

Abstract
α-Synuclein (αS) is the primary protein associated with Parkinson's disease, and it undergoes aggregation from its intrinsically disordered monomeric form to a cross-β fibrillar form. The closely related homolog β-synuclein (βS) is essentially fibril-resistant under cytoplasmic physiological conditions. Toxic gain-of-function by βS has been linked to dysfunction, but the aggregation behavior of βS under altered pH is not well-understood. In this work, we compare fibril formation of αS and βS at pH 7.3 and mildly acidic pH 5.8, and we demonstrate that pH serves as an on/off switch for βS fibrillation. Using αS/βS domain-swapped chimera constructs and single residue substitutions in βS, we localized the switch to acidic residues in the N-terminal and non-amyloid component domains of βS. Computational models of βS fibril structures indicate that key glutamate residues (Glu-31 and Glu-61) in these domains may be sites of pH-sensitive interactions, and variants E31A and E61A show dramatically altered pH sensitivity for fibril formation supporting the importance of these charged side chains in fibril formation of βS. Our results demonstrate that relatively small changes in pH, which occur frequently in the cytoplasm and in secretory pathways, may induce the formation of βS fibrils and suggest a complex role for βS in synuclein cellular homeostasis and Parkinson's disease.
Keywords: computer modeling, fibril, neurodegeneration, nuclear magnetic resonance (NMR), Parkinson disease, protein aggregation, synuclein, beta-synuclein
Introduction
α-Synuclein (αS),5 the primary protein component of intra-cytoplasmic inclusions known as Lewy bodies (LB) in Parkinson's disease (PD), is a 140-residue, predominantly monomeric, intrinsically disordered protein (IDP) (1–6). It is abundant in the cytosol but present in many organelles (7, 8). The monomers of αS can aggregate to soluble oligomers as well as insoluble oligomers and fibrils, which are considered to be associated with the pathogenesis of PD (9). A closely related family member, β-synuclein (βS) that co-localizes with αS, has ∼60% sequence identity with αS but has not been detected in LBs of PD patients (10, 11). Instead, it has been proposed that βS may delay αS fibril formation and ameliorate αS toxicity in vivo by inhibiting αS aggregation (12–14). Furthermore, despite the high sequence similarity, βS itself does not form fibrils in vitro at cytoplasmic pH without facilitating agents (15). However, recent reports highlight a role for a possible toxic gain-of-function of βS in two different model systems (16, 17), and βS is a component of vesicle-like lesions in the hippocampus, whose formation accompanies dementia in PD (18). In addition, two βS mutations, V70M and P123H, were found in sporadic and familial dementia with LBs and have been suggested to be involved in lysosomal pathology (19–21). There is an emerging role for βS in the pathophysiology of PD, but the molecular determinants of this role remain poorly studied.
Understanding the complex relationship of synucleins with neurodegeneration requires consideration of their structures and functions in diverse subcellular environments, beyond the cytoplasm. Although the function of αS is still unclear, membranes and synaptic vesicles are sites of function for αS (22, 23), which may have a role in dopamine transmission, trafficking, and regulation of the vesicular pool (23), and βS may play similar roles (24). During their functional life cycle, synucleins likely experience a wide range of pH microenvironments, including environments more acidic than the neutral pH maintained in the neuronal cytosol (25, 26). Intravesicular pH drops along the endocytic pathway to ∼6 and in late endosomes and lysosomes to as low as 4.5 (25, 27). In addition to its role in vesicular fusion and trafficking, αS is subject to lysosomal degradation (28), and it may enter the slightly alkaline mitochondrial environment (29). Furthermore, pH dysregulation accompanying oxidative stress can result in cytosolic acidification (30), and oxidative stress is a significant contributor to PD pathology (31). Given the ubiquitous presence of synucleins in different cellular compartments, it is crucial to investigate the environmental pH dependence of βS and αS aggregation to understand synuclein homeostasis and its dysregulation in PD.
In their monomeric forms, αS and βS synucleins are model IDPs that exist as unfolded protein ensembles, only transiently adopting small degrees of local secondary structure (2, 13, 32–34). IDP side chains will be exposed to the cellular environment to a significantly greater extent than buried side chains of a folded protein, thereby making them more susceptible to environmental changes but uniquely able to respond rapidly to these changes and perform regulatory roles (35, 36). Subcellular pH differences between organelles may have more profound effects on IDPs, increasing local hydrophobicity as side-chain charge decreases. Altered charge/hydrophobicity balance will therefore change the conformational preferences in a particular region of an IDP and modulate its aggregation propensity (37–40). The distribution of charge and hydrophobicity differs significantly between αS and βS. A comparison of their sequences shows that synuclein proteins, including αS and βS, have a three-domain organization and contain the following (Fig. 1): 1) an N-terminal amphipathic domain of alternating charge and hydrophobicity, 2) a central hydrophobic core domain known as the non-amyloid component (NAC), and 3) an acidic C-terminal domain, which is comparatively more proline-rich and highly charged. αS and βS differ most in their hydrophobic NAC domains where the βS NAC is 11 residue shorter. The N-terminal domains share the highest sequence identity, differing only in six point mutations. The C-terminal domain of βS is more acidic and more proline-rich than αS, and it constitutes a larger percentage of the polypeptide length compared with αS. This distribution of charge/hydrophobicity implies that the pH-dependent behavior of these proteins may be mediated by charge changes in the N-terminal, NAC, and C-terminal domains upon alteration of the environmental pH. Although the NAC region is known to be necessary for aggregation of αS (41), the susceptibility of the N, NAC, and C domains of αS and βS to promote aggregation, and the interdependence of these domains to regulate aggregation in a pH-dependent manner, is not well-understood.
Figure 1.

Sequence comparison of αS and βS. Aligned sequences of βS and αS in a three-domain organization: (amphipathic) terminus, NAC (hydrophobic), and C terminus (acidic) domains. Charged residues are highlighted, and ones with potentially altered pKa values in our experimental pH range are colored as follows: free N terminus, dark blue; histidine, light blue; glutamic acid, dark red; aspartic acid, orange. Identical residues in the aligned position are indicated by a vertical dash between the two sequences. Sequence gaps are indicated by a horizontal dash and a pale yellow color.
Altered microenvironments may also alter the stability, structure, and formation rates of the aggregated state of synucleins, which, in turn, may determine their toxicity (42). αS in a pathogenic fibrillar form exists in parallel, in-register β-sheets (43). A novel Greek-key topology for the αS fibril has been determined by solid-state NMR experiments to a high level of molecular detail (44). Alternative topologies, derived from modeling based on experimentally determined structural restraints, have also been proposed (45), and other experimental results suggest an inherent polymorphism and environmental sensitivity of the αS aggregate morphology (46–48). Environmental changes, such as those in pH, may exert their effects by stabilization of one or more of these fibrillar topologies similar to the effect of familial mutations switching the preference between two different fibril topologies, as recently proposed by Nussinov and co-workers (45). In several amyloidogenic systems, exposure to mildly acidic pH results in fibril formation and/or alters fibril stability and structure (46, 49–53). As the fibrillation reaction is under kinetic control, a small change in the protonation states of key charged residues, leading to the formation of aggregation-competent species, can dramatically alter the reaction rate (51). As Vendruscolo and co-workers (38) have demonstrated for αS, mildly acidic pH (pH 5.2) can contribute to fibrillation acceleration relative to neutral pH by amplification of secondary fragmentation processes. Structurally, altered protonation of side chains has been shown to alter the registry of the fibril structure (46), and may relieve inter- and intra-chain repulsion between like charges, as well as provide additional stability to the fibril in the formation of cooperative side chain-mediated hydrogen bond networks (“polar zippers” (54)).
Here, we investigate the role of mildly acidic pH on the propensity for synuclein fibril formation. We demonstrate the presence of an environmentally sensitive pH mechanism in βS that serves as an on/off switch for fibrillation at mildly acidic physiological pH, thereby modifying the view that βS is non-fibrillogenic. By constructing a set of αS/βS domain-swapped chimeras, we show that although the NAC domain is indeed the strongest determinant of fibrillation propensity, the βS N-terminal domain and its interactions with the βS NAC domain can significantly modulate its fibril formation. Structural studies with NMR and AFM show that there are no pH-dependent conformational changes in the intrinsically disordered monomer but rather that the pH switch may derive from the ability of βS to form fibrils due to a new charge distribution. Computational modeling of βS fibrils indicates that a Greek-key motif observed in αS fibrils can be maintained in models of βS fibrillar structures. Fibril models also reveal two glutamate side chains that may underlie the observed pH-dependent fibrillation of βS, and single substitutions to alanine at these positions lead to significantly altered aggregation behavior. These findings provide insight into the complex interplay of charge and hydrophobicity for fibril formation in different cellular microenvironments, and they suggest that βS may play a multifaceted role in disease and homeostasis in PD.
Results
βS forms fibrils at mildly acidic pH
To investigate the impact of pH on αS and βS fibrillation, we performed thioflavin T (ThT)-based fibrillation assays with purified proteins that were incubated at two different pH values. Fibrillation kinetics of αS and βS measured at near-physiological pH 7.3 and mildly acidic pH 5.8 show that αS fibrillation is accelerated at pH 5.8 relative to neutral pH, whereas βS exhibits a striking pH-dependent on/off fibrillation switch (Fig. 2, A and B); at pH 7.3 βS does not form fibrils, although at pH 5.8 βS forms fibrils with an apparent half-time of greater than 60 h (Fig. 2B). Aggregation of βS has similar (slow) lag and (rapid) elongation phases characteristic of amyloid-like fibril formation by αS and many other fibrillogenic proteins, suggesting that βS aggregates are fibrillar in topology. Indeed, upon visualization of βS aggregates with AFM (Fig. 2, C–F), fibrillar structures similar to αS aggregates at both pH values were observed. Both fibrils have similar overall topologies and heights of ∼5–8 nm.
Figure 2.

pH-dependent ThT fibrillation of αS and βS and AFM imaging. 1 mg/ml αS (A) and βS (B) were incubated at 37 °C in a plate reader, and fibril formation was monitored with a ThT-binding assay in 20 mm MES, 20 mm MOPS, 100 mm NaCl, pH 5.8 (blue), and pH 7.3 (red). Normalized average fluorescence values were used for plotting the curve, and the standard deviations calculated from at least three replicates are indicated by shading in a lighter color. The samples that did not show any fluorescence are indicated by a horizontal line for comparison. AFM imaging was carried out at the end of the fibrillation process when samples that formed fibrils reached the plateau phase. AFM images for αS at low pH 5.8 (C) and high pH 7.3 (D) showed fibrils. βS forms fibrils at pH 5.8 (E) and spherical oligomers (F) at pH 7.3. The oligomers do not exhibit fluorescence in the ThT-binding assay (B; red curve).
We next investigated the molecular determinants of the observed pH-dependent fibril formation by βS by considering elements of the protein that may change their charge when the pH is changed from 7.3 to 5.8 (supplemental Fig. S1). A histidine side chain (theoretical pKa∼6.4), His-65, which is present in the βS NAC region (but not in αS NAC), is a likely candidate. Additionally, a previous study showed that several Asp and Glu residues, distributed throughout the N-terminal, NAC, and C-terminal domains, had altered pKa values in the αS monomeric state (55), suggesting that the titration of one or more of the homologous residues in βS could also impart the observed pH sensitivity for aggregation. The N terminus (with a theoretical pKa of 7.7 ± 0.5 (56)) may have a small charge difference in this pH range, in the monomeric state. Therefore, we investigated the following three potential candidates for the pH-dependent switch: 1) the His-65 residue in the NAC region; 2) the free-N terminus; and 3) the acidic residues in the N-terminal, NAC, and C-terminal domains.
To uncover which of the three sites identified above contribute to the pH-dependent βS fibrillation, two of the sites were made insensitive to pH changes (between pH 5.8 and 7.3); the H65N mutation converts the His at position 65 in the βS NAC region to the polar charge-invariant Asn, which is also the identity of the residue at position 65 in αS (Fig. 3A). We found that this variant exhibited similar on/off pH-dependent fibrillation kinetics compared with the wild-type βS protein, suggesting that the His-65 is not responsible for the pH-dependent switch. Next a pH-insensitive N terminus was designed by enzymatic N-terminal acetylation of the H65N mutant (4), thereby creating a variant that is pH-insensitive both at the N terminus and in the NAC region of βS. Despite simultaneous elimination of possible pH sensitivity at the N terminus and His-65, pH-dependent fibrillation is still observed (Fig. 3B). Therefore, these results suggest that the third putative pH-sensitive element, the series of acidic residues in the N-terminal, NAC, and C-terminal domains, is likely to be the primary determinant of the observed pH sensitivity of βS fibril formation.
Figure 3.
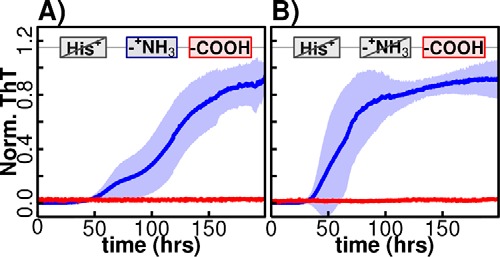
pH-dependent fibril formation of H65N-βS and acetylated H65N-βS. pH-dependent fibril formation for H65N-βS (A) and a variant in which the free N terminus is acetylated in the H65N variant (B) are shown. ThT fibrillation assays for both variants show pH dependence, indicating that neither site is responsible for the observed pH-dependent switch.
Domain-swapped synuclein chimeras reveal the interplay of synuclein domains in βS pH-regulated fibril formation
βS has eight Glu/Asp residues in the N-terminal domain, 19 in the C-terminal domain, and 1 in the NAC domain (Fig. 1) compared with 7, 15, and 2, respectively, in αS. An intractable number of combinations can be generated by swapping these residues between αS and βS. Therefore, to further delineate the contribution of the individual synuclein domains to the observed pH-dependent fibrillation of βS, and to investigate the domain–domain interactions underlying fibrillation of αS and βS, we utilized segmental mutagenesis. A set of eight chimeric proteins that contain segmental domain swaps between the N-terminal amphipathic domains (residues 1:60), the NAC hydrophobic domains (residues 61–95 in αS; residues 61–86 in βS), and the C-terminal acidic domains (residues 96–140 in αS; residues 87–134 in βS) were designed and isolated. The eight chimeric proteins are designated as αS WT (AAA), βS WT (BBB), BBA, ABB, ABA, BAB, BAA, and AAB with the notation A and B indicating the protein from which the domain is taken (αS and βS, respectively), and had lengths that range from 130 to 143 amino acids.
Examination of the fibrillation kinetics of this set of chimeras at the two pH values shows a clear grouping defined by the NAC domains, indicating that the NAC region is a key determinant of fibrillation behavior. Strikingly, the XAX (where X represents an A or B domain in the indicated position) chimeras that contain the αS NAC are always fibrillogenic at both pH values, whereas those containing the βS NAC, XBX are non-fibrillogenic at high pH, but ThT-fluorescent and therefore cross-β structure-positive, with the exception of ABB, at low pH (Fig. 4, A–D). Additionally, XXB chimeras typically have a longer lag time than XXA chimeras suggesting that the βS C-terminal domain may be involved in inhibitory processes (supplemental Fig. S2).
Figure 4.

pH-dependent αS/βS domain-swapped chimera fibrillation. ThT-binding fluorescence assay for chimeras that contain the αS-NAC domain (XAX) at low (A) and high (B) pH and chimeras that contain the βS NAC domain (XBX) at low (C) and high (D) pH are shown. Normalized averages of fluorescence values are used to plot the curves, and the standard deviation calculated from at least three replicates is shown by shading in a lighter color. The chimeras that did not show any fluorescence are indicated by a horizontal line. Fluorescence traces for AAA and BBB constructs are identical those in Fig. 2 but are reproduced here for comparison.
The observed fibrillation trends of chimeras enable a delineation of the contribution of each domain and domain–domain interactions to fibrillation. For example, the contribution of the N-terminal domain to low-pH fibrillation can be investigated by comparing fibrillation of ABB and BBB chimeric proteins. ABB chimeric protein fibrillation is significantly diminished compared with WT βS (BBB) despite having an identical NAC domain (Fig. 4D). AFM-derived images show that βS (BBB) forms robust, αS-like, fibrils at mildly acidic pH, whereas the ABB protein forms spherical globules (Fig. 5, A and B). Replacing the C-terminal domain of the ABB construct with the corresponding αS domain, resulting in the ABA chimera, restores the ability for β-sheet structures (as detected by ThT binding) at pH 5.8. However, the resulting aggregates do not resemble amyloid fibrils (Fig. 5D). In contrast, the chimeric protein BBA forms BBB-like fibrils at pH 5.8, suggesting that the pH-dependent interaction between βS NAC and βS N-terminal domains is a significant contributor to inducing fibrillation at mildly acidic pH (Fig. 5C). In the XAX set, the αS NAC region, on the other hand, is necessary and sufficient for fibrillation at both pH values, and the flanking N- and C-terminal domains modulate its fibrillation to a less striking degree.
Figure 5.
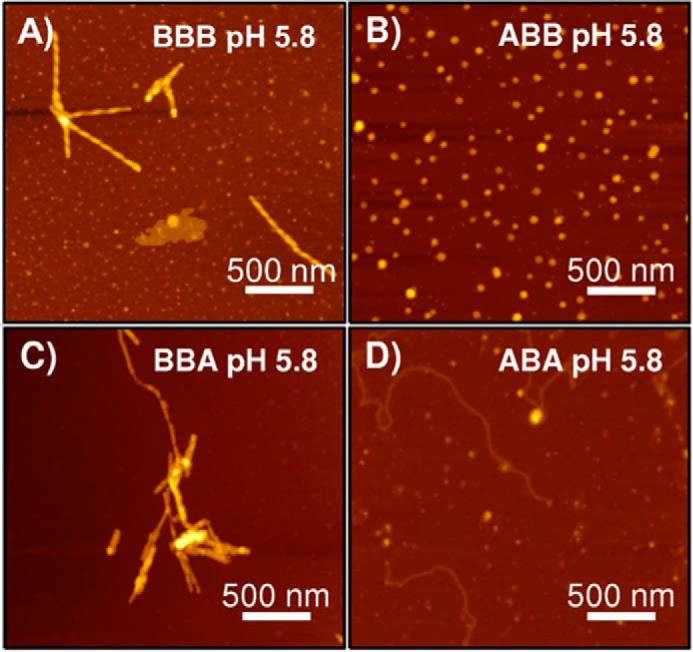
AFM images of low pH 5.8 ABX and BBX fibrils. Non-contact mode AFM images for low pH samples of chimeric constructs with a βS NAC domain in a 2.5 × 2.5 μm spot size are shown. A, WT βS; B, ABB; C, BBA; and D, ABA chimeric proteins. Scale bar, 500 nm.
Taken together, the observed fibrillation behavior of H65N βS mutant, N-terminally acetylated βS (Fig. 3) and XBX chimeric variants (Fig. 4) indicate that 1) the βS NAC region plays a predominant role in fibril formation at pH 5.8, 2) pH-dependent changes in the NAC and N-terminal domains critically modulate the aggregation propensity of βS, and 3) the presence of the βS C terminus consistently slows the aggregation kinetics suggesting that this region plays an inhibitory role in fibril formation.
αS and βS monomer conformational ensembles are largely unperturbed by changing pH
To determine whether the observed pH-dependent aggregation of βS is a result of changes in the conformational ensemble of monomeric βS, pH-dependent structural rearrangements of the monomer were explored by measuring residual dipolar couplings (RDC) and intrachain paramagnetic spin relaxation experiments (PREs) at the two pH values (Fig. 6). 1H-15N HSQC comparison of βS at pH 7.3 and 5.8 shows small chemical shift perturbations primarily at the sites of ionizable side chains, suggesting that the protonation states of these residues are being affected in this experimental pH range (Fig. 6A). Strikingly, no significant pH-dependent rearrangement is observed via the RDCs or PREs for βS or αS (Fig. 6, B–E). In the RDC experiment, which is a measure of order in the monomer, the profile is the same at the two pH values; but notably, the C-terminal domain of βS between residues ∼90 and ∼115 is more ordered than that of αS, consistent with previous results (Fig. 6, B and C) (57–59). In the PRE experiment, a spin label is placed in the NAC domain adjacent to the C-terminal domain to probe transient interactions with the neighboring N- and/or C-terminal domains. αS-90-MTSL and βS-79-MTSL pH-dependent PRE measurements indicate that the monomer conformations are populated differently in αS and βS with the NAC interacting more strongly with the C terminus in αS and the NAC interacting more strongly with the N terminus in βS consistent with previous results (Fig. 6, D and E) (60). The stronger interaction between the βS NAC and N-terminal domain compared with αS may contribute to its greater sensitivity to pH changes as observed by the lack of Tht-fluorescent aggregation of the ABB chimera. Nonetheless, these long-range interactions in the monomer appear to be unperturbed by pH change. Although our results do not rule out the existence of an additional rarely populated conformational state at low pH values that is not detectable by these NMR experiments due to its transient nature, we conclude that αS and βS monomer conformations do not exhibit any significant pH-dependent conformational change. The differences in the fibrillation behavior of βS at low and high pH values, therefore, may arise from a difference in formation rates and/or stabilities of higher-order (oligomeric and/or fibril) species.
Figure 6.
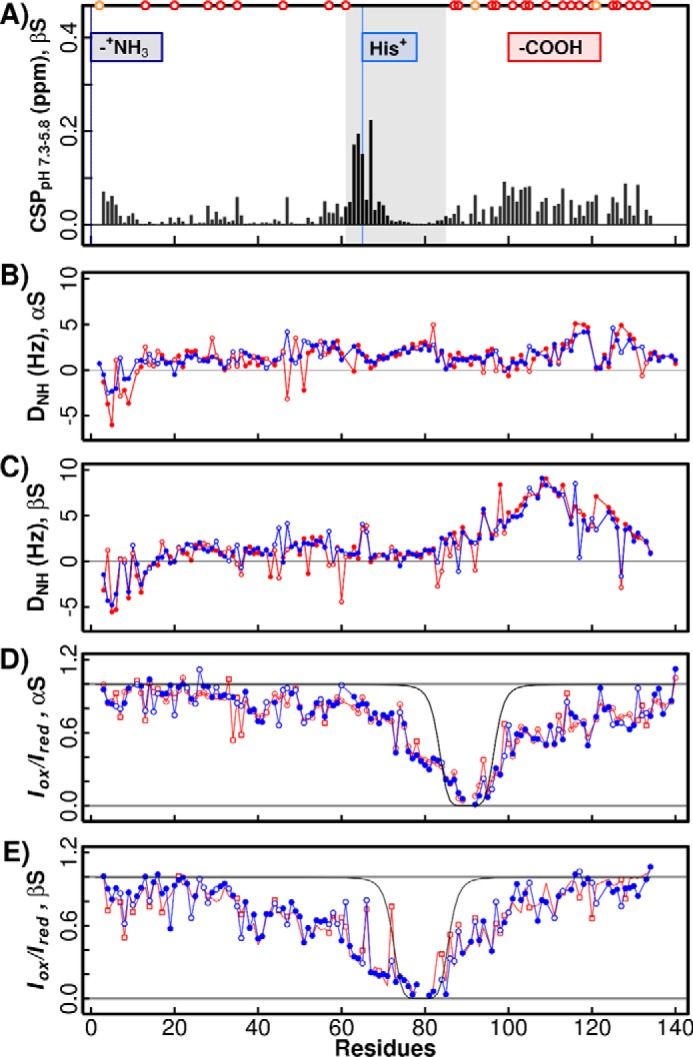
RDCs and intrachain PREs. A, βS chemical shift perturbation across the pH range 7.3–5.8 calculated using the normalized CSP expression ((ΔH)2 + (0.159·Δ15N)2)1/2 (4). Three putative pH-dependent events are indicated as follows: at the free N terminus (dark blue), at His-65 (light blue), and groups of aspartic and glutamic acids (−COOH). Although these acidic residues are concentrated at the C terminus, they are also present in the N-terminal domain indicated as dark red (glutamic) and orange (aspartic acid) circles. pH-dependent amide RDCs are shown for αS in B and βS in C with low pH 5.8 (blue) and high pH 7.3 (red). 1H-15N HSQC-IPAP spectra were collected in the absence or presence of C8E5 aligning media in 20 mm NaPi, 100 mm NaCl at both pH values. pH-dependent intra-chain PRE values are shown for A90C-MTSL-labeled αS in D and A79C-MTSL-labeled βS in E measured in 20 mm NaPi, 100 mm NaCl buffer and after the addition of 10 mm sodium ascorbate. High pH 7.3 PRE values are shown in red and low pH 5.8 values in blue. Open circles indicate points that are overlapped. Black lines are calculated as the expected profile for a random coil chain as in Sung and Eliezer (2).
Modeling the structure of βS fibrils suggests sites of pH sensitivity
Structural studies with AFM (Fig. 2) showed that fibrils formed by βS at low pH adopt overall topologies that are similar to αS fibrils under high-pH conditions. Therefore, we investigated the molecular structure of βS fibrils. Recently, an αS fibril structure was determined using solid-state NMR techniques and was reported to have a Greek-key like architecture (44). Given the similarity between αS fibrils at high and low pH, we used this structure as our starting model to ask if and how βS is capable of adopting and maintaining a similar fibril structure. Using Rosetta software, the aligned βS sequence was threaded onto the backbone structure of fibrils obtained for αS, and the relative stability of βS fibrils was calculated in the context of the Greek-key polymorph using Rosetta Symmetry (61).
We first investigated the stability and structure of the αS fibril structure in Rosetta simulations with and without conformational restraints on the ssNMR-derived structure, to generate a baseline for comparison with βS models. We observed that a funnel-like landscape is obtained in both constrained and unconstrained Rosetta simulations with low r.m.s.d. structures corresponding to the lowest energies (Fig. 7A). The Greek-key type structure is maintained in the low energy models, indicating the existence of a robust funnel around the ssNMR-derived structure in the Rosetta force field (Fig. 7B). Having recapitulated the thermodynamic stability of the αS fibril structure and obtained a baseline for comparison, we next asked whether βS was capable of taking on the same type of Greek-key structure as αS. We first performed a global sequence alignment of αS and βS, and we threaded the corresponding N-terminally aligned residues of βS on the Greek-key motif observed in the αS structure without considering gaps arising due to a shorter length of the βS NAC. This approach required incorporating part of the C-terminal region of βS (residues 87–97) in the threaded model in the αS NAC region. Starting unconstrained simulations from this model resulted in an ensemble that was considerably destabilized and is dominated by structures that deviated significantly from the Greek-key structure (Fig. 7, C and D). In analyzing the models, we observed that the residues needed to form a critical tight turn in the Greek-key structure, which were GAG in αS and were VKR in the threaded starting model; these large side chains are incompatible with the formation of a tight turn in βS. In addition, this threading places the C-terminal residues in a sterically incompatible area of the Greek-key structure. The steric repulsion incurred by packing these side chains likely led to the observed destabilization of the Greek-key structure in this threading.
Figure 7.
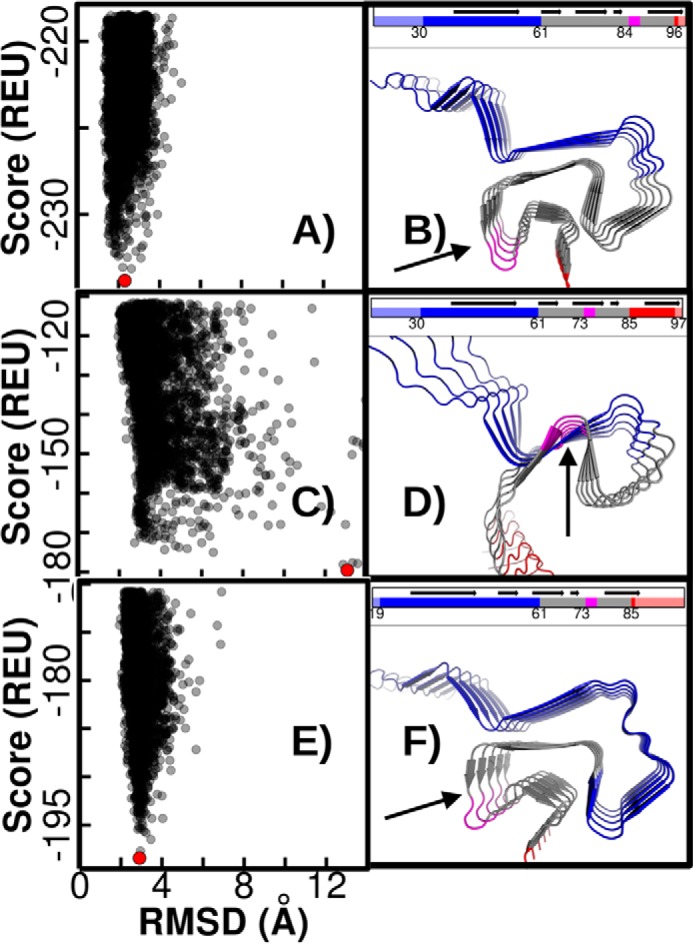
Rosetta energy landscapes for the αS and βS Greek-key architectures. A, Rosetta score (in Rosetta energy units, REU) versus r.m.s.d. (Å) for the ssNMR-derived αS structure showing a robust funnel. The lowest energy structure is indicated by a red point, and in B the lowest energy structure identified in the simulation is pictured. The N terminus is colored blue; the NAC is colored gray; and the C terminus is red. Trailing ends of the N and C termini are shown in light blue and red, respectively. The GAG-turn compatible motif is highlighted in magenta. Rosetta simulations using a threading in which residues 30–97 from βS are N-terminally aligned with corresponding residues from αS show a lack of stable energy funnel, and higher energy values (C) and a large deviation from the Greek-key ssNMR structure (D). A C-terminally aligned threading for the βS(19–86), which incorporates more of the N-terminal domain in the Greek-key, results in a more stable form compared with the βS(30–97) threading. A robust energy funnel, albeit with increased energy values compared with αS, is restored (E), and the lowest energy structure has a low r.m.s.d. from the Greek-key structure (F).
In light of the nascent N–NAC interactions in the βS monomer ensembles and the unlikely involvement of the highly charged C-terminal domain in amyloid, we next aligned the sequences by incorporating a greater number of the N-terminal portion of the βS sequence in the model. This was possible by threading residues 19–86 of βS instead of residues 30–97 into the Greek key. Interestingly, this alignment of the GAG motif corresponds to replacing the 11 “missing” residues of the βS NAC with the residues from βS N-terminal domain. Performing unconstrained Rosetta Symmetric Relax simulations starting from this alignment, we observed that the stability of the Greek-key-containing structures was restored, and the structures of the observed lowest-energy conformations closely resembled the αS fibril topology (Fig. 7, E and F). Although the absolute Rosetta energy values for the βS models are still higher than those obtained for the αS ssNMR structure, a funnel-like shape is observed in both landscapes. Although little structural information is available about the βS fibril to validate or further refine our models, our simulation results indicate that it is possible for βS to adopt a similar amyloid form as αS when the N-terminal domain of βS participates in the core structure of the fibril.
Single amino acid substitutions of key glutamate side chains alter βS fibrillation behavior
Computational analyses, the aggregation behavior of chimera, and greater interactions between the N-terminal and NAC domains in the βS monomer observed by NMR indicated that acidic residues in the N-terminal and NAC domains are key determinants of pH-dependent fibril formation. Further examination of low-energy fibril models, and the sequence differences between βS and αS in the N-terminal and NAC domains led us to hypothesize that Glu-31 and Glu-61 side chains contribute to the observed pH-dependent switch. Glu-31 is present only in βS but not αS (residue 31 is instead a glycine). Glu-61, the first residue position of the NAC domain, is the only buried glutamate side chain in both αS and βS fibril models, and it may experience different microenvironments in both the monomer (Fig. 6) and the fibril states of αS and βS.
To test the contribution of these identified residues (Glu-31 and Glu-61) to pH-dependent fibril formation, these residues were individually substituted to the pH-insensitive alanine. The E31A βS variant showed a significant increase in the lag time for aggregation at low pH, confirming that Glu-31 contributes to the pH-dependent aggregation behavior (Fig. 8A). In contrast, the E61A βS variant lost pH dependence of aggregation and was found to robustly and rapidly aggregate under both low and high pH conditions. AFM characterization showed that aggregates of E61A βS at both pH values adopt a fibril morphology. Thus, strikingly, a single mutation in the βS NAC region (E61A) allows aggregation at high pH with αS-like efficiency (Fig. 8B). These results suggest that glutamate switches exert their effects by both promoting and inhibiting fibril formation in a pH-dependent manner, and they highlight how small changes in charge content can dramatically alter synuclein fibrillation behavior.
Figure 8.

ThT fluorescence and AFM images of βS mutants E31A and E61A at pH 5.8 and pH 7.3. Aggregation kinetics (as detected by ThT fluorescence) of βS mutants E31A (A) and E61A (B) at low pH (blue), high pH (red), and wild-type βS at low pH (black) are shown. At low pH, E31A shows a significantly higher lag time compared with wild-type βS. AFM images of E31A at low pH (C) and high pH (D) show fibrils and oligomers, respectively, similar to wild-type βS (Fig. 2). The E61A substitution abolishes the pH dependence for fibrillation and shows similar lag times at both pH values. Fibril morphologies at low pH (E) and high pH (F) are shown.
The contribution of both identified glutamate residues is consistent with the observed aggregation behavior of chimeric proteins. The chimeric protein ABB, which does not have Glu-31 (αS N-terminal domain has Gly-31), does not form fibrils at mildly acidic pH (Fig. 5 and supplemental S4). Whereas Glu-61 from the NAC region is present in both the ABB chimera and wild-type βS (BBB), NMR characterization of the monomer (Fig. 6) and computational models of fibrils (Fig. 7) indicate that the NAC domain participates in interactions with the N-terminal domain, which differs between these variants. Thus, both identified sites contributing to the pH switch are either absent or have a significantly different microenvironment in the ABB chimera compared with the wild-type βS and BBA chimera, which is consistent with their observed aggregation behavior (Figs. 4 and 5).
Although the presence of contiguous βS N-terminal and NAC domains appears to be the key feature for fibrillation at mildly acidic pH, we note that the ABA chimeric protein, which lacks this feature, does form fibrillar aggregates. The interactions between the N-terminal domain of αS and NAC domain of βS in this chimera are expected to be less fibril-stabilizing compared with BBX chimera, but the lower negative charge of the C-terminal domain in this ABA chimera compared with ABB chimera may relieve inhibition of aggregation to a greater extent and allow formation of ThT-positive oligomers (that are nevertheless distinct from “regular” BBB or AAA fibrils at either pH value; supplemental Fig. S4). The balance between fibrillation-promoting interactions in the N-terminal and NAC domains and other inhibitory interactions utilizing all three domains contributes to fibril formation.
Taken together, our results show that a subtle balance of pH-dependent stabilizing as well as inhibitory intra- and inter-chain interactions in all relevant thermodynamic states, monomer, non-fibrillar aggregates, and mature fibrils, underlies the pH-sensitive aggregation behavior of synucleins mediated by key glutamate residues.
Discussion
The key observation in our study is that mildly acidic pH 5.8 renders the otherwise non-fibrillogenic βS fibrillogenic. At high pH (pH 7.3) the βS protein is non-fibrillogenic as previously demonstrated in the literature (13, 62). However, in contrast to αS, which is fibrillogenic at both pH values, βS fibrillation is effectively controlled by an on/off pH-dependent switch. ThT binding-based fibril formation assays of chimeric constructs, AFM-based imaging, point mutagenesis, and atomic resolution modeling of βS fibrils suggest that pH-dependent interactions mediated by key glutamate side chains and involving the N-terminal and NAC domains of βS are key contributing factors of this observed pH-dependent fibrillation of βS. In addition, pH-dependent effects at acidic residues in the C-terminal domain may also play an important role in fibril formation. Given that several subcellular organelles have mildly acidic pH, our data demonstrate that βS aggregation is highly environment-dependent and suggest that the view of βS as a simple αS aggregation inhibitor, which is not prone to aggregation itself, may need to be reconsidered.
Despite their high sequence similarity, where and how the fibrillation-determining differences between αS and βS reside in their primary sequence and tertiary structures have remained unknown. Their NAC regions are the most disparate in length and sequence, and although deletion of the essential stretch of hydrophobic residues in αS renders it non-fibrillogenic, simply transferring the missing residues from αS to βS has not made the latter fibrillogenic (41, 63, 64). Although swapping other regions of the NAC between the two proteins similarly modulates the aggregation propensity to some extent (63, 65, 66), a complete transfer of fibrillation properties by swapping NAC domains has not been possible. Taken together, previous studies and our results indicate that the formation of βS fibrils is determined not exclusively by its NAC domain but is controlled by interactions between the NAC and other domains, whereas the NAC domain appears to be both necessary and sufficient for αS aggregation.
The set of domain swapped chimeras allowed us to investigate the role of inter-domain interactions in aggregation of βS. The most striking result from comparing the aggregation behavior of low pH ABB (not amyloid-forming) and BBX (fibril-forming) chimeric proteins was that the N-terminal and β-NAC domains together play a key role in the ability to form fibrils at mildly acidic pH. Although the NAC domain is necessary for fibrillation, in βS the N- and C-terminal domains regulate the NAC domain-mediated fibrillation inducing interactions, with interactions between the N-terminal and NAC domains playing a more crucial role in the stabilization of the βS fibril architecture similar to previous observations in αS (67–69). The C-terminal domain, which is highly enriched in negative charges, and is thus expected to be more pH-sensitive, does not appear to provide the required pH-sensitive stabilizing interactions, but may instead contribute to pH-dependent aggregation via C-terminal inhibitory (N-C and NAC-C) intra/inter-chain interactions as evidenced by the differing lag times (but highly similar fibril topology) between all C-terminally swapped chimeras (supplemental Figs. S2 and S4) (AAX, ABX, BAX, BBX) (62, 70), and by the enhanced aggregation propensity at low pH of the ABA chimera compared with ABB. Janowska et al. (62) also suggest an enhanced interaction of the αN and βC domains relative to the αN and αC domains, which could also be a contributing factor to the lack of formation of fibrils by ABB chimera. Further exploration of the ABX monomer conformational ensemble will provide insight into the subtle balance of pH-dependent stabilizing and inhibitory interactions that determine aggregation behavior.
Analysis of the computational models for βS fibril provides insight into the balance of intra- and inter-chain interactions formed by glutamate residues, which may determine the observed pH dependence of βS fibril formation (Fig. 9A). A decrease in pH is expected to lead to greater protonation of glutamate residues involved in interchain fibril-stabilizing interactions, as well as relieve inhibition of aggregation due to charge-mediated interactions. For example, protonated Glu-31 and Glu-61 side chains (at low pH) may themselves form a glutamine-like “polar zipper” (54) interaction (Fig. 9B) or stabilize other polar zippers (Fig. 9C) to promote fibril formation, whereas their deprotonated forms at high pH may inhibit fibrillation by interchain charge repulsion and more favorable intra-chain interactions as observed in the NMR data (Fig. 6). The E31A variant, which has a higher lag time for aggregation compared with wild-type βS (Fig. 8A), is predicted to lack the stabilizing polar zipper interaction (Fig. 9B). Charge-mediated aggregation-inhibitory interactions at high pH, e.g. interchain repulsion, may be relieved in the E61A variant compared with wild-type βS, and pH-independent (in this pH range) polar zipper interaction ladders formed by surrounding polar residues in the N-terminal and NAC domains, e.g. Gln-50 and Gln-62, may sufficiently stabilize the E61A fibril form at both low and high pH. At high pH, the desolvation penalty associated with burying a negatively charged Glu-61 in the fibril core (Fig. 9A) would also be abolished in the E61A variant. Although Rosetta-based homology models of βS fibrils based on Greek-key αS structures appear to be consistent with experimental data, it is possible that other fibril topologies may equally well explain the results. Moreover, the stabilities and morphologies of fibrillation intermediates, and the impact of mutations on these, including in the context of the various αS/βS chimera, need to be considered for a fuller understanding of fibril formation. Structure determination of the fibrils formed by βS and its variants under different pH conditions may provide further insight into these issues.
Figure 9.
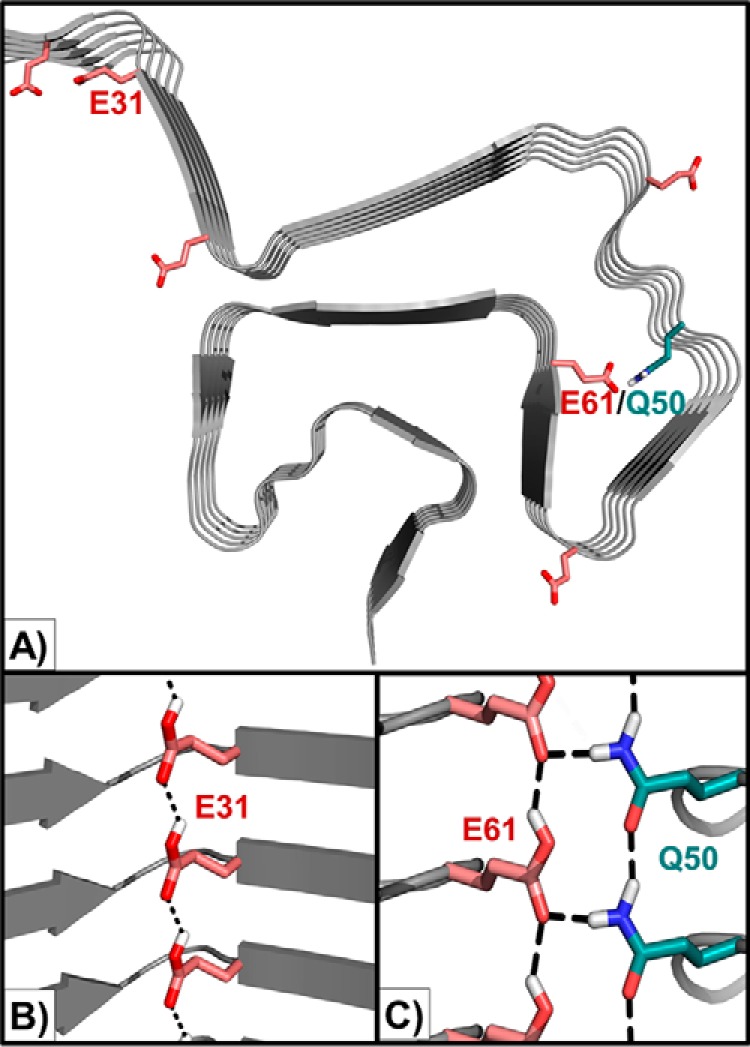
Identified charge-mediated interactions formed by key glutamates in the βS fibril model. A, location of N-terminal glutamate residues (red) and polar side chains in the N-terminal domain in the βS Greek-key fibril model. B, Glu-31 may form a hydrogen-bonded polar zipper ladder in its protonated form (rotameric preferences modeled using glutamine), thereby promoting fibril formation. C, Glu-61 may form a similar ladder and participate in additional interactions with the Gln-50 ladder at low pH. The Gln-50 ladder is predicted to form in a pH-independent manner in the E61A variant.
Based on the results of point mutagenesis at Glu-61 and Glu-31, chimeric mutagenesis, computational modeling of fibrils, and NMR data on the monomer ensembles, we argue that pH-dependent aggregation of synucleins is dependent on a subtle balance of various aggregation-promoting (stabilizing) and -inhibiting effects. The N-terminal and NAC domain interactions, evident in the NMR data (intrachain) and Rosetta models (intra and interchain), serve to stabilize the fibrils in a pH-dependent manner, whereas charge interactions involving negatively charged side chains throughout the protein chains (including C-terminal domain) may serve to inhibit aggregation. The balance between charge-dependent stabilizing interactions in the fibril state and aggregation–inhibitory interactions in the soluble states contributes to fibril formation.
Our results have several implications for the role of βS in disease. They indicate that although βS may serve in a neuroprotective role at cytoplasmic pH by preventing αS aggregation, this inhibition may be relieved by a switch into its own fibrillation in more acidic organelles, like the lysosome, which enhance protein clearance and enable homeostasis. Several recent reports suggest that βS may contribute to a toxic gain-of-function in different cellular organelles in a different way than αS (16, 17). For instance, defects associated with βS are linked with defects in endoplasmic reticulum-Golgi-associated proteins in yeast, which is a mildly acidic subcellular environment. There is also an emerging role for endolysosomal dysfunction in PD, particularly in relation to recent reports of Gaucher's disease, a lysosomal storage disease (71, 72). Given that membranes and vesicles are likely sites of function for synucleins, and also regulate microenvironments associated with acidification, fibril formation by βS in these microenvironments may lead to dysfunction. Our in vitro work provides molecular level insights into βS conformations at mildly acidic pH values and suggests that the toxic gain-of-function observed in vivo may be related to the formation of βS aggregates and/or a loss-of-function of the soluble cytoplasmic pH form of βS (73). Evaluation of the stability and toxicity of the in vitro generated βS fibrils in cell culture and animal models will be useful in further investigating the role of βS in PD.
Experimental procedures
Mutagenesis, expression, and purification
For both the αS N65H and βS H65N as well as all cysteine point mutants, site-directed mutagenesis was performed to exchange the appropriate residues using Invitrogen AccuPrime pfx DNA polymerase (Thermo Fisher Scientific, Waltham, MA) and the appropriate primers (Integrated DNA Technologies, Coralville, IA). All acetylated and non-acetylated protein were expressed and purified, as described previously (4), and verified for the correct weight and purity with ESI-MS (data not shown).
Chimera preparation
To obtain the DNA plasmids required to express chimeras of αS/βS, the Gibson assembly protocol was employed to insert the necessary G-blocks (Integrated DNA Technologies, Coralville, IA) for the AAB, BAB, BBA, and ABA chimeras, and site-directed mutagenesis starting with point mutants K10M of αS and M10K of βS, respectively, was performed for the BAA and ABB chimeras. Expression and purification of unlabeled protein were then performed in the same manner as the wild-type protein and other point mutants used in this study, and the correct molecular weight and purity were confirmed with ESI-MS (data not shown).
HSQC and RDC experiments for αS/βS
αS and βS solutions were prepared to a concentration of 250 μm in the same manner as the wild-type protein and other point mutant used in this study. Briefly, lyophilized protein was dissolved in buffer, passed through a 100K Amicon centrifugal filter, and then concentrated with a 3K Amicon centrifugal filter for 250 mm. These samples were prepared in 20 mm sodium phosphate, 100 mm NaCl, pH 5.8, and 7.3 buffers. 1H-15N HSQC and RDC experiments were prepared and analyzed similarly to previously described protocols (70). We confirm that βS exhibits similar pH-dependent on/off fibrillation behavior in this buffer in supplemental Fig. S3.
MTSL labeling
Site-directed mutagenesis was performed as mentioned before to incorporate cysteine residues at A90. In βS, a cysteine residues were incorporated at A79. To incorporate the nitroxide spin label MTSL (S-(1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothioate), the cysteine mutants were first reduced with a 20 times molar ratio of dithiothreitol (DTT) incubated at 4 °C for 4–6 h to remove cysteine–cysteine disulfide bonds. A GE Healthcare HiPrepTM 26/10 desalting column was used to remove excess DTT. Immediately after protein eluted from the column, a 20× molar excess of MTSL was added, and the reaction was left to proceed on a shaking platform at 4 °C overnight. Excess MTSL was removed by dialyzing against ultrapure (18 megohms/cm) deionized water before lyophilizing and storing the protein at −20 °C. PRE experiments were performed as described previously from lyophilized MTSL-labeled protein (70), 100 μm protein, and 10 mm sodium ascorbate to reduce the spin label.
Thioflavin T assay
Lyophilized protein samples were prepared to a concentration of 1 mg/ml by filtering first with a 100K filter from dissolved lyophilized protein and concentrating and buffer exchanging with a 3K centrifugal filter. Samples were loaded with 20 μm ThT (Acros Organics, Pittsburgh, PA) into 96-well clear bottom plates (Corning, Corning, NY) at a concentration of 70 μm, sealed with Axygen sealing tape (Corning), and shaken at a rate of 600 rpm at 37 °C for at least 200 h. Between 3 and 6 samples were measured for each sample type. A POLAR Star Omega plate reader (BMG Labtech, Cary, NC) was used to monitor the increase in ThT intensity. Protocol is adapted from the literature (74).
AFM protocol
All AFM images were taken on an NX-10 instrument (Park Systems, Suwon, South Korea), using non-contact mode tips (PPP-NCHR, force constant 42 newtons/m; 330 kHz frequency; Nanosensors, Neuchatel, Switzerland). All synuclein samples were obtained after ThioT fluorescence was complete, at a concentration of 70 μm. A 1 × 1-cm2 of freshly cleaved mica (obtained from Ted Pella Inc., Redding, CA) had 20 μl of sample deposited on the surface and was allowed to incubate for 5–10 min while covered. Then the surface of the sample was washed three times with 100 μl of water, and the bottom and edges were dried using filter paper. The surface was allowed to air-dry for 1 h before being imaged. Image processing was conducted using Gwyddion software.
Computational modeling
From the ssNMR-derived structure (Protein Data Bank code 2n0a), the fibril core was isolated by removing the edge chains A, B, H, I, and J, and disordered residues 1–29 and 98–140 from the chains C–G. An idealized symmetric conformation was generated from this modified structure using fibril symmetry (61). A Rosetta Script input .xml file (see supplemental information) was created to mutate the desired residues to match the sequence of the threaded variant. Backbone nitrogen, carbon, and oxygen atoms were constrained to be within 0.2 Å of their initial coordinates in the constrained Relax simulations. The structure was then symmetrically relaxed using the Rosetta Fast relax algorithm in Rosetta 3.6 (61) to generate 1000 output decoys. In unconstrained simulations, the 0.2 Å limit was removed, and symmetric relax was then performed with the Rosetta Fast relax algorithm to produce 4000 decoys.
Author contributions
G. M. M. performed the NMR characterization, contributed to aggregation kinetics, experimental design, data analysis, and manuscript preparation. M. P. O. performed the computational modeling and data analysis, chimera mutagenesis, and protein production and contributed to aggregation kinetics. T. B. A. performed AFM characterization, mutagenesis, and protein production. M. K. J. contributed to experimental design and assisted with chimera mutagenesis. S. D. K. and J. B. contributed to experimental design, data analysis, and preparation of manuscript. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgment
We thank Dr. Ana Monica Nunes for helpful discussions.
Note added in proof
The β-synuclein, pH 5.8, fibrils picture in Fig. 5A was changed from the version that was published as a Paper in Press on July 14, 2017. In addition, the BAB and AAB traces in Fig. 4B were incorrectly labeled in the Papers in Press version. Neither the replacement of Fig. 5 nor the correction of the error in Fig. 4 affects the results or conclusions of this work.
This work was supported in part by National Institutes of Health Grant GM110577 (to J. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
This article contains supplemental Figs. S1–S4 and information.
- αS
- α-synuclein
- βS
- β-synuclein
- LB
- body
- PD
- Parkinson's disease
- IDP
- intrinsically disordered protein
- NAC
- non-amyloid-β component
- PRE
- paramagnetic spin relaxation experiment
- AFM
- atomic force microscopy
- MTSL
- S-(1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothioate
- ThT
- thioflavin T
- r.m.s.d.
- root mean square deviation
- RDC
- residual dipolar coupling
- HSQC
- heteronuclear single quantum coherence
- ssNMR
- solid state NMR.
References
- 1. Spillantini M. G., Schmidt M. L., Lee V. M., Trojanowski J. Q., Jakes R., and Goedert M. (1997) α-Synuclein in Lewy bodies. Nature 388, 839–840 [DOI] [PubMed] [Google Scholar]
- 2. Sung Y. H., and Eliezer D. (2007) Residual structure, backbone dynamics, and interactions within the synuclein family. J. Mol. Biol. 372, 689–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fauvet B., Mbefo M. K., Fares M. B., Desobry C., Michael S., Ardah M. T., Tsika E., Coune P., Prudent M., Lion N., Eliezer D., Moore D. J., Schneider B., Aebischer P., El-Agnaf O. M., et al. (2012) α-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J. Biol. Chem. 287, 15345–15364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kang L., Moriarty G. M., Woods L. A., Ashcroft A. E., Radford S. E., and Baum J. (2012) N-terminal acetylation of α-synuclein induces increased transient helical propensity and decreased aggregation rates in the intrinsically disordered monomer. Protein Sci. 21, 911–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Moriarty G. M., Janowska M. K., Kang L., and Baum J. (2013) Exploring the accessible conformations of N-terminal acetylated α-synuclein. FEBS Lett. 587, 1128–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schwalbe M., Ozenne V., Bibow S., Jaremko M., Jaremko L., Gajda M., Jensen M. R., Biernat J., Becker S., Mandelkow E., Zweckstetter M., and Blackledge M. (2014) Predictive atomic resolution descriptions of intrinsically disordered hTau40 and α-synuclein in solution from NMR and small angle scattering. Structure 22, 238–249 [DOI] [PubMed] [Google Scholar]
- 7. Kim W. S., Kågedal K., and Halliday G. M. (2014) α-Synuclein biology in Lewy body diseases. Alzheimers Res. Ther. 6, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Surguchov A. (2015) Intracellular dynamics of synucleins: “Here, There and Everywhere.” Int. Rev. Cell Mol. Biol. 320, 103–169 [DOI] [PubMed] [Google Scholar]
- 9. Breydo L., Wu J. W., and Uversky V. N. (2012) α-Synuclein misfolding and Parkinson's disease. Biochim. Biophys. Acta 1822, 261–285 [DOI] [PubMed] [Google Scholar]
- 10. Spillantini M. G., Crowther R. A., Jakes R., Hasegawa M., and Goedert M. (1998) α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. U.S.A. 95, 6469–6473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Murphy D. D., Rueter S. M., Trojanowski J. Q., and Lee V. M. (2000) Synucleins are developmentally expressed, and α-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 20, 3214–3220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hashimoto M., Rockenstein E., Mante M., Mallory M., and Masliah E. (2001) β-Synuclein inhibits α-synuclein aggregation: a possible role as an anti-parkinsonian factor. Neuron 32, 213–223 [DOI] [PubMed] [Google Scholar]
- 13. Uversky V. N., Li J., Souillac P., Millett I. S., Doniach S., Jakes R., Goedert M., and Fink A. L. (2002) Biophysical properties of the synucleins and their propensities to fibrillate: inhibition of α-synuclein assembly by β- and γ-synucleins. J. Biol. Chem. 277, 11970–11978 [DOI] [PubMed] [Google Scholar]
- 14. Brown J. W., Buell A. K., Michaels T. C., Meisl G., Carozza J., Flagmeier P., Vendruscolo M., Knowles T. P., Dobson C. M., and Galvagnion C. (2016) β-Synuclein suppresses both the initiation and amplification steps of α-synuclein aggregation via competitive binding to surfaces. Sci. Rep. 6, 36010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yamin G., Munishkina L. A., Karymov M. A., Lyubchenko Y. L., Uversky V. N., and Fink A. L. (2005) Forcing nonamyloidogenic β-synuclein to fibrillate. Biochemistry 44, 9096–9107 [DOI] [PubMed] [Google Scholar]
- 16. Tenreiro S., Rosado-Ramos R., Gerhardt E., Favretto F., Magalhães F., Popova B., Becker S., Zweckstetter M., Braus G. H., and Outeiro T. F. (2016) Yeast reveals similar molecular mechanisms underlying α- and β-synuclein toxicity. Hum. Mol. Genet. 25, 275–290 [DOI] [PubMed] [Google Scholar]
- 17. Taschenberger G., Toloe J., Tereshchenko J., Akerboom J., Wales P., Benz R., Becker S., Outeiro T. F., Looger L. L., Bähr M., Zweckstetter M., and Kügler S. (2013) β-Synuclein aggregates and induces neurodegeneration in dopaminergic neurons. Ann. Neurol. 74, 109–118 [DOI] [PubMed] [Google Scholar]
- 18. Galvin J. E., Uryu K., Lee V. M., and Trojanowski J. Q. (1999) Axon pathology in Parkinson's disease and Lewy body dementia hippocampus contains α-, β-, and γ-synuclein. Proc. Natl. Acad. Sci. U.S.A. 96, 13450–13455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ohtake H., Limprasert P., Fan Y., Onodera O., Kakita A., Takahashi H., Bonner L. T., Tsuang D. W., Murray I. V., Lee V. M., Trojanowski J. Q., Ishikawa A., Idezuka J., Murata M., Toda T., et al. (2004) β-Synuclein gene alterations in dementia with Lewy bodies. Neurology 63, 805–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fujita M., Sugama S., Sekiyama K., Sekigawa A., Tsukui T., Nakai M., Waragai M., Takenouchi T., Takamatsu Y., Wei J., Rockenstein E., Laspada A. R., Masliah E., Inoue S., and Hashimoto M. (2010) A β-synuclein mutation linked to dementia produces neurodegeneration when expressed in mouse brain. Nat. Commun. 1, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wei J., Fujita M., Nakai M., Waragai M., Watabe K., Akatsu H., Rockenstein E., Masliah E., and Hashimoto M. (2007) Enhanced lysosomal pathology caused by β-synuclein mutants linked to dementia with Lewy bodies. J. Biol. Chem. 282, 28904–28914 [DOI] [PubMed] [Google Scholar]
- 22. Snead D., and Eliezer D. (2014) α-Synuclein function and dysfunction on cellular membranes. Exp. Neurobiol. 23, 292–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang T., and Hay J. C. (2015) α-Synuclein toxicity in the early secretory pathway: how it drives neurodegeneration in Parkinson's disease. Front. Neurosci. 9, 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ducas V. C., and Rhoades E. (2012) Quantifying interactions of β-synuclein and γ-synuclein with model membranes. J. Mol. Biol. 423, 528–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Casey J. R., Grinstein S., and Orlowski J. (2010) Sensors and regulators of intracellular pH. Nat. Rev. Mol. Cell Biol. 11, 50–61 [DOI] [PubMed] [Google Scholar]
- 26. Ruffin V. A., Salameh A. I., Boron W. F., and Parker M. D. (2014) Intracellular pH regulation by acid-base transporters in mammalian neurons. Front. Physiol. 5, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sorkin A., and Von Zastrow M. (2002) Signal transduction and endocytosis: close encounters of many kinds. Nat. Rev. Mol. Cell Biol. 3, 600–614 [DOI] [PubMed] [Google Scholar]
- 28. Lopes da Fonseca T., Villar-Piqué A., and Outeiro T. F. (2015) The interplay between α-synuclein clearance and spreading. Biomolecules 5, 435–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rostovtseva T. K., Gurnev P. A., Protchenko O., Hoogerheide D. P., Yap T. L., Philpott C. C., Lee J. C., and Bezrukov S. M. (2015) α-Synuclein shows high affinity interaction with voltage-dependent anion channel, suggesting mechanisms of mitochondrial regulation and toxicity in Parkinson disease. J. Biol. Chem. 290, 18467–18477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Majdi A., Mahmoudi J., Sadigh-Eteghad S., Golzari S. E., Sabermarouf B., and Reyhani-Rad S. (2016) Permissive role of cytosolic pH acidification in neurodegeneration: a closer look at its causes and consequences. J. Neurosci. Res. 94, 879–887 [DOI] [PubMed] [Google Scholar]
- 31. Hwang O. (2013) Role of oxidative stress in Parkinson's disease. Exp. Neurobiol. 22, 11–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sung Y. H., and Eliezer D. (2006) Secondary structure and dynamics of micelle bound β- and γ-synuclein. Protein Sci. 15, 1162–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu K. P., Kim S., Fela D. A., and Baum J. (2008) Characterization of conformational and dynamic properties of natively unfolded human and mouse α-synuclein ensembles by NMR: implication for aggregation. J. Mol. Biol. 378, 1104–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Marsh J. A., Singh V. K., Jia Z., and Forman-Kay J. D. (2006) Sensitivity of secondary structure propensities to sequence differences between α- and γ-synuclein: implications for fibrillation. Protein Sci. 15, 2795–2804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Babu M. M., van der Lee R., de Groot N. S., and Gsponer J. (2011) Intrinsically disordered proteins: regulation and disease. Curr. Opin. Struct. Biol. 21, 432–440 [DOI] [PubMed] [Google Scholar]
- 36. Wright P. E., and Dyson H. J. (2015) Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 16, 18–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Uversky V. N., Gillespie J. R., and Fink A. L. (2000) Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 41, 415–427 [DOI] [PubMed] [Google Scholar]
- 38. Buell A. K., Galvagnion C., Gaspar R., Sparr E., Vendruscolo M., Knowles T. P., Linse S., and Dobson C. M. (2014) Solution conditions determine the relative importance of nucleation and growth processes in α-synuclein aggregation. Proc. Natl. Acad. Sci. U.S.A. 111, 7671–7676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sormanni P., Aprile F. A., and Vendruscolo M. (2015) The CamSol method of rational design of protein mutants with enhanced solubility. J. Mol. Biol. 427, 478–490 [DOI] [PubMed] [Google Scholar]
- 40. Tartaglia G. G., and Vendruscolo M. (2008) The Zyggregator method for predicting protein aggregation propensities. Chem. Soc. Rev. 37, 1395–1401 [DOI] [PubMed] [Google Scholar]
- 41. Giasson B. I., Murray I. V., Trojanowski J. Q., and Lee V. M. (2001) A hydrophobic stretch of 12 amino acid residues in the middle of α-synuclein is essential for filament assembly. J. Biol. Chem. 276, 2380–2386 [DOI] [PubMed] [Google Scholar]
- 42. Villar-Piqué A., Lopes da Fonseca T., Sant'Anna R., Szegö É. M., Fonseca-Ornelas L., Pinho R., Carija A., Gerhardt E., Masaracchia C., Abad Gonzalez E., Rossetti G., Carloni P., Fernández C. O., Foguel D., Milosevic I., Zweckstetter M., Ventura S., and Outeiro T. F. (2016) Environmental and genetic factors support the dissociation between α-synuclein aggregation and toxicity. Proc. Natl. Acad. Sci. U.S.A. 113, E6506–E6515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vilar M., Chou H. T., Lührs T., Maji S. K., Riek-Loher D., Verel R., Manning G., Stahlberg H., and Riek R. (2008) The fold of α-synuclein fibrils. Proc. Natl. Acad. Sci. U.S.A. 105, 8637–8642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tuttle M. D., Comellas G., Nieuwkoop A. J., Covell D. J., Berthold D. A., Kloepper K. D., Courtney J. M., Kim J. K., Barclay A. M., Kendall A., Wan W., Stubbs G., Schwieters C. D., Lee V. M., George J. M., and Rienstra C. M. (2016) Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat. Struct. Mol. Biol. 23, 409–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xu L., Ma B., Nussinov R., and Thompson D. (2017) Familial mutations may switch conformational preferences in α-synuclein fibrils. ACS Chem. Neurosci. 8, 837–849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tycko R. (2014) Physical and structural basis for polymorphism in amyloid fibrils. Protein Sci. 23, 1528–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bousset L., Pieri L., Ruiz-Arlandis G., Gath J., Jensen P. H., Habenstein B., Madiona K., Olieric V., Böckmann A., Meier B. H., and Melki R. (2013) Structural and functional characterization of two α-synuclein strains. Nat. Commun. 4, 2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hoyer W., Antony T., Cherny D., Heim G., Jovin T. M., and Subramaniam V. (2002) Dependence of α-synuclein aggregate morphology on solution conditions. J. Mol. Biol. 322, 383–393 [DOI] [PubMed] [Google Scholar]
- 49. McGlinchey R. P., Jiang Z., and Lee J. C. (2014) Molecular origin of pH-dependent fibril formation of a functional amyloid. Chembiochem 15, 1569–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pfefferkorn C. M., McGlinchey R. P., and Lee J. C. (2010) Effects of pH on aggregation kinetics of the repeat domain of a functional amyloid, Pmel17. Proc. Natl. Acad. Sci. U.S.A. 107, 21447–21452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tipping K. W., Karamanos T. K., Jakhria T., Iadanza M. G., Goodchild S. C., Tuma R., Ranson N. A., Hewitt E. W., and Radford S. E. (2015) pH-induced molecular shedding drives the formation of amyloid fibril-derived oligomers. Proc. Natl. Acad. Sci. U.S.A. 112, 5691–5696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Petkova A. T., Buntkowsky G., Dyda F., Leapman R. D., Yau W. M., and Tycko R. (2004) Solid state NMR reveals a pH-dependent antiparallel β-sheet registry in fibrils formed by a β-amyloid peptide. J. Mol. Biol. 335, 247–260 [DOI] [PubMed] [Google Scholar]
- 53. French K. C., and Makhatadze G. I. (2012) Core sequence of PAPf39 amyloid fibrils and mechanism of pH-dependent fibril formation: the role of monomer conformation. Biochemistry 51, 10127–10136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Perutz M. (1994) Polar zippers: their role in human disease. Protein Sci. 3, 1629–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Croke R. L., Patil S. M., Quevreaux J., Kendall D. A., and Alexandrescu A. T. (2011) NMR determination of pKa values in α-synuclein. Protein Sci. 20, 256–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Grimsley G. R., Scholtz J. M., and Pace C. N. (2009) A summary of the measured pK values of the ionizable groups in folded proteins. Protein Sci. 18, 247–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bernadó P., Bertoncini C. W., Griesinger C., Zweckstetter M., and Blackledge M. (2005) Defining long-range order and local disorder in native α-synuclein using residual dipolar couplings. J. Am. Chem. Soc. 127, 17968–17969 [DOI] [PubMed] [Google Scholar]
- 58. Janowska M. K., and Baum J. (2016) The loss of inhibitory C-terminal conformations in disease associated P123H β-synuclein. Protein Sci. 25, 286–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Marsh J. A., Baker J. M., Tollinger M., and Forman-Kay J. D. (2008) Calculation of residual dipolar couplings from disordered state ensembles using local alignment. J. Am. Chem. Soc. 130, 7804–7805 [DOI] [PubMed] [Google Scholar]
- 60. Allison J. R., Rivers R. C., Christodoulou J. C., Vendruscolo M., and Dobson C. M. (2014) A relationship between the transient structure in the monomeric state and the aggregation propensities of α-synuclein and β-synuclein. Biochemistry 53, 7170–7183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. DiMaio F., Leaver-Fay A., Bradley P., Baker D., and André I. (2011) Modeling symmetric macromolecular structures in Rosetta3. PLoS ONE 6, e20450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Janowska M. K., Wu K. P., and Baum J. (2015) Unveiling transient protein-protein interactions that modulate inhibition of α-synuclein aggregation by β-synuclein, a pre-synaptic protein that co-localizes with α-synuclein. Sci. Rep. 5, 15164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zibaee S., Jakes R., Fraser G., Serpell L. C., Crowther R. A., and Goedert M. (2007) Sequence determinants for amyloid fibrillogenesis of human α-synuclein. J. Mol. Biol. 374, 454–464 [DOI] [PubMed] [Google Scholar]
- 64. Rivers R. C., Kumita J. R., Tartaglia G. G., Dedmon M. M., Pawar A., Vendruscolo M., Dobson C. M., and Christodoulou J. (2008) Molecular determinants of the aggregation behavior of α- and β-synuclein. Protein Sci. 17, 887–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Roodveldt C., Andersson A., De Genst E. J., Labrador-Garrido A., Buell A. K., Dobson C. M., Tartaglia G. G., and Vendruscolo M. (2012) A rationally designed six-residue swap generates comparability in the aggregation behavior of α-synuclein and β-synuclein. Biochemistry 51, 8771–8778 [DOI] [PubMed] [Google Scholar]
- 66. Koo H. J., Lee H. J., and Im H. (2008) Sequence determinants regulating fibrillation of human α-synuclein. Biochem. Biophys. Res. Commun. 368, 772–778 [DOI] [PubMed] [Google Scholar]
- 67. Kang L., Janowska M. K., Moriarty G. M., and Baum J. (2013) Mechanistic insight into the relationship between N-terminal acetylation of α-synuclein and fibril formation rates by NMR and fluorescence. PLoS ONE 8, e75018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Crowther R. A., Jakes R., Spillantini M. G., and Goedert M. (1998) Synthetic filaments assembled from C-terminally truncated α-synuclein. FEBS Lett. 436, 309–312 [DOI] [PubMed] [Google Scholar]
- 69. Xu L., Nussinov R., and Ma B. (2016) Coupling of the non-amyloid-component (NAC) domain and the KTK(E/Q)GV repeats stabilize the α-synuclein fibrils. Eur. J. Med. Chem. 121, 841–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wu K. P., Weinstock D. S., Narayanan C., Levy R. M., and Baum J. (2009) Structural reorganization of α-synuclein at low pH observed by NMR and REMD simulations. J. Mol. Biol. 391, 784–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kett L. R., and Dauer W. T. (2016) Endolysosomal dysfunction in Parkinson's disease: recent developments and future challenges. Mov. Disord. 31, 1433–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. McGlinchey R. P., and Lee J. C. (2013) Emerging insights into the mechanistic link between α-synuclein and glucocerebrosidase in Parkinson's disease. Biochem. Soc. Trans. 41, 1509–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Winklhofer K. F., Tatzelt J., and Haass C. (2008) The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases. EMBO J. 27, 336–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Khurana R., Coleman C., Ionescu-Zanetti C., Carter S. A., Krishna V., Grover R. K., Roy R., and Singh S. (2005) Mechanism of thioflavin T binding to amyloid fibrils. J. Struct. Biol. 151, 229–238 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.