

Abstract
The OK cell line derived from the kidney of a female opossum Didelphis virginiana has proven to be a useful model in which to investigate the unique regulation of ion transport and membrane trafficking mechanisms in the proximal tubule (PT). Sequence data and comparison of the transcriptome of this cell line to eutherian mammal PTs would further broaden the utility of this culture model. However, the genomic sequence for D. virginiana is not available and although a draft genome sequence for the opossum Monodelphis domestica (sequenced in 2012 by the Broad Institute) exists, transcripts sequenced from both species show significant divergence. The M. domestica sequence is not highly annotated, and the majority of transcripts are predicted rather than experimentally validated. Using deep RNA sequencing of the D. virginiana OK cell line, we characterized its transcriptome via de novo transcriptome assembly and alignment to the M. domestica genome. The quality of the de novo assembled transcriptome was assessed by the extent of homology to sequences in nucleotide and protein databases. Gene expression levels in the OK cell line, from both the de novo transcriptome and genes aligned to the M. domestica genome, were compared with publicly available rat kidney nephron segment expression data. Our studies demonstrate the expression in OK cells of numerous PT-specific ion transporters and other key proteins relevant for rodent and human PT function. Additionally, the sequence and expression data reported here provide an important resource for genetic manipulation and other studies on PT cell function using these cells.
Keywords: proximal tubule, opossum, endocytosis, ion transport, transcriptome
ok cells are a spontaneously immortalized cell line derived in 1975 from kidney tissue harvested from a female American opossum Didelphis virginiana (16). While they were originally prepared for studies on the mechanism of X chromosome inactivation, further characterization revealed that OK cells represent an excellent cell culture model in which to investigate proximal tubule (PT) ion transport and membrane trafficking (8, 14, 15, 23). OK cells retain many morphological and functional characteristics of the PT, including a well-differentiated brush border, formation of an epithelial monolayer readily permeable to ions and water, expression of the multiligand receptors megalin and cubilin (5, 39), and high apical endocytic capacity relative to other kidney cell lines (25). In contrast, many of the available kidney cell lines of human and mouse origin lack essential features of the PT, thus limiting their utility as a model system (12, 24, 25). While OK cells (and other PT cell culture models) also exhibit some features typically ascribed to other nephron segments [e.g., a cAMP response to AVP (31, 34) and possible expression of NKCC2 (38)], studies in OK cells have contributed significantly to our understanding of how PT function is regulated and maintained. In particular, this cell line has provided valuable insight into our understanding of transcriptional control of PT cell polarization and differentiation (21), the regulation of PT phosphate reabsorption (4, 23), and the mechanistic basis of cisplatin and aminoglycoside nephrotoxicity (3, 32).
Recent advances in methods to alter protein expression through siRNA and gene editing have expanded the range of experimental approaches to study PT function. However, extending such techniques to OK cells is hampered by the poor characterization of this cell line at the genomic level. Additionally, generating antibodies against opossum proteins remains challenging due to the lack of sequence information. For these reasons, we elected to assemble the OK transcriptome.
Marsupials (metatherians) and placental mammals diverged at least 170 million years ago. The genomic sequence of the South American opossum species Monodelphis domestica (MonDom) was reported in 2007 (22, 29). The existing MonDom genome and transcriptome structure predicts 18,000–20,000 genes. Of these, nearly all have eutherian orthologs and only eight predicted reading frames are thought to encode functional genes without human homologs (22). Since the MonDom reference genome has not been updated and comparison of several mRNAs sequenced from both species showed 3-11% divergence, we created an OK cell line RNA Seq de novo transcriptome assembly. Simultaneously, we also aligned the RNA Seq reads to the M. domesticus genome. These studies yielded gene expression data that enabled comparisons of OK transcripts with recently reported human and rat PT transcriptomes (11, 19).
MATERIALS AND METHODS
Cell culture and sample preparation.
Opossum kidney cells were obtained at passage 30 from Moshe Levi (University of Colorado Health Center). Cells were cultured in DMEM/F12 medium with 10% FBS (Atlanta Biologicals) and 5 mM GlutaMAX (GIBCO). Then, 4 × 105 cells were plated on 12-mm Transwells with 0.4-µm pore polycarbonate membrane inserts (Corning) in a 12-well plate, with 0.5 ml apical medium and 1.0 ml basolateral medium. Cells were collected after five days using Accutase (Sigma), and RNA was extracted using the Ambion PureLink RNA mini kit (ThermoFisher) according to the manufacturer’s protocol. A total of 1 µg of RNA was sent to the University of Pittsburgh Genomics Core for sequencing and library preparation. Library preparation was performed using the TruSEQ Stranded Total RNA Sample Preparation Kit (Illumina, San Diego, CA) according to the manufacturer’s instructions. Following removal of ribosomal RNA, the remaining RNA was fragmented for 8 min, followed by reverse transcription performed. Double-stranded cDNA was subjected to 3′ adenylation and ligation of sequencing adapters. Sequencing was carried out on a NextSeq 500 (Illumina) to generate 75-bp paired-end reads. The loading concentration was 1.6 pM.
Data preprocessing.
Sequencing generated 212,077,093 pairs of reads. Among these, 60 million read pairs were randomly selected using home grown Perl and Shell scripts (6). FASTQC was run on reads data to examine the quality of reads. Reads were trimmed to remove the adaptor sequence and low-quality reads. To minimize the effect of low-quality reads on data analysis, both full and random samples were trimmed at quality setting 13, with random data further trimmed at qualities 10, 15, 20, 25, 30, and 35. Cutadapt v.1.8.3 software was used for adapter and quality trimming. The sequences used for adapter trimming were AGATCGGAAGAGCACACGTCTGAACTCCAGTCA (left reads) and AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT (right reads).
De novo transcriptome assembly.
De novo transcriptome assembly was performed on 60 million randomly selected reads trimmed at base qualities 13, 10, 15, 20, 25, 30, and 35, using Trinity v2.1.1 with strand-specific option (9). The quality of the assembly was assessed using the following metrics: number of genes and transcripts assembled, length of the assemblies, and percent guanine-cytosine. The positive control used for assessing the assembly quality was a mouse paired-end sample (size: 50 million pairs), obtained from a previously published study (10). The above metrics were calculated using the Trinity assembly statistics module. The mouse data were also assembled with K3 to K10. The quality of the assemblies was assessed using the Trinity assembly statistics module, and assemblies at base-quality filter 30 and coverage K3 and K10 were selected for further analysis.
BLAST alignments.
To annotate the assembled transcripts and further analyze their homology to sequences in transcript and protein databases, assemblies were aligned to RefSeq and Swiss-Prot using BLAST v2.2.31 (1). BLASTn (MegaBLAST) was used for nucleotide alignment and BLASTX for protein. For MegaBLAST, word size 16 was selected. Masking of regions in query sequences were avoided with selection –dust no in MegaBLAST and –seg no BLASTX. First best alignment was selected using max_target_seqs 1. BLAST hits to mammalian sequences and e-value ≤1e−4 were considered. The %Transcript coverage and %Transcript shared were calculated for each alignment and were used for further annotations. One output consisted of %Transcript shared >80 and the other, %Transcript covered >40 and %Identity >70. %Transcript shared >80 gave a list of nearly complete assemblies of database transcripts, whereas %Transcript covered >40 and %Identity >70 provided a border list of assemblies, some of which are incomplete assemblies of database transcripts. Blast statistics were calculated by noting the number of contigs with hits to mammals, especially to the closely related genome of M. domestica or to the well-curated genomes of Homo sapiens and Mus musculus.
Assembly of partial and full-length transcripts.
Trinity script analyze_blastPlus_topHit_coverage.pl was used to calculate the percent target hit length, which represents the percentage of the database hit that the assembled contig covers in its alignment. These covered bases can also be mismatches in the alignment. To take care of any discontinuous alignments, Trinity script blast_outfmt6_group_segments.pl was used to combine multiple high-scoring pairs (HSPs). Subsequently, blast_outfmt6_group_segments.tophit_coverage.pl was used to determine the full coverage of the database sequence over all these HSPs and generate histograms. The histograms visualize the overall coverage of the contigs in the assembly, which is useful in determining which Trinity assembly produces the highest number of full-length transcripts. In addition to the percent database transcript coverage calculated using Trinity, the percent transcript bases/amino acid shared (%Transcript shared) was also calculated. This removes the mismatched bases from the calculation to truly represent the %Transcript that each assembled sequence contains. A cutoff of 80% was used to filter for high-quality assemblies.
Mapping to reference M. domestica.
In parallel to the de novo assembly, a trimmed sample of 60 million randomly selected reads was mapped to the reference genome of M. domestica (MonDom5) using the spliced alignment tool TopHat v2.1.0 (13). The reference genome and transcript annotation gtf were downloaded from NCBI RefSeq. Bowtie2 v2.2.7 (17), and TopHat reference indexes were created for NCBI RefSeq annotations. TopHat mapping of reads was calculated on indexes with the settings: –library-type fr-firststrand, mismatch 2, 4, 6, 8, 10, and –read-edit-dist with the same value as mismatch. Alignment at mismatch setting 6 was selected for downstream analysis.
Quantifying transcripts.
Trinity de novo assembled transcripts and alignment Cufflinks v2.2.1 (36) (Cuffmerge) assembled transcripts were quantified for their expression using RSEM 1.2.21 (20). The transcript expression for de novo assembled transcripts was calculated using the Trinity RSEM module align_and_estimate_abundance.pl with the options –est_method RSEM, aln_method bowtie. The gene annotations from BLASTX were later included in the assembled transcript expression output file. Quantification was completed after filtering the complete Trinity assembly for high-quality contigs (>80% amino acids shared with top BLASTx alignment) and also on the canonical set of transcripts, (≥70% identity and 40% database transcript sequence coverage). For Cufflinks (Cuffmerge) assembled transcripts, reference FASTA sequences were provided to RSEM along with the 60 million randomly selected trimmed reads used in the TopHat alignment. Here also aln_method bowtie was used in RSEM. Fragments per kilobase per million (FPKM) values for de novo assembly and alignment methods generated by RSEM were compared with expression data from microdissected rat kidney nephron segments (19).
Hierarchical clustering analysis.
Gene lists to assess whether OK cells express kidney-specific genes and to assess RNA expression patterns relative to the individual nephron segments were obtained from published studies on kidney transcriptomes (11, 19). NCBI Gene was used to obtain gene name orthologs among H. sapiens, Rattus norvegicus, and M. domestica. If no known ortholog was found in R. norvegicus or M. domestica, the gene was excluded from analysis. FPKM values from de novo assembly and alignment were averaged to compensate for differences in quantification between the two assemblies. Any gene with no expression data in both assembly and alignment was removed from analysis. The averaged assembly FPKM was compared with RPKM values from R. norvegicus data (Ref. 19; https://helixweb.nih.gov/ESBL/Database/NephronRNAseq/index.html). FPKM and RPKM values were log2-transformed and then clustered via Euclidean distance using the complete linkage method. Clustering analysis was completed using TIGR Multiple Experiment Viewer (MeV) version 4.6 (28). Because MeV does not log2-transform samples with a 0 expression value, we set the lower color gradient limit to 0.0 to avoid confusion with negative log2-transformed values.
Data set availability.
Sequencing files and assembled sequences from the K3 de novo assembly and alignment to the reference MonDom genome have been deposited in GEO (GSE94443; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE94443).
RESULTS
De novo assembly of the opossum kidney OK cell transcriptome.
RNA was prepared from OK cells cultured on permeable supports to elicit cell differentiation. Under these conditions, OK cells form a polarized monolayer with abundant apical microvilli and primary cilia (Fig. 1). To characterize the OK transcriptome, we used RNA Seq to generate 212 million paired 75-bp reads. Sixty million read pairs were randomly selected from the data and filtered for adapter and base quality. The reads were then assembled, using Trinity v2.1.1 at different kmer coverage settings, followed by different base-quality filters (Supplemental Table S1; supplemental material for this article is available online at the journal website). A mouse de novo assembly was performed in parallel, and the opossum assembly was assessed in comparison to the mouse assembly, which is well characterized (10). The parameters of these Trinity-generated assemblies are shown in Fig. 2. We used a base-quality filter of 30 since higher levels reduced the number of reads and bases available for assembly (Fig. 2A). At this base quality, the guanine-cytosine content plateaus at kmer coverage 3 (K3) (Fig. 2B); with increasing kmer coverage, the number of transcripts decreases from 583,397 at K1 to 59,517 at K10 (Fig. 2C), and contig length increases from N50 of 512 at K1 to 2156 at K10 (Fig. 2D). This indicates that as kmer coverage increases, contig length increases, the number of genes assembled is in a range expected for a higher eukaryotic genome, and the overall assembly resembles the mouse assembly. Both K3 and K10 were selected for subsequent steps in our analysis. For downstream analysis and gene annotation, we modified published methodologies for transcriptome annotations (18) as described in the schematic in Fig. 3.
Fig. 1.

Opossum kidney OK cells form a differentiated monolayer with microvilli and primary cilia characteristic of the proximal tubule (PT). OK cells cultured on permeable supports were fixed, processed for fluorescence staining using rhodamine phalloidin and anti-acetylated tubulin antibody to visualize microvilli (actin) and primary cilia, respectively, and imaged by confocal microscopy. Scale bar = 10 µm.
Fig. 2.

Parameters of sequence assembly. A: reads used for de novo assembly were trimmed using different base-quality score filters and the percent reads and bases remaining with different cutoffs are shown. Trinity was used to construct the transcriptome of Didelphis virginiana using reads trimmed with quality score 30 and a mouse transcriptome as a positive control from Haas et al. (10). Statistics for both assemblies, including the percent guanine-cytosine (%GC) content (B), the number of genes and transcripts reported by Trinity (C), and the N50 of contigs (D) are plotted.
Fig. 3.

Pipeline for transcriptome assembly and analysis. A: single tissue samples were sequenced using NextGen sequencing technology to generate >200 million read pairs. B: 60 million randomly selected pairs were extracted and trimmed for adaptors and bases with quality score <30. C: reads were assembled using 2 methods: Trinity de novo assembly and alignment to the Monodelphis domestica NCBI reference genome (GCF_000002295.2 MonDom5) using TopHat and Cufflinks in the Tuxedo Suite. We tested parameters to determine which provides the most complete de novo assembly. D: de novo assembled contigs were aligned to the nucleotide RefSeq RNA database and to the protein Swiss-Prot database using BLASTn and BLASTx, respectively. The BLAST output against these databases was used to filter contigs that matched to mammals with an e-value <1e−4 and to annotate contigs with gene names. Using the Trinity provided module, we calculated the percent coverage of the top database match for each contig. E: from all assembled contigs, we utilized the various statistics to filter our assemblies, one whose amino acids matches identically to greater than or equal to 80% of their top database hit, and a second less stringent filter of contigs with at %Identity >70% and percent_hit_length >40% to include high-quality but partial transcript assemblies. F: the contigs in the filtered de novo assembly and the alignment to MonDom5 were quantified at the isoform and gene level using RSEM to generate counts, transcripts per million (TPM), and fragments per kilobase per million (FPKM). With the use of FPKM, the expression patterns were compared with published studies of rat kidney tubule expression.
Transcript annotation.
The contigs produced from K3 and K10 were aligned by BLASTn to the RefSeq RNA nucleotide databases. At K3, 52% of the contigs matched RefSeq nucleotide sequences, of which 88.5% were mammalian, 84% specifically from M. domestica (Fig. 4A). At K10, 76% of K10 contigs matched Refseq, of which 91% matched mammalian sequences, 88% specifically from M. domestica (Fig. 4B). For the matches to M. domestica, only 1% of the contigs in both assemblies aligned to validated sequences while the remainder aligned to predicted sequences. This is not surprising as majority of the M. domestica sequences are predicted. After we applied filters of e-value <1e−4 and matches to mammalian sequences, 100,828 de novo contigs from the K3 and 43,773 from K10 remained as best matches to the RefSeq database. Next, we wanted to assess the quality of the de novo assemblies, i.e., whether the de novo transcripts were simply spurious transcripts or could, in fact, code for proteins that have some degree of homology to known proteins.
Fig. 4.

Analysis of BLAST results. Contigs assembled using Trinity for the Didelphis virginiana at kmer coverage settings 3 and 10 were aligned to the RefSeq RNA database using BLASTn (A and B) and to the Swiss-Prot database using BLASTx (C and D). Of the total contigs constructed by Trinity in each assembly, the number of contigs with a BLAST alignment to each respective database is shown above each pie chart. Left: number of contigs from matches with an e-value >1e−4 (blue-region) or e-value ≤1e−4 (green-region). Right: the contigs with e-value ≤1e−4 (green-region) are further broken down into their species distribution, specifically highlighting matches to M. domestica, Homo sapiens, and Mus musculus (human and mouse), other mammals, or those that did not have a top hit to mammals. A and B also denote the number of matches to the predicted M. domestica genome since mostly predicted transcripts are present in the RefSeq RNA database.
Since the M. domestica reference genome is poorly annotated, rarely updated, and consists mostly of predicted transcripts, we did not consider the BLASTn alignments to be sufficient for annotating contigs as protein coding sequences or assigning gene identifiers. Therefore, we aligned all the contigs from both the K3 and K10 assemblies using BLASTx to the Swiss-Prot protein database to evaluate the construction of partial and full-length protein coding transcripts (Fig. 4, C and D). Of the contigs with matches to the protein database, 28% of the K3 assembly had a top match with an e-value <1e−4, of which 94% matched to mammalian proteins, 69% specifically to human and mouse proteins. Comparatively, 52% of the K10 assembly had a top match with an e-value <1e−4, of which 95% matched to sequences in mammals, 71% matched to human and mouse proteins. To explore which assembly produced a more complete set of both partial and full-length coding transcripts, we used Trinity to compute the grouped percent hit coverage of each contig as well as the percentage of bases or amino acids shared with the database result. Table 1 shows a comparison of the number of de novo contigs used as query to BLASTx, the number of database sequences aligned to this query, and the number of database sequences that pass further filters of percentage identity to the query sequences. Only those de novo contigs that align to the database with a high degree of identity (>70%) are likely to represent protein coding sequences with homology to known protein coding sequences and known genes. At K3, 180,731 contigs were queried by BLASTx; 46,741 e-value-filtered de novo contigs matched to 16,465 protein coding sequences, representing 11,895 genes in the SwissProt database matched the query sequences after the e-value filter. Comparatively, from the 53,511 contigs in the K10 assembly queried by BLASTx 26,684 e-value-filtered de novo contigs matched to 13,005 different database coding sequences representing 10,529 genes. After we filtered for contigs that matched over ≥80% of a Swiss-Prot sequence, 4,875 Swiss-Prot coding sequences representing 4,677 database genes were in the K3 assembly, slightly higher than the 4,498 Swiss-Prot sequences and 4,336 Swiss-Prot genes that were in the K10 assembly. For both assemblies, the number of database sequences aligned was only slightly greater than the number of genes, suggesting low isoform diversity.
Table 1.
De novo assembly statistics using different BLASTx alignment cutoffs
| Column A; Mammal Filter and e-Value ≤1e−4 | Column B: ≥80% of a Swiss-Prot Sequence Matched | Column C: ≥40% Swiss-Prot Sequence Covered and ≥70% identity | |
|---|---|---|---|
| K3 assembly: 180,731 contigs with BLASTx result | |||
| De novo contigs constructed | 46,741 | 8,055 | 19,812* |
| Swiss-Prot sequences aligned | 16,465 | 4,875 | 9,510* |
| Swiss-Prot Genes | 11,895 | 4,677 | 8,433* |
| K10 assembly: 53,511 contigs with BLASTx result | |||
| De novo contigs constructed | 26,684 | 6,478 | 13,251 |
| Swiss-Prot sequences aligned | 13,005 | 4,498 | 8,220 |
| Swiss-Prot Genes | 10,529 | 4,336 | 7,576 |
Contigs generated by the Trinity de novo assembly using minimum kmer coverage settings 3 and 10 were aligned using BLASTx to the Swiss-Prot database. For each assembly the table shows the number of contigs that pass the filter denoted by the column heading, the number of Swiss-Prot sequences aligned by the contigs, and the number of Swiss-Prot genes matched in the assembly. Column A: filter for BLASTx matches to sequences in mammals with an e-value <1e−4. Columns B and C: further filtering of the contigs selected by the mammal and e-value filter. Column B: filter for contigs that match identically to ≥80% of the full sequence found in Swiss-Prot. Column C: filter for contigs that cover ≥40% of the Swiss-Prot sequence as determined using a Trinity module and with ≥70% identity to the sequence as reported by BLAST. Details of these filters can be found in materials and methods.
Final assembly chosen for further analysis.
In addition to the coding genes fully or nearly fully constructed in our de novo approach, additional genes in our assembly were represented by lower coverage transcripts or by multiple transcripts that could not be assembled fully by the de novo approach. One such example is the LRP2 gene, which encodes the large (4,655 amino acids in M. domestica) multiligand receptor megalin that plays an essential role in the PT uptake of filtered ligands. In the K3 assembly, 12 transcripts aligned to LRP2, of which 6 match with >70% identity and 4 with near full-length grouped percent hit coverage. This suggests that each of the short transcripts for LRP2 is highly similar to portions of this protein in Swiss Prot and yet does not assemble a full length transcript due to size and low coverage. By relaxing selection criteria from 80% match over a Swiss-Prot sequence to a selection criteria of ≥70% identity and ≥40% grouped percent hit coverage (see materials and methods for details), we aimed to include all partial and full length contigs of high accuracy, such as LRP2. With these criteria, 19,812 de novo contigs from the K3 assembly match 9,510 protein sequences representing 8,433 genes, compared with the 13,521 de novo K10 contigs that match 8,220 protein sequences and 7,576 database genes. Supplemental Table S2 shows the complete list of genes and their BLASTx statistics. Of the ~20,000 to 30,000 genes expressed by a typical mammalian genome, ~30–60% are believed to be expressed by a biological sample (26). Thus the approximate 8,000 genes of both the K3 and K10 assemblies represent a significant fraction of the expressed genes of OK cells. From this BLASTx analysis of the de novo assembly, we conclude that even though the K3 assembly has greater potential for shorter false transcripts, after being filtered for high accuracy and coverage, the K3 assembly is more complete, representing 857 more protein coding genes than the K10. Thus, we used the K3 assembly with a ≥70% identity and ≥40% grouped percent hit coverage filter for our subsequent analysis (highlighted in Table 1).
Alignment of OK cell transcriptome to the M. domestica reference genome.
The same randomly selected trimmed (adaptor and base quality = 30) reads that were used in the de novo assembly were also mapped to the NCBI M. domestica (MonDom5) reference genome using spliced alignment software TopHat v2.1.0. Mapping at mismatches 2 (default), 4, 6, 8, and 10 were tested, and the mismatch 6 alignment was found to have the best balance between sensitivity (%reads mapped) and specificity (reads mapped in exon regions). At this mismatch setting, 44.2% of left reads and 45% of right reads mapped, and of these, 88.8% of left reads and 88.4% of right reads mapped uniquely. Thus, although more than half of the data were removed by this process, the majority of the reads mapped unambiguously.
Comparison of OK cell de novo transcriptome and the alignment to the M. domestica genome.
As described above, the K3 de novo assembly resulted in 19,811 contigs representing 8,432 genes after employing the 70/40 filter. In contrast, the alignment to the MonDom5 reference genome resulted in 38,185 identifiers, 14,952 of which were specifically annotated with a gene name. In addition, 7,678 genes are shared between the K3 assembly and the MonDom5 alignment while 754 are found only in the de novo assembly and 7,274 are found only in the alignment. Due to incomplete annotation of the MonDom5 genome, many identifiers without specific gene names, for example, the LOC IDs, may have gene homologs in other species, making it likely that there is even more shared gene coverage between the de novo assembly and the alignment. To compare gene expression measurements in the two approaches, expression was quantified by RSEM resulting in expression counts, transcripts per million (TPM), or FPKM. A side-by-side comparison of the genes in the de novo assembly and alignment and their expression values are provided in Supplemental Table S2. The expression values of identifiers without specific gene names from the alignment are provided in Supplemental Table S3. With the use of TPM, the genes that were detected by both de novo and the alignment to the reference show concordance in expression with a Pearson correlation r = 0.81. We hypothesize that genes found only by alignment are either filtered out of the de novo assembly due to poor matches to Swiss-Prot protein sequences, have low expression, or are long and difficult to assemble transcripts. Although we did not perform extensive evaluations of gene expression from the two methods, we noted that 5,247 out of 7,274 genes detected only by alignment have TPM values <1 suggesting that majority of these are expressed at low levels and may have been difficult to assemble using the de novo approach. Similarly, a subset of these gene identifiers overlap with contigs that did not meet our de novo assembly filtering criteria suggesting that they are not good matches to proteins in the Swiss-Prot databases. These may be transcripts potentially unique to D. virginiana kidney cells or could point to annotation errors or gaps in the MonDom5. For example, genes such as KRT8, PTMA, and Shfm1 are found only in the de novo assembly while annotations for these are not found in the reference genome.
Comparison of OK cell transcriptome with data from human and rodent kidney studies.
To compare gene expression levels with the expression of known PT proteins identified in previously published studies, we utilized the RSEM-generated FPKM gene expression values for both the de novo assembled genes and from alignment to the MonDom5 (see above). These FPKM values from de novo assembly and alignment were averaged to compensate for differences in found transcripts between the two assemblies. The data for microdissected rat kidney nephron segments were published in a recent study by Lee et al. (19). Of the 17,145 genes in the rat kidney tubule data set, 7,087 are found in our de novo assembly and 11,712 in the alignment to the Monodelphis reference genome. Because of the difference in gene names between species, the number of genes matching to the rat data set is likely to be an underestimate.
We first compared our expression data with that published by Lee et al. (19) via clustering analysis, using a list of genes that distinguish the human PT, distal tubule, and collecting duct (11). Whereas all of these transcripts were highly expressed in their respective human nephron segment, there was sizable variation in their expression levels in both the OK cell line and in rat segments. Approximately one-third of the human PT-specific genes were expressed at low levels or not detected in OK cells and/or rat PT segments (S1, S2, and S3). Additionally, there were differences in expression specificity of some of the human distal tubule and collecting duct-specific genes. For example, two genes expressed differentially in the human kidney had different correlations with the rat kidney. HPN, predominantly expressed in human collecting duct, was expressed in all rat nephron segments, while the GLTPD2, highly expressed in human distal tubule, was not well expressed in any of the rat segments (Fig. 5). Furthermore, gene clustering analysis of the OK and rat nephron gene expression data did not completely segregate the human collecting duct, distal tubule, and PT-specific genes (Fig. 5). Regardless, the majority of transcripts in the human gene list are differentially expressed in rat nephron segments, validating the use of this list to assess nephron segment origin of the OK cell line. With the use of this list, our analysis revealed that gene expression in the OK cell line clustered more closely to the PT (S1, S2, and S3) than to the other rat nephron segments (Fig. 5).
Fig. 5.

Clustering of the OK cell transcriptome and rat nephron segment expression values against human PT-specific genes. A list of genes highly expressed in the human PT, distal tubule, or collecting duct [see Habuka et al. (11)] was used to compare the average FPKM value of the OK de novo assembly and alignment (lane marked “OK cell”) to RPKM values of different rat nephron segments from Lee et al. (18). S1, S1 PT; S2, S2 PT; S3, S3 PT; SDL, short descending limb; LDLOM, long descending limb, outer medulla; LDLIM, long descending limb, inner medulla; tAL, thin ascending limb; mTAL, medullary thick ascending limb; cTAL, cortical thick ascending limb; DCT, distal convoluted tubule; CNT, connecting tubule; CCD, cortical collecting duct; OMCD, outer medullary collecting duct; IMCD, inner medullary collecting duct. The comparison is shown as a clustered heat map with both sample and gene clustering. The specific nephron section with high expression in humans is color coded on the right of the heat map (PT = green, distal tubule (DT) = yellow, and collecting duct (CD) = blue).
Because OK cells are frequently used as a model system to study ion transport in the PT, we next compared our gene expression data with those of the most highly expressed transporter and ion channel genes reported for rat S1, S2, and S3 PT segments (19). Here, our assembly clustered more closely to segments S1 and S2 than to S3 (Fig. 6). The few genes where the OK cell line more closely mimicked S3 expression patterns are demarcated by the green line in Fig. 6. There were also some differences in relative expression of genes between the OK cell line and the rat PT S1 and S2 segments. One cluster contained genes highly expressed in the rat PT that were minimally expressed in OK cells (Fig. 6, blue line). Included in this cluster is SLC27a2, one of three fatty acid transporters from the SLC27 family specifically expressed in rat kidney. The other two genes in this family, SLC27a1 and SLC27a4, share the same substrates as SLC27a2 and all three protein sequences are highly conserved (2). Interestingly, SLC27a1 and SLC27a4 are highly expressed in the OK cell line, but not in the rat PT, suggesting a species difference in kidney expression of this gene family. Several members of the SLC16 family were also found in the cluster of differentially expressed genes. SLC16a2 is an active thyroid hormone transporter with a primary responsibility for cellular uptake of thyroxine, triiodothyronine, reverse triiodothyronine, and diiodothyronine (7). The functional significance of the low expression of SLC16a2, as well as SLC16a11 and SLC16a12, is difficult to assess, since the function and preferred substrates of the transporters in this family are not well characterized. Of note, OK cells respond to thyroid hormone similar to rat kidney (30, 37). Thus minimal expression of SLC16a2 may be sufficient for hormone entry, or another transporter could contribute to uptake in OK cells.
Fig. 6.
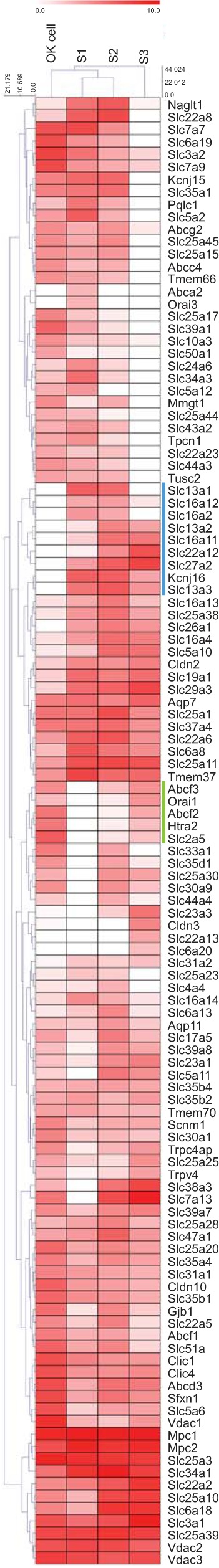
Clustering of the OK cell transcriptome against top transporter and ion channel genes in rat PT segments. The top 100 expressed transporter and ion channel genes for each segment in the rat PT were used to compare the average FPKM value of the OK de novo assembly and alignment to RPKM values of different rat PT segments (see Fig. 5 legend for corresponding abbreviations). The comparison is shown as a clustered heat map with both sample and gene clustering.
The lack of available antibodies against opossum proteins coupled with limited available genomic information has complicated the identification of proteins believed to be important for essential PT function in OK cells. We curated a partial list of such genes and examined their expression and abundance relative to data from rat nephron segments (Fig. 7). Our list included enzymes dipeptidylpeptidase 4 (Dpp4), aminopeptidase N (Anpep), 5′-nucleotidase (Nt5e), and OCRL1 (Ocrl1); transcription factors Hnf1α (Hnf1a) (35) and ZONAB (Ybx3) (21); transporters and channels CLC-5 (Clcn5), sodium/phosphate cotransporter NaPiIIa (Slc34a1), aquaporin 1 (Aqp1), and aquaporin 11 (Aqp11) (33); receptors megalin (Lrp2), cubilin (Cubn), and FcRN (Fcgrt); and endocytic and cellular trafficking proteins (Rab4a, Rab5a, Rab11a, and Rab38); (Ref. 27). While expression of many of these proteins in OK cells has not previously been confirmed, we found transcripts for all of the above proteins in our data set. For these proteins, the OK cell line assembly clustered closest to the S3 PT segment when compared against the data generated in rat (19).
Fig. 7.

OK cell transcriptome assembly is most closely related to the PT based on a curated list of proximal tubule-specific functional genes. The average FPKM value of the OK de novo assembly and alignment and RPKM values of different rat nephron segments (see Fig. 5 legend for corresponding abbreviations) were compared with a curated list of genes relevant to PT function. The comparison is shown as a clustered heat map with both sample and gene clustering.
DISCUSSION
We generated the OK cell line transcriptome using both de novo assembly and alignment to the M. domestica genome from RNA Seq reads and showed that both approaches successfully assemble a transcriptome whose protein coding gene expression pattern is most similar to the rat PT. While both assemblies are successful in capturing the PT phenotype, each has distinctive features and future studies with additional samples and experimental conditions may allow a more complete characterization of the OK transcriptome. The detailed transcriptome of OK cells from D. virginiana is likely different from the M. domestica transcriptome of comparable cells, and a combination approach of de novo assembly and alignment to the M. domestica has provided two resources for studies of this valuable cell line. Future OK studies with additional samples and experimental conditions will enable a more complete characterization of its transcriptome including identification of novel introns, exons, UTRs, long noncoding RNAs, and novel transcripts.
Comparison of the average RNA expression values obtained from these analyses confirmed that OK cells are more closely related to kidney cells in the PT than in other nephron segments. However, we could not determine whether OK cells more closely resemble PT segment S1, S2, or S3. In part, this is because transcript expression data among these three segments is quite similar (18). While there are sure to be species-specific expression differences between M. domestica and R. norvegicus, it is also worth noting that there will be evident differences between a cell line and microdissected tissue. As the OK cell line has been immortalized and undergone continuous passage since removal from the opossum donor, the cells have likely undergone changes in expression patterns. For example, the reduced expression of SLC16a2 in these cells could therefore reflect either a distinctive phenotype of Monodelphis PT or changes from establishment and culturing of the cell line. Nevertheless, our transcriptome and clustering analysis strongly validates the use of the OK cell line as a cell culture model of the PT.
Additional clustering analysis demonstrated that proteins previously implicated in a wide variety of PT functions using other model systems are also expressed in OK cells. While cultured cell lines in general never fully recapitulate protein expression patterns or functions of cells in vivo, our data suggest that OK cells represent a reasonable surrogate for studies of PT ion transport, hormonal regulation, and membrane trafficking. The current greatest limitation in utilizing OK cells for PT research is the lack of sequence information. By further elucidating OK cell transcript sequences and expression values, our work provides a valuable resource for investigators interested in genetic manipulation studies, including siRNA and CRISPR. Furthermore, by providing differences in transcript sequences from M. domestica and other mammals, we can better identify cross-reacting antibodies and other reagents that recognize proteins in the OK cell line. Additionally, our quantitative gene expression data provide a useful baseline for assessing the effects of physiologic stimuli on transcription profiles in the PT in the OK cell line.
GRANTS
This project was supported by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grants R01-DK-101484 and R01-DK-100357 (to O. A. Weisz). M. L. Eshbach was supported in part by NIDDK Grant T32-DK-061296. We are grateful to the Pittsburgh Center for Kidney Research (NIDDK Grant P30-DK-079307) and to the University of Pittsburgh for subsidizing services of the Biomedical Informatics Core and the Genomics Research Core for support.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.L.E., J.L., D.J.H., D.N.F., U.R.C., and O.A.W. conceived and designed research; M.L.E., J.L., and D.J.H. performed experiments; M.L.E., R.S., R.A., J.D.L., and U.R.C. analyzed data; M.L.E., R.S., R.A., D.N.F., J.D.L., U.R.C., and O.A.W. interpreted results of experiments; M.L.E., R.S., R.A., and O.A.W. prepared figures; M.L.E., R.S., R.A., U.R.C., and O.A.W. drafted manuscript; M.L.E., R.S., R.A., J.L., D.J.H., D.N.F., J.D.L., U.R.C., and O.A.W. approved final version of manuscript; R.S., R.A., D.N.F., J.D.L., U.R.C., and O.A.W. edited and revised manuscript.
ACKNOWLEDGMENTS
We are grateful to Barbara Methé for helpful discussions and to the University of Pittsburgh School of Medicine to the Genomics Analysis Core, a data analysis core for School of Medicine investigators.
REFERENCES
- 1.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol 215: 403–410, 1990. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 2.Anderson CM, Stahl A. SLC27 fatty acid transport proteins. Mol Aspects Med 34: 516–528, 2013. doi: 10.1016/j.mam.2012.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Antoine DJ, Srivastava A, Pirmohamed M, Park BK. Statins inhibit aminoglycoside accumulation and cytotoxicity to renal proximal tubule cells. Biochem Pharmacol 79: 647–654, 2010. doi: 10.1016/j.bcp.2009.09.021. [DOI] [PubMed] [Google Scholar]
- 4.Biber J, Hernando N, Traebert M, Völkl H, Murer H. Parathyroid hormone-mediated regulation of renal phosphate reabsorption. Nephrol Dial Transplant 15, Suppl 6: 29–30, 2000. doi: 10.1093/ndt/15.suppl_6.29. [DOI] [PubMed] [Google Scholar]
- 5.Biemesderfer D, Nagy T, DeGray B, Aronson PS. Specific association of megalin and the Na+/H+ exchanger isoform NHE3 in the proximal tubule. J Biol Chem 274: 17518–17524, 1999. doi: 10.1074/jbc.274.25.17518. [DOI] [PubMed] [Google Scholar]
- 6.Francis WR, Christianson LM, Kiko R, Powers ML, Shaner NC, Haddock SH. A comparison across non-model animals suggests an optimal sequencing depth for de novo transcriptome assembly. BMC Genomics 14: 167, 2013. doi: 10.1186/1471-2164-14-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Friesema EC, Jansen J, Heuer H, Trajkovic M, Bauer K, Visser TJ. Mechanisms of disease: psychomotor retardation and high T3 levels caused by mutations in monocarboxylate transporter 8. Nat Clin Pract Endocrinol Metab 2: 512–523, 2006. doi: 10.1038/ncpendmet0262. [DOI] [PubMed] [Google Scholar]
- 8.Gekle M, Knaus P, Nielsen R, Mildenberger S, Freudinger R, Wohlfarth V, Sauvant C, Christensen EI. Transforming growth factor-beta1 reduces megalin- and cubilin-mediated endocytosis of albumin in proximal-tubule-derived opossum kidney cells. J Physiol 552: 471–481, 2003. doi: 10.1113/jphysiol.2003.048074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29: 644–652, 2011. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, MacManes MD, Ott M, Orvis J, Pochet N, Strozzi F, Weeks N, Westerman R, William T, Dewey CN, Henschel R, LeDuc RD, Friedman N, Regev A. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8: 1494–1512, 2013. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Habuka M, Fagerberg L, Hallström BM, Kampf C, Edlund K, Sivertsson Å, Yamamoto T, Pontén F, Uhlén M, Odeberg J. The kidney transcriptome and proteome defined by transcriptomics and antibody-based profiling. PLoS One 9: e116125, 2014. doi: 10.1371/journal.pone.0116125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holthouser KA, Mandal A, Merchant ML, Schelling JR, Delamere NA, Valdes RR Jr, Tyagi SC, Lederer ED, Khundmiri SJ. Ouabain stimulates Na-K-ATPase through a sodium/hydrogen exchanger-1 (NHE-1)-dependent mechanism in human kidney proximal tubule cells. Am J Physiol Renal Physiol 299: F77–F90, 2010. doi: 10.1152/ajprenal.00581.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36, 2013. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kolb HA. Ion channels in opossum kidney cells. Ren Physiol Biochem 13: 26–36, 1990. [DOI] [PubMed] [Google Scholar]
- 15.Komaba S, Coluccio LM. Myosin 1b regulates amino acid transport by associating transporters with the apical plasma membrane of kidney cells. PLoS One 10: e0138012, 2015. doi: 10.1371/journal.pone.0138012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koyama H, Goodpasture C, Miller MM, Teplitz RL, Riggs AD. Establishment and characterization of a cell line from the American opossum (Didelphys virginiana). In Vitro 14: 239–246, 1978. doi: 10.1007/BF02616032. [DOI] [PubMed] [Google Scholar]
- 17.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359, 2012. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee AK, Kulcsar KA, Elliott O, Khiabanian H, Nagle ER, Jones ME, Amman BR, Sanchez-Lockhart M, Towner JS, Palacios G, Rabadan R. De novo transcriptome reconstruction and annotation of the Egyptian rousette bat. BMC Genomics 16: 1033, 2015. doi: 10.1186/s12864-015-2124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee JW, Chou CL, Knepper MA. Deep sequencing in microdissected renal tubules identifies nephron segment-specific transcriptomes. J Am Soc Nephrol 26: 2669–2677, 2015. doi: 10.1681/ASN.2014111067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12: 323, 2011. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lima WR, Parreira KS, Devuyst O, Caplanusi A, N’kuli F, Marien B, Van Der Smissen P, Alves PM, Verroust P, Christensen EI, Terzi F, Matter K, Balda MS, Pierreux CE, Courtoy PJ. ZONAB promotes proliferation and represses differentiation of proximal tubule epithelial cells. J Am Soc Nephrol 21: 478–488, 2010. doi: 10.1681/ASN.2009070698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mikkelsen TS, Wakefield MJ, Aken B, Amemiya CT, Chang JL, Duke S, Garber M, Gentles AJ, Goodstadt L, Heger A, Jurka J, Kamal M, Mauceli E, Searle SM, Sharpe T, Baker ML, Batzer MA, Benos PV, Belov K, Clamp M, Cook A, Cuff J, Das R, Davidow L, Deakin JE, Fazzari MJ, Glass JL, Grabherr M, Greally JM, Gu W, Hore TA, Huttley GA, Kleber M, Jirtle RL, Koina E, Lee JT, Mahony S, Marra MA, Miller RD, Nicholls RD, Oda M, Papenfuss AT, Parra ZE, Pollock DD, Ray DA, Schein JE, Speed TP, Thompson K, VandeBerg JL, Wade CM, Walker JA, Waters PD, Webber C, Weidman JR, Xie X, Zody MC; Broad Institute Genome Sequencing Platform; Broad Institute Whole Genome Assembly Team; Graves JA, Ponting CP, Breen M, Samollow PB, Lander ES, Lindblad-Toh K. Genome of the marsupial Monodelphis domestica reveals innovation in non-coding sequences. Nature 447: 167–177, 2007. doi: 10.1038/nature05805. [DOI] [PubMed] [Google Scholar]
- 23.Murer H, Hernando N, Forster I, Biber J. Proximal tubular phosphate reabsorption: molecular mechanisms. Physiol Rev 80: 1373–1409, 2000. [DOI] [PubMed] [Google Scholar]
- 24.Prozialeck WC, Edwards JR, Lamar PC, Smith CS. Epithelial barrier characteristics and expression of cell adhesion molecules in proximal tubule-derived cell lines commonly used for in vitro toxicity studies. Toxicol In Vitro 20: 942–953, 2006. doi: 10.1016/j.tiv.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 25.Raghavan V, Rbaibi Y, Pastor-Soler NM, Carattino MD, Weisz OA. Shear stress-dependent regulation of apical endocytosis in renal proximal tubule cells mediated by primary cilia. Proc Natl Acad Sci USA 111: 8506–8511, 2014. [Erratum. Proc Natl Acad Sci USA 113: E1587, 2016.] doi: 10.1073/pnas.1402195111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramsköld D, Wang ET, Burge CB, Sandberg R. An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLOS Comput Biol 5: e1000598, 2009. doi: 10.1371/journal.pcbi.1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rangel-Filho A, Lazar J, Moreno C, Geurts A, Jacob HJ. Rab38 modulates proteinuria in model of hypertension-associated renal disease. J Am Soc Nephrol 24: 283–292, 2013. doi: 10.1681/ASN.2012090927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, Sturn A, Snuffin M, Rezantsev A, Popov D, Ryltsov A, Kostukovich E, Borisovsky I, Liu Z, Vinsavich A, Trush V, Quackenbush J. TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34: 374–378, 2003. [DOI] [PubMed] [Google Scholar]
- 29.Samollow PB. The opossum genome: insights and opportunities from an alternative mammal. Genome Res 18: 1199–1215, 2008. doi: 10.1101/gr.065326.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sorribas V, Markovich D, Verri T, Biber J, Murer H. Thyroid hormone stimulation of Na/Pi-cotransport in opossum kidney cells. Pflugers Arch 431: 266–271, 1995. doi: 10.1007/BF00410200. [DOI] [PubMed] [Google Scholar]
- 31.States B, Harris D, Segal S. Differences between OK and LLC-PK1 cells: cystine handling. Am J Physiol Cell Physiol 261: C8–C16, 1991. [DOI] [PubMed] [Google Scholar]
- 32.Takano M, Nakanishi N, Kitahara Y, Sasaki Y, Murakami T, Nagai J. Cisplatin-induced inhibition of receptor-mediated endocytosis of protein in the kidney. Kidney Int 62: 1707–1717, 2002. doi: 10.1046/j.1523-1755.2002.00623.x. [DOI] [PubMed] [Google Scholar]
- 33.Tanaka Y, Watari M, Saito T, Morishita Y, Ishibashi K. Enhanced autophagy in polycystic kidneys of AQP11 null mice. Int J Mol Sci 17: 17, 2016. doi: 10.3390/ijms17121993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Teitelbaum AP, Strewler GJ. Parathyroid hormone receptors coupled to cyclic adenosine monophosphate formation in an established renal cell line. Endocrinology 114: 980–985, 1984. doi: 10.1210/endo-114-3-980. [DOI] [PubMed] [Google Scholar]
- 35.Terryn S, Tanaka K, Lengelé JP, Olinger E, Dubois-Laforgue D, Garbay S, Kozyraki R, Van Der Smissen P, Christensen EI, Courtoy PJ, Bellanné-Chantelot C, Timsit J, Pontoglio M, Devuyst O. Tubular proteinuria in patients with HNF1α mutations: HNF1α drives endocytosis in the proximal tubule. Kidney Int 89: 1075–1089, 2016. doi: 10.1016/j.kint.2016.01.027. [DOI] [PubMed] [Google Scholar]
- 36.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578, 2012. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yusufi AN, Murayama N, Keller MJ, Dousa TP. Modulatory effect of thyroid hormones on uptake of phosphate and other solutes across luminal brush border membrane of kidney cortex. Endocrinology 116: 2438–2449, 1985. doi: 10.1210/endo-116-6-2438. [DOI] [PubMed] [Google Scholar]
- 38.Zaarour N, Demaretz S, Defontaine N, Mordasini D, Laghmani K. A highly conserved motif at the COOH terminus dictates endoplasmic reticulum exit and cell surface expression of NKCC2. J Biol Chem 284: 21752–21764, 2009. doi: 10.1074/jbc.M109.000679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhai XY, Nielsen R, Birn H, Drumm K, Mildenberger S, Freudinger R, Moestrup SK, Verroust PJ, Christensen EI, Gekle M. Cubilin- and megalin-mediated uptake of albumin in cultured proximal tubule cells of opossum kidney. Kidney Int 58: 1523–1533, 2000. doi: 10.1046/j.1523-1755.2000.00314.x. [DOI] [PubMed] [Google Scholar]