOI] [PubMed] [Google Scholar]
- 28. Binici J., and Koch J. (2014) BAG-6, a jack of all trades in health and disease. Cell. Mol. Life Sci. 71, 1829–1837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang Q., Liu Y., Soetandyo N., Baek K., Hegde R., and Ye Y. (2011) A ubiquitin ligase-associated chaperone holdase maintains polypeptides in soluble states for proteasome degradation. Mol. Cell 42, 758–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kao A., Chiu C. L., Vellucci D., Yang Y., Patel V. R., Guan S., Randall A., Baldi P., Rychnovsky S. D., and Huang L. (2011) Development of a novel cross-linking strategy for fast and accurate identification of cross-linked peptides of protein complexes. Mol. Cell. Proteomics 10, M110.002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Upla P., Kim S. J., Sampathkumar P., Dutta K., Cahill S. M., Chemmama I. E., Williams R., Bonanno J. B., Rice W. J., Stokes D. L., Cowburn D., Almo S. C., Sali A., Rout M. P., and Fernandez-Martinez J. (2017) Molecular architecture of the major membrane ring component of the nuclear pore complex. Structure 25, 434–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fernandez-Martinez J., Kim S. J., Shi Y., Upla P., Pellarin R., Gagnon M., Chemmama I. E., Wang J., Nudelman I., Zhang W., Williams R., Rice W. J., Stokes D. L., Zenklusen D., Chait B. T., et al. (2016) Structure and function of the nuclear pore complex cytoplasmic mRNA export platform. Cell 167, 1215–1228.e25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sali A., Berman H. M., Schwede T., Trewhella J., Kleywegt G., Burley S. K., Markley J., Nakamura H., Adams P., Bonvin A. M., Chiu W., Peraro M. D., Di Maio F., Ferrin T. E., Grunewald K., et al. (2015) Outcome of the first wwPDB Hybrid/Integrative Methods Task Force Workshop. Structure 23, 1156–1167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shi Y., Fernandez-Martinez J., Tjioe E., Pellarin R., Kim S. J., Williams R., Schneidman-Duhovny D., Sali A., Rout M. P., and Chait B. T. (2014) Structural characterization by cross-linking reveals the detailed architecture of a coatomer-related heptameric module from the nuclear pore complex. Mol. Cell. Proteomics 13, 2927–2943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shi Y., Pellarin R., Fridy P. C., Fernandez-Martinez J., Thompson M. K., Li Y., Wang Q. J., Sali A., Rout M. P., and Chait B. T. (2015) A strategy for dissecting the architectures of native macromolecular assemblies. Nat. Methods 12, 1135–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sali A., and Blundell T. L. (1993) Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 37. Söding J., Biegert A., and Lupas A. N. (2005) The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Subbotin R. I., and Chait B. T. (2014) A pipeline for determining protein-protein interactions and proximities in the cellular milieu. Mol. Cell. Proteomics 13, 2824–2835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Collart M. A., and Panasenko O. O. (2012) The Ccr4–not complex. Gene 492, 42–53 [DOI] [PubMed] [Google Scholar]
- 40. Silva G. M., Netto L. E., Simões V., Santos L. F., Gozzo F. C., Demasi M. A., Oliveira C. L., Bicev R. N., Klitzke C. F., Sogayar M. C., and Demasi M. (2012) Redox control of 20S proteasome gating. Antioxid. Redox Signal. 16, 1183–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kajava A. V., Gorbea C., Ortega J., Rechsteiner M., and Steven A. C. (2004) New HEAT-like repeat motifs in proteins regulating proteasome structure and function. J. Struct. Biol. 146, 425–430 [DOI] [PubMed] [Google Scholar]
- 42. Bose S., Stratford F. L., Broadfoot K. I., Mason G. G., and Rivett A. J. (2004) Phosphorylation of 20S proteasome α subunit C8 (α7) stabilizes the 26S proteasome and plays a role in the regulation of proteasome complexes by γ-interferon. Biochem. J. 378, 177–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shin K. J., Wall E. A., Zavzavadjian J. R., Santat L. A., Liu J., Hwang J. I., Rebres R., Roach T., Seaman W., Simon M. I., and Fraser I. D. (2006) A single lentiviral vector platform for microRNA-based conditional RNA interference and coordinated transgene expression. Proc. Natl. Acad. Sci. U.S.A. 103, 13759–13764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ran F. A., Hsu P. D., Lin C. Y., Gootenberg J. S., Konermann S., Trevino A. E., Scott D. A., Inoue A., Matoba S., Zhang Y., and Zhang F. (2013) Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154, 1380–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fang L., Wang X., Yamoah K., Chen P. L., Pan Z. Q., and Huang L. (2008) Characterization of the human COP9 signalosome complex using affinity purification and mass spectrometry. J. Proteome Res. 7, 4914–4925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yu C., Huszagh A., Viner R., Novitsky E. J., Rychnovsky S. D., and Huang L. (2016) Developing a multiplexed quantitative cross-linking mass spectrometry platform for comparative structural analysis of protein complexes. Anal. Chem. 88, 10301–10308 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.