

Abstract
5-Oxoproline (OP) is well-known as an enzymatic intermediate in the eukaryotic γ-glutamyl cycle, but it is also an unavoidable damage product formed spontaneously from glutamine and other sources. Eukaryotes metabolize OP via an ATP-dependent 5-oxoprolinase; most prokaryotes lack homologs of this enzyme (and the γ-glutamyl cycle) but are predicted to have some way to dispose of OP if its spontaneous formation in vivo is significant. Comparative analysis of prokaryotic genomes showed that the gene encoding pyroglutamyl peptidase, which removes N-terminal OP residues, clusters in diverse genomes with genes specifying homologs of a fungal lactamase (renamed prokaryotic 5-oxoprolinase A, pxpA) and homologs of allophanate hydrolase subunits (renamed pxpB and pxpC). Inactivation of Bacillus subtilis pxpA, pxpB, or pxpC genes slowed growth, caused OP accumulation in cells and medium, and prevented use of OP as a nitrogen source. Assays of cell lysates showed that ATP-dependent 5-oxoprolinase activity disappeared when pxpA, pxpB, or pxpC was inactivated. 5-Oxoprolinase activity could be reconstituted in vitro by mixing recombinant B. subtilis PxpA, PxpB, and PxpC proteins. In addition, overexpressing Escherichia coli pxpABC genes in E. coli increased 5-oxoprolinase activity in lysates ≥1700-fold. This work shows that OP is a major universal metabolite damage product and that OP disposal systems are common in all domains of life. Furthermore, it illustrates how easily metabolite damage and damage-control systems can be overlooked, even for central metabolites in model organisms.
Keywords: bacterial metabolism, glutamate, glutamine, metabolism, microbiology, 5-oxoproline, metabolite damage, metabolite repair, pyroglutamate
Introduction
5-Oxo-l-proline (OP3; also called pyroglutamate) is the lactam of l-glutamate (Fig. 1A). OP is a familiar intermediate in the mammalian γ-glutamyl cycle (Fig. 1A), which involves synthesis and breakdown of glutathione and has a proposed role in amino acid transport (1). In this cycle the glutamyl moiety of glutathione is enzymatically converted to OP, which is then hydrolyzed to glutamate by an ATP-dependent 5-oxoprolinase (EC 3.5.2.9; henceforth, eukaryote-type OPase) (2, 3) (Fig. 1A). The γ-glutamyl cycle has been most studied in mammals, but similar pathways (and eukaryote-type OPase) occur in plants (4, 5) and fungi (6). This cycle is largely absent from prokaryotes; a few bacteria have homologs of its enzymes and probably have a γ-glutamyl cycle-like pathway to metabolize glutathione (7), but most do not. Furthermore, bacteria such as Bacillus subtilis do not even have glutathione (8).
Figure 1.

OP metabolism and the genomic organization and distribution of OP metabolic genes in prokaryotes. A, OP is formed spontaneously (dashed red arrows) and/or enzymatically (solid red arrows) from glutamine, glutamate, γ-glutamyl phosphate (γ-glutamyl-P), N-terminal glutaminyl or glutamyl residues of peptides (GPCT, glutaminyl-peptide cyclotransferase), and γ-glutamyl-amino acids (γ-glutamyl-AA) via the γ-glutamyl cycle (indicated with curved arrows; AA, amino acid; CG, cysteinyl-glycine; C, cysteine; G, glycine; γEC, γ-glutamyl-cysteine; γ-GCT, γ-glutamylcyclotransferase). Hydrolysis to glutamate is the only way to recycle OP. Green arrows indicate glutathione synthesis reactions of the γ-glutamyl cycle. B, clustering and fusion of the candidate pxpABC OP metabolism genes in representative genomes. The pxpABC genes (colored blue) often cluster with genes encoding pyroglutamyl peptidase (PCP; colored orange), two conserved membrane-spanning proteins (DUF969 and DUF979; colored yellow), an NRAMP family transporter (ycsG; colored pink), and a putative metal chaperone (ycsI; colored light blue). Genes apparently unrelated to OP metabolism are colored white. C, distribution of candidate pxpABC OP metabolism genes and known 5-oxoprolinase genes among a representative set of 70 diverse bacteria and archaea. hyuA and hyuB represent eukaryotic-type 5-oxoprolinase homologs; AI-OP represents ATP-independent 5-oxoprolinase homologs.
Other, less widely recognized routes to OP are nonenzymatic and are present in all organisms. OP is formed via the spontaneous cyclization of the central metabolites glutamine (9), glutamate (10), and γ-glutamyl phosphate (11). Glutamine is particularly susceptible; under physiological conditions it cyclizes to OP at the rate of 10% per day (9). N-terminal glutaminyl and glutamyl residues of polypeptides can likewise spontaneously cyclize to OP residues; this reaction is also catalyzed by glutaminyl-peptide cyclotransferase (12, 13). The enzyme pyroglutamyl peptidase, which occurs in all domains of life, removes N-terminal OP residues, yielding free OP (14). OP is thus a damage product that is spontaneously formed always and everywhere as well as a γ-glutamyl cycle intermediate.
Several lines of evidence show that OP accumulation is deleterious. Its reported effects include growth inhibition in prokaryotes and plants (10, 15) and interference with energy production, lipid synthesis, and antioxidant defenses in mammalian brain (16, 17). Human inborn errors of glutathione metabolism that lead to OP buildup result in metabolic acidosis, hemolytic anemia, neurological problems, and massive urinary excretion of OP (18, 19).
Before this study the only characterized and cloned OPase besides the eukaryote-type one was an enigmatic ATP-independent enzyme from Alcaligenes faecalis (20) that has no sequence similarity to eukaryote-type OPases (21). Although the A. faecalis enzyme has OPase activity in vitro, the equilibrium of the ring-opening reaction that it catalyzes favors OP formation so strongly (Keq = [glutamate]/[OP] = 0.035) (20) that it may not facilitate OP hydrolysis in vivo. The eukaryote-type OPase overcomes the thermodynamic constraint on the ring-opening reaction by coupling it to ATP hydrolysis (2).
Because prokaryotes are doomed to form OP whether or not they have a γ-glutamyl cycle and OP is harmful, we hypothesized that prokaryotes without a eukaryote-type OPase have a different disposal system for OP, most likely a second type of OPase. In advancing this hypothesis, we were encouraged by a series of classical papers (22–25) on a multicomponent ATP-dependent OPase from a Pseudomonas putida strain (now lost). This enzyme, which was never cloned, appears to have been unlike eukaryote-type OPases. We used comparative analysis of prokaryotic genomes to predict that a conserved cluster of three genes encodes a novel OPase, and we validated this prediction by genetic and biochemical methods. This prokaryotic OPase is almost surely the one adumbrated in the classical work on P. putida.
Results
Comparative genomics identifies candidate genes for a novel 5-oxoprolinase
We first used the SEED database and its tools (26) to survey the occurrence of known OPase genes in a representative set of 984 genomes (27). Of these genomes, only 115 have eukaryote-type OPase homologs (encoded by hyuA and hyuB genes that are almost always fused), and only 10 have homologs of the A. faecalis ATP-independent OPase. Both the known types of OPase are thus quite rare in bacteria and archaea. Among the 984 genomes surveyed, 220 encode at least one OP-producing enzyme (e.g. pyroglutamyl peptidase; Fig. 1A), and of these 220, only 16 have eukaryote-type OPase homologs.
Because gene clustering on the chromosome often reflects functional relationships (28), we searched for genes that cluster with genes encoding enzymes that produce OP (Fig. 1A). Three previously uncharacterized genes were found to cluster consistently with the pcp pyroglutamyl peptidase gene in diverse bacteria (Fig. 1B). These genes specify homologs of the fungal lactamase LamB (e.g. B. subtilis ycsF, here renamed prokaryotic 5-oxoprolinase A (pxpA)) and homologs of allophanate hydrolase subunits 1 and 2 (e.g. B. subtilis ycsJ and ycsK, renamed pxpB and pxpC). The pxpABC genes cluster with each other in various arrangements, pxpB and pxbC being sometimes fused (Fig. 1B). The pxpABC genes almost always co-occur; in the 374 genomes that contain at least one gene of the trio, 364 have all three. The pxpABC and eukaryote-type OPase genes are basically inversely distributed and seldom occur together (Fig. 1C), which fits with their having redundant functions. To summarize: clustering of pxpABC with pcp and the inverse distribution with eukaryote-type OPase suggest that the gene trio encodes a novel OPase. In addition, the P. putida OPase was reported to have three distinct protein subunits (23–25), and the P. putida genome includes pxpABC genes (Fig. 1C).
The pxpABC and pcp genes also cluster with genes specifying (i) a putative metal transporter of the NRAMP family, YcsG in B. subtilis, (ii) a putative metal chaperone, YcsI in B. subtilis, and (iii) uncharacterized membrane proteins DUF969 and DUF979 (Fig. 1B). The same four genes also cluster with eukaryote-type OPase genes (supplemental Fig. S1), implying a role in OP metabolism.
B. subtilis pxpABC deletants cannot utilize OP
To assess whether pxpABC genes encode an OPase, we tested the effect of ablating these genes on the ability of B. subtilis to use OP as sole nitrogen source. Wild-type B. subtilis (but not Escherichia coli) can grow on defined medium containing 50 mmOP as the sole nitrogen source (29) but grows more slowly than on ammonium (Fig. 2, A and B). We obtained BKE deletants (30) for each of the pxpABC genes; the deletions were designed to create single-gene knockouts without affecting the expression of neighboring genes in the same operon. The pxpA, pxpB, and pxpC deletants all failed to grow on OP (Fig. 2, A and B). Because B. subtilis pxpABC genes are in an operon with the putative metal transporter ycsG and the putative metal chaperone ycsI (Fig. 1B), which may both relate to OP metabolism (see above), we also tested BKE deletants of these genes. The ycsI deletant grew slightly more slowly than wild type on OP; the ycsG deletant did not grow at all (Fig. 2, A and B). We verified that the BKE deletions do not affect neighboring gene expression by transforming each deletant with plasmid pHCMC04 harboring the coding sequence of the respective deleted gene. In all cases, reintroducing the deleted gene restored the ability to grow on OP (Fig. 2B).
Figure 2.

Growth of B. subtilis on OP depends on pxpA, pxpB, pxpC, and ycsG. A, growth at 37 °C of wild-type B. subtilis (closed circles) and various mutants with OP as the sole nitrogen source. There was no significant difference between growth of pxpA, pxpB, pxpC, or ycsG knock-out cells, which are all represented by open triangles; open squares represent the growth of ycsI knock-out cells. Data represent the mean ± S.E. of three independent cultures; the S.E. bars were smaller than the symbol for all cultures represented by open triangles. B, the indicated strains were grown overnight in LB medium, and washed twice with water, diluted to optical densities (600 nm) of 1.0, 0.2, 0.04, 0.008, and 0.0016, and 10 μl was spotted on medium with OP or ammonium as the sole nitrogen source. In the middle column, each strain harbored an expression plasmid carrying the respective gene that was deleted; the medium also contained 0.1% (w/v) xylose. Wild-type B. subtilis harbored the empty vector. Photographs were taken after 16 h (ammonium) or 72 h (OP) incubation at 37 °C and are representative of three independent experiments.
We also tested the growth of pxp deletants on minimal medium with ammonium as nitrogen source. The pxpA, pxpB, and pxpC deletants all grew less well than wild type on plates (Fig. 2B) or in liquid medium (supplemental Fig. S2). The pxp genes thus affect fitness even when OP is not supplied.
pxpABC deletants accumulate OP
Because OP formation in vivo is inevitable, knocking out the pxpABC genes is expected to cause OP accumulation if these genes indeed encode an OPase. When grown with ammonium as the nitrogen source, OP was readily detected in B. subtilis pxpA, pxpB, and pxpC deletant cells and medium, with the majority of OP accumulating in the medium (Fig. 3A). No OP was detectable in the cells or medium of wild-type cells (Fig. 3A).
Figure 3.
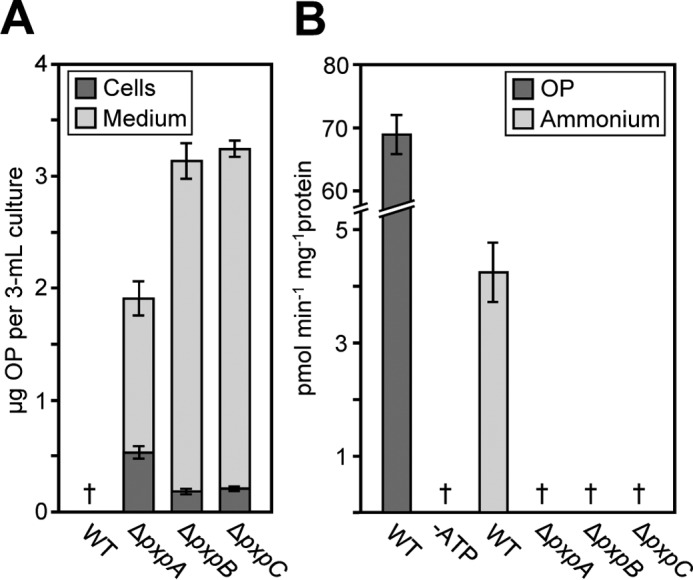
B. subtilis pxpA, pxpB, and pxpC deletant cells accumulated OP and lack 5-oxoprolinase activity. A, the amount of OP accumulated in cells and medium of 3-ml cultures grown with ammonium as nitrogen source to an optical density (600 nm) of 1.43 ± 0.02 (4.3 ± 0.3 × 108 cells). Data are mean ± S.E. of six independent cultures. B, OPase activity in the desalted lysates of wild-type cells grown with OP as the nitrogen source or wild-type and deletant cells grown with ammonium as the nitrogen source. Assays (50 μl) contained 50 mm Tris-HCl, pH 8.0, 10 mm MgCl2, 5 mm ATP (unless indicated otherwise), 2 mm glutamate, and 30 μm OP (0.1 μCi [3H]OP) and were started by adding 25 μl of lysate (10–17 mg of protein ml−1). Assays were incubated at 37 °C for 15 min; glutamate was isolated by Dowex-50 (H+) chromatography and analyzed by scintillation counting. Data are the mean ± S.E. of three independent lysate preparations. †, not detectable (detection limits: 0.01 μg of OP in cells; 0.09 μg of OP in medium; 0.02 pmol min−1 mg−1 protein in assays).
pxpABC mutants lack 5-oxoprolinase activity
To confirm that the pxpABC genes specify OPase activity, we used a radiometric assay (2) to measure OPase activity in cell lysates. Lysates prepared from wild-type B. subtilis cells grown with ammonium as nitrogen source contained readily detectable ATP-dependent OPase activity, but lysates prepared from pxpA, pxpB, or pxpC deletant cells did not (Fig. 3B), indicating that each of these genes is essential for the activity, and therefore, that the enzyme has three different subunits. When OP replaced ammonium as the nitrogen source, the OPase activity of wild-type cells increased 16-fold (Fig. 3B). The apparent Km for OP was determined as 39 ± 6 μm. Deleting the putative metal transporter gene ycsG or the putative metal chaperone gene ycsI did not significantly lower OPase activity (supplemental Fig. S3), indicating that neither of these genes is required for OPase activity.
Reconstitution of 5-oxoprolinase activity
To show that the pxpABC genes encode the subunits of an oligomeric OPase, we measured the OPase activity of recombinant proteins. The pxpB and pxpC genes are sometimes fused (Fig. 1, B and C), and the structure of the PxpBC fusion protein of Thermus thermophilus (PDB code 3ORE) suggests that unfused PxpB and PxpC could interact closely (31). We, therefore, cloned the B. subtilis pxpBC genomic region into pET28B as a unit. pxpBC was cloned so that either PxpB contained an N-terminal His tag and PxpC was untagged, or PxpB was untagged, and PxpC had a C-terminal His-tag. pxpA was cloned into pET28b with either an N- or C-terminal His tag. The proteins were expressed in E. coli and purified to near homogeneity (Fig. 4A). Untagged PxpC co-purified with N-terminally His-tagged PxpB and untagged PxpB co-purified with C-terminally His-tagged PxpC in roughly equal amounts (Fig. 4A), confirming that these two proteins interact strongly. Neither PxpA nor PxpBC alone had detectable OPase activity, but when they were combined in equimolar amounts ATP-dependent OPase activity was observed (Fig. 4A). As this activity was highly unstable (supplemental Fig. S3), kinetic constants could not be determined.
Figure 4.
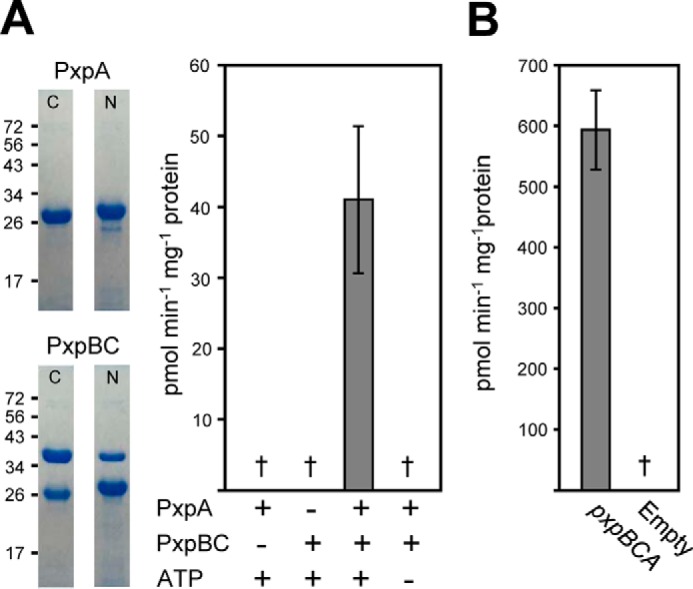
Recombinant B. subtilis and E. coli PxpABC proteins had 5-oxoprolinase activity. A, purified B. subtilis PxpA and PxpBC proteins containing an N- or C-terminal His tag were analyzed by SDS-PAGE and assayed for OPase activity. The positions of SDS-PAGE molecular weight markers (kDa) are indicated. Assays contained 10 μg of PxpBC (PxpB containing an N-terminal His tag), 4.4 μg of PxpA containing a C-terminal His-tag, and 5 mm ATP as indicated as in Fig. 3. B, OPase activity measured in lysates of E. coli cells harboring expression vector containing the E. coli pxpBCA genomic region. Data represent the mean ± S.E. of four purified protein preparations or three independent lysate preparations. †, not detectable (detection limit: 1.0 pmol min−1 mg−1 protein in purified protein assays; 0.3 pmol min−1 mg−1 protein in E. coli lysates).
pxpABC genes from E. coli encode 5-oxoprolinase
To show that pxpABC genes from other bacteria also encode OPase, we tested the genes from E. coli, a Gram-negative bacterium that is taxonomically distant from the Gram-positive B. subtilis. The E. coli pxpBCA genomic region was cloned into pET28b and expressed in E. coli. OPase activity was readily detected in pxpBCA-expressing cell lysates but not in empty-vector control lysates (Fig. 4B). The apparent Km for OP was estimated as 32 ± 3 μm for the E. coli enzyme. Deleting pxpA, pxpB, or pxpC in E. coli slowed growth on minimal medium with ammonium as nitrogen source (supplemental Fig. S2), indicating that, as for B. subtilis, these genes affect fitness in the absence of exogenous OP.
Discussion
Our results provide several lines of evidence that a conserved cluster of three hitherto uncharacterized but widely distributed prokaryotic genes jointly encode a novel ATP-dependent OPase. To recapitulate: (i) these three genes cluster with genes involved in OP metabolism, show an inverse distribution pattern with eukaryotic-type OPase genes, and are induced by OP; (ii) B. subtilis knockouts cannot use OP as nitrogen source, accumulate OP when grown on ammonium as nitrogen source, and lack detectable OPase activity; (iii) recombinant B. subtilis and E. coli enzymes have OPase activity. This evidence led us to name these genes prokaryotic 5-oxoprolinase pxpA, pxpB, and pxpC. These genes almost certainly encode the 3-subunit ATP-dependent OPase that was reported from P. putida more than 30 years ago (22–25). The P. putida enzyme had key features in common with the OPase described here, particularly that two of its subunits co-purified (23–25).
Ever since the discovery of the eukaryote-type OPase in the context of the γ-glutamyl cycle, participation in this cycle has generally been held to be the physiological role of OPases (32, 33). Our results challenge this assumption; pxpA, B, or C knockouts in B. subtilis, which has no glutathione or γ-glutamyl cycle, accumulate OP intracellularly to a concentration of 2.1 mm (based on a cell volume of 4.6 fl; Ref. 34) (Fig. 3A), excrete even more to the medium (Fig. 3A), and show a growth defect (Fig. 2B and supplemental Fig. S2). Clearly, in this case at least, OPase has a damage-control role, i.e. it prevents deleterious accumulation of OP of spontaneous chemical origin. That the growth effects of ablating pxpABC in B. subtilis, and also in E. coli (supplemental Fig. S2), are modest conforms to a pattern noted for other microbial damage-control genes (27, 35–37).
Although the eukaryote-type OPase and the prokaryote-type OPase described here catalyze the same reaction, they share no sequence similarity and differ in quaternary structure (one kind of subunit versus three). They, therefore, presumably represent independent evolutionary solutions to the universal problem of OP disposal. Because the eukaryote-type OPase occurs in α-Proteobacteria and Cyanobacteria (Fig. 1C), from which the progenitors of mitochondria and plastids, respectively, are thought to have come (38), this type of OPase could have an ancient prokaryotic origin. As glutamine, glutamate, and γ-glutamyl phosphate as well as peptides with N-terminal glutaminyl and glutamyl residues were almost surely present in the last universal common ancestor (39), OP has been forming since the dawn of life and long before the γ-glutamyl cycle arose. The ancestral role of both ATP-dependent OPases was, therefore, most likely damage-control, with the eukaryote-type enzyme being later recruited for use in the γ-glutamyl cycle. The old damage-control role presumably persists in eukaryotes today alongside the newer γ-glutamyl cycle role.
B. subtilis can take up OP and use it as a nitrogen source, and comparative genomic evidence suggests that the same may be true of many other bacteria (although not E. coli). OP uptake and utilization would allow such bacteria to exploit a common metabolite damage product. As noted above, bacterial OPase genes cluster with genes specifying the uncharacterized membrane proteins DUF969 and DUF979, which could play a role in OP transport. Prokaryote OPase genes also cluster with a putative metal transporter of the NRAMP family and a putative metal chaperone, suggesting that one or more of the pxp genes has a metal requirement. A close homolog of the NRAMP protein was recently shown to transport manganese (40).
Although our data demonstrate that PxpB and PxpC are subunits of an ATP-dependent OPase, other roles cannot be excluded, and some have been postulated. The B. subtilis pxpB gene product was reported to inhibit the autophosphorylation of kinase A, thus perturbing a phosphorelay signal transduction system involved in sporulation (41, 42). However, the significance of this observation is unclear because inhibition was only observed when PxpB was expressed alone and not when PxpB and PxpC were co-expressed to form a complex, as our data show they normally would be. It has also recently been shown that certain fused pxpB and pxpC family genes, which cluster strongly with a urea carboxylase gene, are involved in urea catabolism and encode an enzyme with allophanate hydrolase activity (40, 43). Allophanate hydrolase activity has only been demonstrated in a fused PxpBC homolog (43), but it is possible that other PxpB and PxpC proteins have dual roles in OP and urea metabolism. Additionally, the prokaryote-type OPase genes of Ralstonia solanacearum were shown to be required for pathogenesis in tomato (44).
Although OP had long been known as a damage product of glutamine, glutamate, and γ-glutamyl phosphate (9–11), the discovery of eukaryote-type OPase in the context of the γ-glutamyl cycle probably favored the general view, mentioned above, that OP comes mainly from this cycle (1, 45). If so, it illustrates how a “metabolic pathway-centric” view of metabolism (46, 47) can divert attention away from metabolite damage and the mechanisms that counter it, in this case enabling three very common damage-control genes to “hide in plain sight” in the genomes of two iconic model bacteria as these genomes were sequenced in 1997 (48, 49).
Experimental procedures
Comparative genomics
Sequences were taken from GenBankTM or the SEED database (26). Comparative analysis of 984 representative genomes was performed with SEED and its tools; the full results of the analysis are available in the SEED subsystem named “pyroglutamate.”
Bacterial strains and culture conditions
Wild-type B. subtilis strain 168 and BKE mutants (30) BKE04050 (pxpA; ycsF), BKE04060 (ycsG), BKE04070 (ycsI), BKE04080 (pxpB; kipI), and BKE04090 (pxpC; kipA) were obtained from the Bacillus Genetic Stock Center. All BKE mutant loci were back-crossed into wild-type B. subtilis 168 by transforming 5 μg of isolated mutant DNA into wild-type cells (50) and selecting for recombinants on LB agar plates containing 1 μg ml−1 erythromycin and 25 μg ml−1 lincomycin. The presence of the BKE cassette in the expected locus was verified (supplemental Fig. S4). For growth and functional complementation tests and growing samples for OP analysis and enzyme assays, the medium contained 8 mm K2HPO4, 4.4 mm KH2PO4, 30 mm NH4Cl, 2 mm MgSO4, 0.6 mm MgCl2, 27 mm glucose, 0.3 mm sodium citrate, 0.25 mm l-tryptophan, 0.1 mm FeCl3, 50 μm CaCl2, 5 μm MnCl2, 12 μm ZnCl2, 2.5 μm CuCl2, 2.5 μm CoCl2, and 2.5 μm Na2MoO4. For medium with OP as the sole N source, NH4Cl was replaced with 50 mm OP (neutralized with NaOH), and glucose was replaced with 30 mm sodium succinate. Solid media contained 0.8% (w/v) agarose. For growth tests, cells from overnight cultures in LB medium were washed twice with water and used to inoculate 3 ml of the appropriate medium to an optical density (600 nm) of 0.05 and grown at 37 °C with shaking at 250 rev/min. For functional complementation studies, strains were transformed with the appropriate plasmid (50) and grown as indicated. Cultures for OP analysis were grown in the same way, but when cells reached an optical density of 1.43 ± 0.02 (4.29 ± 0.06 × 108 total cells) cells and medium were harvested by centrifugation at 21,000 × g for 20 s and immediately frozen in liquid N2 and stored at −80 °C. For lysate preparation, cultures were grown in a similar way except that the volume was 100 ml and growth was for 7 h (when ammonium was the nitrogen source) or 48 h (when OP was the nitrogen source) until the optical density (600 nm) reached ∼2.0. Cells were collected by centrifugation at 8000 × g for 5 min and frozen in liquid nitrogen and stored at −80 °C. Note that E. coli could not be used to test the effect of pxpABC deletions on OP utilization because wild-type E. coli was found not to use OP as sole nitrogen source.
Expression constructs
Supplemental Table S1 lists the PCR primers used. All constructs were sequence-verified. Purified genomic DNA from B. subtilis strain 168 or E. coli strain MG1655 was used as the template in PCR reactions. To create constructs for functional complementation studies, B. subtilis pxpA (Uniprot ID: P42963), pxpB (Uniprot ID: P60495), pxpC (Uniprot ID: Q7WY77), ycsG (Uniprot ID: P42964), and ycsI (Uniprot ID: P42966) coding sequences were PCR-amplified, digested with SpeI and BamHI, and ligated into pHCMC04. To create constructs for expression of N- or C-terminally His-tagged proteins, B. subtilis pxpA and pxpBC were PCR-amplified, treated with PciI and XhoI or with NdeI and XhoI, respectively, and ligated into pET28b. The E. coli pxpBCA (ybgJKL; Uniprot IDs: P0AAV4, P75745, P75746) genomic region was PCR-amplified, treated with XbaI and XhoI, and ligated into pET28b.
Protein expression and purification
For protein expression in E. coli, 1 ml of overnight culture of E. coli strain BL21 (DE3) RIPL harboring the appropriate expression vector was used to inoculate 100 ml of LB medium containing 50 μg ml−1 kanamycin, which was grown at 37 °C with shaking until the optical density (600 nm) reached ∼0.8. At that point, cultures were cooled to 22 °C, supplemented with isopropyl-β-d-thiogalactoside and ethanol (final concentrations 0.5 mm and 4% v/v, respectively), and incubated for a further 20 h at 22 °C before harvesting cells. All B. subtilis and E. coli lysates (except when used for protein purification) were prepared by resuspending the pellet (from 50 ml of culture) in 0.2–0.5 ml of ice-cold extraction buffer (100 mm KCl, 50 mm Tris-HCl, pH 8.0, 5 mm tris(2-carboxyethyl)phosphine (TCEP)) and sonicating (Fisher Ultrasonic Dismembrator, model 150E) for 5 × 3 s pulses at 70% power, cooling on ice for 30–60 s between pulses. The resulting lysates were centrifuged at 10,000 × g for 10 min, and the supernatant was collected. B. subtilis lysates were desalted with extraction buffer using PD MiniTrap G-25 columns (GE Healthcare) according to the manufacturer's recommendations before assaying. Protein was determined by dye-binding (51). For protein purification, pellets were resuspended in 3 ml of ice-cold lysis buffer (50 mm Tris-HCl, pH 8.0, 300 mm KCl, 10 mm imidazole, 5 mm TCEP) and sonicated and centrifuged as above. The resulting supernatant was applied to 0.25 ml of nickel-nitrilotriacetic acid superflow resin (Qiagen) columns, which were washed and eluted according to the manufacturer's protocol; the wash and elution buffers were the same as the lysis buffer but contained 25 mm and 200 mm imidazole, respectively. Purified proteins were passed through PD-10 columns (GE Healthcare) equilibrated with 100 mm KCl, 50 mm Tris-HCl, pH 8.0, 5 mm TCEP, and 10% (v/v) glycerol and concentrated to 2–20 mg/ml with Amicon Ultra-4 10,000 NMWL centrifugal filters (Millipore). Aliquots (5–10 μl) were snap-frozen in liquid nitrogen and stored at −80 °C.
Enzyme assays
Assays (50 μl) contained 50 mm Tris-HCl, pH 8.0, 100 mm KCl, 5 mm ATP (in 10 mm MgCl2), 5 mm l-glutamate (lysates only), 0.1 μCi [3H]OP [2,3,4-3H]pyroglutamic acid (supplied by Moravek at a specific activity of 2.3 Ci mmol−1), and up to 1 mm unlabeled OP and were started by adding 4.4–10 μg of purified protein or 10–35 μg of total protein from bacterial lysates. Assays were incubated at 37 °C for 10–20 min, then 0.45 ml of carrier solution (0.1 mm OP, 0.1 mm l-glutamate) was added, and the reactions were loaded onto 0.5-ml Dowex-50 (H+) columns. The columns were washed with 4 ml of water to remove OP; l-glutamate was then eluted with 1.5 ml of 3 m NH4OH, and eluates were analyzed by scintillation spectroscopy.
OP analysis
Samples were thawed on ice (for medium samples, 200 μl was aliquoted to a fresh 1.5-ml Eppendorf tube). Extraction was conducted by adding 1.0 ml of cold 3:1 methanol:water containing l-glutamine-[2,3,3,4,4-D5] (for estimating extraction efficiency and to monitor spontaneous cyclization of glutamine to OP) to each sample tube and vortexing for 20 s, then sonicating for 5 min. After a 30-min freeze at −20 °C, samples were centrifuged for 5 min at 14,000 × g, and 475 μl of the resulting supernatant was transferred to a fresh 1.5 ml Eppendorf tube and evaporated to dryness with a Labconco CentriVap Concentrator. The dried material was resuspended with 100 μl of 80:20 acetonitrile:water with internal standards and analyzed by liquid chromatography mass spectrometry (LC-MS). Samples and solvents were kept on ice whenever possible to limit conversion of endogenous glutamate and glutamine to OP. Chromatography was performed on an Agilent 1290 Infinity LC System. Samples were maintained at 4 °C in the autosampler, and 3 μl of sample material was injected onto an Acquity UPLC BEH Amide column (150 × 2.1 mm; 1.7 μm) coupled to a VanGuard BEH Amide precolumn (5 × 2.1 mm; 1.7 μm) (Waters) kept at 45 °C. Mobile phase A consisted of 10 mm ammonium formate with 0.125% formic acid in water; mobile phase B consisted of 95:5 acetonitrile:10 mm ammonium formate in water (v/v) with 0.125% formic acid. Target metabolites were eluted from the column using the following gradient: 0 to 0.5 min, 90% B; 0.5 to 1.0 min, linear gradient to 75% B; 1.0 to 5.0 min, linear gradient to 60% B with 0.5 min hold; 5.5 to 5.8 min, linear gradient to 30% B with a 1.2 min hold followed by return to initial conditions at 7.2 min. Re-equilibration time was 2.8 min, and flow rate was held at 0.40 ml/min throughout the method. Metabolites were detected with a SCIEX TripleTOF 6600 mass spectrometer equipped with a SCIEX DuoSpray electrospray ionization source. MS data were acquired in negative ionization polarity; full-scan data were acquired from mass range m/z 50–500 with a 500-ms period cycle time and 150-ms accumulation time; OP (m/z 128.03) was quantified using product scan data at −25 eV collision energy and 100 ms accumulation time. Additional MS parameters were as follows: curtain gas, 25 p.s.i.; ion source gas 1, 50 p.s.i.; ion source gas 2, 60 p.s.i.; ion spray voltage floating, −4.5 kV; temperature, 350 °C; declustering potential, 100 V. Mass calibration was maintained via automatic injection of a premixed atmospheric pressure chemical ionization negative calibration solution delivered by a calibration delivery system. Target analytes were identified based on retention time and accurate mass matching to authentic standards; quantification was based on standard curves of the authentic standards.
Author contributions
T. D. N. and A. D. H. conceived the project. T. D. N, A. D. H., and V. de C.-L. made comparative genomics analyses. T. D. N., M. E.-S., and O. F. performed metabolic profiling. T. D. N performed all other experimental work. T. D. N. and A. D. H. wrote the paper.
Supplementary Material
Acknowledgments
We thank Daniel Ziegler and the Bacillus Genetic Stock Center for providing B. subtilis strains and vectors and for advice.
This work was supported by National Science Foundation Grant number MCB-1153413 and by an endowment from the C. V. Griffin, Sr. Foundation. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Table S1 and Figs. S1–S4.
- OP
- 5-oxoproline
- OPase
- 5-oxoprolinase
- TCEP
- tris(2-carboxyethyl)phosphine.
References
- 1. Orlowski M., and Meister A. (1970) The γ-glutamyl cycle: a possible transport system for amino acids. Proc. Natl. Acad. Sci. U.S.A. 67, 1248–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Griffith O. W., Bridges R. J., and Meister A. (1978) Evidence that the γ-glutamyl cycle functions in vivo using intracellular glutathione: effects of amino acids and selective inhibition of enzymes. Proc. Natl. Acad. Sci. U.S.A. 75, 5405–5408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Van der Werf P., Orlowski M., and Meister A. (1971) Enzymatic conversion of 5-oxo-l-proline (l-pyrrolidone carboxylate) to l-glutamate coupled with cleavage of adenosine triphosphate to adenosine diphosphate, a reaction in the γ-glutamyl cycle. Proc. Natl. Acad. Sci. U.S.A. 68, 2982–2985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Paulose B., Chhikara S., Coomey J., Jung H. I., Vatamaniuk O., and Dhankher O. P. (2013) A γ-glutamyl cyclotransferase protects Arabidopsis plants from heavy metal toxicity by recycling glutamate to maintain glutathione homeostasis. Plant Cell 25, 4580–4595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ohkama-Ohtsu N., Oikawa A., Zhao P., Xiang C., Saito K., and Oliver D. J. (2008) A γ-glutamyl transpeptidase-independent pathway of glutathione catabolism to glutamate via 5-oxoproline in Arabidopsis. Plant Physiol. 148, 1603–1613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kumar A., and Bachhawat A. K. (2010) OXP1/YKL215c encodes an ATP-dependent 5-oxoprolinase in Saccharomyces cerevisiae: functional characterization, domain structure, and identification of actin-like ATP-binding motifs in eukaryotic 5-oxoprolinases. FEMS Yeast Res. 10, 394–401 [DOI] [PubMed] [Google Scholar]
- 7. Okada T., Suzuki H., Wada K., Kumagai H., and Fukuyama K. (2006) Crystal structures of γ-glutamyltranspeptidase from Escherichia coli, a key enzyme in glutathione metabolism, and its reaction intermediate. Proc. Natl. Acad. Sci. U.S.A. 103, 6471–6476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Newton G.L., Rawat M., La Clair J.J., Jothivasan V.K., Budiarto T., Hamilton C.J., Claiborne A., Helmann J.D., and Fahey R.C. (2009) Bacillithiol is an antioxidant thiol produced in Bacilli. Nat. Chem. Biol. 5, 625–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tritsch G. L., and Moore G. E. (1962) Spontaneous decomposition of glutamine in cell culture media. Exp. Cell Res. 28, 360–364 [DOI] [PubMed] [Google Scholar]
- 10. Park C. B., Lee S. B., and Ryu D. D. (2001) l-Pyroglutamate spontaneously formed from l-glutamate inhibits growth of the hyperthermophilic archaeon Sulfolobus solfataricus. Appl. Environ. Microbiol. 67, 3650–3654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Orlowski M., and Meister A. (1971) Partial reactions catalyzed by γ-glutamylcysteine synthetase and evidence for an activated glutamate intermediate. J. Biol. Chem. 246, 7095–7105 [PubMed] [Google Scholar]
- 12. Schilling S., Wasternack C., and Demuth H. U. (2008) Glutaminyl cyclases from animals and plants: a case of functionally convergent protein evolution. Biol. Chem. 389, 983–991 [DOI] [PubMed] [Google Scholar]
- 13. Carrillo D. R., Parthier C., Jänckel N., Grandke J., Stelter M., Schilling S., Boehme M., Neumann P., Wolf R., Demuth H. U., Stubbs M. T., and Rahfeld J. U. (2010) Kinetic and structural characterization of bacterial glutaminyl cyclases from Zymomonas mobilis and Myxococcus xanthus. Biol. Chem. 391, 1419–1428 [DOI] [PubMed] [Google Scholar]
- 14. Awadé A., Cleuziat P., Gonzalès T., and Robert-Baudouy J. (1992) Characterization of the pcp gene encoding the pyrrolidone carboxyl peptidase of Bacillus subtilis. FEBS Lett. 305, 67–73 [DOI] [PubMed] [Google Scholar]
- 15. Pedrotti E. L., Jay-Allemand C., Doumas P., and Cornu D. (1994) Effect of autoclaving amino acids on in vitro rooting response of wild cherry shoot. Scientia Horticulturae 57, 89–98 [Google Scholar]
- 16. Silva A. R., Silva C. G., Ruschel C., Helegda C., Wyse A. T., Wannmacher C. M., Wajner M., and Dutra-Filho C. S. (2001) l-Pyroglutamic acid inhibits energy production and lipid synthesis in cerebral cortex of young rats in vitro. Neurochem. Res. 26, 1277–1283 [DOI] [PubMed] [Google Scholar]
- 17. Pederzolli C. D., Sgaravatti A. M., Braum C. A., Prestes C. C., Zorzi G. K., Sgarbi M. B., Wyse A. T., Wannmacher C. M., Wajner M., and Dutra-Filho C. S. (2007) 5-Oxoproline reduces non-enzymatic antioxidant defenses in vitro in rat brain. Metab. Brain Dis. 22, 51–65 [DOI] [PubMed] [Google Scholar]
- 18. Ristoff E., and Larsson A. (2007) Inborn errors in the metabolism of glutathione. Orphanet. J. Rare Dis. 2, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sass J. O., Gemperle-Britschgi C., Tarailo-Graovac M., Patel N., Walter M., Jordanova A., Alfadhel M., Barić I., Çoker M., Damli-Huber A., Faqeih E. A., García Segarra N., Geraghty M. T., Jåtun B. M., Kalkan Uçar S., et al. (2016) Unravelling 5-oxoprolinuria (pyroglutamic aciduria) due to bi-allelic OPLAH mutations: 20 new mutations in 14 families. Mol. Genet. Metab. 119, 44–49 [DOI] [PubMed] [Google Scholar]
- 20. Nishimura A., Ozaki Y., Oyama H., Shin T., and Murao S. (1999) Purification and characterization of a novel 5-oxoprolinase (without ATP-hydrolyzing activity) from Alcaligenes faecalis N-38A. Appl. Environ. Microbiol. 65, 712–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nishimura A., Oyama H., Hamada T., Nobuoka K., Shin T., Murao S., and Oda K. (2000) Molecular cloning, sequencing, and expression in Escherichia coli of the gene encoding a novel 5-oxoprolinase without ATP-hydrolyzing activity from Alcaligenes faecalis N-38A. Appl. Environ. Microbiol. 66, 3201–3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Van der Werf P., and Meister A. (1974) Isolation of 5-oxoprolinase from a prokaryote. Biochem. Biophys. Res. Commun. 56, 90–96 [DOI] [PubMed] [Google Scholar]
- 23. Seddon A. P., Li L. Y., and Meister A. (1984) Resolution of 5-oxo-l-prolinase into a 5-oxo-l-proline-dependent ATPase and a coupling protein. J. Biol. Chem. 259, 8091–8094 [PubMed] [Google Scholar]
- 24. Seddon A. P., and Meister A. (1986) Trapping of an intermediate in the reaction catalyzed by 5-oxoprolinase. J. Biol. Chem. 261, 11538–11543 [PubMed] [Google Scholar]
- 25. Li L. Y., Seddon A. P., and Meister A. (1988) Interaction of the protein components of 5-oxoprolinase. Substrate-dependent enzyme complex formation. J. Biol. Chem. 263, 6495–6501 [PubMed] [Google Scholar]
- 26. Overbeek R., Begley T., Butler R. M., Choudhuri J.V., Chuang H. Y., Cohoon M., de Crécy-Lagard V., Diaz N., Disz T., Edwards R., Fonstein M., Frank E. D., Gerdes S., Glass E. M., Goesmann A., et al. (2005) The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 33, 5691–5702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Niehaus T. D., Gerdes S., Hodge-Hanson K., Zhukov A., Cooper A. J., ElBadawi-Sidhu M., Fiehn O., Downs D. M., and Hanson A. D. (2015) Genomic and experimental evidence for multiple metabolic functions in the RidA/YjgF/YER057c/UK114 (Rid) protein family. BMC Genomics 16, 382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hanson A. D., Pribat A., Waller J. C., and de Crécy-Lagard V. (2009) “Unknown” proteins and “orphan” enzymes: the missing half of the engineering parts list and how to find it. Biochem. J. 425, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bochner B. R., Gadzinski P., and Panomitros E. (2001) Phenotype microarrays for high-throughput phenotypic testing and assay of gene function. Genome Res. 11, 1246–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Koo B.M., Kritikos G., Farelli J.D., Todor H., Tong K., Kimsey H., Wapinski I., Galardini M., Cabal A., Peters J. M., Hachmann A. B., Rudner D. Z., Allen K. N., Typas A., and Gross C. A. (2017) Construction and analysis of two genome-scale deletion libraries for Bacillus subtilis. Cell Syst. 4, 291–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jacques D. A., Langley D. B., Kuramitsu S., Yokoyama S., Trewhella J., and Guss J. M. (2011) The structure of TTHA0988 from Thermus thermophilus, a KipI-KipA homologue incorrectly annotated as an allophanate hydrolase. Acta Crystallogr. D. Biol. Crystallogr. 67, 105–111 [DOI] [PubMed] [Google Scholar]
- 32. Mazelis M., and Creveling R. K. (1978) 5-Oxoprolinase (l-pyroglutamate hydrolase) in higher plants: partial purification and characterization of the wheat germ enzyme. Plant Physiol. 62, 798–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jaspers C. J., Gigot D., and Penninckx M. J. (1985) Pathways of glutathione degradation in the yeast Saccharomyces cerevisiae. Phytochemistry 24, 703–707 [Google Scholar]
- 34. Yu A. C., Loo J. F., Yu S., Kong S. K., and Chan T. F. (2014) Monitoring bacterial growth using tunable resistive pulse sensing with a pore-based technique. Appl. Microbiol. Biotechnol. 98, 855–862 [DOI] [PubMed] [Google Scholar]
- 35. Marbaix A. Y., Noël G., Detroux A. M., Vertommen D., Van Schaftingen E., and Linster C. L. (2011) Extremely conserved ATP- or ADP-dependent enzymatic system for nicotinamide nucleotide repair. J. Biol. Chem. 286, 41246–41252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lambrecht J. A., Flynn J. M., and Downs D. M. (2012) Conserved YjgF protein family deaminates reactive enamine/imine intermediates of pyridoxal 5′-phosphate (PLP)-dependent enzyme reactions. J. Biol. Chem. 287, 3454–3461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Peracchi A., Veiga-da-Cunha M., Kuhara T., Ellens K. W., Paczia N., Stroobant V., Seliga A. K., Marlaire S., Jaisson S., Bommer G. T., Sun J., and Huebner K., and Linster C. L., et al. (2017) Nit1 is a metabolite repair enzyme that hydrolyzes deaminated glutathione. Proc. Natl. Acad. Sci. U.S.A. 114, E3233–E3242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gray M. W. (2017) Lynn Margulis and the endosymbiont hypothesis: 50 years later. Mol. Biol. Cell 28, 1285–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ouzounis C. A., Kunin V., Darzentas N., and Goldovsky L. (2006) A minimal estimate for the gene content of the last universal common ancestor: exobiology from a terrestrial perspective. Res. Microbiol. 157, 57–68 [DOI] [PubMed] [Google Scholar]
- 40. Juttukonda L. J., Chazin W. J., and Skaar E. P. (2016) Acinetobacter baumannii coordinates urea metabolism with metal import to resist host-mediated metal limitation. MBio. 7, e01475–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang L., Grau R., Perego M., and Hoch J. A. (1997) A novel histidine kinase inhibitor regulating development in Bacillus subtilis. Genes Dev. 11, 2569–2579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jacques D. A., Langley D. B., Hynson R. M., Whitten A. E., Kwan A., Guss J. M., and Trewhella J. (2011) A novel structure of an antikinase and its inhibitor. J. Mol. Biol. 405, 214–226 [DOI] [PubMed] [Google Scholar]
- 43. Lin Y., Boese C. J., and St Maurice M. (2016) The urea carboxylase and allophanate hydrolase activities of urea amidolyase are functionally independent. Protein Sci. 25, 1812–1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang Y., Kiba A., Hikichi Y., and Ohnishi K. (2011) prhKLM genes of Ralstonia solanacearum encode novel activators of hrp regulon and are required for pathogenesis in tomato. FEMS Microbiol. Lett. 317, 75–82 [DOI] [PubMed] [Google Scholar]
- 45. Meister A. (1973) On the enzymology of amino acid transport. Science 180, 33–39 [DOI] [PubMed] [Google Scholar]
- 46. Golubev A. G. (1996) The other side of metabolism. Biokhimiia 61, 2018–2039 [PubMed] [Google Scholar]
- 47. Golubev A., Hanson A. D., and Gladyshev V. N. (2017) Non-enzymatic molecular damage as a prototypic driver of aging. J. Biol. Chem. 292, 6029–6038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Blattner F. R., Plunkett G. 3rd, Bloch C. A., Perna N. T., Burland V., Riley M., Collado-Vides J., Glasner J. D., Rode C. K., Mayhew G. F., Gregor J., Davis N. W., Kirkpatrick H. A., Goeden M. A., Rose D. J., Mau B., and Shao Y. (1997) The complete genome sequence of Escherichia coli K-12. Science 277, 1453–1462 [DOI] [PubMed] [Google Scholar]
- 49. Kunst F., Ogasawara N., Moszer I., Albertini A. M., Alloni G., Azevedo V., Bertero M. G., Bessières P., Bolotin A., Borchert S., Borriss R., Boursier L., Brans A., Braun M., and Brignell S. C., et al. (1997) The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature 390, 249–256 [DOI] [PubMed] [Google Scholar]
- 50. Harwood C. R., and Cutting S. M. (1990) Molecular Biological Methods for Bacillus, pp. 27–74, John Wiley & Sons Ltd, Chichester, England [Google Scholar]
- 51. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.