

Abstract
Sterol regulatory element-binding proteins (SREBPs) in the fission yeast Schizosaccharomyces pombe regulate lipid homeostasis and the hypoxic response under conditions of low sterol or oxygen availability. SREBPs are cleaved in the Golgi through the combined action of the Dsc E3 ligase complex, the rhomboid protease Rbd2, and the essential ATPases associated with diverse cellular activities (AAA+) ATPase Cdc48. The soluble SREBP N-terminal transcription factor domain is then released into the cytosol to enter the nucleus and regulate gene expression. Previously, we reported that Cdc48 binding to Rbd2 is required for Rbd2-mediated SREBP cleavage. Here, using affinity chromatography and mass spectrometry experiments, we identified Cdc48-binding proteins in S. pombe, generating a list of many previously unknown potential Cdc48-binding partners. We show that the established Cdc48 cofactor Ufd1 is required for SREBP cleavage but does not interact with the Cdc48-Rbd2 complex. Cdc48-Ufd1 is instead required at a step prior to Rbd2 function, during Golgi localization of the Dsc E3 ligase complex. Together, these findings demonstrate that two distinct Cdc48 complexes, Cdc48-Ufd1 and Cdc48-Rbd2, are required for SREBP activation and low-oxygen adaptation in S. pombe.
Keywords: ATPases associated with diverse cellular activities (AAA), E3 ubiquitin ligase, hypoxia, membrane transport, Schizosaccharomyces pombe, transcription regulation, Cdc48, SREBP, VCP, p97
Introduction
Sterol regulatory element-binding proteins (SREBP)2 are basic helix-loop-helix–leucine zipper transcription factors that regulate sterol homeostasis in many eukaryotes. In Schizosaccharomyces pombe, there are two SREBP homologs, Sre1 and Sre2. Sre1 regulates both sterol homeostasis and low oxygen adaptation (1). Sre1 and Sre2 are translated as precursors, existing as two-pass transmembrane proteins in the endoplasmic reticulum (ER). When ergosterol levels decrease, SREBP cleavage-activating protein (Scp1) brings Sre1 to the Golgi for cleavage and membrane release of the N-terminal transcription factor domain into the cytosol (1). Sre2 lacks the C-terminal domain that binds Scp1; therefore, Sre2 is instead constitutively transported to the Golgi and cleaved (1). The biological role of Sre2 is largely unknown, although it has been shown to repress flocculation (2).
Large-scale genetic screens using the S. pombe deletion collection and methyl methanesulfonate (MMS) mutagenesis identified seven genes (dsc1–dsc5, cdc48, and rbd2) that are required for activation of both Sre1 and Sre2 (3–6). Dsc1–Dsc5 are subunits of a Golgi-resident E3 ligase complex (the Dsc E3 ligase complex), which is homologous to the Saccharomyces cerevisiae Tul1 E3 ligase that has a recently ascribed function in vacuolar quality control (3, 7). Rbd2 is a Golgi-resident rhomboid protease that is required for cleavage of SREBPs at the Golgi. Based on these findings, our current model is that the Dsc E3 ligase complex ubiquitinylates SREBP to target it to Rbd2 for cleavage. However, Rbd2 may not be the only protease involved as it is predicted to cleave the SREBP in the second transmembrane segment or luminal loop, whereas the active N-terminal transcription factor terminates before the first transmembrane segment (6). Indeed, in Aspergillus nidulans, where this system is conserved, a signal peptide peptidase provides the second cleavage of the SREBP homolog SrbA, indicating a requirement for two proteases in that species (8). However, the homologous peptidase is not required for SREBP cleavage in S. pombe (6). Alternatively, the cytosolic N terminus could be mechanically extracted from the membrane before it can act as a transcription factor. In the mammalian system, Fleig et al. (9) showed that Cdc48 acts downstream of substrate cleavage by the rhomboid protease RHBDL4 during ERAD.
cdc48 is the fission yeast homolog of mammalian VCP (also known as p97), a highly conserved gene that codes for an essential AAA+ ATPase making up 1% of total cellular protein (10). Cdc48 is a separase, using the energy from ATP hydrolysis for mechanical remodeling of ubiquitinylated substrates (11). This function is required for many cellular processes, including ERAD, autophagy, endosomal trafficking, chromosome-associated degradation, ribosomal quality control, membrane fusion, and cell cycle control (10–12). Cdc48 exists as a homohexamer, with each monomer consisting of an N-terminal domain (N domain), a D1 domain, a D2 domain, and a C-terminal domain (C domain). Each D1 and D2 domain has an ATP-binding site and hydrolysis site, for a total of 12 ATP-binding sites in the hexamer. ATP binding to D1 is required for ATP hydrolysis by the D2 domain, whereas ATP hydrolysis by D2 accounts for the majority of ATP hydrolyses by Cdc48 (13). Substrate binding inhibits ATP hydrolysis in the D1 domain but stimulates ATP hydrolysis in the D2 domain, resulting in an overall increase in Cdc48 ATP hydrolysis upon substrate binding (14). Recent in vitro work suggests that substrate unfolding occurs via ATP-driven transport of ubiquitinylated substrate through the central pore of the Cdc48 hexamer, after which the substrate is partially deubiquitinylated and released (14). A series of Cdc48 cofactors target this ubiquitous protein to specific substrates and regulate its ATPase activity (11, 15). Autosomal dominant disease mutations clustered at the interface between the human VCP N and D1 domains change the balance of cofactors binding to VCP (16, 17). These mutations lead to degenerative neuronal disorders, including familial ALS, parkinsonism, and Inclusion Body Myopathy associated with Paget disease of bone and Frontotemporal Dementia (IBMPFD) (17–24).
Previous studies regarding the role of cdc48 in SREBP cleavage highlight its versatility. Dsc5, the fission yeast homolog of FAF1, contains a canonical Cdc48-binding UBX domain that allows Cdc48 to bind the Dsc E3 ligase, but this domain is not required for SREBP cleavage (4). This suggests that the interaction between Cdc48 and the Dsc E3 ligase is not required for SREBP activation. What role this interaction plays in cells is unknown. Additionally, recent studies showed that Cdc48 binds Rbd2 through the SHP domain at the C terminus of Rbd2 and that loss of this interaction inhibits SREBP cleavage (6). Rbd2 is not stably associated with the Dsc E3 ligase; therefore, Cdc48 may serve to shuttle ubiquitinylated SREBP from the Dsc E3 ligase to Rbd2. Indeed, experiments suggest that Cdc48 serves as a substrate adaptor between Rbd2 and SREBP because the block in SREBP cleavage due to a failure to form the Rbd2-Cdc48 complex can be overcome by overexpression of Rbd2 (6).
The exact function of Cdc48 at Rbd2 is unknown, and Cdc48 may have additional roles in the pathway before or after cleavage of substrate by Rbd2. To determine whether cdc48 performs additional roles during SREBP activation, we extended our analysis of cdc48 point mutants in S. pombe (4). Here, we report identification of two new alleles of cdc48 that impact SREBP cleavage. Five cdc48 mutants show defects in both Sre1 and Sre2 cleavage but had minimal impact on Cdc48 protein level or cellular growth rate. Affinity chromatography followed by mass spectrometry experiments identified Cdc48-binding proteins in S. pombe, revealing many novel interacting proteins. Of these binding partners, we demonstrate that the substrate-recruiting Cdc48 cofactor Ufd1 is required for SREBP cleavage. Importantly, we find that the Cdc48-Ufd1 complex is functionally distinct from the previously described Cdc48-Rbd2 complex. Instead, we show that Cdc48-Ufd1 acts prior to Cdc48-Rbd2, during Golgi localization of the Dsc E3 ligase complex. Therefore, Cdc48 plays multiple, distinct roles in SREBP cleavage.
Results
Identification of cdc48 alleles
cdc48 is required for SREBP cleavage in fission yeast (4, 6), but whether this multipurpose enzyme has additional functions in the SREBP pathway is unknown. Further analysis of our four cdc48 mutants revealed that two of the four showed relatively weak SREBP cleavage defects that are not suitable for dissecting the role of Cdc48 during SREBP cleavage (4). To expand our set of cdc48 alleles and to identify other genes involved in SREBP activation, we revisited our MMS mutagenesis screen and performed additional linkage analysis on uncharacterized mutants (4). As reported previously, this screen was performed by generating a reporter strain in which Sre1-binding sites were in the promoter of the ura4 gene, such that when Sre1 is functional, orotidine 5′-phosphate decarboxylase (Ura4) is produced (4). This reporter strain was mutagenized with either 0.024 or 0.012% MMS, and the resulting isolates were plated on 5-fluoroorotic acid (5-FOA), which is converted to the toxic 5-fluorouracil by Ura4, thereby selecting for isolates with deficient Sre1 activity. The majority of surviving mutants (86%) from the original screen phenocopied sre1 deletion and were sensitive to cobalt chloride (CoCl2), a hypoxia mimetic (Fig. 1A).
Figure 1.

Identification of cdc48 alleles. A, flow chart of results from two MMS mutagenesis screens (0.024 and 0.012% (w/v)). Red numbers denote the number of isolates in each category. Numbers in parentheses are the number of isolates with mutations confirmed by sequencing (see supplemental Table S1 for specific mutations). Aside from the first box, all numbers are pooled from the two MMS dosages. B, comparison of gene-coding sequence size with number of isolated mutants in MMS mutagenesis screen. Best-fit line calculated against non-essential genes (filled markers), R2 = 0.87. C, table of remade cdc48 alleles showing amino acid changes in S. pombe, corresponding to homologous amino acids in humans and corresponding allele from our previous report (4). D, line diagram of S. pombe cdc48, where the orange box indicates the N domain to which the majority of cofactors bind. The blue box indicates the minor ATPase D1 domain required for hexamer formation. The green box indicates the major ATPase D2 domain required for force generation. Yellow boxes in both domains indicate sequences essential for ATP binding and hydrolysis. The gray box indicates the C domain to which a minority of cofactors bind. Red asterisks indicate the locations of our cdc48 point mutations.
Cobalt-sensitive mutants were transformed with plasmids expressing sre1+ and scp1+ to identify these frequent mutants. For mutants not rescued by these plasmids, we performed linkage analysis by mating uncharacterized mutants with known SREBP pathway mutants and tested whether the resulting spores grew on CoCl2. Matings from which no spores grew on CoCl2 indicated linkage of the uncharacterized mutant to the known gene, most likely because the uncharacterized mutation was also within the known gene. In many cases, we identified the precise mutation in these strains by PCR amplification and sequencing of the gene of interest. After this analysis, 196 of the 305 cobalt chloride-sensitive mutants (64%) were assigned to linkage groups, although 109 strains remain to be tested (Fig. 1A). The specific mutations in the sequenced genes are available in supplemental Table S1. The only new gene identified in this search was the essential translation initiation RNA helicase sum3, which was identified via whole genome sequencing of two linked mutagenized strains. Further analysis indicated that the sum3 mutants were not rescued by Sre1N expression and had intermediate Sre1 cleavage defects (data not shown). We concluded that the role of sum3 in this pathway was likely a non-specific stress response, and we did not study it further.
As expected for non-essential genes, we observed a linear relationship between the number of strains isolated and the length of the coding sequence (Fig. 1B). Notably, cdc48 and sum3 are essential and lie below the line, with fewer strains identified than would be expected for the given coding region length. In other words, the gene sequence that can be viably mutated is smaller for essential genes than non-essential genes. Analysis of the number of strains per gene found through the MMS screen indicated that the screen could identify non-essential genes larger than 750 base pairs (Fig. 1B). An additional 1100 S. pombe non-essential genes fall below this threshold, indicating that additional non-essential and essential genes required for Sre1 activity may remain unidentified.
This new analysis identified two cdc48 point mutations (A366D and R764C) in addition to the four previously described (Fig. 1C) (4). To further examine these mutants, we compared the S. pombe cdc48 point mutants to the human VCP sequence, to which the fission yeast protein shares 70% identity. All six mutated fission yeast cdc48 residues are identical in humans (Fig. 1C). These six mutations are dispersed throughout this essential protein and are not clustered in three-dimensional space (Fig. 1D and data not shown). Importantly, none of these cdc48 mutants are temperature-sensitive (data not shown). Additionally, none are found in the N domain, which is the major site of cofactor binding (11). One of these mutants, cdc48-8, is in the known ATP hydrolysis region of the D1 domain (Fig. 1D).
SREBP activation requires cdc48
Cdc48 is a versatile protein that may function in multiple steps of SREBP activation. Our previous studies of cdc48 mutants used the original isolates from the MMS mutagenesis screen (4). To evaluate and directly compare the cdc48 alleles, we recreated all six cdc48 mutations in a wild-type, non-mutagenized strain background. These strains were generated by homologous recombination, and all strains, including the isogenic wild type, were marked with the nourseothricin resistance gene (see “Experimental procedures”). Because these strains are in a non-mutagenized, marked background, we gave them the new allele numbers cdc48-5 through cdc48-10 to differentiate them from those previously studied (Fig. 1C).
To characterize these new mutant strains, we first assayed growth on CoCl2. We observed no growth defect for cdc48-7 and complete CoCl2 growth defects for the other five mutants (Fig. 2A). To compare Sre1N production in all cdc48 mutant strains, we assayed Sre1 cleavage by Western blotting. We cultured wild-type and the indicated cdc48 mutant strains for 0 or 4 h in the absence of oxygen. We then probed whole-cell lysates with anti-Sre1N, which detects both the full-length ER membrane-bound precursor form (P) as well as the cleaved N-terminal transcription factor form (N). Wild-type cells accumulated Sre1N after 4 h of growth under low oxygen (Fig. 2, B and C, lanes 1 and 2). The cdc48 mutant strains showed a range of Sre1N production defects, from complete blocks (cdc48-8, -9, and -10) to partial defects (cdc48-5 and -6) (Fig. 2, B and C, lanes 3–14). Additionally, we examined cleavage of an N-terminally FLAG-tagged Sre2 model substrate, Sre2-MS, which recapitulates Sre2 constitutive activation (25). The cdc48 mutant strains showed Sre2-MS cleavage defects consistent with their Sre1 cleavage defects, as had been previously shown for cdc48-4 (Fig. 2, D and E) (25). Notably, we observed no CoCl2 growth, Sre1, or Sre2-MS cleavage defects in cdc48-7, which is inconsistent with our previously published results (Fig. 2, B and C, lanes 7 and 8, and D, lanes 7 and 8) (4). This may be due to the highly mutagenized background of the original mutant and validates our decision to remake these strains in a non-mutagenized strain. Therefore, we conclude that cdc48-7 is a mutant allele with no effect on SREBP cleavage and serves as a wild-type control. Together, these data indicate that cdc48 is required for SREBP cleavage in S. pombe at a step that is common to both Sre1 and Sre2 activation.
Figure 2.

SREBP cleavage requires cdc48. A, wild-type cells or the indicated mutants (5000 cells) were grown on rich medium plus or minus CoCl2 for 2 or 10 days, respectively. B, Western blots, probed with monoclonal anti-Sre1 IgG (5B4) and polyclonal anti-Dsc5 IgG (for loading), of lysates treated with alkaline phosphatase for 1 h from wild-type cells and the indicated cdc48 mutants grown for 0 or 4 h in the absence of oxygen. P and N denote precursor and cleaved N-terminal transcription factor forms, respectively. Asterisk denotes non-specific band. C, quantification of Sre1 N terminus from B of two biological replicates each denoted by different marker symbols. The quantity of Sre1N was normalized to Dsc5 for loading and then to the WT 4-h sample (lane 2) for comparison between blots. D, Western blots, probed with monoclonal anti-FLAG M2 and polyclonal anti-Dsc5 IgG (for loading), of lysates treated with alkaline phosphatase for 1 h from wild-type cells and the indicated cdc48 mutants containing a plasmid expressing sre2-MS (+) or the empty vector (−) grown in the presence of oxygen. P and N denote precursor and cleaved N-terminal transcription factor forms, respectively. E, quantification of Sre2-MS cleavage from D of three biological replicates each denoted by different marker symbols. The quantity of precursor and N terminus were normalized first to empty vector and then to Dsc5 for loading. Percent cleavage in each sample was calculated by dividing the normalized quantity of N terminus by the total signal (N+P). Error bars are 1 S.D. (**, p < 0.01 versus WT by two-tailed Student's t test).
cdc48 mutations do not exhibit major defects in Cdc48 expression or cell growth
To better understand how these cdc48 mutations affect SREBP cleavage, we characterized their effects on Cdc48 protein accumulation and cell growth. To assay Cdc48 expression, we probed whole-cell lysates from each strain with anti-Cdc48 polyclonal antibody (Fig. 3A). Despite some statistically significant changes, all mutants had levels of Cdc48 protein similar to the wild type, and expression levels did not show a correlation with strength of SREBP cleavage defect (Fig. 3B). This is consistent with the fact that cdc48 is an essential gene, and large changes in expression may not be tolerated. These data suggest that differential expression is not the cause of the observed SREBP cleavage defects.
Figure 3.

cdc48 mutants have minimal effect on Cdc48 expression or growth rate. A, Western blottings, probed with polyclonal anti-Cdc48 IgG and polyclonal anti-Dsc5 IgG (for loading), of lysates from wild-type cells and the indicated cdc48 mutants. B, quantification of A from three biological replicates, each indicated by a different marker shape, normalized for loading to Dsc5 and then normalized to wild-type cells for comparison among blots. Error bars are 1 S.D. (*, p < 0.05; **, p < 0.01 versus WT by two-tailed Student's t test). C, indicated strains were grown in liquid culture for 12 h. Cell density was measured by absorbance at 600 nm every 3 h. Doubling times were calculated using 3- and 9-h optical density readings using the formula: doubling time = (culture time × log(2))/log(final OD) − log(initial OD). Doubling times are displayed as mean ± S.D. from four biological replicates as indicated by marker shape. (*, p < 0.05; **, p < 0.01 versus WT by two-tailed Student's t test).
To further examine the overall cellular effects of these cdc48 point mutations, we assayed growth rate. We grew wild-type and cdc48 mutants in liquid culture in the presence of oxygen for 12 h and calculated the doubling time. Of the six cdc48 mutants, only cdc48-9 had a dramatically slower doubling time than wild-type cells under normoxic conditions (Fig. 3C). Because the cdc48-8, cdc48-9, and cdc48-10 mutants have equally strong blocks in SREBP cleavage but different growth rates, it is unlikely that the change in growth rate is responsible for the block in SREBP cleavage. These results suggest that the effects of our cdc48 mutations on essential Cdc48 activities are minimal.
Identification of Cdc48-binding proteins in S. pombe
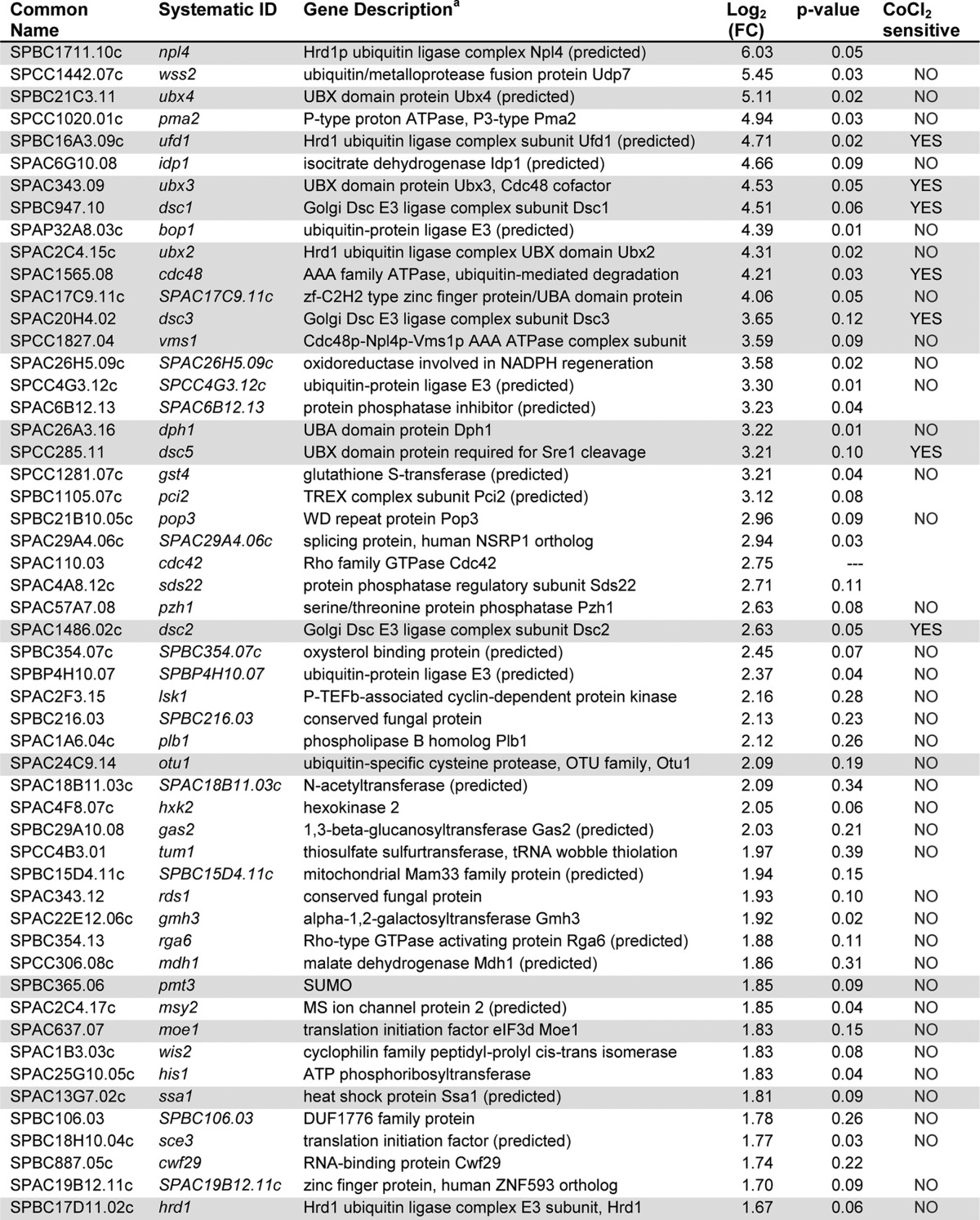
It is well-established in the literature that a complex network of cofactors direct Cdc48 to specific targets. However, we did not identify any Cdc48 cofactors that are required for SREBP cleavage in this MMS mutagenesis or a previous screen of non-essential genes (Fig. 1A) (5). Identification and assessment of Cdc48 cofactors could clarify the roles for Cdc48 in SREBP cleavage. To identify Cdc48 cofactors in fission yeast, we purified Cdc48 using a C-terminal 5xFLAG tag and quantitatively identified all bound proteins using tandem mass tag mass spectrometry (26). Tagging Cdc48 with 5xFLAG did not alter Cdc48 function as judged by the ability to accumulate Sre1N to wild-type levels under low oxygen (Fig. 4A, lane 2 versus 4). Purification of Cdc48–5xFLAG with anti-FLAG-conjugated magnetic beads recovered the majority of Cdc48 protein, and no Cdc48 was isolated in the untagged strain (Fig. 4B, lanes 5 and 6). Quantitative mass spectrometry analysis of the bound fractions from wild-type and Cdc48–5xFLAG lysates identified 2283 proteins, of which 52 were enriched in the Cdc48–5xFLAG sample at least two standard deviations from the mean (fold change ≥3.2) (Fig. 4C, red dots, and Table 1). The full list of identified proteins and peptides is available in supplemental Table S2. Of note, Dsc1, Dsc2, Dsc3, and Dsc5 were highly enriched in the Cdc48-bound population (Table 1). This result is consistent with our published observation that Cdc48 binds the Dsc E3 ligase through the Dsc5-UBX domain independent of any role in SREBP cleavage (4). Interestingly, Rbd2 was not identified as a Cdc48-binding protein. This could either be due to an inability to detect Rbd2 peptides by mass spectrometry or the reported transient nature of the Cdc48-Rbd2 interaction (6).
Figure 4.
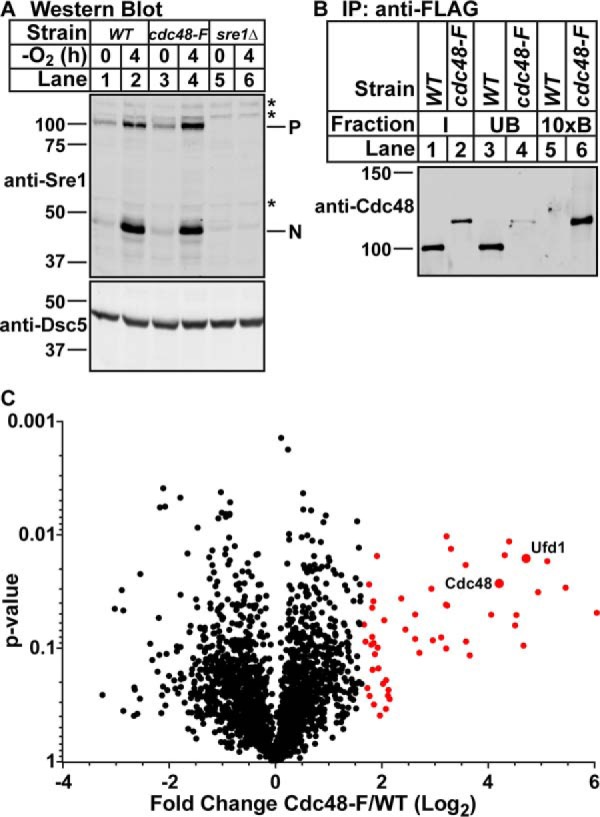
Identification of Cdc48-binding proteins in S. pombe. A, Western blottings, probed with monoclonal anti-Sre1 IgG (5B4) and polyclonal anti-Dsc5 IgG (for loading), of lysates treated with alkaline phosphatase for 1 h from wild-type cells, cdc48-5xFLAG (cdc48-F) cells, or sre1Δ cells grown for 0 or 4 h in the absence of oxygen. P and N denote precursor and cleaved N-terminal transcription factor forms, respectively. Asterisks denote non-specific bands. B, Cdc48 was immunoprecipitated (IP) from wild-type or cdc48-5xFLAG cells using monoclonal anti-FLAG M2 IgG as described under “Experimental procedures.” Input, unbound, and 10-fold enriched bound fractions were analyzed by Western blotting using polyclonal anti-Cdc48 IgG. The blot is representative of four biological replicates. C, volcano plot of all proteins identified during TMT mass spectrometry of Cdc48–5xFLAG bound proteins. Points in red are proteins with enrichment >2 S.D. from the mean in the Cdc48–5xFLAG versus wild-type samples. p values were calculated using two-way ANOVA with quantile normalization for three biological replicates.
Table 1.
Cdc48-binding proteins in S. pombe
Cdc48-binding proteins, identified by TMT mass spectrometry, of three biological replicates, are enriched at least 2 S.D. from the mean. Rows highlighted in gray are known Cdc48-binding proteins.
a Descriptions were obtained from PomBase (www.pombase.org; please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site) (50) with some additional hand editing.
To determine which Cdc48-binding proteins were required for SREBP processing, we tested deletions of the non-essential genes in S. pombe for growth on CoCl2 (27). In addition, we created deletions of those genes not present in the Bioneer deletion collections and also tested temperature-sensitive mutants of the essential cofactor ufd1 (kind gift of the Boddy Lab) (28). We were unable to test a number of essential genes due to lack of availability of temperature-sensitive mutations and a failure to knock them down. Results of the CoCl2 growth assays can be found in Table 1. Interestingly, of all the genes tested only the ufd1 point mutants and ubx3Δ were sensitive to CoCl2 and not previously known components of the SREBP pathway. Both of these genes code for known Cdc48 cofactors (Table 1, gray rows). Upon further examination, the CoCl2 growth defect in ubx3Δ cells was minor, and we observed minor effects on Sre1 cleavage (data not shown). Therefore, we focused our analysis on Ufd1.
Ufd1 is a Cdc48 cofactor required for SREBP cleavage
Ufd1 is an established Cdc48 cofactor with homologs from yeast to humans (29, 30). Together with the cofactor Npl4, it is thought to direct Cdc48 to ERAD functions (31, 32). To test the requirement for ufd1 in SREBP activation, we tested three temperature-sensitive mutations in this essential gene using strains generated by the Boddy Lab (28). These wild-type and ufd1 mutant strains express ufd1-13xMyc from the endogenous locus. ufd1-7 and ufd1-8 have mutations in the conserved UFD1 domain, whereas ufd1-5 and ufd1-7 have mutations outside of that domain, near the C terminus (Fig. 5A) (28). We assayed growth of these wild-type and ufd1 mutant strains on CoCl2 and observed complete growth defects in the mutant strains consistent with our results in Table 1 (Fig. 5B). We observed decreased growth at semi-permissive temperature on rich medium (30 °C) (Fig. 5B) and in a liquid culture growth assay (Fig. 5C). These results are consistent with the previously published report that these strains are temperature-sensitive (28). To directly measure effects of the ufd1 mutations on SREBP activation, we assayed Sre1 and Sre2-MS cleavage in each strain. ufd1 mutants were deficient for Sre1 cleavage induction by low oxygen and had defects in Sre2-MS cleavage (Fig. 5, D–G). Notably, ufd1-5 and ufd1-7 had complete blocks in Sre2-MS cleavage, whereas ufd1-8 had a partial block indicating an incomplete defect. Importantly, all ufd1 mutants had normal Sre1 precursor levels, indicating no defect in Sre1 expression in these strains (Fig. 5D). These data suggest that ufd-5 and ufd-7 are the strongest mutants but that all three have significant effects on SREBP cleavage. Therefore, we concluded that ufd1 is a new component of the SREBP activation pathway.
Figure 5.

Ufd1 is a Cdc48 cofactor required for SREBP cleavage. A, table of ufd1 alleles used in this study. B, wild-type cells or the indicated mutants (5000 cells) were grown on rich medium plus or minus CoCl2 for 10 days at 30 °C. C, indicated strains were grown in liquid culture for 12 h at 30 °C. Cell density was measured by absorbance at 600 nm every 3 h. Doubling times were calculated using 3- and 9-h optical density readings using the formula: doubling time = (culture time × log(2))/log(final OD) −log(initial OD). Doubling times are displayed as mean ± S.D. for three biological replicates as indicated by marker shape. (**, p < 0.01 versus WT by two-tailed Student's t test). D, Western blottings, probed with monoclonal anti-Sre1 IgG (5B4) and polyclonal anti-Dsc5 IgG (for loading), of lysates treated with alkaline phosphatase for 1 h from wild-type cells, sre1Δ, or the indicated ufd1 mutants grown for 0 or 4 h in the absence of oxygen. P and N denote precursor and cleaved N-terminal transcription factor forms, respectively. Asterisk denotes non-specific band. E, quantification of Sre1 N terminus from D of two biological replicates each denoted by different marker symbols. The quantity of Sre1N was normalized to Dsc5 for loading and then to the WT 4 h sample (lane 2) for comparison between blots. F, Western blottings, probed with monoclonal anti-FLAG M2 and polyclonal anti-Dsc5 IgG (for loading), of lysates treated with alkaline phosphatase for 1 h from wild-type cells and the indicated ufd1 mutants containing a plasmid expressing sre2-MS (+) or the empty vector (−) grown in the presence of oxygen. P and N denote precursor and cleaved N-terminal transcription factor forms, respectively. The blot is representative of three biological replicates. G, quantification of Sre2-MS cleavage from F of three biological replicates each denoted by different marker symbols. The quantity of precursor and N terminus were normalized first to empty vector and then to Dsc5 for loading. Percent cleavage in each sample was calculated by dividing the normalized quantity of N terminus by the total signal (N+P). Error bars are 1 S.D. (*, p < 0.05; **, p < 0.01 versus WT by two-tailed Student's t test).
Cdc48-Ufd1 complex formation is altered in cdc48 and ufd1 mutants
Cdc48-Ufd1 complex formation is essential for Cdc48 function in cellular processes such as ERAD (31, 32). To examine how ufd1 mutations affect Cdc48-Ufd1 complex formation, we assayed Cdc48-Ufd1 binding. Immunopurification of Ufd1–13xMyc recovered less Cdc48 in ufd1 mutant cells compared with ufd1 wild-type cells (Fig. 6A, lanes 11–14). Notably, all ufd1 point mutants had lower Ufd1–13xMyc protein than wild-type cells in the input, although Cdc48 levels were normal (Fig. 6A, lanes 2–5). Given that ufd1 is essential, this suggests that reduced Ufd1–13xMyc expression may cause the observed growth defects and temperature sensitivity of these mutants (Fig. 5, B and C). However, after normalizing for Ufd1 levels, the amount of Cdc48 bound to mutant Ufd1–13xMyc was also significantly decreased compared with wild type (Fig. 6B), suggesting a defect in Cdc48-Ufd1 complex formation in these strains.
Figure 6.

Cdc48-Ufd1 complex formation is altered in ufd1 and cdc48 mutants. A and C, Nonidet P-40-solubilized membrane protein was prepared from WT (−), ufd1–13xmyc (WT), or the indicated ufd1 mutant cells, and Ufd1–13xMyc was immunoprecipitated (IP) with anti-Myc monoclonal IgG as described under “Experimental procedures.” Equal quantities of input, unbound, and bound fractions were analyzed by immunoblotting using polyclonal antibodies against Cdc48 and Myc. B and D, quantification of Cdc48 pulldown from A or C of four (A) or five (C) biological replicates each denoted by different marker symbols. The quantity of Cdc48 in the bound fractions was normalized to the quantity of Myc in the bound fractions and then to the WT sample for comparison between blots. Error bars are 1 S.D. (*, p < 0.05; **, p < 0.01 versus WT by two-tailed Student's t test).
To determine whether our cdc48 mutations also cause a defect in Cdc48-Ufd1 complex formation, we again assayed Cdc48-Ufd1 binding by immunopurification from strains expressing the seven cdc48 mutants in a wild-type ufd1-13xmyc background. Interestingly, mutant Cdc48 binding to Ufd1-13xMyc increased compared with wild type, with the exception of cdc48-7, which phenocopied wild-type cdc48 (Fig. 6, C and D). These data suggest that these mutations increase Cdc48-Ufd1 binding and that the cdc48 and ufd1 mutants do not have the same effects on Cdc48-Ufd1 complex formation, despite resulting in the same SREBP cleavage phenotype.
SREBP activation requires Cdc48-Ufd1 during Dsc E3 ligase Golgi localization
SREBP activation requires ER-to-Golgi transport of SREBP, the Dsc E3 ligase, and Rbd2, as well as Dsc E3 ligase activity and cleavage by Rbd2. Defects in any of these steps would result in the cleavage defect observed in cdc48 and ufd1 mutants. We previously demonstrated that Cdc48 is required for Rbd2 cleavage of SREBPs in the Golgi. Rbd2 binds Cdc48 through its C-terminal SHP domain, and disruption of Rbd2-Cdc48 binding blocks cleavage (6). To determine whether Ufd1 is involved in the function of Cdc48 at Rbd2, we tested whether Ufd1 physically associates with Cdc48 when bound to Rbd2. We performed an in vitro pulldown experiment from wild-type and ufd1-13xmyc lysates using GST-tagged Dsc5 UBX domain, GST-tagged Rbd2 SHP domain, or GST alone. As expected, both GST-UBX and GST-SHP efficiently purified Cdc48 from lysates (Fig. 7A, lanes 9–14) (6). However, only GST-UBX also purified Ufd1–13xMyc (Fig. 7A, lane 12 versus 14). The ability of Dsc5 UBX domain to purify Ufd1 suggests that a Cdc48-Ufd1 complex may interact with the Dsc E3 ligase in an uncharacterized function independent of SREBP cleavage (4). The fact that Ufd1–13xMyc is not bound to Cdc48 when Cdc48 is bound to the Rbd2-SHP domain suggests that Ufd1 is not part of the Cdc48-Rbd2 complex required for SREBP cleavage. These data indicate that two distinct Cdc48 complexes, Cdc48-Ufd1 and Cdc48-Rbd2, are required for SREBP activation in fission yeast.
Figure 7.

SREBP cleavage requires Cdc48-Ufd1 during Dsc E3 ligase Golgi localization. A, recombinant GST-HA-V5 control (GST), GST-Dsc5 UBX (Dsc5(323–425), UBX), and GST-Rbd2 C terminus (Rbd2(200–251), SHP) were bound to GST magnetic beads and incubated with S. pombe cytosol from wild-type cells (−) or ufd1–13xmyc cells (+). Input, unbound, and 10-fold enriched bound fractions were probed with monoclonal anti-Myc IgG, polyclonal anti-Cdc48 IgG, and monoclonal anti-GST IgG. The blot is representative of five replicates. B and D, Western blottings, probed with polyclonal anti-Sre2 IgG and polyclonal anti-Dsc5 IgG (for loading), of lysates treated with alkaline phosphatase for 1 h from wild-type, dsc1Δ (1Δ), or the indicated cdc48 (B) or ufd1 (D) mutant cells. rbd2 (+) or rbd2Δ (−) genotype is indicated. P and N denote precursor and cleaved N-terminal transcription factor forms, respectively. C and E, quantification of Sre2 precursor from B and D of three (B) or four (D) biological replicates each denoted by different marker symbols. The quantity of precursor was normalized to Dsc5 for loading and then to WT rbd2+ (lane 1) for comparison between blots. Error bars are 1 S.D. (*, p < 0.05; **, p < 0.01 versus WT rbd2+ by two-tailed Student's t test). Note, C, lane 3, has only two replicates, and therefore p values could not be calculated. F, Western blotting, probed with polyclonal anti-Dsc1 IgG, of Nonidet P-40-solubilized membrane protein from wild-type, dsc2Δ, dsc5ΔUBX (ΔUBX), rbd2Δ, or the indicated cdc48 or ufd1 mutant cells. M and I indicate mature and intermediate glycosylated forms, respectively. The blot is representative of three biological replicates. G, quantification of Dsc1 from F of three biological replicates each denoted by different marker symbols. The quantity of the mature form was divided by total Dsc1 signal for percent mature, allowing comparison between lanes and blots. Error bars are 1 S.D. (*, p < 0.05; **, p < 0.01 versus WT by two-tailed Student's t test).
To determine whether Cdc48-Ufd1 is required prior to SREBP cleavage by Rbd2, we performed a genetic epistasis test that takes advantage of a useful rbd2Δ phenotype. In rbd2Δ cells, uncleaved SREBP precursor proteins do not accumulate as in wild-type cells but instead are degraded (6). This degradation requires both the Dsc E3 ligase and the proteasome, and we proposed that it results from degradation of the ubiquitinylated SREBP precursor when Rbd2 cleavage does not occur. In contrast, all dsc deletions, cdc48, and ufd1 mutants have wild-type SREBP precursor levels (Figs. 2, B and D, and 5, D and F) (3, 4). Because these phenotypes are opposing, we can create double mutants between rbd2Δ and other pathway components and perform epistasis tests to determine their order of action in this SREBP activation pathway.
As shown previously, in wild-type cells endogenous Sre2 is constitutively cleaved, and primarily the N-terminal form was detected (Fig. 7, B–E, lane 1). In contrast, rbd2 deletion resulted in a failure to cleave Sre2 without a corresponding increase in Sre2 precursor, resulting in a minimal Sre2 signal (Fig. 7, B and C, lane 2) (6). As expected, rbd2Δ dsc1Δ cells accumulated Sre2 precursor (Fig. 7, B and C, lane 3) because the Dsc E3 ligase acts before Rbd2 during SREBP activation and is required for precursor degradation by the proteasome (6). This is consistent with our model that the SREBP is ubiquitinylated by the Dsc E3 ligase before transfer to Rbd2 for cleavage.
In cdc48 mutant cells, Sre2 cleavage defects ranged from none (cdc48-7), partial (cdc48-5 and -6), to strong (cdc48-8, -9, and -10) (Fig. 7, B and C), consistent with observed Sre1 and Sre2-MS defects (Fig. 2). When the cdc48 mutants were combined with rbd2Δ, the Sre2 precursor was retained in the strongest mutants (cdc48-8, -9, and -10) and partially retained in the weaker mutants (cdc48-5 and -6) (Fig. 7, B and C, lanes 3–14). This is consistent with the magnitude of the SREBP cleavage defect in each cdc48 allele (Fig. 2, B–E). These results demonstrate that cdc48 functions prior to rbd2. Likewise, ufd1 rbd2Δ double mutants also accumulated Sre2 precursor, confirming that Ufd1 and Cdc48 act before Sre2 cleavage by Cdc48-Rbd2 (Fig. 7, D and E, lane 2 versus lanes 6 and 8). The ufd1-5 rbd2Δ combination could not be tested because we were unable to generate this strain by mating or homologous recombination. Together, these data suggest that distinct Cdc48 complexes play multiple roles during SREBP cleavage, one of which is prior to Rbd2 cleavage at the Golgi.
Cdc48-Ufd1 is required for SREBP activation prior to cleavage by Rbd2, but it is unclear what role it performs at that stage. To test which step of SREBP cleavage is defective in cdc48 and ufd1 mutants, we assayed Dsc E3 ligase localization in these cells by taking advantage of the fact that the Dsc1 subunit is glycosylated in the Golgi. Glycosylation causes a change in apparent molecular weight on an SDS-polyacrylamide gel when dsc1–dsc4 are deleted, when the Dsc E3 ligase is catalytically inactivated, or when the cognate E2, ubc4, is inactivated (33).
To examine Dsc E3 ligase localization, we subjected microsomal protein preparations from cdc48 and ufd1 mutant cells to SDS-PAGE and probed for Dsc1. As reported previously, wild-type cells showed primarily a higher molecular weight (Golgi) Dsc1 band representing 55% maturely glycosylated, whereas dsc2Δ cells showed only a lower molecular weight (ER) Dsc1 band representing 23% maturely glycosylated (Fig. 7, F and G, lane 1 versus 8) (33). Consistent with the wild-type Sre1 cleavage observed in dsc5-ΔUBX cells, deletion of the UBX domain also had no effect on Dsc1 glycosylation (Fig. 7F, lane 9). Importantly, rbd2 deletion also showed a wild-type Dsc1 glycosylation pattern, indicating that Rbd2 is not required for Dsc1 Golgi localization (Fig. 7F, lane 10). When we examined Dsc1 glycosylation in the strongest cdc48 and ufd1 mutant strains (cdc48-8, -9, and -10 and ufd1–5 and -7), we observed accumulation of the lower molecular weight form and between 25 and 35% maturely glycosylated, phenocopying dsc2Δ (Fig. 7, F and G, lanes 5–7, 12, and 13). cdc48-7 had wild-type Dsc1 glycosylation, whereas cdc48-5 and -6 and ufd1–8 had partial Dsc1 glycosylation defects with 38–48% maturely glycosylated form, consistent with their SREBP cleavage effects (Fig. 7, F and G, lanes 2–4 and 14). Because neither the dsc5-ΔUBX nor the rbd2Δ cells showed Dsc1 glycosylation defects, the impacts of the cdc48 mutants on Dsc1 glycosylation are likely not due to Cdc48 binding to either of these proteins. These data suggest that the Cdc48-Ufd1 complex is required for Dsc E3 ligase Golgi localization in a role separate from the binding of Cdc48 to Dsc5 or Rbd2.
Discussion
Our previous efforts to discover components of the S. pombe SREBP activation pathway identified the AAA+ ATPase Cdc48 but failed to identify any Cdc48 cofactors. Here, we analyzed six cdc48 point mutations and performed mass spectrometry identification of Cdc48-binding proteins to further elucidate the role of Cdc48 and any cofactors in SREBP activation. These data add to a growing body of evidence supporting multiple interactions between Cdc48 and SREBP pathway components. This evidence leads to a model in which the Dsc E3 ligase only traffics from the ER to the Golgi when it is a functional E3 ligase and when Cdc48 and Ufd1 are active. Sre1 is retained in the ER by Scp1 under high sterol conditions, but under low sterol conditions Sre1-Scp1 traffics to the Golgi where it encounters the Dsc E3 ligase and Rbd2-Cdc48 for cleavage and release of the N-terminal transcription factor (Fig. 8).
Figure 8.

SREBP activation mechanism in S. pombe. Model outlining the steps of SREBP transport and cleavage in the Golgi, incorporating the two roles of Cdc48 at Dsc E3 ligase Golgi localization and Rbd2.
This updated model is supported by several new observations in this study. First, in this report we used cdc48 and ufd1 point mutants to show that the Cdc48 cofactor Ufd1 is required for SREBP cleavage and that Cdc48-Ufd1 act at an early step in the activation pathway, namely Golgi localization of the Dsc E3 ligase complex (Fig. 7). cdc48 and ufd1 mutants show defects in Dsc1 glycosylation, suggesting that the Dsc E3 ligase is trapped in the ER where it is unable to participate in SREBP activation (Fig. 7F). Importantly, in all cases, the strength of the Dsc1 glycosylation defect is consistent with the strength of the SREBP cleavage defect, indicating that the Dsc E3 ligase localization defect causes the SREBP cleavage defect.
In our characterization of the ufd1 mutants, we observed growth and Ufd1 expression defects (Figs. 5, B and C, and 6A). This is likely due to the temperature-sensitive nature of these strains, but it may have contributed to the SREBP cleavage defects (28). These ufd1 mutants also pulled down less Cdc48 protein than wild-type Ufd1–13xMyc, even when controlling for Ufd1 expression (Fig. 6, A and B). Of the three ufd1 mutants, only ufd1-7 has a mutation in the predicted Cdc48-binding region (aa 219–235 based on homology to S. cerevisiae), and none are mutants in the predicted Npl4-binding region (aa 251–266) (28, 32). ufd1-7 and ufd1-8 also have mutations in the predicted polyubiquitin-binding site (34). The ufd1 mutations are not clustered in three-dimensional space (data not shown). It may be that these mutants more generally destabilize the protein and that results in a defect in Cdc48-Ufd1 binding. This supports our hypothesis that it is the Cdc48-Ufd1 complex that is required for SREBP cleavage.
Interestingly, ufd1 was not identified in our MMS screen (Fig. 1B). This may be due to the fact that ufd1 is an essential gene with a coding sequence of only 1029 bp, likely too small to come through our screen (Fig. 1B). Similarly, the Ufd1-binding partner Npl4 is also essential and could have been missed in our screen. Although the literature suggests that Ufd1 usually is in complex with the binding partner Npl4 when bound to Cdc48, we were unable to generate temperature-sensitive mutants or tagged versions of npl4, and therefore we cannot comment on its role in SREBP activation.
The specific function for Cdc48-Ufd1 during Dsc E3 ligase Golgi localization is unknown. Our previous work showed that the Dsc E3 ligase enzymatic function is also required for proper Golgi localization (33). One attractive hypothesis is that both Dsc E3 ligase function and Cdc48 are required for the same process, which permits traffic to the Golgi. Perhaps, the Dsc E3 ligase and Cdc48 act to ubiquitinylate and degrade a repressor protein, whose removal is required for Dsc E3 ligase packaging into COPII vesicles. This would provide a quality control mechanism to ensure that the Dsc E3 ligase is functional before being shipped to its final destination.
The second role for Cdc48 in the SREBP activation pathway is during SREBP cleavage by the rhomboid protease Rbd2 (Fig. 8). We previously showed that Cdc48 interacts with Rbd2 through the Rbd2 SHP domain and that this interaction is required for SREBP cleavage (6). The function performed by Cdc48 at Rbd2 is unknown but could be as a substrate adaptor, as an activator of Rbd2 enzymatic activity, and/or to release the cleaved SREBP N terminus into the cytosol. Here, we show that both cdc48 and ufd1 mutants are epistatic to rbd2Δ and retain the Sre2 precursor in the absence of rbd2 (Fig. 7, B–E). This phenocopies dsc1Δ rbd2Δ and supports the requirement for Cdc48-Ufd1 in Dsc E3 ligase function. Furthermore, we showed that the Rbd2-SHP domain binds to Cdc48 but not Ufd1 in vitro (Fig. 7A). Although it is possible that Ufd1 interacts with Rbd2 at a location separate from the SHP domain, this suggests that Ufd1 cannot interact with Rbd2 through Cdc48. Together, these data indicate that Cdc48-Ufd1 is distinct from Cdc48-Rbd2 and that two Cdc48 complexes are required for the SREBP activation pathway.
Finally, we previously published the interaction between Cdc48 and the Dsc E3 ligase through the Dsc5 UBX domain (4). This interaction is not required for SREBP activation. However, it could play a role in a non-SREBP function of the Dsc E3 ligase in the Golgi, such as Golgi quality control. Indeed, there are homologs of the Dsc proteins in S. cerevisiae, despite a lack of SREBP homologs in that organism (35). In that system, the homologous Tul1 E3 ligase regulates turnover of certain vacuolar membrane proteins in budding yeast during vacuolar quality control (7). These three related roles for Cdc48 highlight the versatility of this powerful molecular machine and the necessity of cofactors to target it to specific substrates.
How the cdc48 point mutations characterized in this study affect Cdc48 function is unknown. The mutations are spread throughout the D1 and D2 domains, are notably absent from the N domain, and are not clustered in three-dimensional space. Two of the mutations (E325K and A366D) are in or near the ATP-binding region of the D1 domain. Although the enzymatic consequence of a lysine mutation at Glu-325 is untested, the homologous residue has been extensively studied in both budding yeast and mammalian cells and has effects on the ATP hydrolysis rate of the D1 domain (14, 36). Interestingly, a report in fission yeast independently isolated the E325K mutation as a cold-sensitive suppressor of the temperature-sensitive cdc48-353 (G338D) mutation (37). The authors concluded that E325K likely alters Cdc48 structure in a way that is compensatory to the effects of cdc48-353. Therefore, fission yeast cdc48-8 (E325K), and possibly cdc48-9 (A366D), may produce structural changes that impact Cdc48 ATPase activity.
Because our cdc48 mutants are distributed throughout the sequence and do not cluster in three-dimensional space, the mutations are unlikely to directly impact a single binding interaction with a specific cofactor or substrate to cause the observed SREBP cleavage defects. However, we found in the cdc48 mutants that binding between Cdc48 and the substrate-recruiting cofactor Ufd1 was increased compared with wild type (Fig. 6, C and D). This is surprising given that the ufd1 mutants decreased Cdc48-Ufd1 binding (Fig. 6, A and B). Other groups have shown that mutation of VCP Glu-305 to glutamine or alanine reduced the rate of ATP hydrolysis and substrate unfolding (14, 38). We predict that mutation of the homologous residue Glu-325 (cdc48-8) would have a similar effect. Furthermore, another study showed a completely hydrolysis-defective VCP had increased Ufd1 binding in vivo (18). Because our remaining cdc48 mutants showed increased binding to Ufd1–13xMyc like cdc48-8, this may indicate that all of the cdc48 mutants have ATP hydrolysis defects. Such a defect in the ATPase cycle could lead to the observed Dsc E3 ligase localization and SREBP cleavage defects. This would be consistent with our hypothesis that Cdc48 needs to unfold a Dsc-ubiquitylated substrate (such as a repressor) before the Dsc E3 ligase can traffic to the Golgi. Future in vitro studies are needed to examine the interaction between Cdc48, Ufd1, and any substrates in our cdc48 mutants.
Interestingly, although cdc48 is an essential gene, none of these mutations is lethal, suggesting that they have relatively minor functional defects. However, we observe strong defects in SREBP cleavage in many of the mutants (Fig. 2). This may indicate synthetic effects between the roles for Cdc48 during Dsc E3 ligase Golgi localization and Rbd2 cleavage. The impacts of deficient Cdc48 activity on each step of the pathway may be relatively minor, but together they strongly affect SREBP cleavage. Therefore, the requirement for Cdc48 at multiple steps may make SREBP activation uniquely sensitive to alterations in Cdc48 function.
Given this finding in fission yeast, we wondered whether VCP plays a role in mammalian SREBP activation. Although there is no evidence that VCP is required for Golgi localization of the Site-1 and Site-2 proteolytic machinery, VCP is required for two other aspects of sterol homeostasis. First, VCP and Ufd1 are involved in ubiquitination, membrane extraction, and degradation of the major sterol-synthesis enzyme HMG-CoA reductase in the presence of excess sterols (39, 40). Additionally, VCP binds the Dsc5 homolog, UBXD8, for extraction and degradation of the SREBP inhibitor Insig under low sterol conditions (41). Therefore, mammalian VCP has multiple roles during maintenance of sterol homeostasis. Fission yeast has HMG-CoA reductase and Insig homologs (Hmg1 and Ins1, respectively), but Hmg1 is not regulated by degradation (42). Therefore, it is unlikely that Cdc48 is involved in the regulation of Hmg1 activity in fission yeast in an analogous mechanism to that seen in mammals. The stability of Ins1 has not been well-characterized; therefore, it remains to be determined whether Cdc48 regulates that aspect of the pathway.
In contrast to our cdc48 mutants, the VCP mutations believed to cause diseases such as IBMPFD are all clustered in the N and D1 domains at the N-D1 interface (ClinVar). These mutations increase ATPase activity and alter cofactor binding, resulting in defects in autophagy (especially clearance of damaged mitochondria), lysosomal degradation, and endosomal trafficking (16, 17, 19, 21, 23). The disease mechanism for these VCP mutations is related to mitochondrial uncoupling and a decrease in cellular ATP followed by a failure to clear these damaged mitochondria (20, 24, 43, 44). It is difficult to imagine a way in which our cdc48 point mutations are having a similar effect in S. pombe, with the downstream result being a defect in SREBP activation at the Golgi. Creating our mutations in human cell culture to examine their effects may provide additional insight into VCP function and is a topic for future studies.
In addition to describing a novel role for Cdc48-Ufd1 during SREBP cleavage, this study generated two valuable resources for future studies of this pathway and low oxygen adaptation in fission yeast. Through the MMS screen, we report point mutations in sum3, sre1, scp1, Dsc E3 ligase subunits, cdc48, and rbd2 that exhibit low oxygen adaptation defects (Fig. 1A and supplemental Table 1). These mutants will provide insight into the molecular function of these proteins in this and other pathways. Additionally, the mass spectrometry data contain a number of leads for future studies. 18 of the 51 proteins enriched at least two standard deviations from the mean in the Cdc48-bound fraction of our Cdc48–5xFLAG pulldown were known Cdc48 cofactors in S. pombe and/or other species (Table 1). In addition to Ufd1 and many known Cdc48 cofactors, four Dsc E3 ligase complex members were identified as Cdc48-binding proteins, consistent with our previously published results that Cdc48 interacts with the Dsc E3 ligase by binding to the Dsc5 UBX domain (4). These data validate our data set and suggest that the 33 unexpected binding proteins are very interesting candidates for future study. Included in this list are three predicted E3 ligases and a ubiquitin-metalloprotease fusion protein (Table 1). Interestingly, mammalian VCP was shown to interact with a large number of E3 ligases through UBX domain-containing proteins, including the Dsc5 homologs UBXD8 and FAF1 (45). These proteins are therefore likely Cdc48-binding proteins. The remaining candidates may be unidentified components of Cdc48-interacting complexes or possibly substrates. Further analysis of the interaction between Cdc48 and these binding partners may identify new pathways requiring Cdc48 function.
Together, these results highlight the ubiquitous value of Cdc48/VCP separase activity in organisms from yeast to humans. As careful structural and biochemical studies of Cdc48/VCP rapidly advance our understanding of Cdc48 mechanical function, future work will need to turn toward understanding the regulation of numerous cofactor interactions with Cdc48, as it is these interactions that ultimately define the fate of Cdc48 and the essential pathways in which it functions.
Experimental procedures
Materials
General chemicals and materials were obtained from Sigma or Fisher. Other sources include the following: yeast extract, peptone, and agar from BD Biosciences; brefeldin A, Igepal CA-630 (Nonidet P-40), cobalt(II) chloride, amino acid supplements, 1× protease inhibitors (PI) (10 μg/ml leupeptin, 5 μg/ml pepstatin A, 0.5 μm PMSF), acid-washed glass beads (425–600 μm), and FLAG M2 monoclonal IgGs (F1804 and F3165) from Sigma; alkaline phosphatase (catalog no. 713023) and complete EDTA-free PI from Roche Applied Sciences; oligonucleotides from Integrated DNA Technologies; IRDye donkey anti-rabbit and donkey anti-mouse from Li-Cor; Myc monoclonal 9E10 IgG and GST monoclonal IgG from Santa Cruz Biotechnology, Inc; Myc polyclonal IgG from Millipore (catalog no. 06-549); prestained protein standards from Bio-Rad; Dynabeads coupling kit from Life Technologies, Inc. (14311D); protein G magnetic beads from New England Biolabs (catalog no. S1430S); glutathione HiCap matrix beads from Qiagen; MagneGST beads and Trypsin/Lys-C mix (catalog no. V5073) from Promega; and 5-fluoroorotic acid (catalog no. F595000) from Thermo Fisher.
Strains and media
Yeast strains are described in Table 2. Strain PEY1516 was generated as described previously using seven tandem copies of the Tf2-1 sterol-regulatory element to drive ura4+ reporter gene activation (46). S. pombe was cultured to exponential phase at 30 °C in rich YES medium (0.5% (w/v) yeast extract plus 3% (w/v) glucose supplemented with 225 μg/ml each of uracil, adenine, leucine, histidine, and lysine) or Edinburgh minimal medium plus supplements (EMM) (20 g/liter glucose, 225 mg/liter each of uracil, adenine, leucine, histidine, and lysine) unless otherwise indicated. YES + CoCl2 medium was prepared by dissolving cobalt(II) chloride in H2O and adding to a final concentration of 1.6 mm in YES medium.
Table 2.
S. pombe strain list
Antibodies
Rabbit polyclonal antibody anti-Sre1 IgG (aa 1–260) was generated using a standard protocol as described previously (1). Briefly, antigen was expressed in Escherichia coli and affinity-purified by an N-terminal polyhistidine tag. Sre1-specific antibodies were isolated from rabbit serum by affinity to the polyhistidine-tagged Sre1 antigen. Specificity of this antibody was assayed by loss of immunoreactivity in an sre1Δ strain. We generated rabbit polyclonal antibody anti-Sre2 IgG (aa 1–426) using a standard protocol as described above for anti-Sre1 (1).
We generated monoclonal antibody 5B4 IgG1κ to Sre1 (aa 1–260) as described previously using recombinant protein that was purified from E. coli by nickel-affinity chromatography (Qiagen) and injected into BALB/c mice (5). Antibody specificity was tested by immunoblotting against S. pombe extracts from cells overexpressing sre1.
Generation of polyclonal antibodies to Dsc E3 ligase complex members (Dsc1 and Dsc5) was described previously (3, 4). Hexa-histidine-tagged recombinant protein antigens, Dsc1 (aa 20–319) and Dsc5 (aa 251–427), were purified from E. coli using nickel-nitrilotriacetic acid (Qiagen). Antisera were generated by Covance using a standard protocol. Dsc1 antibody was affinity-purified by passing over a column containing Dsc1 N terminus (aa 1–300) coupled to agarose beads (AminoLink Plus immobilization kit, Thermo Scientific) according to the manufacturer's protocol as described previously (33). Antiserum to Cdc48 was the kind gift of R. Hartmann-Petersen (University of Copenhagen) (47).
Yeast mutagenesis and selection
S. pombe were mutagenized using (MMS as described previously (4). Briefly, PEY1516 cells were grown to a cell density of 1 × 107 cells/ml, washed in sterile water, and resuspended at a concentration of 1 × 108 cells/ml in EMM containing 0.024 or 0.012% (w/v) MMS. Cultures were then grown for 3 h at room temperature. Cells were washed with sterile water and resuspended in sterile water at a density of 5 × 106 cells/ml. Cells were plated onto selection medium (SM: EMM, 2% (w/v) agar, 20 g/liter glucose, 225 mg/liter each of adenine, leucine, and histidine, 50.25 mg/liter uracil, 0.1% (w/v) 5-FOA, 0.2 mm CoCl2) at a density of 5 × 105 cells/plate. Plates were wrapped in foil and incubated at 30 °C. Colonies were picked and streaked onto SM. Single colonies were then patched onto SM and then streaked onto YES + CoCl2. CoCl2-sensitive isolates were then streaked from the SM patch a second time onto SM to isolate single colonies.
Mutant identification
Mutant genes were identified as described previously (4). To test whether strains contained mutations in sre1 and scp1, CoCl2-sensitive isolates were cotransformed with plasmids expressing sre1+ and scp1+ from the cauliflower mosaic virus promoter or the empty vector control plasmids pSLF101 and pSLF102 (48), respectively, and then screened again for CoCl2 sensitivity. Strains whose growth on CoCl2 was not rescued by plasmids expressing sre1 and scp1 were evaluated by Western blotting for Sre1 cleavage. Strains that exhibited deficient Sre1 cleavage were mated to dsc1-5Δ, cdc48-8, rbd2Δ, sre1Δ, and scp1Δ cells. A suspension of spores from the mating was plated on YES or YES + CoCl2. The inability to recover CoCl2-resistant spores indicated tight linkage between the mutations. To identify the nucleotide mutations in each gene, genomic DNA was prepared, the full coding sequence of the relevant locus was PCR-amplified, and the coding region was sequenced.
Recreation of cdc48 point mutations
cdc48 point mutations were created in a non-mutagenized wild-type KGY425 background by generating a plasmid containing cdc48 (bp 4–2504) followed by the NatMX6 marker. This wild-type plasmid was mutated using site-directed mutagenesis to create plasmids with the cdc48 mutations of interest. Plasmids were digested to release the cdc48-NatMX6 sequence, and the cdc48 fragment was transformed into KGY425 wild-type yeast for homologous recombination. Successful transformants were selected by nourseothricin resistance (Nat) and tested for sensitivity to CoCl2. An isogenic wild-type cdc48 strain with the nourseothricin resistance cassette was also generated. All mutant strains were then confirmed by sequencing of genomic DNA as described above. Because these strains were generated in a non-mutagenized background and contained nourseothricin resistance, they were assigned allele numbers cdc48-5–cdc48-10 to distinguish them from the mutagenized versions published in Stewart et al. (4).
SREBP cleavage assay
For Sre1 cleavage, cells were grown in YES medium to exponential phase inside an InVivo2 400 hypoxic work station (Biotrace, Inc.) at 30 °C. For Sre2 and Sre2-MS cleavage, cells were grown in YES medium (Sre2) or EMM medium minus leucine (Sre2-MS) in normoxic conditions at 30 °C.
Whole-cell lysis
Cells were harvested for protein extraction and immunoblotting by the whole-cell lysis method as described previously, unless otherwise indicated (49). Briefly, cell pellets were resuspended in 1.85 m NaOH, 7.4% (v/v) β-mercaptoethanol, and incubated on ice for 10 min. Trichloroacetic acid (TCA) was added to a final concentration of 30% (w/v), and the samples were again incubated on ice. Lysate was centrifuged at 20,000 × g for 10 min at 4 °C, and the pellet was washed with cold acetone. Samples were centrifuged at 20,000 × g for 5 min at 4 °C and dried completely under vacuum. Pellets were resuspended in SDS lysis buffer (1% SDS, 150 mm NaCl, 50 mm Tris-HCl, pH 8.0, 1× protease inhibitors) and sonicated for 10 s. Protein was quantified using the BCA protein assay (Pierce). For alkaline phosphatase treatment, 20–50 μg of lysate were diluted at least 1:2 in 50 mm Tris-HCl, pH 8.0, and then alkaline phosphatase was added to 20% (v/v). Samples were incubated at 37 °C for 1 h, then 1× loading dye was added (30 mm Tris-HCl, 3% SDS, 5% glycerol, 0.004% bromphenol blue, 2.5% 2-mercaptoethanol). Entire phosphatase-treated sample was loaded onto SDS-polyacrylamide gels, and consistent loading was confirmed following electroblotting by staining the membrane with Ponceau S. Blots were imaged using the Odyssey CLx infrared imaging system (LI-COR Biosciences). Protein signal was quantified using the LI-COR Biosciences Image Studio software to box the bands of interest, normalized to blot background, and then normalized to the indicated loading control.
Cdc48–5xFLAG affinity chromatography
Exponentially growing cells (1 × 1010) were pelleted and then washed with H2O. Cell pellets were resuspended in 15 ml of cold B88 buffer (20 mm HEPES, pH 7.2, 150 mm KOAc, 5 mm Mg(OAc)2, 250 mm sorbitol) + 1× protease inhibitors, 1× Complete EDTA-free PI. Cells were lysed by high pressure emulsifier (EmulsiFlex-C-3, Avestin); lysate was centrifuged for 10 min at 100,000 × g to pellet insoluble material, and the supernatant was saved. Monoclonal FLAG antibody F3165 (Sigma) was conjugated to Dynabeads using the Dynabeads coupling kit (Life Technologies, Inc., 14311D) to a final concentration of 10 μg of antibody/1 mg of beads. FLAG-conjugated Dynabeads (1.5 mg) were washed with IP buffer (B88 + 1× PI, 1× Complete EDTA-free PI, 0.5% Triton X-100). Lysate (25 mg) was diluted in IP buffer to 550 μl of total volume, and Triton X-100 concentration was returned to 0.5%. Lysate (50 μl) was saved as input, and 500 μl was incubated with the FLAG-Dynabeads for 30 min at 4 °C. Beads were collected by magnet, and the unbound fraction was reserved. Beads were washed three times in 200 μl of IP buffer and transferred to a new tube on the last wash to minimize background. The bound fraction was eluted into SDS lysis buffer + 1× PI at 95 °C for 5 min and transferred to a new tube. Aliquots of all fractions were run on SDS-PAGE and silver-stained to analyze pulldowns before sending total bound fraction for mass spectrometry analysis.
TMT labeling and proteomics analysis
Protein samples from three biological replicates of wild-type and Cdc48–5xFLAG samples subjected to anti-FLAG purification were adjusted to pH 8.0 using 500 mm triethylammonium bicarbonate (TEAB) added dropwise. Cysteine residues were reduced using 0.5 μg/μl DTT for 1 h at 60 °C and then alkylated with 1 μg/μl iodoacetamide for 30 min in the dark. Samples were precipitated with 8× volume of 10% TCA/acetone at −20 °C for 5 h, centrifuged at maximum speed for 10 min to remove supernatant, and then washed with 8× acetone at −20 °C for 20 min. Samples were centrifuged at maximum speed to remove supernatant, and protein pellets were air-dried. For protein digestion, samples were incubated with 2 μg trypsin/LysC mix in 50 μl of 400 mm TEAB at 37 °C for 4 h. After proteolysis, samples were labeled with amine-reactive 6-plex TMT reagents (Thermo Fisher Scientific) dissolved in 41 μl of anhydrous acetonitrile at room temperature for 1 h. Labels are as follows: 1_ctr, 126; 1_exp, 129; 2_ctr, 130; 2_exp, 127; 3_ctr, 131; 3_exp, 128. After labeling, 8 μl of 5% (v/v) hydroxylamine was added to quench the reaction. After labeling, all samples were mixed and dried, resulting in a total protein quantity of 140 μg.
The combined mixture of TMT 6-plex labeled tryptic peptides was reconstituted in 2 ml of basic reverse phase (bRP) solvent A (10 mm TEAB, pH 8.5), then pH checked, and fractionated over 8 min on a XBridge C18 column, 5 μm, 2.1 × 100 mm analytical column (Waters), with a XBridge C18 Guard column, 5 μm, 2.1 × 10 mm (Waters), using an Agilent HPLC system containing 1100 series binary pump, 1200 series UV detector, and a 1200 series micro-fraction collector. Fractionation of peptides were carried out by a linear gradient (starting at 16 min) between solvent A and solvent B (10 mm TEAB in 90% acetonitrile), flow rate 250 μl/min. 12 flow-through fractions between 1 and 20 min were not used for MS/MS. 84 × 225 μl of bRP fractions collected between 20 and 95 min (0–100% solvent B) were recombined into 28 fractions for MS/MS analysis.
Each bRP fraction was completely dried and then reconstituted in 2% (v/v) acetonitrile, 0.1% (v/v) formic acid, and 50% was loaded on a 75-μm × 2.5-cm ODS-A C18 HPLC column trap (YMC) at 600 nl/min 0.1% formic acid (solvent A). Peptides were fractionated by reverse-phase HPLC on a 75-μm × 100-mm ProntoSil C18H reverse-phase column (5 μm, 120Å, Bischoff Chromatography) at 300 nl/min using a 2–10% solvent B (90% acetonitrile in 0.1% formic acid) gradient over the first 2 min, then up to 25% B by 55 min, 45% B by 67 min, and 100% B by 75 min. Eluting peptides were sprayed through 1-μm emitter tip (New Objective) at 2.0 kV directly into an LTQ Orbitrap Velos mass spectrometer in FTFT (Thermo Fisher Scientific) interfaced with nano-Acquity LC system (Waters). Survey scans (full MS) were acquired from 350 to 1800 m/z. Precursor ions were individually isolated with 15 ppm tolerance between 2.5 and 5.5 Da and fragmented (MS/MS) using an HCD activation collision energy of 35 and dynamic exclusion of 30 s. Fragment ions were isolated with 0.03 Da tolerance. Precursor and the fragment ions were analyzed at resolution 30,000 and 15,000, respectively.
Protein quantification
MS/MS spectra (.RAW) were analyzed via Proteome Discoverer software (version 1.4 Thermo Fisher Scientific) using 3Nodes (extracted, processed by MS2Processor, and PD1.4), with Mascot (version 2.2, Matrix Science) using the RefSeq2012 Complete Database with concatenated decoy database specifying S. pombe species (5020 entries). Fixed modifications of TMT 6-plex of N termini and carbamidomethyl on Cys, and variable modifications of TMT 6-plex on Lys and oxidation of Met, were allowed. One missed cleavage was allowed. 80,498 PSMs were matched to 16,290 peptides. The peptide identifications and reporter ions with the highest Mascot score for the same peptide-matched spectrum from the different extraction methods were processed within the Proteome Discoverer to identify peptides with a confidence threshold 1% false discovery rate. Peptides were grouped by mass and sequence into 2355 protein groups, considering only PSMs with ΔCn better than 0.15. Spectra were assessed using principal component analysis, quantile-quantile plots, and box and whisker plots using Partek statistical software (Genomic Solutions). Based on that analysis, spectral intensities were subjected to logarithmic transformation to more closely approximate a normal distribution. The relative protein abundance was obtained by taking the median value of all reporter ion intensities assigned to that protein. Protein abundance values were quantile-normalized prior to averaging of the three biological replicates and fold change calculation between the FLAG-tagged experiment and the untagged control. p values were calculated by two-way ANOVA because principal component analysis showed evidence of significant batch effects among our three biological replicates.
Ufd1–13xMyc immunoprecipitation
Exponentially growing cells (2.5 × 108) were subjected to coimmunoprecipitation with anti-Myc antibody. Briefly, cell pellets were resuspended in 200 μl of Nonidet P-40 lysis buffer (50 mm HEPES, pH 6.8, 50 mm KOAc, 2 mm MgOAc, 1 mm CaCl2, 200 mm sorbitol, 1 mm NaF, 0.3 mm Na3VO4, 1% (w/v) Nonidet P-40, 2× PI, 1× Complete EDTA-free PI). Cells were lysed by bead beating for 10 min at 4 °C, and then beads were washed with 800 μl of Nonidet P-40 lysis buffer to collect all lysate. The lysate was incubated with rotation for 40 min at 4 °C and then centrifuged for 10 min at 100,000 × g to pellet insoluble material. Supernatant protein (2–3 mg) was incubated with 4 μg of anti-Myc antiserum for 15 min and then incubated with 80 μl of protein G-magnetic beads overnight at 4 °C. Beads were washed three times with Nonidet P-40 lysis buffer, and the bound fraction was eluted into SDS lysis buffer + 1× PI and 1× loading dye (30 mm Tris-HCl, 3% SDS, 5% glycerol, 0.004% bromphenol blue, 2.5% 2-mercaptoethanol) at 95 °C for 5 min.
GST in vitro pulldown
Exponentially growing KGY425 (WT) and NBY3824 (ufd1-13xmyc) cells (3.0 × 108) were collected and washed with H2O. Cell pellets were resuspended in 500 μl of cold B88 buffer + 1× protease inhibitors and lysed using glass beads for 20 min at 4 °C. Beads were washed with 400 μl of B88 buffer, and lysate was centrifuged at 47,000 rpm for 10 min at 4 °C. Supernatant was saved as cleared lysate.
Recombinant GST-HA-V5, GST-Dsc5-UBX, and GST-Rbd2-SHP were purified from E. coli (4.0 × 1010) after 4-h induction with IPTG. Bacterial pellets were resuspended in 5 ml of PBS, pH 7.2, and sonicated for 3 min total, with 15 s on and 59 s off. Triton X-100 was added to a final concentration of 1% (v/v), and lysate was rotated for 30 min at 4 °C. Lysate was centrifuged at 15,000 × g for 20 min at 4 °C. Supernatant was saved as unpurified protein. For purification, 500 μl of glutathione Hi-Cap matrix beads (Qiagen) were washed three times with 2.5 ml of PBS-EW wash buffer (50 mm NaH2PO4, 150 mm NaCl, pH 7.2, 1 mm EDTA, 1 mm DTT) and centrifuged 4000 × g for 1.5 min after each wash. Cell lysate was added to beads and nutated for 1 h at 4 °C. Beads were centrifuged at 4000 × g for 1.5 min and washed two times with 1.3 ml of PBS-EW. Beads were washed 1× with 1.25 ml of wash buffer (50 mm Tris, pH 8.0, 0.4 m NaCl, 0.1% Triton X-100, 1 mm DTT). Elution buffer (250 μl) (50 mm Tris, pH 8.0, 0.4 m NaCl, 0.1% Triton X-100, 1 mm DTT, 50 mm reduced glutathione) was added to beads and incubated for 10 min at 4 °C. Beads were centrifuged 4000 × g for 1.5 min; eluate was removed, and beads were incubated 2× in elution buffer for 5 min each, saving eluates separately. SDS lysis buffer and loading dye were added to aliquots of eluates and intermediate steps. These samples were run on an SDS-polyacrylamide gel and Coomassie-stained to evaluate purity and concentration.
For pulldown, 25 μl of MagneGST beads (Promega) were washed three times with 1 ml of PBS, pH 7.2, and then blocked for 1 h with PBS + 5% BSA at 4 °C. Beads were washed once with PBS, and then 50 μg of purified GST-tagged protein was added, and volume was brought up to 500 μl with PBS before binding for 1 h at 4 °C. Beads were collected by magnet and washed three times with 1 ml of PBS. Beads were washed once with 1 ml of binding buffer (B88, 0.2% Nonidet P-40, 1× protease inhibitors). Cytosol (250 μg) and binding buffer were added to beads to a final volume of 200 μl and incubated for 30 min at 4 °C. Beads were collected by magnet, and unbound fraction was saved before washing beads three times with 1 ml of binding buffer. Beads were resuspended in 100 μl of SDS lysis buffer and 1× loading dye and boiled at 95 °C for 5 min to elute bound proteins.
Dsc1 glycosylation assay
Exponentially growing cells (2.5 × 108) were collected and resuspended in 300 μl of B88 buffer + 1× protease inhibitors, 1× Complete EDTA-free PI. Cells were lysed using glass beads for 12 min at 4 °C and then centrifuged for 5 min at 500 × g to clear cell debris. The supernatant was centrifuged at 20,000 × g for 20 min, and the pelleted membranes were resuspended in 100 μl of B88 buffer with 1% Nonidet P-40 (v/v) and then sonicated for 5 s. Membranes were solubilized at 4 °C for 1 h; samples were centrifuged at 20,000 × g for 20 min, and the supernatant was collected as detergent-solubilized membrane. Loading dye was added, and samples were incubated at 37 °C for 30 min before running an SDS-polyacrylamide gel. At no time were samples boiled, as Dsc1 aggregates at high temperature. % mature was calculated by quantifying the higher molecular weight band and the lower molecular weight band on the LiCor and dividing the high molecular weight signal by the total signal.
Author contributions
R. B. designed and conducted the experiments, analyzed the results, and wrote the manuscript. D. R. and S. R. conducted preliminary experiments. E. V. S. performed the MMS mutagenesis screen. D. R. and J. H. analyzed results of the MMS mutagenesis screen. P. J. E. conceived of the project, designed experiments, and wrote the manuscript. All authors reviewed the results and approved the final version of this manuscript.
Supplementary Material
Acknowledgments
We thank the following for help and advice: Dr. M. N. Boddy (Scripps Research Institute) for the ufd1 strains; Dr. R. Hartmann-Petersen (University of Copenhagen) for the Cdc48 antibody; Dr. J. Burg for generation of the cdc48 homologous recombination plasmid; The Johns Hopkins Mass Spectrometry Core for Mass Spectrometry; The Johns Hopkins Microarray facility for data analysis; and S. Zhao and other members of the Espenshade laboratory.
This work was supported by National Institutes of Health Grants HL077588 (to P. J. E.) and GM007445 (to R. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Tables S1–S2.
- SREBP
- sterol regulatory element-binding protein
- ER
- endoplasmic reticulum
- MMS
- methyl methanesulfonate
- ERAD
- ER-associated degradation
- IBMPFD
- inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia
- 5-FOA
- 5-fluoroorotic acid
- PI
- protease inhibitor
- EMM
- Edinburgh Minimal Medium
- YES
- yeast extract plus supplements
- aa
- amino acid
- TMT
- tandem mass tag
- TEAB
- triethylammonium bicarbonate
- bRP
- basic reverse phase
- ANOVA
- analysis of variance
- IPTG
- isopropyl 1-thio-β-d-galactopyranoside
- PSM
- peptide spectrum match.
References
- 1. Hughes A. L., Todd B. L., and Espenshade P. J. (2005) SREBP pathway responds to sterols and functions as an oxygen sensor in fission yeast. Cell 120, 831–842 [DOI] [PubMed] [Google Scholar]
- 2. Kwon E. J., Laderoute A., Chatfield-Reed K., Vachon L., Karagiannis J., and Chua G. (2012) Deciphering the transcriptional-regulatory network of flocculation in Schizosaccharomyces pombe. PLoS Genet. 8, e1003104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stewart E. V., Nwosu C. C., Tong Z., Roguev A., Cummins T. D., Kim D. U., Hayles J., Park H. O., Hoe K. L., Powell D. W., Krogan N. J., and Espenshade P. J. (2011) Yeast SREBP cleavage activation requires the Golgi Dsc E3 ligase complex. Mol. Cell 42, 160–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stewart E. V., Lloyd S. J., Burg J. S., Nwosu C. C., Lintner R. E., Daza R., Russ C., Ponchner K., Nusbaum C., and Espenshade P. J. (2012) Yeast sterol regulatory element-binding protein (SREBP) cleavage requires Cdc48 and Dsc5, a ubiquitin regulatory X domain-containing subunit of the Golgi Dsc E3 ligase. J. Biol. Chem. 287, 672–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Burr R., Stewart E. V., Shao W., Zhao S., Hannibal-Bach H. K., Ejsing C. S., and Espenshade P. J. (2016) Mga2 transcription factor regulates an oxygen-responsive lipid homeostasis pathway in fission yeast. J. Biol. Chem. 291, 12171–12183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hwang J., Ribbens D., Raychaudhuri S., Cairns L., Gu H., Frost A., Urban S., and Espenshade P. J. (2016) A Golgi rhomboid protease Rbd2 recruits Cdc48 to cleave yeast SREBP. EMBO J. 35, 2332–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li M., Koshi T., and Emr S. D. (2015) Membrane-anchored ubiquitin ligase complex is required for the turnover of lysosomal membrane proteins. J. Cell Biol. 211, 639–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bat-Ochir C., Kwak J. Y., Koh S. K., Jeon M. H., Chung D., Lee Y. W., and Chae S. K. (2016) The signal peptide peptidase SppA is involved in sterol regulatory element-binding protein cleavage and hypoxia adaptation in Aspergillus nidulans. Mol. Microbiol. 100, 635–655 [DOI] [PubMed] [Google Scholar]
- 9. Fleig L., Bergbold N., Sahasrabudhe P., Geiger B., Kaltak L., and Lemberg M. K. (2012) Ubiquitin-dependent intramembrane rhomboid protease promotes ERAD of membrane proteins. Mol. Cell 47, 558–569 [DOI] [PubMed] [Google Scholar]
- 10. Yeung H. O., Kloppsteck P., Niwa H., Isaacson R. L., Matthews S., Zhang X., and Freemont P. S. (2008) Insights into adaptor binding to the AAA protein p97. Biochem. Soc. Trans. 36, 62–67 [DOI] [PubMed] [Google Scholar]
- 11. Meyer H., Bug M., and Bremer S. (2012) Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol. 14, 117–123 [DOI] [PubMed] [Google Scholar]
- 12. Verma R., Oania R. S., Kolawa N. J., and Deshaies R. J. (2013) Cdc48/p97 promotes degradation of aberrant nascent polypeptides bound to the ribosome. eLife 2, e00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nishikori S., Esaki M., Yamanaka K., Sugimoto S., and Ogura T. (2011) Positive cooperativity of the p97 AAA ATPase is critical for essential functions. J. Biol. Chem. 286, 15815–15820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bodnar N. O., and Rapoport T. A. (2017) Molecular Mechanism of Substrate Processing by the Cdc48 ATPase Complex. Cell 169, 722–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang X., Gui L., Zhang X., Bulfer S. L., Sanghez V., Wong D. E., Lee Y., Lehmann L., Lee J. S., Shih P. Y., Lin H. J., Iacovino M., Weihl C. C., Arkin M. R., Wang Y., and Chou T. F. (2015) Altered cofactor regulation with disease-associated p97/VCP mutations. Proc. Natl. Acad. Sci. U.S.A. 112, E1705–E1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Buchberger A. (2010) Control of ubiquitin conjugation by cdc48 and its cofactors. Subcell. Biochem. 54, 17–30 [DOI] [PubMed] [Google Scholar]
- 17. Schuetz A. K., and Kay L. E. (2016) A Dynamic molecular basis for malfunction in disease mutants of p97/VCP. eLife 5, e20143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Erzurumlu Y., Kose F. A., Gozen O., Gozuacik D., Toth E. A., and Ballar P. (2013) A unique IBMPFD-related P97/VCP mutation with differential binding pattern and subcellular localization. Int. J. Biochem. Cell Biol. 45, 773–782 [DOI] [PubMed] [Google Scholar]
- 19. Watts G. D., Wymer J., Kovach M. J., Mehta S. G., Mumm S., Darvish D., Pestronk A., Whyte M. P., and Kimonis V. E. (2004) Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 36, 377–381 [DOI] [PubMed] [Google Scholar]
- 20. Kimura Y., Fukushi J., Hori S., Matsuda N., Okatsu K., Kakiyama Y., Kawawaki J., Kakizuka A., and Tanaka K. (2013) Different dynamic movements of wild-type and pathogenic VCPs and their cofactors to damaged mitochondria in a Parkin-mediated mitochondrial quality control system. Genes Cells 18, 1131–1143 [DOI] [PubMed] [Google Scholar]
- 21. Johnson J. O., Mandrioli J., Benatar M., Abramzon Y., Van Deerlin V. M., Trojanowski J. Q., Gibbs J. R., Brunetti M., Gronka S., Wuu J., Ding J., McCluskey L., Martinez-Lage M., Falcone D., Hernandez D. G., et al. (2010) Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron 68, 857–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ju J. S., Fuentealba R. A., Miller S. E., Jackson E., Piwnica-Worms D., Baloh R. H., and Weihl C. C. (2009) Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J. Cell Biol. 187, 875–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Niwa H., Ewens C. A., Tsang C., Yeung H. O., Zhang X., and Freemont P. S. (2012) The role of the N domain in the ATPase activity of the mammalian AAA ATPase p97/VCP. J. Biol. Chem. 287, 8561–8570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim N. C., Tresse E., Kolaitis R. M., Molliex A., Thomas R. E., Alami N. H., Wang B., Joshi A., Smith R. B., Ritson G. P., Winborn B. J., Moore J., Lee J. Y., Yao T. P., Pallanck L., et al. (2013) VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron 78, 65–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cheung R., and Espenshade P. J. (2013) Structural requirements for sterol regulatory element-binding protein (SREBP) cleavage in fission yeast. J. Biol. Chem. 288, 20351–20360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thompson A., Schäfer J., Kuhn K., Kienle S., Schwarz J., Schmidt G., Neumann T., Johnstone R., Mohammed A. K., and Hamon C. (2003) Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 75, 1895–1904 [DOI] [PubMed] [Google Scholar]
- 27. Kim D. U., Hayles J., Kim D., Wood V., Park H. O., Won M., Yoo H. S., Duhig T., Nam M., Palmer G., Han S., Jeffery L., Baek S. T., Lee H., Shim Y. S., et al. (2010) Analysis of a genome-wide set of gene deletions in the fission yeast Schizosaccharomyces pombe. Nat. Biotechnol. 28, 617–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nie M., Aslanian A., Prudden J., Heideker J., Vashisht A. A., Wohlschlegel J. A., Yates J. R. 3rd., and Boddy M. N. (2012) Dual recruitment of Cdc48 (p97)-Ufd1-Npl4 ubiquitin-selective segregase by small ubiquitin-like modifier protein (SUMO) and ubiquitin in SUMO-targeted ubiquitin ligase-mediated genome stability functions. J. Biol. Chem. 287, 29610–29619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Meyer H. H., Shorter J. G., Seemann J., Pappin D., and Warren G. (2000) A complex of mammalian ufd1 and npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. EMBO J. 19, 2181–2192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wolf D. H., and Stolz A. (2012) The Cdc48 machine in endoplasmic reticulum associated protein degradation. Biochim. Biophys. Acta 1823, 117–124 [DOI] [PubMed] [Google Scholar]
- 31. Ye Y., Meyer H. H., and Rapoport T. A. (2003) Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. J. Cell Biol. 162, 71–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bruderer R. M., Brasseur C., and Meyer H. H. (2004) The AAA ATPase p97/VCP interacts with its alternative cofactors, Ufd1-Npl4 and p47, through a common bipartite binding mechanism. J. Biol. Chem. 279, 49609–49616 [DOI] [PubMed] [Google Scholar]
- 33. Raychaudhuri S., and Espenshade P. J. (2015) Endoplasmic reticulum exit of Golgi-resident defective for SREBP cleavage (Dsc) E3 ligase complex requires its activity. J. Biol. Chem. 290, 14430–14440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Park S., Isaacson R., Kim H. T., Silver P. A., and Wagner G. (2005) Ufd1 exhibits the AAA-ATPase fold with two distinct ubiquitin interaction sites. Structure 13, 995–1005 [DOI] [PubMed] [Google Scholar]
- 35. Reggiori F., and Pelham H. R. (2002) A transmembrane ubiquitin ligase required to sort membrane proteins into multivesicular bodies. Nat. Cell Biol. 4, 117–123 [DOI] [PubMed] [Google Scholar]
- 36. Chou T. F., Bulfer S. L., Weihl C. C., Li K., Lis L. G., Walters M. A., Schoenen F. J., Lin H. J., Deshaies R. J., and Arkin M. R. (2014) Specific inhibition of p97/VCP ATPase and kinetic analysis demonstrate interaction between D1 and D2 ATPase domains. J. Mol. Biol. 426, 2886–2899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Marinova I. N., Engelbrecht J., Ewald A., Langholm L. L., Holmberg C., Kragelund B. B., Gordon C., Nielsen O., and Hartmann-Petersen R. (2015) Single site suppressors of a fission yeast temperature-sensitive mutant in cdc48 identified by whole genome sequencing. PLoS One 10, e0117779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Blythe E. E., Olson K. C., Chau V., and Deshaies R. J. (2017) Ubiquitin- and ATP-dependent unfoldase activity of P97/VCP*NPLOC4*UFD1L is enhanced by a mutation that causes multisystem proteinopathy. Proc. Natl. Acad. Sci. U.S.A. 114, E4380–E4388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cao J., Wang J., Qi W., Miao H. H., Wang J., Ge L., DeBose-Boyd R. A., Tang J. J., Li B. L., and Song B. L. (2007) Ufd1 is a cofactor of gp78 and plays a key role in cholesterol metabolism by regulating the stability of HMG-CoA reductase. Cell Metab. 6, 115–128 [DOI] [PubMed] [Google Scholar]
- 40. Hartman I. Z., Liu P., Zehmer J. K., Luby-Phelps K., Jo Y., Anderson R. G., and DeBose-Boyd R. A. (2010) Sterol-induced dislocation of 3-hydroxy-3-methylglutaryl coenzyme A reductase from endoplasmic reticulum membranes into the cytosol through a subcellular compartment resembling lipid droplets. J. Biol. Chem. 285, 19288–19298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lee J. N., Zhang X., Feramisco J. D., Gong Y., and Ye J. (2008) Unsaturated fatty acids inhibit proteasomal degradation of Insig-1 at a postubiquitination step. J. Biol. Chem. 283, 33772–33783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Burg J. S., Powell D. W., Chai R., Hughes A. L., Link A. J., and Espenshade P. J. (2008) Insig regulates HMG-CoA reductase by controlling enzyme phosphorylation in fission yeast. Cell Metab. 8, 522–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nalbandian A., Llewellyn K. J., Kitazawa M., Yin H. Z., Badadani M., Khanlou N., Edwards R., Nguyen C., Mukherjee J., Mozaffar T., Watts G., Weiss J., and Kimonis V. E. (2012) The homozygote VCP(R(1)(5)(5)H/R(1)(5)(5)H) mouse model exhibits accelerated human VCP-associated disease pathology. PLoS One 7, e46308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bartolome F., Wu H. C., Burchell V. S., Preza E., Wray S., Mahoney C. J., Fox N. C., Calvo A., Canosa A., Moglia C., Mandrioli J., Chiò A., Orrell R. W., Houlden H., Hardy J., et al. (2013) Pathogenic VCP mutations induce mitochondrial uncoupling and reduced ATP levels. Neuron 78, 57–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Alexandru G., Graumann J., Smith G. T., Kolawa N. J., Fang R., and Deshaies R. J. (2008) UBXD7 binds multiple ubiquitin ligases and implicates p97 in HIF1α turnover. Cell 134, 804–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hughes B. T., and Espenshade P. J. (2008) Oxygen-regulated degradation of fission yeast SREBP by Ofd1, a prolyl hydroxylase family member. EMBO J. 27, 1491–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hartmann-Petersen R., Wallace M., Hofmann K., Koch G., Johnsen A. H., Hendil K. B., and Gordon C. (2004) The Ubx2 and Ubx3 cofactors direct Cdc48 activity to proteolytic and nonproteolytic ubiquitin-dependent processes. Curr. Biol. 14, 824–828 [DOI] [PubMed] [Google Scholar]
- 48. Forsburg S. L. (1993) Comparison of Schizosaccharomyces pombe expression systems. Nucleic Acids Res. 21, 2955–2956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Burr R., Stewart E. V., and Espenshade P. J. (2017) Coordinate regulation of yeast sterol regulatory element-binding protein (SREBP) and Mga2 transcription factors. J. Biol. Chem. 292, 5311–5324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wood V., Harris M. A., McDowall M. D., Rutherford K., Vaughan B. W., Staines D. M., Aslett M., Lock A., Bahler J., Kersey P. J., and Oliver S. G. (2011) PomBase: a comprehensive online resource for fission yeast. Nucleic Acids Res. 10.1093/nar/gkr853 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.