

Abstract
Bladder outlet obstruction (BOO) triggers inflammation in the bladder through the NLRP3 inflammasome. BOO also activates fibrosis, which is largely responsible for the decompensation of the bladder in the chronic state. Because fibrosis can be driven by inflammation, we have explored a role for NLRP3 (and IL-1β produced by NLRP3) in the activation and progression of BOO-induced fibrosis. Female rats were divided into five groups: 1) control, 2) sham, 3) BOO + vehicle, 4) BOO + the NLRP3 inhibitor glyburide, or 5) BOO + the IL-1β receptor antagonist anakinra. Fibrosis was assessed by Masson’s trichrome stain, collagen secretion via Sirius Red, and protein localization by immunofluorescence. BOO increased collagen production in the bladder, which was blocked by glyburide and anakinra, clearly implicating the NLRP3/IL-1β pathway in fibrosis. The collagen was primarily found in the lamina propria and the smooth muscle, while IL-1 receptor 1 and prolyl 4-hydroylase (an enzyme involved in the intracellular modification of collagen) both localized to the urothelium and the smooth muscle. Lysyl oxidase, the enzyme involved in the final extracellular assembly of mature collagen fibrils, was found to some extent in the lamina propria where its expression was greatly enhanced during BOO. In vitro studies demonstrated isolated urothelial cells from BOO rats secreted substantially more collagen than controls, and collagen expression in control cultures could be directly stimulated by IL-1β. In summary, NLRP3-derived-IL-1β triggers fibrosis during BOO, most likely through an autocrine loop in which IL-1β acts on urothelia to drive collagen production.
Keywords: inflammasomes, fibrosis, urinary bladder, bladder outlet obstruction, inflammation
bladder outlet obstruction (BOO) secondary to benign prostatic hyperplasia (BPH) is a common condition in older men. Indeed, more than 80% of men older than 70 yr have at least some degree of BOO caused by BPH and most of them experience voiding and/or storage symptoms that range from bothersome to debilitating (2, 17, 18, 29, 35). The typical patient presenting in the early stage of the condition will likely complain of classic obstructive symptoms: hesitancy, weak stream, straining, and difficulty emptying the bladder. The majority of men will be started on α-blockers to relax the bladder neck with or without a 5-α-reductase inhibitor to shrink the prostate. Few will opt for surgical deobstruction such as a transurethral resection of the prostate (TURP) at this point due to the potential complications such as impotence and the pharmacotherapy is considered effective in reducing symptoms. Pharmacotherapy, however, does not completely eliminate the obstruction, and therefore, a variety of pathological insults are repeated with each filling and voiding cycle. This includes enhanced stretching of the bladder wall during the filling phase, which reduces blood flow and creates a mild hypoxic environment (12). When the patient attempts to void, abnormally high pressure is experienced as the detrusor works against the obstruction. Finally, the reduction in bladder size following a voiding cycle restores normal blood flow and results in reperfusion of the bladder tissue. Thus a patient with BPH/BOO experiences repeated exposure to transient high pressures, stretch, and mild hypoxia/reperfusion cycles. It is very clear in animal models that BOO brings about an inflammatory state (5, 31, 33), most likely through some combination of pressure, stretch, and mild hypoxia/reperfusion. This inflammation leads to demonstrable changes (such as increased urinary frequency) reflective of the irritative voiding symptoms experienced by patients (such as increased urinary frequency, urgency, and urge incontinence). In humans, these irritative symptoms can be more devastating to quality of life than is usually seen from the initial obstructive symptoms.
When BOO is left untreated or undertreated, two critical events occur that can result in a decompensated bladder that ends in bladder failure. The first is a loss of innervation, and the second is the onset of fibrosis. In these studies, we focus on the events triggering fibrosis in an animal model of BOO. In an organ whose proper function depends on its compliance to cyclically relax in the storage phase and contract in the voiding phase, it is easy to envision how a fibrotic reduction of pliability can make the organ essentially nonfunctional. Understanding the pathway from obstruction to fibrosis using animal models may help identify targets for pharmaceutical intervention and ensure that patients who live with a degree of obstruction never progress to the end stages of bladder dysfunction. Successful correlation of our studies with clinical realities may also explain why surgical treatments such as a transurethral resection of the prostate have poor success rates in rendering late-stage patients symptom free.
There is widespread consensus that inflammation is present in the bladder during BOO where it likely plays a detrimental role. Our laboratory recently demonstrated a central role for the NLRP3 inflammasome in triggering both the inflammation and dysfunctional voiding in a rat model of acute BOO (14). Inflammasomes are sensors of the innate immune system that recognize damage-associated molecular patterns (DAMPS) released from damaged cells or pathogen-associated molecular patterns (PAMPS) associated with invading pathogens. The best studied of the inflammasomes is NLRP3, which recognizes both DAMPS and PAMPS. There are a number of mechanisms by which inflammasomes can be activated (13, 25, 36, 41), but the result is activation of caspase-1, which cleaves pro-IL-1β to IL-1β (and pro-IL-18 to IL-18). These mature cytokines are released through a lytic process known as pyroptosis and function as proinflammatory cytokines to precipitate the inflammatory response.
Working with the knowledge that NLRP3 is expressed in bladder urothelial cells (15) and that glyburide is an in vivo inhibitor of NLRP3 (26), we found that NLRP3 is activated in the acute period of BOO and leads directly to inflammation and bladder hypertrophy (14). Urodynamics demonstrated that inhibition of NLRP3 did not affect the elevated voiding pressure or decreased flow rate seen in the BOO animals, as expected with a persistent physical obstruction. However, irritative voiding parameters (decreased voided volume and increased urinary frequency) were diminished with glyburide, exhibiting the importance of NLRP3 to voiding dysfunction during BOO. Glyburide also allowed a longer duration of voiding, a result we attributed to blocking inflammation in the detrusor muscle, which permits it to maintain a longer, stronger contraction than its inflamed counterpart. However, it is possible that reduced fibrosis, a direct effect of reduced inflammation, contributes to maintaining a more compliant bladder wall. In the present study we examined the possible role of NLRP3 in collagen deposition at the same time point, 12 days after obstruction, where irritative voiding is present. We have utilized glyburide, an NLRP3 inhibitor, and anakinra, an IL-1β receptor antagonist, to determine the importance of NLRP3. We utilized control and BOO urothelial cells in culture to measure collagen secretion and localized enzymes related to collagen synthesis to various cell layers in the bladder.
MATERIALS AND METHODS
Animals, treatments, and surgery.
All animal protocols were approved by the Institutional Animal Care and Use Committee at Duke University Medical Center and performed in accordance with the guidelines in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Studies used age-matched female Sprague-Dawley rats of ≈200 g and 45–50 days old (Harlan, Prattville AL) as in previous literature (14). Female rats are the standard for BOO based on ease of surgery, lack of tortuosity, and short length of the urethra. Animals were randomly divided into five groups: 1) control group: no treatments or surgery; 2) sham-operated group: during surgery the suture encircling the urethra was tied loosely; 3) BOO rats treated with vehicle (1 ml of 40% ethanol); 4) BOO rats treated with glyburide (10 mg/kg in 1 ml 40% ethanol po); or 5) BOO rats treated with anakinra (25 mg/kg ip).
To prepare glyburide, 5 mg/ml was resuspended in 100% ethanol and incubated at 56°C. Once dissolved, it was diluted (2:3) with PBS and immediately administered. We have shown that the resulting dose (10 mg/kg) is effective in suppressing NLRP3 activity in the bladder urothelium in vivo (14, 15). Anakinra (Swedish Orphan Biovitrum, Stockholm, Sweden) was obtained from Duke University Pharmacy and was received as prefilled glass syringes containing 100 mg of anakinra in 0.67 ml of 10 mM citric acid (pH 6.5), 6.5 mM EDTA, 0.8 mM polysorbate 80, and 140 mM NaCl. This solution was diluted 1:6 in PBS to allow sufficient volume for injection into the rat.
Rats undergoing surgery were given medications ≈20–24 h before surgery and again 1–2 h before surgery. Thereafter, medications were administered daily for 11 days with death on day 12.
Surgical anesthesia was provided as ketamine hydrochloride (90 mg/kg) and xylazine (10 mg/kg ip) with preemptive analgesic (carprofen, 5 mg/kg ip). To create BOO, a transurethral catheter (P50 tubing; outer diameter: 1 mm) was inserted and the proximal urethra exposed through a low, vertical midline abdominal incision. A suture (5–0 silk) was tied around the urethra (securely for BOO, loosely for sham). The catheter was removed, and the abdominal wall and skin were closed (5–0 PGA).
Bladder hypertrophy.
At death, bladders were excised, emptied, and weighed.
Histology and collagen quantitation.
Bladders were submersed in 10% neutral buffered formalin and incubated overnight at 4°C. Bladders were transferred to 70% ethanol and stored at 4°C until processed by histology core facilities (either Department of Pathology or Surgery) at Duke University Medical Center. They were paraffin embedded and sectioned in a transverse fashion (5 µm) from the lower third of the bladder and mounted onto slides. Sections were then stained with Masson’s trichrome stain (MTS) using standard techniques. Collagen expression was quantitated in a manner similar to Altuntas et al. (3). Stained slides were imaged at ×10 using Zen software (Carl Zeiss, Oberkochen, Germany) and a Zeiss Axio Imager microscope with motorized stage in the Duke Microscopy Core. For each slide, the entire section was imaged using overlapping micrographs and then automatically stitched into large image files (1–5 GB) by the software. A calibration bar was inserted, and the image was exported as a TIFF file. The TIFF files were imported into NIS-Elements (Nikon, Tokyo, Japan) and calibrated using the calibration bar. With the use of a hue intensity spectrum, a binary layer was created corresponding to the blue shading of collagen staining by MTS and the area of that binary quantitated by the software (representing total collagen area in all the tissue). Likewise, the spectrum defining the binary layer was increased to include all color (except white) and the area of that binary was recorded (representing total area of all the tissue). Next, a region of interest was drawn to exclude any debris of adjacent tissue not obviously part of the bladder as well as the lumen (representing the bladder area). Finally, the total area of collagen inside this region of interest was calculated and divided by the total area of the region of interest. The result was multiplied by 100 to give a measure of percent collagen.
Immunohistochemistry.
Paraffin sections were subjected to immunocytochemistry using standard techniques including antigen retrieval with citrate (pH 6.0). Primary antibodies were mouse anti-rat prolyl 4-hydroxylase (β-subunit) (P4H; 1:50 dilution; Cat. No. MAB2073; EMD Millipore, Temecula, CA), mouse anti-human lysyl oxidase (LOX; 1:50 dilution; Cat. No. SC-373995; Santa Cruz Biotechnology, Santa Cruz, CA), and rabbit anti-human IL-1R (1:50 dilution; Cat. No. SC-393998; Santa Cruz Biotechnology). Secondary antibodies were sheep anti-rabbit IgG-Texas red (1:100 dilution; Cat. No. ab6793; Abcam, Cambridge, MA) and goat anti-mouse IgG-Alexa Flour 488 (1:500 dilution; Cat. No. 1030-30; Southern Biotech, Birmingham, AL).
Urothelial isolation, cell culture, and collagen analysis.
Urothelial cells were isolated using a protocol modified from Kloskoski et al. (24). Briefly, rat bladders were inverted onto an 18-gauge blunt-tip needle fastened to a 5-ml syringe filled with sterile PBS and taped to a dissection board. After inversion, a purse-string suture was placed in the bladder neck and the bladder was inflated with PBS. The suture was then cinched and the bladder slid off the needle. The knot was secured, and the inflated, inverted bladder was placed into 5 ml of collagenase P [1 mg/ml in complete F-12K medium (F-12K medium supplemented with 10% fetal bovine serum, 1 µM nonessential amino acids, 10 µg/ml insulin, 5 µg/ml transferrin, 6.7 ng/ml selenium, 100 U/ml penicillin, 100 µg/ml streptomycin, and 1 µg/ml hydrocortisone)]. The vessel was sealed and shaken at 37°C for 1 h with intense rocking. The bladder was removed and scraped lightly. The scrapings were combined with the solution in the vessel and shaken for an additional 15 min. Thereafter, the cells were pelleted, washed, and counted.
For culture, cells were plated at 50,000 cells/well in 100 µl complete F-12K medium. For studies of control vs. BOO, cells were incubated at 37°C and 5% CO2-95% for 48 h. For studies of IL-1β-stimulated collagen secretion, cells were incubated for 24 h followed by the addition of IL-1β (recombinant rat IL-1β in complete F-12K media; Cat. No. 10788–518; VWR, Radnor, PA) at a final concentration of 250 ng/ml for a further 24 h. Wells containing media only served as controls. Media were collected and frozen at −80°C until thawed and analyzed using the Sircol Collagen Assay (a Sirius Red-based assay; Biocolor) according to the manufacturer’s recommendations.
Statistical analysis.
Assessment of bladder weights and percentage of collagen in tissue sections were performed with a one-way ANOVA followed by a Student-Newman-Keuls post hoc test. Analysis of collagen secretion in vitro was performed with a Student’s t-test. Results were considered significantly different if P < 0.05.
RESULTS
As shown in Fig. 1, bladders from sham-operated rats displayed a small but nonsignificant increase in bladder weight (proxy for inflammation) compared with controls whereas BOO triggered a significant increase (≈3-fold) when animals were treated with vehicle alone. Both glyburide and anakinra prevented the increase in bladder weight to levels not significantly different from control or sham values.
Fig. 1.
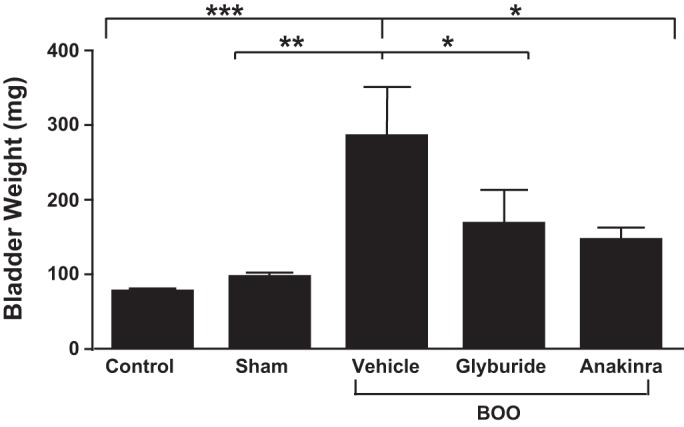
Bladder outlet obstruction (BOO) causes an increase in bladder weight that is blocked by glyburide and anakinra. Rats were obstructed for 12 days and given daily doses of the NLRP3 inhibitor glyburide or the IL-1β antagonist anakinra as described in materials and methods. Control, sham-operated, and BOO rats given vehicle served as controls; after 12 days, animals were killed and bladder wet weights were taken. Results are the means ± SE; n = 6, 8, 5, 5, and 5, respectively. Brackets above the graph indicate group comparisons and their level of significance. All groups were compared using a one-way ANOVA followed by a Student-Newman-Keuls post hoc test. Differences between any two groups not indicated by brackets were not significant. *P < 0.05, **P < 0.01, ***P < 0.005.
Tissue sections from the lower one-third of the bladder were used to calculate the percent of the section that stained positive for collagen. Figure 2A shows a typical cross section from a control bladder stained with MTS, which stains collagen blue. Collagen is primarily deposited in the lamina propria and between the muscle fibrils with little deposition in the urothelial layer. The distribution was similar in the cross sections for all groups (data not shown). For quantitation, the collagen is false colored (yellow in Fig. 2B) and the area quantitated. This is divided by the total area minus adherent tissue (colored purple in Fig. 2C) and multiplied by 100. As shown in Fig. 3, collagen deposition was slightly, but significantly, increased in sham-operated rats but increased further in response to BOO. Both glyburide and anakinra-treated animals had collagen levels not significantly different from controls.
Fig. 2.

Masson’s trichrome stain (MTS) staining and image analysis. A: representative bladder cross section stained with MTS. The entire section has been visualized through a series of overlapping tiles that were stitched together by the Zen software into 1 image. B: same section depicted in A in which a hue spectrum was used to identify the blue-colored collagen. A binary layer, corresponding to the appropriate hue of collagen, was then created on top of the original image and colored yellow. The software (Nikon Elements) can then report the area of the binary. C: similar to B except the hue spectrum was increased to include all color except white and deliver a measurement of total area. (Notice the region of interest was also chosen to exclude superfluous adjacent tissue.)
Fig. 3.
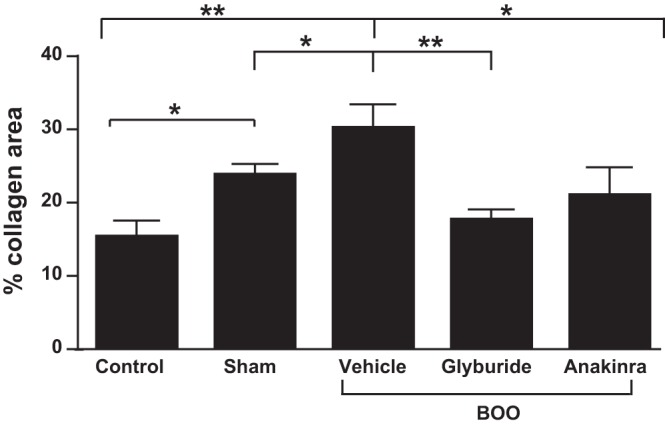
BOO causes an increase in collagen expression in 12 days that is blocked by glyburide and anakinra. The groups are the same used in Fig. 1 and described in materials and methods. Results are the means ± SE; n = 6, 8, 5, 6, and 5, respectively. All groups were compared using a one-way ANOVA followed by a Student-Newman-Keuls post hoc test. Brackets above the graph indicate group comparisons and their level of significance. All groups were compared, and differences between any two groups not indicated by brackets were not significant. *P < 0.05, **P < 0.01.
To determine the source of collagen, sections were stained with an antibody to P4H, an enzyme that modifies collagen before secretion. Thus, unlike collagen, which is synthesized inside cells and then deposited outside, P4H should only be present inside cells producing collagen. As shown in Fig. 4, P4H is expressed in controls primarily in the urothelia and detrusor with little or no expression in the lamina propria. A similar distribution was seen in BOO bladders although the urothelial layer was hyperplastic, consistent with many prior reports of BOO histology.
Fig. 4.
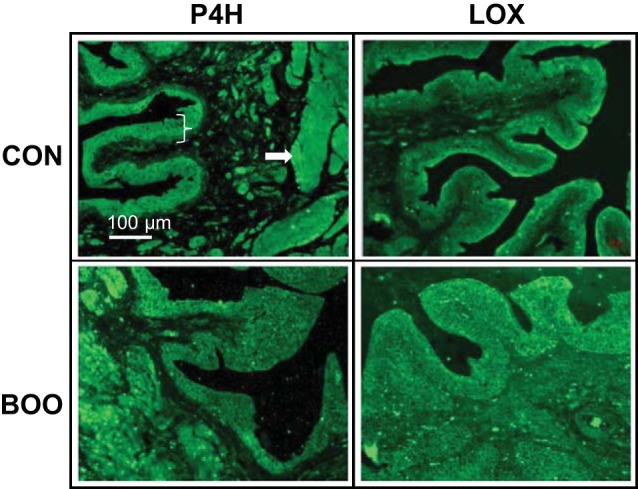
Immunofluorescence staining of bladder sections for prolyl 4-hydroxylase (P4H) and lysyl oxidase (LOX). Tissue sections were stained with antibodies to P4H (which modifies collagen before secretion) or LOX (which cross links mature collagen fibrils in the extracellular environment) (both at 1:50 dilution) as described. Bracket indicates the urothelial layer while the arrow points to the detrusor muscle.
MTS staining (collagen) was primarily in the lamina propria (Fig. 2), although surprisingly a marker of collagen synthesis (P4H) was virtually nonexistent in this layer. To address this apparent discrepancy we stained for LOX, an enzyme that is secreted and cross links collagen to form intact fibrils. As shown in Fig. 4, LOX was expressed in the urothelia (the likely source of transcription/translation) and detrusor (not shown) with low expression in the lamina propria. However, localization to the lamina propria was greatly enhanced in bladders from BOO rats, consistent with enhanced collagen deposition in this layer. It is not clear from these studies if the lamina propria LOX is being produced de novo in this layer or if it has diffused from the urothelial and detrusor layers as the collagen has done.
One outstanding question is the source of enhanced collagen production during BOO. Simple morphological observation (Fig. 4) indicates there are more urothelia cells present during BOO. This result is consistent with long-established results. In addition to more cells available to produce collagen, individual cells may produce more collagen. To assess this possibility, cells from control and BOO rats were cultured and collagen levels in the media analyzed after 48 h. As shown in Fig. 5, urothelial cells from BOO rats produced significantly more collagen than control cells.
Fig. 5.
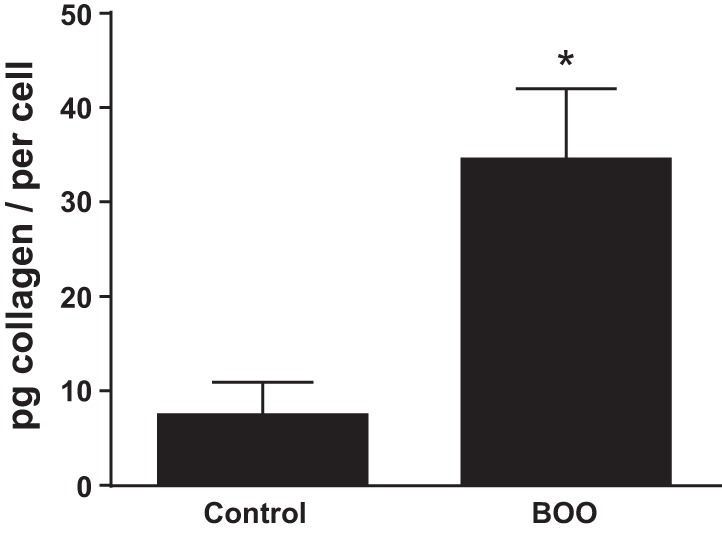
Urothelia from BOO rats secrete more collagen in vitro than cells from control rats. Urothelia were isolated and placed in culture for 48 h. Collagen secreted into the culture media was then quantitated by Sircol Collagen Assay (a Sirius Red-based assay; Biocolor). Results are the means ± SE; n = 3. Differences between groups where assessed by Student’s t-test. *P < 0.05.
We have shown (Fig. 3) that an IL-1 receptor antagonist, anakinra, blocks collagen deposition during BOO, clearly indicating a role for IL-1β in stimulating collagen production. In other cell types, IL-1β can directly stimulate collagen secretion (38). To assess this possibility in the bladder we first stained control bladders for interleukin-1 receptor 1 (IL-1R1). As shown in Fig. 6, IL-1R1 was primarily expressed in the urothelia and detrusor with little or no staining in the lamina propria, an expression pattern reminiscent of P4H. Indeed, when sections were colabeled for P4H and IL-1R1 and the images overlaid (Fig. 6, right), positive cells almost exclusively expressed both molecules. To determine if IL-1β may directly stimulate collagen production in the urothelia, cells from control bladders were cultured overnight and then treated with 250 ng/ml IL-1β for an additional 24 h. As shown in Fig. 7, IL-1β significantly stimulated collagen secretion by isolated urothelial cells.
Fig. 6.

Coimmunofluorescence staining of bladder sections for IL-1R1 and P4H. Tissue sections from bladders were stained with antibodies to IL-1R1 and P4H (both at 1:50 dilution) as described. Secondary antibodies were conjugated to Texas red and Alexa Flour 488 as described. Staining was visualized, and at right is an overlay of the two stains where yellow indicates colocalization. It should be noted that this is the same section used in Fig. 4, left, and the P4H stain is the exact image.
Fig. 7.
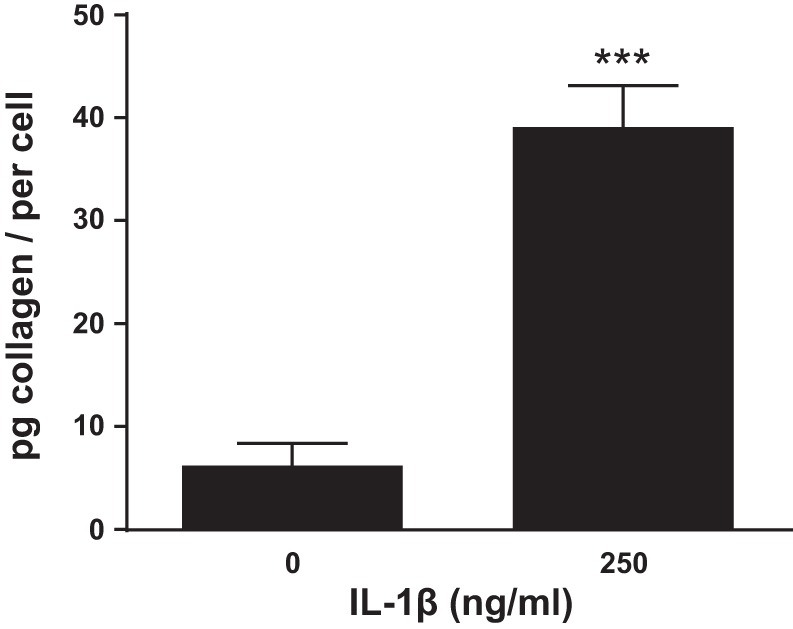
IL-1β stimulates collagen production from control urothelia in vitro. Urothelia were isolated and placed in culture for 24 h and then treated with 250 ng/ml IL-1β for an additional 24 h. Collagen secreted into the culture media was then quantitated by Sircol Collagen Assay (a Sirius Red-based assay; Biocolor). Results are the means ± SE; n = 4. Differences between groups where assessed by Student’s t-test. ***P < 0.005.
DISCUSSION
Inflammation and fibrosis have long been known to be sequelae of BOO, although the mechanisms of their activation and the specific effects that can be directly attributed to them have not been clear. While we have previously identified activation of the NLRP3 inflammasome to be central in the inflammation and irritative voiding produced by BOO (14), herein we focused on the process of fibrosis, as this is responsible for the long-term decrease in bladder compliance and function that leads to irreversible functional decompensation. In this study, we have shown that the NLRP3/IL-1β pathway is critical to the occurrence of fibrosis in the bladder. Moreover, there appears to be an autocrine loop in the urothelia whereby IL-1β produced following the activation of NLRP3 binds the IL-1 receptor on neighboring cells to trigger collagen secretion directly (Fig. 8).
Fig. 8.

Illustration depicting a proposed IL-1β autocrine loop during BOO-induced fibrosis. BOO is associated with repeated bouts of high pressures, increased stretch and mild hypoxia/reperfusion. These insults release damage-associated molecular patterns (DAMPS), which trigger the formation of the NLRP3 inflammasome and the activation of caspase-1 in urothelia. Caspase-1 matures IL-1β, which is released by a lytic process known as pyroptosis. IL-1β can then bind to IL-1R1 on neighboring cells where it triggers collagen synthesis, modification, and ultimately secretion and the formation of mature fibrils. ASC: Apoptosis-Associated Speck-Like Protein Containing CARD.
In previous studies with the same models and with the same treatment groups (with the exception of the anakinra-treated group that was introduced in this current work), we have used more direct indexes of inflammation (Evan’s blue extravasation, histology) (14) rather than simply bladder weight. The judicious use of animals in research dictates that since scientific knowledge would not be gained from repeating these multiple end points, bladder weight was the sole indicator of inflammation in the current study. Likewise, Kanno et al. (23) demonstrated that BOO-induced inflammation and bladder weight gain were decreased during BOO using a mouse in which IL-1β had been knocked out, suggesting that bladder weight is a fair proxy for inflammation in this group as well. However, it is worth noting that hypertrophy of the bladder wall is to be expected during BOO as the bladder works harder to overcome the outflow resistance. Perhaps the muscle hypertrophy explains the consistent (albeit insignificant) increase in weight even in the presence of glyburide (14 and Fig. 1), anakinra (Fig. 1), or knockout of IL-1β (23). Additionally, noninflammasome/IL-1β inflammatory pathways could be involved as well, though this contribution appears to be small.
Although other extracellular matrix proteins can contribute to fibrosis, collagen is by far the most prevalent one in the bladder. Thus it is the primary determinant of compliance and its level of expression correlates to the fibrotic state. While there are at least 16 types of collagen, MTS stains all of them making its use preferable to immunocytochemistry. Our results show that BOO significantly increases the area of MTS staining, which was blocked by both glyburide and anakinra, implicating the role of the NLRP3/IL-1β pathway in collagen deposition and showing an absolute relationship between the inflammasome and fibrosis in the bladder. Such a relationship is far from exclusive to the bladder, since the inflammasome/IL-1β pathway has been implicated in fibrosis in many tissues and disorders including the liver (11), lung (27), kidney (30), and cystic fibrosis (16). In fact, it is estimated that 45% of deaths in the Western world can now be attributed to diseases where fibrosis plays a major etiological role (42).
Not only did our study demonstrate a link between NLRP3 inflammasome and fibrosis, but it clarified the cell types involved in the synthesis/processing of collagen and the location of collagen deposition. Previous studies have found that both urothelia and bladder smooth muscle are sources of collagen (4), and our results confirm that using immunocytochemical staining for P4H. P4H is an enzyme that hydroxylates the proline moieties in procollagen, which promotes the formation of a stable triple helix of procollagen. Thus P4H is believed to be a key enzyme in the regulation of procollagen synthesis (8, 34) and many studies have documented correlations between P4H expression and collagen synthesis in vitro and in vivo (1, 32, 37). Although immunohistochemistry does not allow a quantitative comparison, other studies have shown the P4H is upregulated in the detrusor (9, 28) during BOO and P4H inhibiters improve bladder function and fibrosis in BOO (9). Interestingly, although our MTS staining (Fig. 2) indicates the majority of collagen is found within the lamina propria, our P4H staining was very sparse in this layer (Fig. 4), although not absent.
P4H is a marker of procollagen modification and not an indicator of the formation of mature collagen fibrils, which takes place extracellularly. Thus we sought another marker to help clarify the lamina propria deposition of collagen. LOX catalyzes the final role in collagen modification by oxidizing lysines and hyroxylysines. This eventually results in the formation of covalent cross links that assemble and stabilize the collagen fibrils. Staining for this enzyme was modest in the lamina propria of control rats but strongly increased in the lamina propria from BOO rats, consistent with the observed increased collagen deposition in this space. It should be noted that collagen deposition around smooth muscle fibrils is also likely to play an important role in BOO-induced fibrosis.
The present results with glyburide show the importance of the NLRP3 inflammasome in inducing fibrosis while the result with anakinra suggests it is a direct result of IL-1β and not some other effect or product (i.e., IL-18) of the inflammasome. While we have used glyburide to inhibit the NLRP3 inflammasome in our previous study of BOO (14), to our knowledge anakinra has not been used previously in studies of this disorder. The ability of anakinra to block fibrosis to the same degree as direct inhibition of NLRP3 with glyburide suggests that the fibrotic effects are mediated downstream by IL-1β. IL-1β is known to be profibrogenic in several models (6, 7, 21, 22) and has been shown to directly stimulate collagen (38) and fibronectin (39) production in several cell types. Thus we explored the possibility that IL-1β may mediate collagen production in urothelial cells. We found the receptor for IL-1β, IL-1R1, was present in the urothelial layer and smooth muscle and coexpressed with P4H. Moreover, when urothelia cells from control bladders were stimulated with IL-1β they secreted approximately eightfold more collagen. Thus, as illustrated in Fig. 8, there appears to be an autocrine loop in the urothelia that mediates fibrosis, at least in part. It is known that BOO damages cells, most likely through a combination of increased pressure and stretch as well as repeated bouts of mild hypoxia/reperfusion. This results in the release of DAMPS, which activate NLRP3 to produce IL-1β (14). IL-1β is then released to act at the urothelial surface where it binds to IL-1R1. Binding to the receptor then triggers the release of procollagen. Additional experiments are needed to determine if IL-1β actually stimulates collagen synthesis and/or P4H modification rather than just stimulating release. Most of the procollagen released then diffuses into the lamina propria where it is cross linked through the activity of LOX.
Our simplistic model depicted in Fig. 8 incorporates the findings of this study but does not attempt to assimilate it with other known fibrogenic pathways such as transforming growth factor-β1 (TGF-β1). TGF-β1 is regarded as a major driver of fibrosis in most tissues, including the bladder (19, 20). Not only may it have direct effects on the secretion of extracellular components by the urothelium, it has also been shown to trigger the epithelial-to-mesenchymal transition of porcine urothelial cells into fibroblast-like cells that produce collagen (19). How the NLRP3/IL-1β pathway relates to TGF-β1 is a subject for future studies, but IL-1β can stimulate production of TGF-β1 in a rat hepatic stellate cell line (40) and has been shown to drive the epithelial-to-mesenchymal transition and myofibroblast differentiation via a TGF-β1 dependent mechanism in a rat kidney cell line (10). As such, the answer could be as simple as a second autocrine loop added to Fig. 8 whereby IL-1β stimulates secretion of TGF-1β to act back on its receptor and trigger collagen secretion. The results presented in this paper do not preclude such a scenario. Interestingly, TGF-β mRNA levels were increased in BOO mice while this increase was not apparent in IL-1β KO mice (23).
Therapeutics to target the NLRP3 inflammasome or its downstream products could serve as a useful adjunct to current pharmacotherapeutic regimens where persistent obstruction leads to irreversible fibrotic changes. Primarily, this could carry the benefit of preventing end stage bladder decompensation and possibly renal failure. Secondarily, in patients whose enlarging prostates cause worsening obstructive symptoms, the health of the bladder would be preserved so that de-obstructing procedures would have higher success rates in returning them to a proper state of urinary function.
GRANTS
This research was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant R01-DK-103534 (to J. T. Purves) and intramural funds from Division of Urology, Department of Surgery, Duke University Medical Center.
DISCLOSURES
J. T. Purves reports grants and personal fees from Allergan Inc., the scope of which is outside the submitted work.
AUTHOR CONTRIBUTIONS
F.M.H. and J.T.P. conceived and designed research; F.M.H., S.J.S., H.J., and V.G. performed experiments; F.M.H. and S.J.S. analyzed data; F.M.H., S.J.S., and J.T.P. interpreted results of experiments; F.M.H. prepared figures; F.M.H. drafted manuscript; F.M.H. and S.J.S. edited and revised manuscript; F.M.H., S.J.S., and J.T.P. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Julie Fuller and the Substrate Services Core and Research Support Services in the Department of Surgery as well as the Histology Core in the Department of Pathology, both at Duke University Medical Center, for help with histological embedding, sectioning and staining. We also thank Brian Inouye for critical reading of the manuscript.
REFERENCES
- 1.Abergel RP, Pizzurro D, Meeker CA, Lask G, Matsuoka LY, Minor RR, Chu ML, Uitto J. Biochemical composition of the connective tissue in keloids and analysis of collagen metabolism in keloid fibroblast cultures. J Invest Dermatol 84: 384–390, 1985. doi: 10.1111/1523-1747.ep12265471. [DOI] [PubMed] [Google Scholar]
- 2.Akino H, Gobara M, Okada K. Bladder dysfunction in patients with benign prostatic hyperplasia: relevance of cystometry as prognostic indicator of the outcome after prostatectomy. Int J Urol 3: 441–447, 1996. doi: 10.1111/j.1442-2042.1996.tb00573.x. [DOI] [PubMed] [Google Scholar]
- 3.Altuntas CZ, Daneshgari F, Izgi K, Bicer F, Ozer A, Sakalar C, Grimberg KO, Sayin I, Tuohy VK. Connective tissue and its growth factor CTGF distinguish the morphometric and molecular remodeling of the bladder in a model of neurogenic bladder. Am J Physiol Renal Physiol 303: F1363–F1369, 2012. doi: 10.1152/ajprenal.00273.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baskin L, Howard PS, Macarak E. Effect of physical forces on bladder smooth muscle and urothelium. J Urol 150: 601–607, 1993. doi: 10.1016/S0022-5347(17)35560-X. [DOI] [PubMed] [Google Scholar]
- 5.Bjorling DE, Wang ZY, Bushman W. Models of inflammation of the lower urinary tract. Neurourol Urodyn 30: 673–682, 2011. doi: 10.1002/nau.21078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borthwick LA, Wynn TA, Fisher AJ. Cytokine mediated tissue fibrosis. Biochim Biophys Acta 1832: 1049–1060, 2013. doi: 10.1016/j.bbadis.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bujak M, Frangogiannis NG. The role of IL-1 in the pathogenesis of heart disease. Arch Immunol Ther Exp (Warsz) 57: 165–176, 2009. doi: 10.1007/s00005-009-0024-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cardinale GJ, Udenfriend S. Prolyl hydroxylase. Adv Enzymol Relat Areas Mol Biol 41: 245–300, 1974. [DOI] [PubMed] [Google Scholar]
- 9.Chung JM, Jung MJ, Lee SJ, Lee SD. Effects of prolyl 4-hydroxylase inhibitor on bladder function, bladder hypertrophy and collagen subtypes in a rat model with partial bladder outlet obstruction. Urology 80: 1390.e7–1390.e12, 2012. doi: 10.1016/j.urology.2012.07.019. [DOI] [PubMed] [Google Scholar]
- 10.Fan JM, Huang XR, Ng YY, Nikolic-Paterson DJ, Mu W, Atkins RC, Lan HY. Interleukin-1 induces tubular epithelial-myofibroblast transdifferentiation through a transforming growth factor-beta1-dependent mechanism in vitro. Am J Kidney Dis 37: 820–831, 2001. doi: 10.1016/S0272-6386(01)80132-3. [DOI] [PubMed] [Google Scholar]
- 11.Gieling RG, Wallace K, Han YP. Interleukin-1 participates in the progression from liver injury to fibrosis. Am J Physiol Gastrointest Liver Physiol 296: G1324–G1331, 2009. doi: 10.1152/ajpgi.90564.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greenland JE, Brading AF. Urinary bladder blood flow changes during the micturition cycle in a conscious pig model. J Urol 156: 1858–1861, 1996. doi: 10.1016/S0022-5347(01)65553-8. [DOI] [PubMed] [Google Scholar]
- 13.Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 21: 677–687, 2015. doi: 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hughes FM Jr, Hill HM, Wood CM, Edmondson AT, Dumas A, Foo WC, Oelsen JM, Rac G, Purves JT. The NLRP3 inflammasome mediates inflammation produced by bladder outlet obstruction. J Urol 195: 1598–1605, 2016. doi: 10.1016/j.juro.2015.12.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hughes FM Jr, Vivar NP, Kennis JG, Pratt-Thomas JD, Lowe DW, Shaner BE, Nietert PJ, Spruill LS, Purves JT. Inflammasomes are important mediators of cyclophosphamide-induced bladder inflammation. Am J Physiol Renal Physiol 306: F299–F308, 2014. doi: 10.1152/ajprenal.00297.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iannitti RG, Napolioni V, Oikonomou V, De Luca A, Galosi C, Pariano M, Massi-Benedetti C, Borghi M, Puccetti M, Lucidi V, Colombo C, Fiscarelli E, Lass-Flörl C, Majo F, Cariani L, Russo M, Porcaro L, Ricciotti G, Ellemunter H, Ratclif L, De Benedictis FM, Talesa VN, Dinarello CA, van de Veerdonk FL, Romani L. IL-1 receptor antagonist ameliorates inflammasome-dependent inflammation in murine and human cystic fibrosis. Nat Commun 7: 10791, 2016. doi: 10.1038/ncomms10791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Irwin DE, Milsom I, Hunskaar S, Reilly K, Kopp Z, Herschorn S, Coyne K, Kelleher C, Hampel C, Artibani W, Abrams P. Population-based survey of urinary incontinence, overactive bladder, and other lower urinary tract symptoms in five countries: results of the EPIC study. Eur Urol 50: 1306–1314, 2006. doi: 10.1016/j.eururo.2006.09.019. [DOI] [PubMed] [Google Scholar]
- 18.Irwin DE, Milsom I, Kopp Z, Abrams P, Artibani W, Herschorn S. Prevalence, severity, and symptom bother of lower urinary tract symptoms among men in the EPIC study: impact of overactive bladder. Eur Urol 56: 14–20, 2009. doi: 10.1016/j.eururo.2009.02.026. [DOI] [PubMed] [Google Scholar]
- 19.Islam SS, Mokhtari RB, El Hout Y, Azadi MA, Alauddin M, Yeger H, Farhat WA. TGF-β1 induces EMT reprogramming of porcine bladder urothelial cells into collagen producing fibroblasts-like cells in a Smad2/Smad3-dependent manner. J Cell Commun Signal 8: 39–58, 2014. doi: 10.1007/s12079-013-0216-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang X, Chen Y, Zhu H, Wang B, Qu P, Chen R, Sun X. Sodium tanshinone IIA sulfonate ameliorates bladder fibrosis in a rat model of partial bladder outlet obstruction by inhibiting the TGF-β/Smad pathway activation. PLoS One 10: e0129655, 2015. doi: 10.1371/journal.pone.0129655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones LK, O’Sullivan KM, Semple T, Kuligowski MP, Fukami K, Ma FY, Nikolic-Paterson DJ, Holdsworth SR, Kitching AR. IL-1RI deficiency ameliorates early experimental renal interstitial fibrosis. Nephrol Dial Transplant 24: 3024–3032, 2009. doi: 10.1093/ndt/gfp214. [DOI] [PubMed] [Google Scholar]
- 22.Kamari Y, Shaish A, Vax E, Shemesh S, Kandel-Kfir M, Arbel Y, Olteanu S, Barshack I, Dotan S, Voronov E, Dinarello CA, Apte RN, Harats D. Lack of interleukin-1α or interleukin-1β inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J Hepatol 55: 1086–1094, 2011. doi: 10.1016/j.jhep.2011.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanno Y, Mitsui T, Kitta T, Moriya K, Tsukiyama T, Hatakeyama S, Nonomura K. The inflammatory cytokine IL-1β is involved in bladder remodeling after bladder outlet obstruction in mice. Neurourol Urodyn 35: 377–381, 2016. doi: 10.1002/nau.22721. [DOI] [PubMed] [Google Scholar]
- 24.Kloskowski T, Uzarska M, Gurtowska N, Olkowska J, Joachimiak R, Bajek A, Gagat M, Grzanka A, Bodnar M, Marszałek A, Drewa T. How to isolate urothelial cells? Comparison of four different methods and literature review. Hum Cell 27: 85–93, 2014. doi: 10.1007/s13577-013-0070-y. [DOI] [PubMed] [Google Scholar]
- 25.Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell 157: 1013–1022, 2014. doi: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 26.Lamkanfi M, Mueller JL, Vitari AC, Misaghi S, Fedorova A, Deshayes K, Lee WP, Hoffman HM, Dixit VM. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol 187: 61–70, 2009. doi: 10.1083/jcb.200903124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lasithiotaki I, Giannarakis I, Tsitoura E, Samara KD, Margaritopoulos GA, Choulaki C, Vasarmidi E, Tzanakis N, Voloudaki A, Sidiropoulos P, Siafakas NM, Antoniou KM. NLRP3 inflammasome expression in idiopathic pulmonary fibrosis and rheumatoid lung. Eur Respir J 47: 910–918, 2016. doi: 10.1183/13993003.00564-2015. [DOI] [PubMed] [Google Scholar]
- 28.Lee SD, Akbal C, Miseeri R, Jung C, Rink R, Kaefer M. Collagen prolyl 4-hydroxylase is up-regulated in an acute bladder outlet obstruction. J Pediatr Urol 2: 225–232, 2006. doi: 10.1016/j.jpurol.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 29.Levin RM, Yu HJ, Kim KB, Longhurst PA, Wein AJ, Damaser MS. Etiology of bladder dysfunction secondary to partial outlet obstruction. Calcium disregulation in bladder power generation and the ability to perform work. Scand J Urol Nephrol Suppl 184: 43–50, 1997. [PubMed] [Google Scholar]
- 30.Masood H, Che R, Zhang A. Inflammasomes in the pathophysiology of kidney diseases. Kidney Dis (Basel) 1: 187–193, 2015. doi: 10.1159/000438843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Metcalfe PD, Wang J, Jiao H, Huang Y, Hori K, Moore RB, Tredget EE. Bladder outlet obstruction: progression from inflammation to fibrosis. BJU Int 106: 1686–1694, 2010. doi: 10.1111/j.1464-410X.2010.09445.x. [DOI] [PubMed] [Google Scholar]
- 32.Mio K, Yamashita M, Odake Y, Tamai H, Takada K. Coenzyme A stimulates collagen production in cultured fibroblasts; possible mechanisms in enzymatic and gene expression. Arch Dermatol Res 293: 522–531, 2001. doi: 10.1007/PL00007467. [DOI] [PubMed] [Google Scholar]
- 33.Oka M, Fukui T, Ueda M, Tagaya M, Oyama T, Tanaka M. Suppression of bladder oxidative stress and inflammation by a phytotherapeutic agent in a rat model of partial bladder outlet obstruction. J Urol 182: 382–390, 2009. doi: 10.1016/j.juro.2009.02.104. [DOI] [PubMed] [Google Scholar]
- 34.Pihlajaniemi T, Myllylä R, Kivirikko KI. Prolyl 4-hydroxylase and its role in collagen synthesis. J Hepatol 13, Suppl 3: S2–S7, 1991. doi: 10.1016/0168-8278(91)90002-S. [DOI] [PubMed] [Google Scholar]
- 35.Platz EA, Smit E, Curhan GC, Nyberg LM Jr, Giovannucci E. Prevalence of and racial/ethnic variation in lower urinary tract symptoms and noncancer prostate surgery in U.S. men. Urology 59: 877–883, 2002. doi: 10.1016/S0090-4295(01)01673-9. [DOI] [PubMed] [Google Scholar]
- 36.Sutterwala FS, Haasken S, Cassel SL. Mechanism of NLRP3 inflammasome activation. Ann N Y Acad Sci 1319: 82–95, 2014. doi: 10.1111/nyas.12458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takahashi Y, Takahashi S, Shiga Y, Yoshimi T, Miura T. Hypoxic induction of prolyl 4-hydroxylase alpha (I) in cultured cells. J Biol Chem 275: 14139–14146, 2000. doi: 10.1074/jbc.275.19.14139. [DOI] [PubMed] [Google Scholar]
- 38.Tiggelman AM, Boers W, Linthorst C, Sala M, Chamuleau RA. Collagen synthesis by human liver (myo)fibroblasts in culture: evidence for a regulatory role of IL-1 beta, IL-4, TGF beta and IFN gamma. J Hepatol 23: 307–317, 1995. [PubMed] [Google Scholar]
- 39.Vesey DA, Cheung C, Cuttle L, Endre Z, Gobe G, Johnson DW. Interleukin-1beta stimulates human renal fibroblast proliferation and matrix protein production by means of a transforming growth factor-beta-dependent mechanism. J Lab Clin Med 140: 342–350, 2002. doi: 10.1067/mlc.2002.128468. [DOI] [PubMed] [Google Scholar]
- 40.Wang H, Liu S, Wang Y, Chang B, Wang B. Nod-like receptor protein 3 inflammasome activation by Escherichia coli RNA induces transforming growth factor beta 1 secretion in hepatic stellate cells. Bosn J Basic Med Sci 16: 126–131, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wen H, Miao EA, Ting JP. Mechanisms of NOD-like receptor-associated inflammasome activation. Immunity 39: 432–441, 2013. doi: 10.1016/j.immuni.2013.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol 4: 583–594, 2004. doi: 10.1038/nri1412. [DOI] [PMC free article] [PubMed] [Google Scholar]