

Abstract
Oxidative stress impacts normal cellular function leading to the pathogenesis of various diseases including pulmonary illnesses. Protein arginine methyltransferase 4 (PRMT4) is critical for normal lung alveolar epithelial cell development; however, the regulation of PRMT4 within such pulmonary diseases has yet to be elucidated. Using biochemical approaches, we uncovered that peroxide (H2O2) treatment decreases PRMT4 protein stability in murine lung epithelial (MLE12) cells to impede cell migration. Protein kinase glycogen synthase kinase 3β (GSK-3β) interacts with PRMT4 and catalyzes PRMT4 T132 phosphorylation that protects PRMT4 from ubiquitin proteasomal degradation. H2O2 downregulates GSK-3β to reduce PRMT4 at protein level. PRMT4 promotes cell migration and H2O2 degrades PRMT4 to inhibit lung epithelial cell migration. These observations demonstrate that oxidative stress destabilizes PRMT4 via GSK-3β signaling to impede lung epithelial cell migration that may hinder the lung repair and regeneration process.
Keywords: protein arginine methyltransferase 4, PRMT4, glycogen synthase kinase 3β, GSK-3β, phosphorylation, ubiquitin and proteasome degradation, oxidative stress, migration
oxidative stress occurs when the body cannot properly regulate free radical production or harmful intermediates through antioxidant defenses. Exposure to the excessive endogenous and exogenous reactive oxygen species (ROS) alters crucial signal transduction pathways, which are exclusively involved in the pathogenesis of a variety of diseases including pulmonary diseases (8, 32, 40). Epigenetics governs gene transcriptional activity in all of life processes, yet oxidative stress can disrupt epigenetic enzymes that change chromatin architecture, histone posttranslational modification, or DNA methylation (9, 22, 38). For example, cigarette smoking promotes protein turnover of histone deacetylase 2 (HDAC2) by phosphorylation and subsequent ubiquitination (18, 24, 37). Another histone deacetylase, sirtuin 1 (SIRT1), is also reduced in chronic obstructive pulmonary disease (COPD) patients (17). Inactivation of HDAC2 exacerbates lung epithelial inflammation by unleashing proinflammatory gene transcription in COPD patients (30). Emerging evidence indicates an increased histone acetylation in patients effected by asthma (5). Studies discovered that several miRNA species are responsible for idiopathic pulmonary fibrosis pathogenesis (29). In addition, epigenetic enzymes take part in the pathogenesis of acute lung injury (ALI). Bacterial infection alters epigenetic enzymes to reprogram host defense, affect cell survival, and impact proliferation (13, 46, 47).
Protein arginine N-methyltransferase 4/coactivator-associated arginine methyltransferase 1 (PRMT4/CARM1), a type I methyltransferase enzyme in protein arginine methyltransferase family, is involved in crucial life processes including gene transcription, proliferation, cell cycle, DNA splicing, and development (4, 7, 10, 12, 16, 27). PRMT4 catalyzes the addition of mono- or asymmetric arginine residues to the guanidino nitrogens of arginyl residues in histone and nonhistone protein substrates. Coordinated with the action of histone acetyltransferase p300 and p160, PRMT4-dependent asymmetric dimethylation of histone H3 at Arg-17 (H3R17me2a) activates gene transcription (4). PRMT4 catalyzes methylation of a range of nonhistone proteins including p300 to exert its diverse biological functions in NF-κB-mediated inflammation, p53-related signal transduction, mRNA maturation, proliferation, and cell cycle (7, 10, 12, 16, 27). PRMT4 also functions in cell migration. PRMT4 catalyzes BAF155 methylation to regulate breast cancer cell migration and functions in proper slow myosin heavy chain localization in muscle development (3, 41). It is yet to be studied if PRMT4 affects lung epithelial cell migration, which is involved in lung epithelial cell repair and regeneration after injury.
PRMT4 has been determined as critical for normal lung development in rodent models. PRMT4 knockout mice can cause neonatal death and developmental defects in respiratory system such as increased immature alveolar type II cells and lack of type I cells, as well as smaller alveolar air space (27). Moreover, asymmetric dimethylarginine is increased in asthma (35). Yet, the degradation metabolite for PRMT4 catalyzed methylation, asymmetrical dimethylarginine, acts as an endogenous inhibitor of nitride oxide synthase, which is now a risk marker in a variety of life-threatening diseases (44). These include end-stage renal diseases, liver insufficiency, heart failure, diabetes, pre-eclampsia, atherosclerotic complication, asthma, and chronic pulmonary diseases (34, 36, 44). Nevertheless, the mechanism(s) of how PRMT4 is regulated in oxidative stress-related pulmonary diseases is largely unknown.
In this study, we report that oxidative stress reduces PRMT4 protein stability via protein kinase GSK-3β. Under normal conditions, GSK-3β phosphorylates PRMT4 at T132, and that phosphorylation stabilizes PRMT4 by ablating its ubiquitination and later protein degradation. Oxidative stress reduces GSK-3β protein level that results in PRMT4 ubiquitination and proteasome degradation. Reduced PRMT4 protein stability impedes cell migration that is required in injured lung epithelial cell repair and regeneration.
MATERIALS AND METHODS
Cell lines and reagents.
Murine lung epithelial cell line MLE12 cells were cultured with HITES medium (500 ml of DMEM/F12, 2.5 mg of insulin, transferrin, sodium selenite, 2.5 mg of transferrin, 10 μM hydrocortisone, 10 μM β-estradiol, 10 mM HEPES, and 2 mM l-glutamine) supplemented with 10% of FBS, as previously described (49). PRMT4 expression plasmid was purchased from GeneCopoeia (Rockville, MD). PRMT4 (catalog no. 12495s), GSK-3β (catalog no. 9832s), and phospho-threonine (catalog no. 9381s) were from Cell Signaling (Danvers, MA). Phospho-serine (catalog no. ab9332) antibody was from Abcam. H2O2 and β-actin antibodies were from Sigma-Aldrich (St. Louis, MO). The concentration of H2O2 used in the experiments is in the range of 50–300 μM, which fits in perspective of in vivo oxidative stress in pathophysiological conditions (15, 26). Ubiquitin antibody (catalog no. sc-166553) and γ-tubulin antibody were from Santa Cruz Biotechnology (Santa Cruz, CA). TOPO pcDAN3.1/V5 directional plasmid and V5 antibody (catalog no. P/N46-1157) were purchased from Invitrogen (Waltham, MA). PRMT4 and GSK-3β short hairpin RNA lenti-viral constructs were from Origene (Rockville, MD). The protein synthesis inhibitor cycloheximide (CHX; 10 μM) and the proteasome reversible inhibitor MG132 (20 μM) were from Calbiochem (Billerica, MA). All the materials used in the experiments are commercially available in the highest quality.
Coimmunoprecipitation and immunoblotting analyses.
Coimmunoprecipitation (co-IP) and immunoblotting were performed as previously described (47). Briefly, PRMT4 protein and its interaction partner(s) were immunoprecipitated from MLE12 cell lysates. Cells were rinsed with cold PBS buffer and lysed with co-IP buffer [150 mM NaCl, 50 mM Tris·HCl, 1 mM EDTA, 2 mM dithiothreitol (DTT), 0.5% Triton X-100 (vol/vol), and 1:1,000 protease inhibitor cocktail (vol/vol)]. Cell lysates containing 1 mg of total protein were incubated with 2 μg of PRMT4 antibody for 2 h. Immunoprecipitates of PRMT4-recognized proteins were incubated with protein A/G beads for an additional hour. The precipitates were washed with co-IP buffer for three times and applied to immunoblotting analyses. For immunoblotting, cell lysates were prepared by brief sonication in buffer A [150 mM NaCl, 50 mM Tris, 1.0 mM EDTA, 0.5% Triton X-100 (vol/vol), 2 mM DTT, 1:1,000 protease inhibitor cocktail, 0.025% sodium azide, and 1 mM phenylmethylsulfonyl fluoride pH 7.4] at 4°C. The cell lysates were separated by SDS-PAGE. Immunoblotting images were acquired with a Kodak image 2000 system.
Molecular cloning and site-directed mutagenesis.
The full-length coding region cDNA of PRMT4 (GeneCopoeia, Rockville, MD) was amplified by PCR using following forward and reverse primers: forward: 5′-CACCATGGCAGCGGCGGCAGCG-3′; and reverse: 5′-ACTCCCATAGTGCATGGTGTTG-3′. The DNA was cloned into TOPO pcDNA3.1/His-V5 directional expression plasmid, and the accuracy of the cloned gene was verified by sequencing. Site-directed mutagenesis of PRMT4 was obtained as previously described (47). The primers used in site-directed mutagenesis are as follows: T132A: forward: 5′-CCTGTCGGGGCCACGCACTGGAGCGCTCTGTG-3′ and reverse: 5′-CACAGAGCGCTCCAGTGCGTGGCCCCGACAGG-3′; T132C: forward: 5′-CCTGTCGGGGCCACTGCCTGGAGCGCTCTGTG-3′ and reverse: 5′-CACAGAGCGCTCCAGGCAGTGGCCCCGACAGG-3′; T132D: forward: 5′-CCTGTCGGGGCCACGACCTGGAGCGCTCTGTG-3′ and reverse: 5′-CACAGAGCGCTCCAGGTCGTGGCCCCGACAGG-3′; and S136A: forward: 5′- CCACACACTGGAGCGCGCTGTGTTCAGTGAGCGG and reverse: CCGCTCACTGAACACAGCGCGCTCCAGTGTGTGG. The mutations were verified by sequencing for the accuracy.
In vitro kinase assay.
MLE cells were rinsed with cold PBS buffer, sonicated in IP buffer, and then centrifuged (12,000 g, 10 min). The supernatant were incubated with 2 μg of GSK-3β antibody for overnight. The mixtures were then incubated with protein A/G beads for 2 h. The beads were washed twice with kinase buffer [1 × PBS, 10 mM DTT, 10 mM MgCl2, 10% glycerol (vol/vol), and 1:1,000 phosphatase inhibitor (vol/vol)]. Wild-type (WT) PRMT4, T132A, and S136A of mutant PRMT4 were synthesized with TnT Quick Coupled Transcription/Translation System separately. The immunoprecipitates were then incubated with the recombinant proteins in kinase buffer (contains 10 mM ATP) for 1 h at 25°C. These samples were applied to immunoblotting analyses.
Plasmid transfection.
Plasmids were introduced into MLE12 cells using electroporation. Briefly, 1,000,000 cells in their exponent growth stage were suspended with 3 μg of plasmid DNA in 100 μl of transfection buffer (20 mM HEPES in PBS, pH 7.4). Electroporation was executed with a nuclear transfection apparatus (Amaxa Biosystems, Gaithersburg, MD) with a preset program T-013 in an electroporation cuvette. The cells were then transferred into six-well plates for culture with 2 ml of HITES medium for 48 h. Short hairpin RNA constructs were introduced into the cells with same approach and incubated for 72 h.
Quantitative reverse transcription polymerase chain reaction.
Quantitative (q)RT-PCR was conducted as previously described (13). Briefly, total cellular RNA was isolated from MLE12 cells using TRI reagent (Invitrogen, Carlsbad, CA) following the protocol of manufacturer. The mRNA was digested with DNase I for 30 min to rule out genomic DNA contamination. cDNA was synthesized from isolated total RNA with a SYBR Select Master Mix (Thermo Fisher Scientific, San Jose, CA). Prmt4-specific primers were designed based on the NCBI mouse mRNA and genome DNA sequence database as follows: forward: 5′-CAACAGCGTCCTCATCCAG-3′ and reverse: 5′-CTCTGTCCGCTCACTGAAC-3′. qPCR reactions were conducted using a Bio-Rad C1000 thermo cycler at a two-step program (95°C for 10 s, and 55°C for 15 s for 40 cycles). Gapdh mRNA level was used as control to standardize Prmt4 mRNA expression.
Wound-healing assays.
MLE12 cells were transfected with a plasmid encoding PRMT4 protein, PRMT shRNA constructs, or an empty vector control, respectively. Cells were grown to 90% confluence in six-well culture plates for 24 h and scratched using a pipette tip to produce a wound. After another 24 h of culture, the wound healing was visualized under light microscopy and the images were acquired with EVOS FL Auto microscope (Life Technologies, Waltham, MA). The recovered area in wound healing was calculated using ImageJ software. Healing speed was normalized by the recovered area in PRMT4 overexpression group in a period of 24 h.
Transwell assay.
MLE12 cell migration was assayed using a Transwell migration kit from Trevigen (Gaithersburg, MD) as described previously (6). Briefly, 50 μl of MLE12 cells (containing 5 × 104 cells) that had been overexpressed or knocked down with PRMT4 were added to the top chamber, and 150 μl of HITES medium without FBS were added to the lower chamber. Low and high concentrations of H2O2 (50 and 100 μM, respectively) were added into the medium of each well. After 18 h of culture, the cells that had migrated into the lower chamber were dissociated with cell dissociation/calcein-acetoxymethyl (calcein-AM) ester and transferred into an assay plate. Relative fluorescence was determined using a fluorescence 96-well microplate reader with 485-nm excitation and 520-nm emission wavelengths. The percentage of cell migration was calculated using migrated cells vs total cells inoculated into the top chamber.
Statistical analysis.
All results were analyzed with one-way ANOVA for multiple groups or a Student’s t-test was used between experimental groups. P < 0.05 was considered to be statistically significant, and P ≤ 0.01 was considered to be highly significant. Data are presented as means ± SD from three independent experiments.
RESULTS
Oxidative stress reduces PRMT4 protein stability in MLE12 cells.
It is reported that oxidative stress differentially deregulates PRMT family members in fibroblast WI-38 cells (23). We investigated the protein stability of PRMT4 under oxidative stress in lung epithelial cells. PRMT4 immunoblotting analyses show that H2O2 decreased PRMT4 protein in a time-dependent manner (Fig. 1, A and B). H2O2 treatment (200 μM) remarkably reduced PRMT4 protein in MLE12 cells at 4 h. Notably, H2O2 downregulates the protein kinase GSK-3β at the protein level as well. To confirm this observation, we conducted a H2O2 concentration course study. PRMT4 and GSK-3β immunoblotting results show that 200 μM H2O2 effectively reduced PRMT4 and GSK-3β protein levels in MLE12 cells (Fig. 1, C and D). These results demonstrate that H2O2 reduces PRMT4 as well as GSK-3β proteins in MLE12 cells.
Fig. 1.

Oxidative stress downregulates protein arginine methyltransferase 4 (PRMT4) protein stability. A: time course study of H2O2 in murine lung epithelial (MLE12) cells. MLE12 cells were treated with 200 μM H2O2 for different time durations. The cell lysates were analyzed with PRMT4 and glycogen synthase kinase 3β (GSK-3β) immunoblotting, and β-actin was used as a loading control. B: the densitometry results of A were plotted. C: concentration course study of H2O2 in MLE12 cells. MLE12 cells were treated with diverse concentration of H2O2 for 4 h. PRMT4, GSK-3β, and β-actin immunoblotting was conducted. D: the densitometry results of C were plotted. E: MLE12 cells were treated with 200 μM H2O2 for 4 h, and total RNA was isolated and analyzed with quantitative RT-PCR. Results are representative of 3 independent experiments. *P < 0.05 or **P ≤ 0.01 vs. corresponding control.
Both altered gene transcription and protein stability attribute to the availability of a protein in living cells. To test if decreased PRMT4 protein level is due to gene transcription, we measured Prmt4 mRNA level in the above treated cells. qRT-PCR results show that Prmt4 mRNA levels were comparable in H2O2 untreated and treated MLE12 cells (Fig. 1E). This observation suggests that H2O2-mediated PRMT4 protein reduction may be a result of altered protein stability.
H2O2 triggers PRMT4 proteasomal degradation.
The rate of turnover, on the one hand, determines the stability of a protein. The proteasome is the major machinery that turns over most of the cellular proteins. We are interested to test if the proteasome is involved in the instability of PRMT4. Immunoblotting results show that protein synthesis inhibitor CHX treatment, as compared with that of H2O2 only (Fig. 2, A and B), accelerated H2O2-induced PRMT4 instability (Fig. 2, C and D). CHX enhanced H2O2-mediated GSK-3β reduction at protein level as well (Fig. 2, C and D). In the presence of 200 μM H2O2, the half-life of PRMT4 is ~1.5 h (Fig. 2, E and F). We then tried to rescue H2O2-induced PRMT4 instability by the reversible proteasome inhibitor MG132. As expected, MG132 successfully rescued H2O2-decreased PRMT4 at the protein level (Fig. 2, G and H). Interestingly, MG132 inhibited H2O2-mediated GSK-3β protein instability too, suggesting that GSK-3β may be an important factor relevant to PRMT4 protein instability. Overall, these observations indicate that oxidative stress decreases PRMT4 protein stability via proteasomal degradation. Furthermore, protein kinase GSK-3β may be important to modulate PRMT4 protein stability under oxidative stress.
Fig. 2.

H2O2-induced PRMT4 instability is via proteasomal degradation. A–F: time and concentration studies of PRMT4 degradation under H2O2. MLE12 cells were treated with H2O2 with a range of concentrations in the absence (A and B) or presence of cycloheximide (CHX) for 4 h (C and D), or cells were treated with CHX for various time durations in the presence of 200 μM H2O2 (E and F). The cell lysates were subjected to PRMT4, GSK-3β, and γ-tubulin immunoblotting analysis. The densitometry results of A, C, and E were plotted, and the half-life of the proteins were calculated (B, D, and F). G: MLE12 cells were treated with diverse concentrations of H2O2 (0, 100, 200, and 300 μM) in the presence of MG132 (20 μM) for 4 h. Cell lysates were subjected to PRMT4, GSK-3β, and γ-tubulin immunoblotting analysis. H: the densitometry results of G were plotted. Results are representative of 3 independent experiments *P < 0.05 or **P ≤ 0.01 vs. corresponding control.
GSK-3β interacts with PRMT4 and catalyzes PRMT4 phosphorylation at T132.
Protein kinase GSK-3β is reported to play an important role in signal transduction under oxidative stress (31, 42). We analyzed the primary sequence of PRMT4 and found that PRMT4 contains a potential GSK-3β phosphorylation site(s), 132-TXXXS. We first assayed if PRMT4 interacts with GSK-3β. Co-IP studies show that GSK-3β was detected in PRMT4 immunoprecipitates (Fig. 3A, top), indicating that GSK-3β associates with PRMT4.
Fig. 3.

GSK-3β interacts with PRMT4 and catalyzes PRMT4 phosphorylation at T132. A: cell lysates were applied with PRMT4 immunoprecipitation (IP) study. PRMT4 precipitates were analyzed with GSK-3β and PRMT4 antibodies subsequently. B and C: GSK-3β in vitro phosphorylation of PRMT4 at T132. Wild-type (WT), T132A, and S136A PRMT4 mutant recombinants were synthesized with TnT expression system and in vitro phosphorylation assay was conducted with enzymatic active GSK-3β. Samples were analyzed with phospho-threonine antibody (B) or phospho-serine antibody (C). The GSK-3β signal in IgG lanes in B and C may be from TnT in vitro protein synthetic system. D: PRMT4 was expressed in GSK-3β-silenced cells. V5 immunoprecipitates were analyzed with phosphor-threonine and V5 immunoblotting. The inputs were analyzed with GSK-3β and β-actin immunoblotting. E: WT and T132A mutant PRMT4 were expressed in MLE12 cells, respectively. V5 immunoprecipitates were analyzed with phospho-threonine. Inputs were analyzed with V5 immunoblotting. F: T132A mutant PRMT4 was coexpressed with GSK-3β in MLE12 cells for 48 h. V5 immunoprecipitates were analyzed with phospho-threonine and V5 immunoblotting, respectively. The inputs were analyzed with GSK-3β and β-actin immunoblotting. Results are representative of 3 independent experiments.
GSK-3β is a protein kinase that catalyzes serine/threonine phosphorylation in various proteins. To investigate if GSK-3β catalyzes PRMT4 phosphorylation at 132-TXXXS, we replaced T132 or S136 with alanine and the synthesized WT and mutant PRMT4 recombinants were used for GSK-3β dependent phosphorylation assay. Results from phospho-threonine immunoblotting indicate that GSK-3β catalyzed WT but not T132A mutant PRMT4 phosphorylation at threonine residue(s) (Fig. 3B). Results from phospho-serine immunoblotting show that GSK-3β catalyzed both WT and S136A mutant PRMT4 phosphorylation (Fig. 3C). These results suggest that T132 but not S136 is the GSK-3β catalyzed phosphorylation site within 132-TXXXS motif. Co-IP results show that PRMT4 phosphorylation was readily detectable via phospho-threonine immunoblotting in MLE12 cells (Fig. 3D, top left). Knockdown of GSK-3β (Fig. 3D, bottom) remarkably reduced PRMT4 phosphorylation (Fig. 3D, top right). Further immunoprecipitation results show that the phosphorylation level of T132A mutant is reduced than that of WT PRMT4 (Fig. 3E, top). Ectopic expression of GSK-3β in MLE12 cells (Fig. 3F, bottom) did not change T132A mutant PRMT4 phosphorylation level (Fig. 3F, top). Overall, these results indicate that GSK-3β catalyzes PRMT4 phosphorylation at T132.
T132 phosphorylation status determines PRMT4 ubiquitination.
Since GSK-3β catalyzed PRMT4 phosphorylation at T132, we attempted to identify if T132 phosphorylation status is relevant to PRMT4 polyubiquitination, a signal for PRMT4 proteasome degradation. Immunoblotting analyses of the V5 immunoprecipitates shows that GSK-3β overexpression reduced the WT but not T132A mutant PRMT4 ubiquitination level (Fig. 4, A and B), suggesting that GSK-3β-catalyzed T132 phosphorylation protects PRMT4 from ubiquitination and proteasomal degradation. We then constructed PRMT4 mutants by replacement of T132 with a cysteine (dephosphorylation mimic) or an aspartic acid (phosphorylation mimic). Results from ubiquitin immunoblotting analyses of the immunoprecipitates show that the T132C mutant was more ubiquitinated as compared with the WT and T132D mutant PRMT4 (Fig. 4, C and D). These results indicate that GSK-3β-catalyzed T132 phosphorylation protects PRMT from ubiquitination. Dephosphorylation of T132 promotes PRMT4 ubiquitination thus triggers PRMT4 proteasomal degradation.
Fig. 4.

T132 phosphorylation status determines PRMT4 ubiquitination. A and B: WT and T132A mutant PRMT4 were coexpressed with GSK-3β for 48 h. V5 immunoprecipitates were analyzed with ubiquitin immunoblotting. C and D: WT, T132C, and T132D mutant PRMT4 were introduced into MLE12 cells for 48 h, respectively. V5 immunoprecipitates were analyzed with ubiquitin and V5 immunoblotting. Results are representative of 3 independent experiments. *P < 0.05 or **P ≤ 0.01 vs. corresponding control.
GSK-3β regulates PRMT4 protein stability.
To further study the role of GSK-3β in PRMT4 protein stability, we observed PRMT4 protein stability in the presence or absence of GSK3β. Immunoblotting results show that PRMT4 was a liable protein with a half-life of ~3.5 h (Fig. 5, A and B). Overexpression of GSK-3β stabilized PRMT4 from degradation in MLE12 cells (Fig. 5, C and D). In contrast, knockdown of GSK-3β by shRNA accelerated PRMT4 degradation (Fig. 5, E–H). These results indicate that GSK-3β mediated phosphorylation stabilizes PRMT4 from degradation.
Fig. 5.

GSK-3β regulates PRMT4 protein stability. Empty vectors (A), GSK-3β expression (C), scramble (E), and shRNA (G) constructs were introduced into MLE12 cells, respectively. Cells were then treated with protein synthesis inhibitor CHX (20 μM) for different time intervals as indicated. PRMT4, GSK-3β, and β-actin immunoblotting was conducted. B, D, F, and H: the densitometry results of A, C, E, and G were plotted, respectively. Results are representative of 3 independent experiments. **P ≤ 0.01 vs. corresponding control.
Since oxidative stress downregulates both PRMT4 and GSK-3β protein levels and GSK-3β-catalyzed phosphorylation determines the protein stability of PRMT4, it is reasonable to hypothesize that H2O2-mediated PRMT4 protein degradation is via GSK-3β. Immunoblotting results show that H2O2 induced PRMT4 degradation in empty vector groups (Fig. 6, A and B). Overexpression of GSK-3β successfully rescued H2O2-mediated decrease of PRMT4 (Fig. 6, C and D). These results indicate that GSK-3β is critical in oxidative stress-mediated PRMT4 degradation.
Fig. 6.

GSK-3β rescues oxidative stress-decreased PRMT4 at protein level. Empty vector (A) or GSK-3β constructs (C) were introduced into MLE12 cells for 48 h. H2O2 was added into the cells at variant amounts as indicated. Cell lysates were immunoblotting analyzed with PRMT4, GSK-3β, and γ-tubulin antibodies, respectively. B and D: the densitometry results of A and C were plotted. C: GSK-3β protein were ectopically expressed in MLE12 cells for 48 h. Results are representative of 3 independent experiments. *P < 0.05 or **P ≤ 0.01 vs. corresponding control.
H2O2-mediated PRMT4 degradation impairs lung epithelial cell migration.
Oxidative stress is one of the major etiological causes for pulmonary diseases. Therefore, we tested if PRMT4 contributes to lung epithelial cell migration under oxidative stress. Lung epithelial progenitor cells including type II cells resided in the distal bronchiole relocate into the alveoli to regenerate and repair the injured type I alveolar cells (1, 25). MLE12 cells, which reserve some features of type II cells, were used in migration assays. Results from wound-healing study show that overexpression of PRMT4 did accelerate wound-healing process (Fig. 7, A and B). Knockdown of PRMT4 by shRNA reduced the process of wound healing as compared with that of control cells (Fig. 7, A and B). The effects of PRMT4 overexpress and knockdown were analyzed with immunoblotting (Fig. 7C). This observation suggests that the cellular level of PRMT4 is relevant to MLE12 cell migration. To confirm this observation, we conducted Transwell assays to determine the role of PRMT4 in lung epithelial cell migration under oxidative stress. Results show that H2O2 treatment inhibited lung epithelial cell migration (Fig. 7D). As predicted, overexpression of PRMT4 successfully rescued H2O2-inhibited cell migration as compared with that of untreated cells. Knockdown of PRMT4 by shRNA showed a tendency of aggravated inhibition of migration, possibly because H2O2 treatment already exhausted PRMT4 in the cells (Fig. 7C). We compared the inhibition effects on migration between a lower concentration (50 μM) and a higher concentration (100 μM) of H2O2 and identified that a low concentration (50 μM) of H2O2 is enough to inhibit cell migration. A lower concentration (50 μM) of H2O2 was enough to reduce PRMT4 and GSK-3β at protein levels in a longer time of treatment (18 h) (Fig. 7, E and F). We further knocked down or ectopically expressed GSK-3β to observe cell migration. Knockdown of GSK-3β impaired cell migration, and ectopic expression of GSK-3β augmented cell migration (Fig. 7, G and H). Ectopic expression of PRMT4 rescued the impaired cell migration by GSK-3β knockdown, indicating that GSK-3β-mediated cell migration is, in part, via PRMT4 signaling. These results indicate that PRMT4 plays an important role in oxidative stress-impaired lung epithelial cell migration that may impede lung epithelial cell repair and regeneration.
Fig. 7.

Oxidative stress inhibits PRMT4-mediated lung epithelial cell migration. A: PRMT4 was ectopically expressed or knocked down with PRMT4-specific shRNA constructs in MLE12 cells for 24 h. Wound-healing assay was conducted as described in materials and methods. Images were acquired with light microscopy. Scar bar = 400 μm. B: the results of A were plotted in a bar graph. C: the cells used in A were analyzed with PRMT4 immunoblotting. D: PRMT4 overexpressed and knockdown cells were treated with 50 or 100 μM H2O2 in Transwells for 18 h. Migrated cells were assayed and the percentage of migration was plotted in a bar graph. E and F: above cells were analyzed with PRMT4 immunoblotting. G and H: MLE12 cells were transfected with, GSK-3β, shRNA against GSK-3β, or PRMT4 constructs as indicated. G and H: immunoblotting analysis (G) and Transwell assay (H) were conducted in the presence of 100 μM H2O2. Results are representative of 3 independent experiments. *P < 0.05 or **P ≤ 0.01 vs. corresponding control.
DISCUSSION
Our central finding in this study is that oxidative stress downregulates the histone and nonhistone protein arginine methylation enzyme PRMT4 at the protein level via an impaired GSK-3β-mediated phosphorylation to impede lung epithelial cell migration (Fig. 8). Pulmonary oxidative stress, including both endogenous and exogenous resources, takes part in the pathogenesis of diverse pulmonary diseases such as asthma, COPD, fibrosis, and lung cancers (2). One of the molecular mechanisms is that pulmonary oxidative stress deregulates histone modification enzymes. Increasing evidence demonstrates that pulmonary oxidative stress deregulates cellular functions at the epigenetic level (38). Here we identified that oxidative stress impedes cell migration through degradation of the protein arginine methylation enzyme PRMT4. H2O2 (200 μM) accelerated PRMT4 for degradation, and the half-life of PRMT4 was shortened from 3.5 to 1.5 h by more than 2.3-fold (Figs. 2 and 5). H2O2 treatment demolishes GSK3β at protein level, which reduces PRMT4 phosphorylation and subsequently accelerates PRMT4 protein degradation. Reduced PRMT4 under oxidative stress impedes on cell migration. Ectopic expression of PRMT4 rescues oxidative stress-depressed cell migration in lung epithelial cells. Type II cells, progenitor cells of type I alveolar cells, relocalize from the distal bronchiole to the alveoli to repair and regenerate alveolar epithelia after injury (1, 25). These observations emphasize that PRMT4 plays an important role in the pathogenesis of oxidative stress-mediated pulmonary diseases.
Fig. 8.
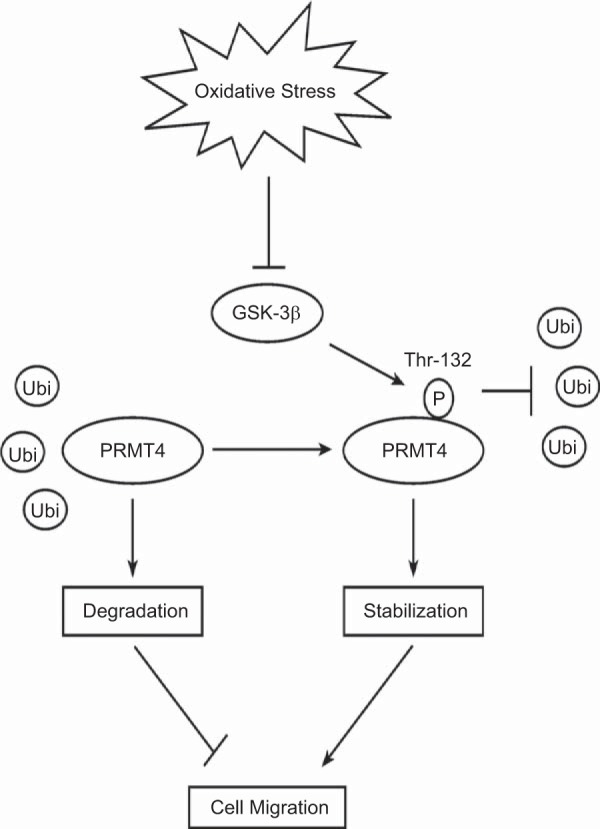
Schematic presentation of oxidative stress modulation of cell migration via GSK-3β/PRMT4 signaling. GSK-3β phosphorylates T132 within PRMT4 to stabilize PRMT4 protein from ubiquitin proteasomal degradation. Oxidative stress downregulates GSK-3β, which leads to PRMT4 degradation. Decreased PRMT4 protein impedes cell migration, which may affect repair and regeneration process after lung injury.
As a histone and nonhistone protein arginine methylation enzyme, PRMT4 functions in various life processes that include, but are not limited to, gene transcriptional activation, proliferation, and inflammation (10, 28, 33). It has been reported that oxidative stress differentially deregulates PRMT family members in a variety of cell types. For example, H2O2 downregulates PRMT1, 4, and 6 proteins to modulate asymmetric dimethylation in fibroblast WI-38 cells (23). H2O2 downregulates PRMT1 and 4 in retinal pigment epithelial cells as well (20). In contrast, high glucose concentration increases PRMT4 to cause cell death in human retinal pigment epithelial cells (21). The molecular mechanism(s) of how oxidative stress differentially regulates PRMT in distinct cell lines remains to be elucidated or uncovered. Here we identified that the protein kinase GSK-3β is critical in regulation of PRMT4 protein stability. GSK-3β associates with PRMT4 to catalyze PRMT4 T132 phosphorylation (Fig. 3). GSK-3β-dependent phosphorylation is essential for maintenance of PRMT4 protein stability. GSK-3β-mediated phosphorylation at T132 prevents PRMT4 from ubiquitination and subsequent degradation.
It is reported that oxidative stress inhibits GSK-3β activity, one way is to phosphorylate GSK-3β to become an inactive form (31, 42). Our observation shows that H2O2 decreases GSK-3β at the protein level in lung epithelial cells. GSK-3β, a key posttranslational modification kinase, is exclusively involved in the regulation of many cellular signal transduction pathways in acute and chronic pulmonary disease. One such role is regulation of protein degradation. Protein destined for degradation is first flagged or labeled with a posttranslational modification to facilitate the subsequent ubiquitination. Such labeling signals include phosphorylation, acetylation, methylation, or other posttranslational modifications. GSK-3β phosphorylates the substrates through the consensus motif S/TXXXS/T, which is also identified as a docking site by many E3 ubiquitin ligases (14, 43). For example, one F-box E3 ubiquitin ligase, SCFFbxw1, specifically recognizes this site to degrade a surfactant synthetic enzyme in lung epithelial cells (45). Interestingly, PRMT4 contains this motif, 132TXXXS, and GSK-3β catalyzes T132 phosphorylation as well. It is possible that PRMT4 ubiquitin proteasomal degradation is via an F-box E3 ubiquitin ligase. Of note, GSK-3β-mediated phosphorylation in PRMT4 stabilizes the protein from ubiquitination, rendering that GSK3β-mediated phosphorylation can also act as an anti-ubiquitination signal.
Ubiquitin proteasome system turns over majority of cellular proteins in living cells. In coordination with gene transcription, ubiquitin proteasomal degradation machinery stringently governs protein availability to assure smooth execution of diverse cellular functions. Histone modification enzymes are protein substrates under stringent surveillance of ubiquitin proteasomal system (48). Deregulated ubiquitin proteasomal degradation of histone modification enzymes has been documented in a vast range of pathophysiological settings including pulmonary diseases (45, 47, 48). One study involves demonstrated deregulation of PRMT4 at the protein level without definition of a specific molecular mechanism (19). Here we add another paradigm that oxidative stress triggers PRMT4 ubiquitination and thereafter proteasomal degradation via impaired GSK-3β signaling. Oxidative stress reduces GSK-3β that impinges on PRMT4 T132 phosphorylation. A PRMT4 dephosphorylation mimic mutant is prone to ubiquitination compared with that of WT and phosphorylation mimic mutant PRMT4. In addition, the reversible proteasome inhibitor MG132 abrogates H2O2-triggered PRMT4 degradation. Protein ubiquitination for proteasomal degradation is an enzymatic cascade involves three ubiquitination enzymes called E1 ubiquitin activation enzyme, E2 ubiquitin conjugation enzyme, and E3 ubiquitin ligase. Among the enzymes, E3 ubiquitin ligase is substrate specific for substrate recognition. The E3 ubiquitin ligase that is specific for PRMT4 ubiquitination has yet to be screened. The studies on the detailed molecular mechanism(s) of how PRMT4 is ubiquitinated and proteasomal degraded are ongoing.
In conclusion, we demonstrate in this study that GSK-3β is critical in the maintenance of PRMT4 protein stability in lung epithelial cells. GSK-3β associates with PRMT4 and promotes PRMT4 phosphorylation at T132. Lack of GSK-3β results in dephosphorylation of PRMT4 at T132 that elevates PRMT4 ubiquitination and proteasomal degradation. Finally, the oxidative reagent H2O2 downregulates GSK-3β protein to destabilize PRMT4 by ubiquitination and proteasome degradation, which impairs lung epithelial cell migration to hinder the process of repair and regeneration after lung injury.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant R01 HL-125435 (to C. Zou).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
C.Z. conceived and designed research; X.L., Y.L., J.L., and M.Z. performed experiments; X.L. and C.Z. analyzed data; X.L. and C.Z. interpreted results of experiments; X.L. and C.Z. prepared figures; X.L. and C.Z. drafted manuscript; Y.L. and C.Z. edited and revised manuscript. X.L., Y.L., J.L., M.Z., and C.Z. approved final version of manuscript.
REFERENCES
- 1.Anversa P, Kajstura J, Leri A, Loscalzo J. Tissue-specific adult stem cells in the human lung. Nat Med 17: 1038–1039, 2011. doi: 10.1038/nm.2463. [DOI] [PubMed] [Google Scholar]
- 2.Bargagli E, Olivieri C, Bennett D, Prasse A, Muller-Quernheim J, Rottoli P. Oxidative stress in the pathogenesis of diffuse lung diseases: a review. Respir Med 103: 1245–1256, 2009. doi: 10.1016/j.rmed.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 3.Batut J, Duboé C, Vandel L. The methyltransferases PRMT4/CARM1 and PRMT5 control differentially myogenesis in zebrafish. PLoS One 6: e25427, 2011. doi: 10.1371/journal.pone.0025427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol Cell 33: 1–13, 2009. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhavsar P, Ahmad T, Adcock IM. The role of histone deacetylases in asthma and allergic diseases. J Allergy Clin Immunol 121: 580–584, 2008. doi: 10.1016/j.jaci.2007.12.1156. [DOI] [PubMed] [Google Scholar]
- 6.Chen W, Xiong S, Li J, Li X, Liu Y, Zou C, Mallampalli RK. The ubiquitin E3 ligase SCF-FBXO24 recognizes deacetylated nucleoside diphosphate kinase A to enhance its degradation. Mol Cell Biol 35: 1001–1013, 2015. doi: 10.1128/MCB.01185-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng D, Côté J, Shaaban S, Bedford MT. The arginine methyltransferase CARM1 regulates the coupling of transcription and mRNA processing. Mol Cell 25: 71–83, 2007. doi: 10.1016/j.molcel.2006.11.019. [DOI] [PubMed] [Google Scholar]
- 8.Cheresh P, Kim SJ, Tulasiram S, Kamp DW. Oxidative stress and pulmonary fibrosis. Biochim Biophys Acta 1832: 1028–1040, 2013. doi: 10.1016/j.bbadis.2012.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chervona Y, Arita A, Costa M. Carcinogenic metals and the epigenome: understanding the effect of nickel, arsenic, and chromium. Metallomics 4: 619–627, 2012. doi: 10.1039/c2mt20033c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Covic M, Hassa PO, Saccani S, Buerki C, Meier NI, Lombardi C, Imhof R, Bedford MT, Natoli G, Hottiger MO. Arginine methyltransferase CARM1 is a promoter-specific regulator of NF-kappaB-dependent gene expression. EMBO J 24: 85–96, 2005. doi: 10.1038/sj.emboj.7600500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El Messaoudi S, Fabbrizio E, Rodriguez C, Chuchana P, Fauquier L, Cheng D, Theillet C, Vandel L, Bedford MT, Sardet C. Coactivator-associated arginine methyltransferase 1 (CARM1) is a positive regulator of the cyclin E1 gene. Proc Natl Acad Sci USA 103: 13351–13356, 2006. doi: 10.1073/pnas.0605692103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellis B, Kaercher L, Snavely C, Zhao Y, Zou C. Lipopolysaccharide triggers nuclear import of Lpcat1 to regulate inducible gene expression in lung epithelia. World J Biol Chem 3: 159–166, 2012. doi: 10.4331/wjbc.v3.i7.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fuchs SY, Spiegelman VS, Kumar KG. The many faces of beta-TrCP E3 ubiquitin ligases: reflections in the magic mirror of cancer. Oncogene 23: 2028–2036, 2004. doi: 10.1038/sj.onc.1207389. [DOI] [PubMed] [Google Scholar]
- 15.Halliwell B, Clement MV, Long LH. Hydrogen peroxide in the human body. FEBS Lett 486: 10–13, 2000. doi: 10.1016/S0014-5793(00)02197-9. [DOI] [PubMed] [Google Scholar]
- 16.Hassa PO, Covic M, Bedford MT, Hottiger MO. Protein arginine methyltransferase 1 coactivates NF-kappaB-dependent gene expression synergistically with CARM1 and PARP1. J Mol Biol 377: 668–678, 2008. doi: 10.1016/j.jmb.2008.01.044. [DOI] [PubMed] [Google Scholar]
- 17.Hwang JW, Chung S, Sundar IK, Yao H, Arunachalam G, McBurney MW, Rahman I. Cigarette smoke-induced autophagy is regulated by SIRT1-PARP-1-dependent mechanism: implication in pathogenesis of COPD. Arch Biochem Biophys 500: 203–209, 2010. doi: 10.1016/j.abb.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ito K, Ito M, Elliott WM, Cosio B, Caramori G, Kon OM, Barczyk A, Hayashi S, Adcock IM, Hogg JC, Barnes PJ. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med 352: 1967–1976, 2005. doi: 10.1056/NEJMoa041892. [DOI] [PubMed] [Google Scholar]
- 19.Kim D, Lim S, Park M, Choi J, Kim J, Han H, Yoon K, Kim K, Lim J, Park S. Ubiquitination-dependent CARM1 degradation facilitates Notch1-mediated podocyte apoptosis in diabetic nephropathy. Cell Signal 26: 1774–1782, 2014. doi: 10.1016/j.cellsig.2014.04.008. [DOI] [PubMed] [Google Scholar]
- 20.Kim DI, Park MJ, Choi JH, Kim IS, Han HJ, Yoon KC, Park SW, Lee MY, Oh KS, Park SH. PRMT1 and PRMT4 regulate oxidative stress-induced retinal pigment epithelial cell damage in SIRT1-dependent and SIRT1-independent manners. Oxid Med Cell Longev 2015: 617919, 2015. doi: 10.1155/2015/617919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim DI, Park MJ, Lim SK, Choi JH, Kim JC, Han HJ, Kundu TK, Park JI, Yoon KC, Park SW, Park JS, Heo YR, Park SH. High-glucose-induced CARM1 expression regulates apoptosis of human retinal pigment epithelial cells via histone 3 arginine 17 dimethylation: role in diabetic retinopathy. Arch Biochem Biophys 560: 36–43, 2014. doi: 10.1016/j.abb.2014.07.021. [DOI] [PubMed] [Google Scholar]
- 22.Lawless MW, O’Byrne KJ, Gray SG. Oxidative stress induced lung cancer and COPD: opportunities for epigenetic therapy. J Cell Mol Med 13, 9A: 2800–2821, 2009. doi: 10.1111/j.1582-4934.2009.00845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lim Y, Lee E, Lee J, Oh S, Kim S. Down-regulation of asymmetric arginine methylation during replicative and H2O2-induced premature senescence in WI-38 human diploid fibroblasts. J Biochem 144: 523–529, 2008. doi: 10.1093/jb/mvn097. [DOI] [PubMed] [Google Scholar]
- 24.Marwick JA, Kirkham PA, Stevenson CS, Danahay H, Giddings J, Butler K, Donaldson K, Macnee W, Rahman I. Cigarette smoke alters chromatin remodeling and induces proinflammatory genes in rat lungs. Am J Respir Cell Mol Biol 31: 633–642, 2004. doi: 10.1165/rcmb.2004-0006OC. [DOI] [PubMed] [Google Scholar]
- 25.McQualter JL, Yuen K, Williams B, Bertoncello I. Evidence of an epithelial stem/progenitor cell hierarchy in the adult mouse lung. Proc Natl Acad Sci USA 107: 1414–1419, 2010. doi: 10.1073/pnas.0909207107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nowak D, Antczak A, Krol M, Pietras T, Shariati B, Bialasiewicz P, Jeczkowski K, Kula P. Increased content of hydrogen peroxide in the expired breath of cigarette smokers. Eur Respir J 9: 652–657, 1996. doi: 10.1183/09031936.96.09040652. [DOI] [PubMed] [Google Scholar]
- 27.O’Brien KB, Alberich-Jordà M, Yadav N, Kocher O, Diruscio A, Ebralidze A, Levantini E, Sng NJ, Bhasin M, Caron T, Kim D, Steidl U, Huang G, Halmos B, Rodig SJ, Bedford MT, Tenen DG, Kobayashi S. CARM1 is required for proper control of proliferation and differentiation of pulmonary epithelial cells. Development 137: 2147–2156, 2010. doi: 10.1242/dev.037150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pal S, Sif S. Interplay between chromatin remodelers and protein arginine methyltransferases. J Cell Physiol 213: 306–315, 2007. doi: 10.1002/jcp.21180. [DOI] [PubMed] [Google Scholar]
- 29.Pandit KV, Milosevic J, Kaminski N. MicroRNAs in idiopathic pulmonary fibrosis. Transl Res 157: 191–199, 2011. doi: 10.1016/j.trsl.2011.01.012. [DOI] [PubMed] [Google Scholar]
- 30.Rajendrasozhan S, Yao H, Rahman I. Current perspectives on role of chromatin modifications and deacetylases in lung inflammation in COPD. COPD 6: 291–297, 2009. doi: 10.1080/15412550903049132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schäfer M, Goodenough S, Moosmann B, Behl C. Inhibition of glycogen synthase kinase 3 beta is involved in the resistance to oxidative stress in neuronal HT22 cells. Brain Res 1005: 84–89, 2004. doi: 10.1016/j.brainres.2004.01.037. [DOI] [PubMed] [Google Scholar]
- 32.Schünemann HJ, Muti P, Freudenheim JL, Armstrong D, Browne R, Klocke RA, Trevisan M. Oxidative stress and lung function. Am J Epidemiol 146: 939–948, 1997. doi: 10.1093/oxfordjournals.aje.a009220. [DOI] [PubMed] [Google Scholar]
- 33.Schurter BT, Koh SS, Chen D, Bunick GJ, Harp JM, Hanson BL, Henschen-Edman A, Mackay DR, Stallcup MR, Aswad DW. Methylation of histone H3 by coactivator-associated arginine methyltransferase 1. Biochemistry 40: 5747–5756, 2001. doi: 10.1021/bi002631b. [DOI] [PubMed] [Google Scholar]
- 34.Scott JA, Duongh M, Young AW, Subbarao P, Gauvreau GM, Grasemann H. Asymmetric dimethylarginine in chronic obstructive pulmonary disease (ADMA in COPD). Int J Mol Sci 15: 6062–6071, 2014. doi: 10.3390/ijms15046062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scott JA, Gauvreau GM, Grasemann H. Asymmetric dimethylarginine and asthma. Eur Respir J 43: 647–648, 2014. doi: 10.1183/09031936.00080313. [DOI] [PubMed] [Google Scholar]
- 36.Scott JA, Grasemann H. Asymmetric dimethylarginine: a disease marker for asthma? Chest 144: 367–368, 2013. doi: 10.1378/chest.13-0480. [DOI] [PubMed] [Google Scholar]
- 37.Sundar IK, Yao H, Rahman I. Oxidative stress and chromatin remodeling in chronic obstructive pulmonary disease and smoking-related diseases. Antioxid Redox Signal 18: 1956–1971, 2013. doi: 10.1089/ars.2012.4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Valavanidis A, Vlachogianni T, Fiotakis K, Loridas S. Pulmonary oxidative stress, inflammation and cancer: respirable particulate matter, fibrous dusts and ozone as major causes of lung carcinogenesis through reactive oxygen species mechanisms. Int J Environ Res Public Health 10: 3886–3907, 2013. doi: 10.3390/ijerph10093886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Eeden SF, Sin DD. Oxidative stress in chronic obstructive pulmonary disease: a lung and systemic process. Can Respir J 20: 27–29, 2013. doi: 10.1155/2013/509130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang L, Zhao Z, Meyer MB, Saha S, Yu M, Guo A, Wisinski KB, Huang W, Cai W, Pike JW, Yuan M, Ahlquist P, Xu W. CARM1 methylates chromatin remodeling factor BAF155 to enhance tumor progression and metastasis. Cancer Cell 25: 21–36, 2014. doi: 10.1016/j.ccr.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xing HY, Cai YQ, Wang XF, Wang LL, Li P, Wang GY, Chen JH. The cytoprotective effect of hyperoside against oxidative stress is mediated by the Nrf2-ARE signaling pathway through GSK-3β inactivation. PLoS One 10: e0145183, 2015. doi: 10.1371/journal.pone.0145183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu C, Kim NG, Gumbiner BM. Regulation of protein stability by GSK3 mediated phosphorylation. Cell Cycle 8: 4032–4039, 2009. doi: 10.4161/cc.8.24.10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zakrzewicz D, Zakrzewicz A, Preissner KT, Markart P, Wygrecka M. Protein arginine methyltransferases (PRMTs): promising targets for the treatment of pulmonary disorders. Int J Mol Sci 13: 12383–12400, 2012. doi: 10.3390/ijms131012383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zou C, Butler PL, Coon TA, Smith RM, Hammen G, Zhao Y, Chen BB, Mallampalli RK. LPS impairs phospholipid synthesis by triggering beta-transducin repeat-containing protein (beta-TrCP)-mediated polyubiquitination and degradation of the surfactant enzyme acyl-CoA:lysophosphatidylcholine acyltransferase I (LPCAT1). J Biol Chem 286: 2719–2727, 2011. doi: 10.1074/jbc.M110.192377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zou C, Chen Y, Smith RM, Snavely C, Li J, Coon TA, Chen BB, Zhao Y, Mallampalli RK. SCF(Fbxw15) mediates histone acetyltransferase binding to origin recognition complex (HBO1) ubiquitin-proteasomal degradation to regulate cell proliferation. J Biol Chem 288: 6306–6316, 2013. doi: 10.1074/jbc.M112.426882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zou C, Li J, Xiong S, Chen Y, Wu Q, Li X, Weathington NM, Han S, Snavely C, Chen BB, Mallampalli RK. Mortality factor 4 like 1 protein mediates epithelial cell death in a mouse model of pneumonia. Sci Transl Med 7: 311ra171, 2015. doi: 10.1126/scitranslmed.aac7793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zou C, Mallampalli RK. Regulation of histone modifying enzymes by the ubiquitin-proteasome system. Biochim Biophys Acta 1843: 694–702, 2014. doi: 10.1016/j.bbamcr.2013.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zou C, Synan MJ, Li J, Xiong S, Manni ML, Liu Y, Chen BB, Zhao Y, Shiva S, Tyurina YY, Jiang J, Lee JS, Das S, Ray A, Ray P, Kagan VE, Mallampalli RK. LPS impairs oxygen utilization in epithelia by triggering degradation of the mitochondrial enzyme Alcat1. J Cell Sci 129: 51–64, 2016. doi: 10.1242/jcs.176701. [DOI] [PMC free article] [PubMed] [Google Scholar]