

This CT scan-based study of airway morphometry in the cystic fibrosis (CF) postnatal period is unique, as analogous studies in humans are greatly limited for ethical and technical reasons. Findings such as reduced airway lumen area and irregular caliber suggest that airway growth and development are CF transmembrane conductance regulator-dependent and that airway growth defects may contribute to CF lung disease pathogenesis.
Keywords: cystic fibrosis, porcine, computed tomography, airway distensibility
Abstract
Mutations in the gene encoding the cystic fibrosis (CF) transmembrane conductance regulator (CFTR) anion channel cause CF. The leading cause of death in the CF population is lung disease. Increasing evidence suggests that in utero airway development is CFTR-dependent and that developmental abnormalities may contribute to CF lung disease. However, relatively little is known about postnatal CF airway growth, largely because such studies are limited in humans. Therefore, we examined airway growth and lung volume in a porcine model of CF. We hypothesized that CF pigs would have abnormal postnatal airway growth. To test this hypothesis, we performed CT-based airway and lung volume measurements in 3-wk-old non-CF and CF pigs. We found that 3-wk-old CF pigs had tracheas of reduced caliber and irregular shape. Their bronchial lumens were reduced in size proximally but not distally, were irregularly shaped, and had reduced distensibility. Our data suggest that lack of CFTR results in aberrant postnatal airway growth and development, which could contribute to CF lung disease pathogenesis.
NEW & NOTEWORTHY This CT scan-based study of airway morphometry in the cystic fibrosis (CF) postnatal period is unique, as analogous studies in humans are greatly limited for ethical and technical reasons. Findings such as reduced airway lumen area and irregular caliber suggest that airway growth and development are CF transmembrane conductance regulator-dependent and that airway growth defects may contribute to CF lung disease pathogenesis.
mutations in the gene encoding the cystic fibrosis (CF) transmembrane conductance regulator (CFTR) anion channel cause CF. The leading cause of death in the CF population is lung disease. Increasing evidence suggests that the airways of infants and young children with CF are abnormal. Anatomically, CF tracheas have irregular shape, reduced caliber, and increased airway smooth muscle mass (18, 21). Functionally, as many as two-thirds of infants with CF have air trapping on computed tomography (CT) imaging (25), and young children with CF have significantly higher rates of tracheomalacia (10). Newborn (< 24-h-old) CF pigs largely recapitulate these phenotypes. Anatomically, newborn CF pig tracheas have reduced caliber and irregular shape (18). Functionally, newborn CF pigs have increased levels of air trapping and elevated airway resistance (2).
These observations of very young humans with CF and newborn CF pigs raise the possibility that abnormal airway development contributes to CF lung disease pathogenesis. However, relatively little is known of CF airway growth and development, largely because such studies are extremely limited in humans. Hence, we examined airway growth in CF pigs. We hypothesized that postnatal airway growth and development would be abnormal in CF. We used quantitative chest CT, which included lung and airway measurements, to test our hypothesis in 3-wk-old non-CF and CF pigs. CT was our primary analysis tool, because it permits quantitative morphological analysis, is relatively noninvasive, and is commonly used clinically. We found that, despite no significant difference in lung size between non-CF and CF pigs, airway growth was reduced in 3-wk-old CF pigs. Abnormal airway growth was associated with irregularly shaped airways that had reduced distensibility. These findings provide insight into the role of CFTR in airway growth and provide a basis of comparison for future longitudinal studies involving various interventions, including therapies that restore CFTR function.
MATERIALS AND METHODS
Animals
The University of Iowa Institutional Animal Care and Use Committee approved all procedures involving the use of animals. Meconium ileus is highly penetrant in CF pigs (28) and is lethal if not corrected. Thus, to alleviate the meconium ileus phenotype, the 3-wk-old CF pig cohort was produced, as previously described, by expression of porcine CFTR cDNA under the control of the intestinal fatty acid-binding protein promoter in CF pigs (CFTR−/−;TgFABP>pCFTR) (28). We previously showed that these “gut-corrected” CF pigs express CFTR in the intestine, but not in the airways. Similar to other newborn CF pigs, they lack CFTR-mediated anion transport and exhibit tracheal size reduction, tracheal smooth muscle abnormalities, and abnormal-appearing tracheal cartilage rings (28). Non-CF and CF pigs were cared for as previously described (20, 26, 28). For the newborn pig studies, non-CF and CF pigs were generated from mating of CFTR+/− pigs (22, 23, 26). To investigate lung and airway growth from birth to 3 wk of age, some data [tracheal cross-sectional area (CSA) and lung volume; see Fig. 2] were from a previously published newborn (<24 h old) pig cohort (2). Since the newborn and 3-wk-old pigs were from two different cohorts of animals, the respective group sizes are not identical.
Fig. 2.
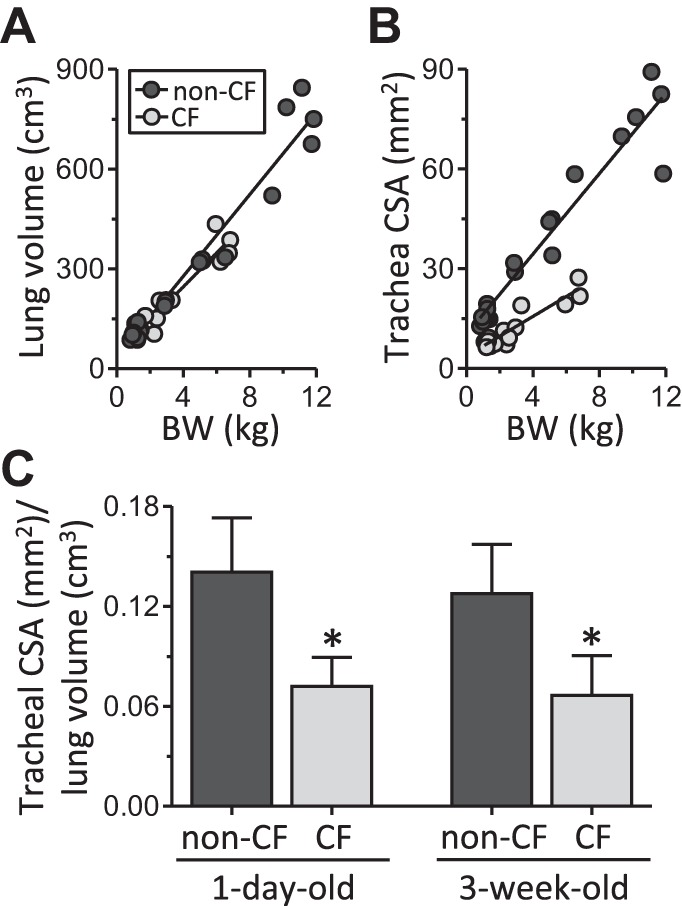
Lung and tracheal size in CF pigs. A: lung volumes obtained from CT scan data sets and plotted as a function of body weight (BW); linear correlation coefficients (r) = 0.97 and 0.96 for non-CF and CF, respectively. B: tracheal lumen cross-sectional area (CSA) obtained from CT scan data sets and plotted as a function of body weight; r = 0.96 and 0.93 for non-CF and CF, respectively. C: tracheal area-to-lung volume ratio assessed by dividing tracheal lumen CSA by lung volume for 1-day-old (newborn) and 3-wk-old non-CF and CF pigs. All data and images obtained from CT scans were acquired at 25-cmH2O airway pressure. For A and B, each symbol represents data from an individual animal. For newborn pigs, n = 8 for both non-CF and CF; for 3-wk-old pigs, n = 11 for non-CF and n = 9 for CF. Previously published data from newborn non-CF and CF pigs (2) are plotted for comparison with data from older pigs. Body weight was similar between genotypes in the newborn cohort: 1.13 ± 0.19 and 1.31 ± 0.19 kg for non-CF and CF, respectively (P > 0.05). Values are means ± SD. *P < 0.05.
CT Scan Acquisition
The 3-wk-old non-CF and CF animals were prospectively CT-scanned using a scanning protocol similar to that previously used for newborn non-CF and CF pigs (2). Briefly, before scanning, pigs were anesthetized, underwent a tracheostomy, and then were paralyzed. After a lung recruitment maneuver, animals were scanned at 0- and 25-cmH2O airway pressure. A Siemens Definition Flash CT scanner was used, and scanning parameters were as follows: 120 kVp, 190 mA, slice thickness 0.6 mm, slice spacing 0.3 mm, and reconstruction kernel B60f. All CT scans were 512 × 512 voxels in the transverse plane, and each had ~500 slices. The newborn non-CF and CF pig scans used for this study were retrospectively analyzed from scans acquired in an earlier study (2).
Quantitative CT Scan Analysis
Lung volume and airway measurements.
Lung volume and airway morphology were quantified using Apollo software (VIDA Diagnostics, Coralville, IA). Lung volume was obtained from automated segmentation. Airway measurements were made perpendicular to the airway’s centerline and included lumen CSA and airway circularity for both the trachea and airway segments of the left and right mainstem bronchi. We quantified airway lumen circularity, a unitless measure of how “circular” a two-dimensional shape is (e.g., airway lumen cross sections) and defined mathematically as follows: circularity = 4π(area/perimeter2). Tracheal measurements were obtained midway between the tracheal bronchus and the carina. The tracheal bronchus, typically not present in humans, is present in all pigs and branches directly off the trachea. Tracheostomy tube placement prohibited measurement of the more-proximal trachea.
Similar to previous studies (1, 7), airway distensibility was defined as 100 × [(airway lumen area at 25 cmH2O – airway lumen area at 0 cmH2O)/(airway lumen area at 0 cmH2O)] and was obtained for airway segments of the right mainstem bronchus and left mainstem bronchus, which we could visualize on CT at both 0- and 25-cmH2O airway pressure. Airway distensibility coefficient of variation was calculated on a per-animal basis and was defined as follows: STDEV(x)/AVG(x), where x equals all bronchial airway segment distensibility measures within an animal.
Scoring CT scans for tracheal airway smooth muscle bundles.
Three reviewers, working independently and blinded to genotype, scored CT scans for the presence, circumferential location, and size of tracheal airway smooth muscle bundles on CT for both newborn and 3-wk-old non-CF and CF pigs. Scans were scored at four tracheal locations: just above the tracheal bronchus, just below the tracheal bronchus, halfway between the tracheal bronchus and the main carina, and just above the main carina. Endotracheal tube placement prohibited scoring at more-proximal tracheal locations. The presence of a bundle was indicated by a positive score from at least two of three reviewers and defined as a protuberance into the lumen space. If a bundle was identified, its circumferential location and width were recorded. Circumferential location was assessed as “angle” within the trachea, where the anterior aspect was 0°, the right 90°, the posterior 180°, and the left 270°. Bundle width was measured using the ImageJ tape-measure tool. Average bundle angle refers to the average angle measured across all locations within an animal and across all three scorers; average bundle width was similarly calculated.
Histopathology
One newborn non-CF and one newborn CF trachea were dissected, and sections were taken at select locations and chemically fixed in 10% neutral buffered formalin. Histological cross sections were routinely prepared from these specimens (18, 19) and stained with Masson’s trichrome.
Statistical Analysis
Values are means ± SD. A two-tailed unpaired Student’s t-test was used for parametric data, and a Mann-Whitney test was used for nonparametric data. Linear regression analysis was used to determine the linear correlation coefficient. P < 0.05 was considered statistically significant.
RESULTS
Three-Week-Old CF Pigs Have Decreased Tracheal Lumen Area
To determine if postnatal airway or lung size was abnormal in CF pigs, we performed CT-based airway and lung measurements in non-CF (21 ± 6 days old, 7.4 ± 3.5 kg body wt) and CF (17 ± 4 days old, 4.4 ± 2.0 kg body wt) pigs (Fig. 1). On average, 3-wk-old CF pigs weighed less than non-CF (P < 0.05) pigs. In both genotypes, lung volume and tracheal lumen CSA linearly correlated with body weight (Fig. 2, A and B). From birth to 3 wk of age, the relationship between lung volume and body weight was not significantly different between non-CF and CF pigs (Fig. 2A). However, for a given body weight, CF pigs had reduced tracheal lumen size (Fig. 2B).
Fig. 1.
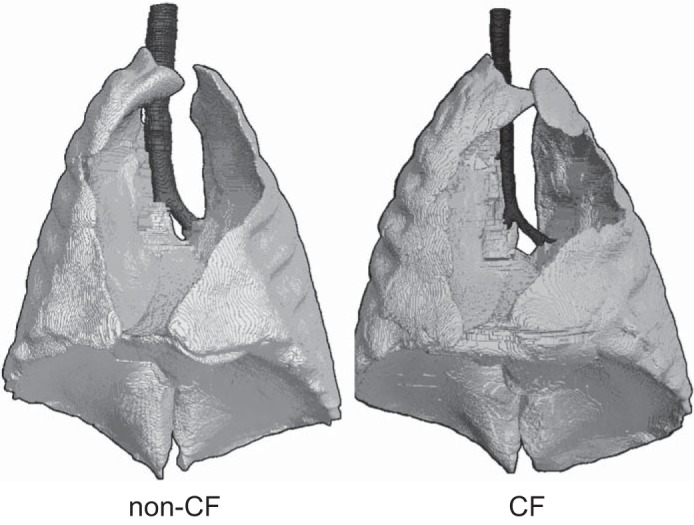
Three-dimensional volumetric CT scan reconstructions of 3-wk-old non-cystic fibrosis (CF) and CF pig lungs at 25-cmH2O airway pressure.
In absolute terms, lung volumes were smaller in some CF than non-CF pigs. However, because CF pigs weighed less than non-CF pigs and lung volume and tracheal lumen area were linearly correlated to body weight, we also report our findings for 3-wk-old pigs normalized to body weight. In 3-wk-old non-CF and CF pigs, lung volumes were similar (64.3 ± 7.8 and 61.6 ± 11.1 cm3/kg), but tracheal lumen area was significantly reduced in CF pigs (8.1 ± 1.7 vs. 4.0 ± 1.0 mm2/kg, P < 0.05). In CF pigs, tracheal lumens were also irregularly shaped, and tracheal wall circularity score (circularity score = 1.0 for a perfect circle) was reduced (0.972 ± 0.010 and 0.964 ± 0.009 in non-CF and CF, respectively, P < 0.05). Thus, compared with non-CF pigs, CF pigs had reduced tracheal area and a reduced ratio of trachea to lung size that was present at birth and persisted into the postnatal period (Fig. 2C). These airway and lung volume measurements are summarized in Table 1.
Table 1.
Trachea and lung size from newborn and 3-wk-old non-CF and CF pigs
| Fold Change from Birth to 3 Weeks |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Age | n | Body Wt, kg | Trachea CSA, mm2 | Lung Vol, cm3 | Trachea CSA/Lung Vol, mm2/cm3 | Lung Vol, cm3/cm3 | Trachea CSA, mm2/mm2 | Trachea CSA/Body Wt, mm2/kg | Lung Vol/Body Wt, cm3/kg |
| Non-CF | |||||||||
| <24 h | 8 | 1.1 ± 0.2 | 15.6 ± 2.1 | 115 ± 22 | 0.14 ± 0.03 | 14.1 ± 1.9 | 103 ± 17 | ||
| 21 ± 6 days | 11 | 7.4 ± 3.5 | 56.2 ± 21.1 | 480 ± 244 | 0.13 ± 0.03 | 4.17 | 3.60 | 8.1 ± 1.7 | 64 ± 8 |
| CF | |||||||||
| <24 h | 8 | 1.3 ± 0.2 | 7.7 ± 0.9 | 112 ± 23 | 0.07 ± 0.02 | 6.0 ± 1.1 | 85 ± 12 | ||
| 17 ± 4 days | 9 | 4.4 ± 2.0 | 16.5 ± 6.7 | 263 ± 113 | 0.07 ± 0.02 | 2.34 | 2.14 | 4.0 ± 1.0 | 62 ± 11 |
Values are means ± SD. CSA, cross-sectional area. Some previously published data from newborn non-CF and CF pigs (2) are included for comparison with data from older pigs.
A Tracheal Smooth Muscle Bundle Is Present in Newborn and Three-Week-Old CF Pigs
Prior histological studies found prominent bundlelike aggregates of tracheal smooth muscle in newborn CF, but not non-CF, pigs (18). Based on our preliminary review of the CT images, we hypothesized that we could visualize these bundles on CT (Fig. 3A) and that they would persist through 3 wk of age. The presence of a tracheal bundle or radiopaque tracheal wall protrusion was noted in nearly all CF pigs: eight of eight newborn and seven of nine 3-wk-old pigs. In contrast, a tracheal bundle was observed in almost no non-CF pigs: one of eight newborn and one of eleven 3-wk-old pigs (Fig. 3B). Bundles were most apparent in the cross section just below the tracheal bronchus (Fig. 3B) and were circumferentially located on the posterior, right side (Fig. 3, C and D). Bundle width was greater in 3-wk-old than newborn CF pigs (Fig. 3E) and was significantly correlated with body weight (Fig. 3F). To verify the CT scan findings, we performed a histological assessment of one newborn non-CF and one CF pig trachea, which confirmed the smooth muscle cellular composition and the location of the bundle within the trachea (Fig. 4). These results are consistent with our earlier findings (18), which demonstrate that loss of CFTR leads to abnormal development of these prominent tracheal smooth muscle bundles.
Fig. 3.

CT imaging assessment of tracheal smooth muscle bundles. A: 1-day-old (newborn) and 3-wk-old non-CF and CF tracheal cross-sectional slices from CT. Arrowheads denote protrusion of airway smooth muscle bundle into lumen air space. R, right; L, left. Scale bar = 5 mm. B: presence of tracheal protrusion or bundle by location for 1-day-old and 3-wk-old CF pigs. Percentage of animals positively scored for the presence of a bundle protrusion on CT is shown for each of the 4 locations. C: airway smooth muscle bundle circumferential angle and width measurements. D: bundle angle in 1-day-old (1 do) and 3-wk-old (3 wo) CF pigs. E: bundle width in 1-day-old and 3-wk-old CF pigs. F: statistically significant positive linear correlation between airway smooth muscle bundle width as measured on CT and pig body weight; r = 0.84 (P < 0.05). Data from 1-day-old and 3-wk-old CF pigs are plotted. Each symbol represents data from an individual animal. All data and images obtained from CT scans were acquired at 25-cmH2O airway pressure. For newborn pigs, n = 8 for both non-CF and CF; for 3-wk-old pigs, n = 11 for non-CF and n = 9 for CF. Values are means ± SD. *P < 0.05.
Fig. 4.
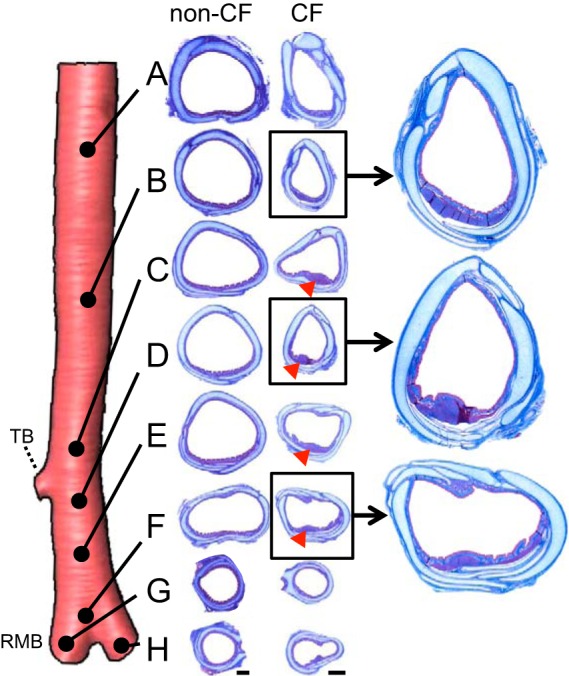
Histological evidence of tracheal smooth muscle bundles in CF pigs. Histology was obtained from 8 specific tracheal and bronchial locations in a newborn non-CF and a newborn CF pig. CF pig trachea had an airway smooth muscle bundle that protruded into the lumen air space (most evident at locations C, D, E, and F; red arrowheads), while the non-CF pig trachea did not. Scale bar = 1 mm for non-CF and CF. TB, tracheal bronchus; RMB, right mainstem bronchi. Masson’s trichrome was used for histological staining.
Proximal Bronchial Area Is Reduced in Three-Week-Old CF Pigs
The reduced tracheal lumen size and abnormal shape raised the following question: Does the aberrant airway size extend into the bronchi? To answer this question, we measured airway lumen CSA, segment-by-segment, up and down the left and right mainstem bronchi. An airway segment was defined as the portion between adjacent bifurcations. In the monopodial branching porcine airway tree, all these airway segments are of the same airway order (analogous to generations in dichotomously branching trees). CF airways exhibited a unique pattern of size reduction (Figs. 5 and 6A). The greatest airway lumen size reduction in CF was in the most-proximally measured airway segments (e.g., segments 1 and 2). This size reduction lessened with progression down the left and right main bronchi, such that, in the most distally measured bronchial segments (e.g., segments 12 and 13), there was virtual size equivalence between non-CF and CF (Figs. 5 and 6A). Normalization of bronchial lumen area to lung volume resulted in a similar pattern of reduction (Fig. 6B). Finally, airway lumen circularity was variable between bronchial airway segments for both genotypes (Fig. 6C) but, on average, was significantly reduced in CF pigs (0.950 ± 0.012 and 0.939 ± 0.006 in non-CF and CF, respectively, P < 0.05; Fig. 6C). In contrast to the CF trachea, we did not observe lumen-protruding smooth muscle bundles on CT scan images of CF pig bronchi.
Fig. 5.
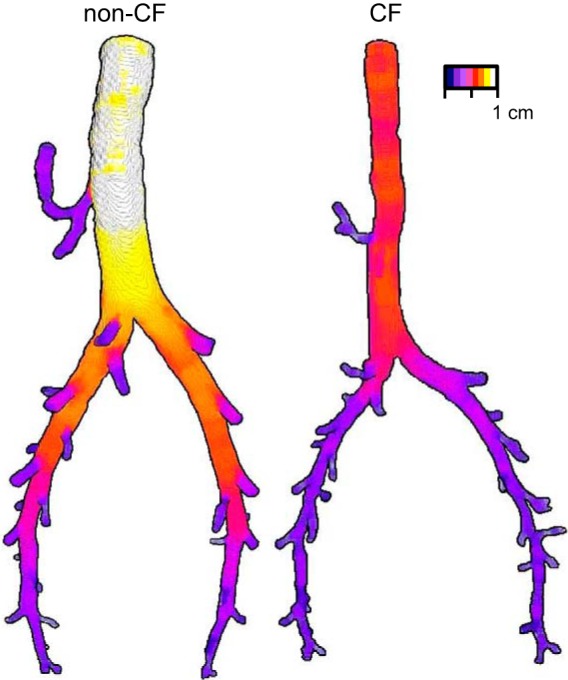
Proximal airway size reduction in right and left mainstem bronchi of 3-wk-old CF pigs. Airway lumen trees from a non-CF and a CF pig are colored according to airway diameter: white corresponds to the widest diameter (non-CF trachea) and dark purple to the smallest measured airways. Animals were age- and weight-matched as well as possible: non-CF pigs were 16 days old and 6.52 kg, and CF pigs were 15 days old and 6.82 kg. Airway trees were generated from CT scans acquired at 25-cmH2O airway pressure and have been digitally pared down to highlight trachea, right mainstem bronchus, and left mainstem bronchus. Scale bar is approximate.
Fig. 6.
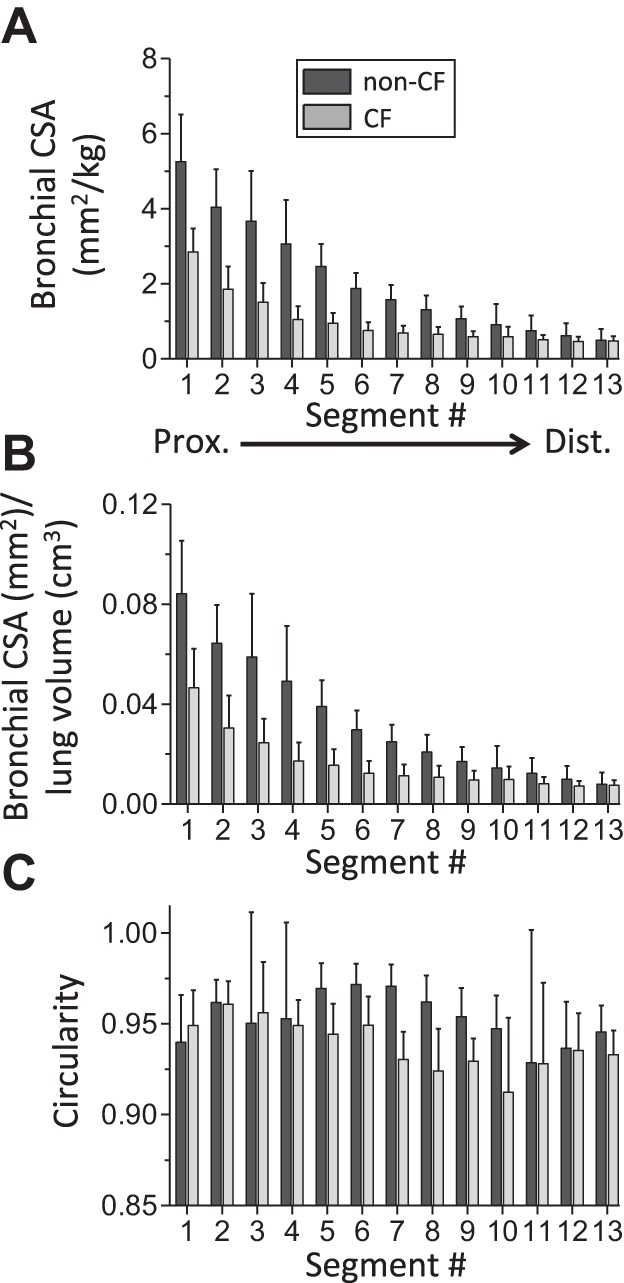
Bronchial airway morphometry in 3-wk-old CF pigs. A: airway lumen CSA normalized to body weight for bronchial airway segments. Airway segments were defined as the portion of airway from 1 airway branch point to the next. Segments are numbered from most-proximal (1) to most-distal (13). B: bronchial lumen area-to-lung volume ratio (bronchial lumen CSA ÷ lung volume). C: inner wall circularity for individual bronchial airway segments. Data are based on measurements from CT scans acquired at 25-cmH2O airway pressure. Data for each segment (n = 9–11 for 3-wk-old non-CF pigs and n = 7–9 for 3-wk-old CF pigs) are average values from corresponding right and left mainstem bronchial segments. Values are means ± SD.
Bronchial Distensibility Is Decreased in Three-Week-Old CF Pigs
Since CF pigs have airway size and shape abnormalities at 25-cmH2O airway pressure, airway smooth muscle bundles, and abnormal airway smooth muscle physiology (8), we hypothesized that airway distensibility, the airway’s ability to change in size in response to distending pressure, would be reduced in 3-wk-old CF pigs. To measure distensibility, we acquired CT scans at 0- and 25-cmH2O airway pressure and defined airway distensibility as follows: 100 × [(airway lumen area at 25 cmH2O – airway lumen area at 0 cmH2O)/(airway lumen area at 0 cmH2O)] (Fig. 7A). Tracheal distensibility was similar between genotypes (Fig. 7, A and B), but bronchial distensibility was reduced in CF pigs (Figs. 7C). In non-CF pigs, bronchial distensibility tended to increase distally in the bronchus (Fig. 7C). The opposite was observed in CF pigs: bronchial distensibility tended to decrease from proximal to distal airway segments (Fig. 7C). Furthermore, there was relatively more segment-to-segment variation in bronchial distensibility within an individual CF pig than in non-CF pigs. This finding was reflected in a greater coefficient of variation for distensibility between bronchial segments within individual CF pigs (Fig. 7D).
Fig. 7.
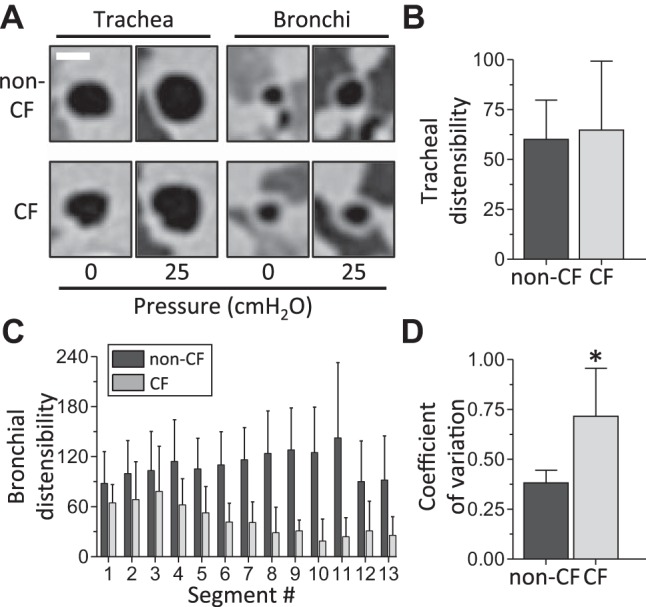
Airway distensibility in 3-wk-old non-CF and CF pigs. A: representative images of CT scan slices from trachea and bronchi of 3-wk-old non-CF and CF pigs at 0- and 25-cmH2O airway pressure. Scale bar = 5 mm. B: tracheal distensibility for 3-wk-old non-CF (n = 11) and CF (n = 8) pigs. C: distensibility for individual bronchial segments. Segments are numbered from most-proximal (1) to most-distal (13). Data for each segment (n = 6–11 for 3-wk-old non-CF pigs and n = 5–9 for 3-wk-old CF pigs) are average values from corresponding right and left mainstem bronchial segments. D: average coefficient of variation for bronchial distensibility in non-CF and CF pig bronchi. Coefficient of variation for bronchial distensibility was calculated for all measured bronchial segments within an individual animal and then averaged for each genotype (n = 11 for 3-wk-old non-CF pigs and n = 9 for 3-wk-old CF pigs). Airway distensibility was calculated as follows: 100 × [(airway lumen area at 25 cmH2O – airway lumen area at 0 cmH2O)/(airway lumen area at 0 cmH2O)]. Values are means ± SD. *P < 0.05.
DISCUSSION
Increasing evidence suggests that loss of CFTR leads to airway developmental defects, which, in turn, may contribute to CF lung disease pathogenesis (2, 4, 10, 14, 18, 30). Because studies in humans with CF are limited, we assessed airway growth and development in CF pigs. In CF pigs, reduced tracheal and bronchial growth, irregularly shaped airways, decreased airway distensibility, and large tracheal smooth muscle bundles were apparent on CT imaging.
Airways Are Small in Three-Week-Old CF Pigs
In 3-wk-old CF pigs, airway size reduction predominantly affected the proximally measured airways. A similar pattern was previously observed in newborn CF pigs (2) and in limited studies of older CF pigs (3). Developmental defects are also present in airways of CF mice and rats (4, 14, 30). Although developmental studies in young humans with CF are limited, several lines of evidence suggest that abnormal airway development and growth may be features of human CF. 1) In an analysis of previously published histological data from infants (<2 wk old) with CF, tracheal lumen area was significantly reduced (18). 2) Review of chest CT images revealed irregularly shaped tracheas in young (<2-yr-old) children with CF (18). These observations are consistent with our findings in newborn and older CF pigs. 3) About 15% of infants with CF have tracheomalacia compared with ~0.05% of healthy infants, suggesting that impaired CFTR function affects airway structure (10). 4) CT-based airway measurements in young (average age 1.3 yr) children with CF revealed smaller-sized airways (15). While luminal obstruction by mucus accumulation or airway wall thickening was speculated to be the underlying etiology in this study, the size reduction could reflect decreased airway lumen growth, such as that in CF pigs. 5) Functional defects that could be related to abnormal airway development and growth, including air trapping, are frequently found in young children with CF, often in the absence of airway infection and inflammation (11, 25). Thus, findings from both humans with CF and CF animal models suggest that CFTR plays a role in airway development and growth.
Our findings in CF pigs could have a number of potential implications. 1) Reduced airway growth, especially the central airways, would increase both airway resistance and turbulent airflow, likely leading to expiratory airflow limitation and increased work of breathing (24). 2) Future studies of airway and lung development, especially during the fetal period, in CF pigs could provide additional insight into the differing regulation and genetic control of airway and lung parenchymal growth. While the current study was not designed to determine the underlying mechanism(s) for abnormal airway growth in CF pigs, there are several possible etiologies, including reduced fetal lung liquid secretion, abnormal airway smooth muscle function (8, 16), and disrupted airway cartilage development. 3) The unique pattern of airway size reduction may be related to the temporal order of proximal vs. distal airway formation in utero. In several species, it is known that CFTR mRNA is expressed at greater levels during early than late gestation (5, 17, 29). Thus it is possible that CFTR is more important early in gestation, when large proximal airways are formed, than later in gestation, when smaller airways are formed (2).
Bronchial Distensibility Is Reduced in CF Pigs
Airway distensibility was reduced in 3-wk-old CF pig bronchi, but not trachea. This effect was most apparent in distally measured bronchial segments, where airway size differences between non-CF and CF pigs were less pronounced. In humans with CF, tracheal distensibility has been examined in a limited number of studies and found to be either similar between non-CF and CF (13) or increased in CF (6). Differences in disease stage, study participant age, sample size, and measurement technique likely account for the discrepant findings.
From this study we are unable to determine the cause of this unique pattern of bronchial distensibility. However, one possible contributor is alteration of CF pig airway smooth muscle function. We know from prior studies that CF pig airway smooth muscle exists in a hypercontractile-like state relative to non-CF pig airway smooth muscle (8). We also know that in healthy airways, after normalization for airway size, small airways have relatively more airway smooth muscle than larger airways. Thus the airway-size-dependent distensibility pattern we observed in CF pigs may reflect a combination of these two phenomena. In support of this notion, using CT-based airway measurements, we recently investigated airway distensibility in humans with CF bearing the G551D-CFTR mutation (1). While we did not compare non-CF with CF, we found that acute restoration of CFTR function with ivacaftor increased airway distensibility in humans with CF (1). Interestingly, this effect was most significant for the distal airways. In our current study we investigated CFTR−/− pigs; thus we were unable to determine if increasing CFTR function changed airway distensibility. Functionally, the normally protective bronchodilatory effects of a deep inspiration could be decreased in a less-distensible CF airway, further worsening airflow obstruction (12).
Advantages and Limitations
This study has a number of advantages and limitations. Advantages are as follows. 1) The CF pig recapitulates hallmark characteristics of human CF lung disease (26, 27). 2) Our study benefited from standardized, paired 0- and 25-cmH2O airway pressure, high-resolution chest CT scans with detailed airway measurements. 3) Studies in humans with CF, especially infants and children, are hampered by a lack of non-CF, healthy controls. Here, we assessed CFTR-dependent development by comparison of non-CF and CF pigs. 4) We were able to study CF-related changes at early time points that are not feasible in humans with CF. Limitations are as follows. 1) Airway cartilage was not visible on CT. Nevertheless, 3-wk-old CF pigs likely have airway cartilage defects, given documented tracheal ring abnormalities in newborn CF pigs (18), CF mice (4), CF rats (30), and humans with CF (9). 2) We focused our airway assessment on the trachea and right and left mainstem bronchi. Although results for other airways may differ, newborn CF pigs also have proximal, but not more-distal, airway size reduction in the left and right mainstem bronchi, as well as conducting airways of the tracheal lobe (2). 3) Limitations in CT scanner resolution may reduce accuracy of more-distal airway measurements. Similarly, changes in airway wall thickness or mucus accumulation could also affect the airway measurements. However, our observation of the pattern of airway size reduction in 3-wk-old CF pigs largely duplicates that observed using micro- (very-high-resolution) CT imaging in the newborn CF pig lung (2), which lacks airway wall thickening and mucus accumulation, suggesting the pattern of airway size reduction we observed to be true. 4) Our lung measurements only focused on total lung volume. It is possible that growth rates of other parts of the lung differed between non-CF and CF or that air space enlargement could differ between genotypes, although non-CF and CF pigs have similar alveolar surface area at birth (2). 5) We were unable to determine if the postnatal airway growth defects were due to continued loss of CFTR after birth or were primarily due to effects of in utero CFTR loss that persisted after birth. 6) The 3-wk-old non-CF and CF pigs were age-matched as closely as possible. Although they were age-matched to non-CF pigs, the 3-wk-old CF pigs had reduced body weight. Similarly, humans with CF commonly gain weight at reduced rates. Thus, although we age-matched our pigs and normalized to body weight for some of the analyses, there may exist differences in “developmental age” both within and between cohorts.
In conclusion, our data demonstrate that loss of CFTR function impairs postnatal airway growth. These findings suggest that abnormalities in infants and children with CF, including reduced airway lumen and circularity (18), air trapping (11, 15, 25), and tracheomalacia (10), could, in part, be caused by abnormal airway growth and development due to loss of CFTR.
GRANTS
This work was supported by National Institutes of Health Grants P01 HL-051670, P01 HL-091842, T32 HL-007638, T32 GM-007337, and DP2 HL-117744; the Cystic Fibrosis Foundation (CFF); and the CFF Mucociliary Clearance Consortium. D. A. Stoltz was supported by the Gilead Sciences Research Scholars Program in Cystic Fibrosis.
DISCLOSURES
D. A. Stoltz and M. J. Welsh receive royalties from the University of Iowa Research Foundation related to a licensing agreement with Exemplar Genetics for gene-modified pigs.
AUTHOR CONTRIBUTIONS
R.J.A., M.H.A.A., J.Z., M.J.W., D.K.M., and D.A.S. conceived and designed research; R.J.A., M.H.A.A., D.C.B., D.P.C., N.D.G., P.J.T., L.S.P., M.R.S., M.J.H., J.D.M., and D.A.S. performed experiments; R.J.A., M.H.A.A., and D.A.S. analyzed data; R.J.A., M.H.A.A., E.A.H., J.Z., M.J.W., D.K.M., and D.A.S. interpreted results of experiments; R.J.A., D.K.M., and D.A.S. prepared figures; R.J.A. and D.A.S. drafted manuscript; R.J.A., M.H.A.A., D.C.B., D.P.C., N.D.G., P.J.T., L.S.P., M.R.S., M.J.H., J.D.M., E.A.H., J.Z., M.J.W., D.K.M., and D.A.S. edited and revised manuscript; R.J.A., M.H.A.A., D.C.B., D.P.C., N.D.G., P.J.T., L.S.P., M.R.S., M.J.H., J.D.M., E.A.H., J.Z., M.J.W., D.K.M., and D.A.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank B. Hilkin, S. Horgen, T. Mayhew, P. McCray, L. Ostedgaard, L. Reznikov, K. Webber, and the University of Iowa Office of Animal Resources for discussions, excellent assistance, and advice.
REFERENCES
- 1.Adam RJ, Hisert KB, Dodd JD, Grogan B, Launspach JL, Barnes JK, Gallagher CG, Sieren JP, Gross TJ, Fischer AJ, Cavanaugh JE, Hoffman EA, Singh PK, Welsh MJ, McKone EF, Stoltz DA. Acute administration of ivacaftor to people with cystic fibrosis and a G551D-CFTR mutation reveals smooth muscle abnormalities. JCI Insight 1: e86183, 2016. doi: 10.1172/jci.insight.86183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adam RJ, Michalski AS, Bauer C, Abou Alaiwa MH, Gross TJ, Awadalla MS, Bouzek DC, Gansemer ND, Taft PJ, Hoegger MJ, Diwakar A, Ochs M, Reinhardt JM, Hoffman EA, Beichel RR, Meyerholz DK, Stoltz DA. Air trapping and airflow obstruction in newborn cystic fibrosis piglets. Am J Respir Crit Care Med 188: 1434–1441, 2013. doi: 10.1164/rccm.201307-1268OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ballard ST, Evans JW, Drag HS, Schuler M. Pathophysiologic evaluation of the transgenic Cftr “gut-corrected” porcine model of cystic fibrosis. Am J Physiol Lung Cell Mol Physiol 311: L779–L787, 2016. doi: 10.1152/ajplung.00242.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonvin E, Le Rouzic P, Bernaudin JF, Cottart CH, Vandebrouck C, Crié A, Leal T, Clement A, Bonora M. Congenital tracheal malformation in cystic fibrosis transmembrane conductance regulator-deficient mice. J Physiol 586: 3231–3243, 2008. doi: 10.1113/jphysiol.2008.150763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Broackes-Carter FC, Mouchel N, Gill D, Hyde S, Bassett J, Harris A. Temporal regulation of CFTR expression during ovine lung development: implications for CF gene therapy. Hum Mol Genet 11: 125–131, 2002. doi: 10.1093/hmg/11.2.125. [DOI] [PubMed] [Google Scholar]
- 6.Brooks LJ. Tracheal size and distensibility in patients with cystic fibrosis. Am Rev Respir Dis 141: 513–516, 1990. doi: 10.1164/ajrccm/141.2.513. [DOI] [PubMed] [Google Scholar]
- 7.Brown RH, Wizeman W, Danek C, Mitzner W. Effect of bronchial thermoplasty on airway distensibility. Eur Respir J 26: 277–282, 2005. doi: 10.1183/09031936.05.00006605. [DOI] [PubMed] [Google Scholar]
- 8.Cook DP, Rector MV, Bouzek DC, Michalski AS, Gansemer ND, Reznikov LR, Li X, Stroik MR, Ostedgaard LS, Abou Alaiwa MH, Thompson MA, Prakash YS, Krishnan R, Meyerholz DK, Seow CY, Stoltz DA. Cystic fibrosis transmembrane conductance regulator in sarcoplasmic reticulum of airway smooth muscle. Implications for airway contractility. Am J Respir Crit Care Med 193: 417–426, 2016. doi: 10.1164/rccm.201508-1562OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diwakar A, Adam RJ, Michalski AS, Tamegnon MM, Fischer AJ, Launspach JL, Horan RA, Kao SC, Chaloner K, Meyerholz DK, Stoltz DA. Sonographic evidence of abnormal tracheal cartilage ring structure in cystic fibrosis. Laryngoscope 125: 2398–2404, 2015. doi: 10.1002/lary.25255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fischer AJ, Singh SB, Adam RJ, Stoltz DA, Baranano CF, Kao S, Weinberger MM, McCray PB Jr, Starner TD. Tracheomalacia is associated with lower FEV1 and Pseudomonas acquisition in children with CF. Pediatr Pulmonol 49: 960–970, 2014. doi: 10.1002/ppul.22922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hall GL, Logie KM, Parsons F, Schulzke SM, Nolan G, Murray C, Ranganathan S, Robinson P, Sly PD, Stick SM, Berry L, Garratt L, Massie J, Mott L, Poreddy S, Simpson S, Simpson S; AREST CF . Air trapping on chest CT is associated with worse ventilation distribution in infants with cystic fibrosis diagnosed following newborn screening. PLoS One 6: e23932, 2011. doi: 10.1371/journal.pone.0023932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kapsali T, Permutt S, Laube B, Scichilone N, Togias A. Potent bronchoprotective effect of deep inspiration and its absence in asthma. J Appl Physiol (1985) 89: 711–720, 2000. [DOI] [PubMed] [Google Scholar]
- 13.Lebecque P, Liistro G, Veriter C, Stănescu D. Tracheal distensibility in cystic fibrosis. Eur Respir J 9: 770–772, 1996. doi: 10.1183/09031936.96.09040770. [DOI] [PubMed] [Google Scholar]
- 14.Livraghi-Butrico A, Kelly EJ, Wilkinson KJ, Rogers TD, Gilmore RC, Harkema JR, Randell SH, Boucher RC, O’Neal WK, Grubb BR. Loss of Cftr function exacerbates the phenotype of Na+ hyperabsorption in murine airways. Am J Physiol Lung Cell Mol Physiol 304: L469–L480, 2013. doi: 10.1152/ajplung.00150.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martínez TM, Llapur CJ, Williams TH, Coates C, Gunderman R, Cohen MD, Howenstine MS, Saba O, Coxson HO, Tepper RS. High-resolution computed tomography imaging of airway disease in infants with cystic fibrosis. Am J Respir Crit Care Med 172: 1133–1138, 2005. doi: 10.1164/rccm.200412-1665OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McCray PB., Jr Spontaneous contractility of human fetal airway smooth muscle. Am J Respir Cell Mol Biol 8: 573–580, 1993. doi: 10.1165/ajrcmb/8.5.573. [DOI] [PubMed] [Google Scholar]
- 17.McCray PB Jr, Wohlford-Lenane CL, Snyder JM. Localization of cystic fibrosis transmembrane conductance regulator mRNA in human fetal lung tissue by in situ hybridization. J Clin Invest 90: 619–625, 1992. doi: 10.1172/JCI115901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meyerholz DK, Stoltz DA, Namati E, Ramachandran S, Pezzulo AA, Smith AR, Rector MV, Suter MJ, Kao S, McLennan G, Tearney GJ, Zabner J, McCray PB Jr, Welsh MJ. Loss of cystic fibrosis transmembrane conductance regulator function produces abnormalities in tracheal development in neonatal pigs and young children. Am J Respir Crit Care Med 182: 1251–1261, 2010. doi: 10.1164/rccm.201004-0643OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meyerholz DK, Stoltz DA, Pezzulo AA, Welsh MJ. Pathology of gastrointestinal organs in a porcine model of cystic fibrosis. Am J Pathol 176: 1377–1389, 2010. doi: 10.2353/ajpath.2010.090849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ostedgaard LS, Meyerholz DK, Chen JH, Pezzulo AA, Karp PH, Rokhlina T, Ernst SE, Hanfland RA, Reznikov LR, Ludwig PS, Rogan MP, Davis GJ, Dohrn CL, Wohlford-Lenane C, Taft PJ, Rector MV, Hornick E, Nassar BS, Samuel M, Zhang Y, Richter SS, Uc A, Shilyansky J, Prather RS, McCray PB Jr, Zabner J, Welsh MJ, Stoltz DA. The ΔF508 mutation causes CFTR misprocessing and cystic fibrosis-like disease in pigs. Sci Transl Med 3: 74ra24, 2011. doi: 10.1126/scitranslmed.3001868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Regamey N, Ochs M, Hilliard TN, Mühlfeld C, Cornish N, Fleming L, Saglani S, Alton EW, Bush A, Jeffery PK, Davies JC. Increased airway smooth muscle mass in children with asthma, cystic fibrosis, and non-cystic fibrosis bronchiectasis. Am J Respir Crit Care Med 177: 837–843, 2008. doi: 10.1164/rccm.200707-977OC. [DOI] [PubMed] [Google Scholar]
- 22.Rogers CS, Hao Y, Rokhlina T, Samuel M, Stoltz DA, Li Y, Petroff E, Vermeer DW, Kabel AC, Yan Z, Spate L, Wax D, Murphy CN, Rieke A, Whitworth K, Linville ML, Korte SW, Engelhardt JF, Welsh MJ, Prather RS. Production of CFTR-null and CFTR-ΔF508 heterozygous pigs by adeno-associated virus-mediated gene targeting and somatic cell nuclear transfer. J Clin Invest 118: 1571–1577, 2008. doi: 10.1172/JCI34773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rogers CS, Stoltz DA, Meyerholz DK, Ostedgaard LS, Rokhlina T, Taft PJ, Rogan MP, Pezzulo AA, Karp PH, Itani OA, Kabel AC, Wohlford-Lenane CL, Davis GJ, Hanfland RA, Smith TL, Samuel M, Wax D, Murphy CN, Rieke A, Whitworth K, Uc A, Starner TD, Brogden KA, Shilyansky J, McCray PB Jr, Zabner J, Prather RS, Welsh MJ. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science 321: 1837–1841, 2008. doi: 10.1126/science.1163600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sheel AW, Guenette JA, Yuan R, Holy L, Mayo JR, McWilliams AM, Lam S, Coxson HO. Evidence for dysanapsis using computed tomographic imaging of the airways in older ex-smokers. J Appl Physiol (1985) 107: 1622–1628, 2009. doi: 10.1152/japplphysiol.00562.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sly PD, Brennan S, Gangell C, de Klerk N, Murray C, Mott L, Stick SM, Robinson PJ, Robertson CF, Ranganathan SC; Australian Respiratory Early Surveillance Team for Cystic Fibrosis (AREST-CF) . Lung disease at diagnosis in infants with cystic fibrosis detected by newborn screening. Am J Respir Crit Care Med 180: 146–152, 2009. doi: 10.1164/rccm.200901-0069OC. [DOI] [PubMed] [Google Scholar]
- 26.Stoltz DA, Meyerholz DK, Pezzulo AA, Ramachandran S, Rogan MP, Davis GJ, Hanfland RA, Wohlford-Lenane C, Dohrn CL, Bartlett JA, Nelson GA 4th, Chang EH, Taft PJ, Ludwig PS, Estin M, Hornick EE, Launspach JL, Samuel M, Rokhlina T, Karp PH, Ostedgaard LS, Uc A, Starner TD, Horswill AR, Brogden KA, Prather RS, Richter SS, Shilyansky J, McCray PB Jr, Zabner J, Welsh MJ. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci Transl Med 2: 29ra31, 2010. doi: 10.1126/scitranslmed.3000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stoltz DA, Meyerholz DK, Welsh MJ. Origins of cystic fibrosis lung disease. N Engl J Med 372: 351–362, 2015. doi: 10.1056/NEJMra1300109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stoltz DA, Rokhlina T, Ernst SE, Pezzulo AA, Ostedgaard LS, Karp PH, Samuel MS, Reznikov LR, Rector MV, Gansemer ND, Bouzek DC, Abou Alaiwa MH, Hoegger MJ, Ludwig PS, Taft PJ, Wallen TJ, Wohlford-Lenane C, McMenimen JD, Chen JH, Bogan KL, Adam RJ, Hornick EE, Nelson GA 4th, Hoffman EA, Chang EH, Zabner J, McCray PB Jr, Prather RS, Meyerholz DK, Welsh MJ. Intestinal CFTR expression alleviates meconium ileus in cystic fibrosis pigs. J Clin Invest 123: 2685–2693, 2013. doi: 10.1172/JCI68867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trezise AE, Chambers JA, Wardle CJ, Gould S, Harris A. Expression of the cystic fibrosis gene in human foetal tissues. Hum Mol Genet 2: 213–218, 1993. doi: 10.1093/hmg/2.3.213. [DOI] [PubMed] [Google Scholar]
- 30.Tuggle KL, Birket SE, Cui X, Hong J, Warren J, Reid L, Chambers A, Ji D, Gamber K, Chu KK, Tearney G, Tang LP, Fortenberry JA, Du M, Cadillac JM, Bedwell DM, Rowe SM, Sorscher EJ, Fanucchi MV. Characterization of defects in ion transport and tissue development in cystic fibrosis transmembrane conductance regulator (CFTR)-knockout rats. PLoS One 9: e91253, 2014. doi: 10.1371/journal.pone.0091253. [DOI] [PMC free article] [PubMed] [Google Scholar]