

Abstract
Nerve growth factor (NGF), a member of the neurotrophin family, is known to regulate the development and survival of a select population of neurons through the binding and activation of the TrkA receptor. Elevated levels of NGF have been associated with painful pathologies such as diabetic neuropathy and fibromyalgia. However, completely inhibiting the NGF signal could hold significant side effects, such as those observed in a genetic condition called congenital insensitivity to pain and anhidrosis (CIPA). Previous methods of screening for NGF‐inhibitors used labeling techniques which have the potential to alter molecular interactions. SPR spectroscopy and NGF‐dependent cellular assays were utilized to identify a novel NGF‐inhibitor, BVNP‐0197 (IC 50 = 90 nmol/L), the first NGF‐inhibitor described with a high nanomolar NGF inhibition efficiency. The present study utilizes molecular modeling flexible docking to identify a novel binding domain in the loop II/IV cleft of NGF.
Keywords: NGF, pain, small molecule, SPR, TrkA
Abbreviations
- ATCC
American type culture collection
- FBS
fetal bovine serum
- NGF
nerve growth factor
- NGF
Nerve growth factor
- SPR
surface plasmon resonance
- TLC
thin layer chromatograph
- TNF
anti‐tumor necrosis factor
Introduction
Elevated levels of nerve growth factor (NGF) have been implicated in several chronic pain syndromes such as osteoarthritis (Kc et al. 2016) and diabetic neuropathy (Malerba et al. 2015). Experimental evidence has shown that NGF is released by several cell types including mast cells (Bienenstock et al. 1987; Skaper et al. 2001), lymphocytes (Torcia et al. 1996), and monocytes/macrophages (Bracci‐Laudiero et al. 2005) in response to tissue inflammation. Interestingly, NGF has also been found to produce hyperalgesia when administered in several animal species (Brodie 1995; Hao et al. 2000; Cahill et al. 2003). These pain‐related behavioral responses to NGF in animals manifest within minutes, and can last anywhere from several hours to days depending on the dose (Lewin et al. 1994; Zahn et al. 2004). Subcutaneous injection of NGF into the forearm of healthy human adults induced localized allodynia and hypersensitivity within minutes, lasting for several hours (Dyck et al. 1997). In addition, small intravenous NGF doses in healthy human adults are responsible for widespread deep pain and tenderness which persists for several days (Svensson et al. 2003). The evidence of upregulated NGF in painful pathological conditions, in addition to the evidence that NGF causes pain in humans and in animals, have led to the rational for developing therapeutics based on the inhibition of NGF activity.
A growing body of evidence suggests that an anti‐hyperalgesia effect can be observed with pharmacological interference of NGF–TrkA interactions in several neuropathic pain models (Beglova et al. 2000; Hefti et al. 2006; Wild et al. 2007). Monoclonal anti‐NGF antibodies, such as Tanezumab, have been used as NGF sequestering therapy. Tanezumab binds to NGF with high selectivity thus blocking NGF–TrkA interactions and inhibiting the signaling of sensory neurons for the perception of pain (Schnitzer et al. 2011). Despite the early clinical success seen by Tanezumab, a clinical hold was placed on the drug during Phase III trials when several individuals developed joint damage, which progressed to a stage where joint replacement was necessary. Even with the apparent successes in the antibody therapeutics, there are still potential drawbacks such as delivery challenges, potential for autoimmune responses, capacity for production and financial considerations (Samaranayake et al. 2009). Therefore, the generation of small molecule antagonists which have the ability to selectively disrupt NGF–TrkA interactions may have significant therapeutic advantage.
A series of novel nonpeptidic small molecules have been demonstrated to inhibit binding of NGF to TrkA. Compounds such as ALE‐0540 (Owolabi et al. 1999), PD 90780 (Colquhoun et al. 2004), Ro 08‐2750 and (Niederhauser et al. 2000) have been shown to inhibit NGF‐TrkA signal transduction pathways in vitro. However, the mechanisms by which these described small molecules exert their inhibitory effect remains speculative (Eibl et al. 2012). Historically, the identification of small molecule NGF‐inhibitors resulted from high‐throughput receptor‐binding assays. However, recent advances in the understanding of the structural biology of NGF–TrkA interactions have allowed for rational development of novel small molecules. PQC 083 is one example of a small molecule inhibitor that was developed to target a specific region on NGF to alter TrkA binding (Eibl et al. 2013a). With newly identified crystal structures explaining the interactions during NGF‐TrkA binding (Wehrman et al. 2007), small molecules have been developed to alter the molecular topology of NGF to inhibit TrkA binding.
Determining how potential therapeutic drugs modulate analyte–ligand interactions and bind to target molecules will help determine strategies for developing future therapeutics. One such technique for investigating the strength and rate of biomolecular interactions is surface plasmon resonance (SPR) spectroscopy (Cooper 2002). SPR is advantageous over other techniques because it monitors biomolecular interactions in real time and is label‐free, eliminating the need for fluorescent reporter molecules or radioisotope tags (Mir and Shinohara 2013). Not only is this advantageous in saving time during labeling and reducing resources, but more importantly it eliminates tags which can alter the molecular interactions (Fraser et al. 2014).
In the present study, we use a combination of molecular modeling and SPR to identify a series of novel small molecule analogs with specificity for NGF that inhibit binding to TrkA. Our theoretical flexible docking experiments revealed a novel‐binding domain in the loop II/IV cleft of NGF where a series of analogs bind to inhibit NGF signaling. SPR spectroscopy was also utilized to characterize novel small molecule interactions with NGF and their inhibitory effect on TrkA interactions. Analogs also demonstrated efficient inhibitory activity of NGF in vitro, using an NGF‐dependent PC12 assay. Guided by receptor docking, we were able to identify BVNP‐0197 as a small molecule capable of inhibiting NGF in vitro with greater potency than previously reported NGF inhibitors. The therapeutic potential of previously reported compounds in modulating NGF signaling is still quite low as they bind to NGF with micromolar affinity. The increase in inhibitory potential and target binding affinity described by BVNP‐0197 to NGF could possibly translate into a therapeutic option with reduced dosing and prolonged biological effects compared to other small molecules described.
Materials and Methods
Molecular modeling
Molecular modeling and in silico docking of bivalent naphthalimide compounds to NGF (RCSB Protein Data Bank ID 1BET) (McDonald et al. 1991) were completed using Sybyl‐X 2.1.1 software. The structure of NGF was prepared for docking, using the Biopolymer suite of Sybyl‐X 2.1.1. Co‐structures were deleted, hydrogens were added and the appropriate formal charges on the C‐ and N‐ termini were added. The structure was optimized using the MMFF94 molecular mechanical force field. Flexible docking of each compound was performed, using the Surflex‐Doc suite incorporated into Sybyl‐X 2.1.1 (Jain 1996). The docking protomol was generated to include the residues of the loop II/IV cleft (Glu41‐Phe49 and Gln96‐Trp99) with a bloat factor of 0 and a threshold value of 0.5. For the Surflex‐Doc function, the angstroms to expand the search grid was set at 6 and the maximum confirmations per fragment was set to 20. Docking to proNGF (RCSB Protein Data Bank ID 3IJ2) (Feng et al. 2010) was completed as described above with a docking protomol generated with residues (Gly33‐Cys39 and Gln90‐Cys100) at the loop II/IV cleft.
SPR spectroscopy
Mouse NGF (Cedarlane, Burlington; Canada) was coupled using the amine coupling method. Sensor Chip CM5 was activated using 0.4 mol/L EDC/0.1 mol/L NHS at a flow rate of 10 μL/min for 7 min. NGF was dissolved in a HBS‐EP buffer, pH 4.5, to yield a 10 μg/mL solution. Following activation, the NGF solution was injected over the activated sensor chip surface at a flow rate of 10 μL/min for 9 min. After the injection, the surface was washed with 1 mol/L NaCl to remove any uncoupled or noncovalently bound material from the surface. The excess hydroxysuccinimidyl groups on the surface were deactivated with 1M ethanolamine hydrochloride, pH 8.5, at a flow rate of 10 μL/min for 7 min. The surface of a reference flow cell was activated with 0.4 mol/L EDC/0.1 mol/L NHS and then deactivated with 1 mol/L ethanolamine, with respective flow rates and times.
Bivalent naphthalimide chemical synthesis
All chemicals and solvents were obtained from commercial sources without further purification. Mass spectra were recorded using Waters (Micromass ZQ) equipped with a Thermo Betasil C18 (150 × 2.1 mm, 3μ). 1H NMR spectra were recorded on a Varian AS500 instrument. Chromatography on silica gel columns were performed, using Merck silica gel 60 (230‐400 mesh), and analytical thin layer chromatography (TLC) was conducted on a glass plate coated with a 0.25 cm thin layer of silica gel 60F254.
General procedure for naphthalimide derivatives
Under a nitrogen atmosphere, the primary amine (1 equiv) and sodium acetate (1 equiv) were added to a stirred solution of 1,4,5,8‐naphthalene tetracarboxylic dianhydride (1 equiv) in glacial acetic acid. The reaction mixture was refluxed and the progress of the reaction was monitored by TLC. Upon completion of the reaction if the resulting solution was clear, the solvent was evaporated under reduced pressure and the residual solid was reprecipitated and/or recrystalized from appropriate solvent(s). If the reaction mixture contained precipitate then the mixture was cooled to room temperature and the solid was removed by filtration. The filtrate was concentrated and then diluted with water to precipitate the crude intermediate product. The crude product was dissolved in ethanol and heated to reflux for 10 min, cooled to room temperature, and filtered to remove any solid. The filtrate was concentrated under reduced pressure to get intermediate product A/B, which was dried in vacuum for 24 h and used for the next steps (Fig. 1). Under a nitrogen atmosphere, the appropriate amine (1 equiv) and sodium acetate (1 equiv) or substituted diaminobenzene (1 equiv) was added to a stirred solution of intermediate compound A (1 equiv) in glacial acetic acid. The reaction was refluxed for 10 h and then cooled to room temperature. The solid obtained was filtered and washed with distilled water or dilute acid. It was reprecipitated and/or recrystalized from the appropriate solvents. The solid final product C/D was dried under a vacuum (Fig. 1).
Figure 1.
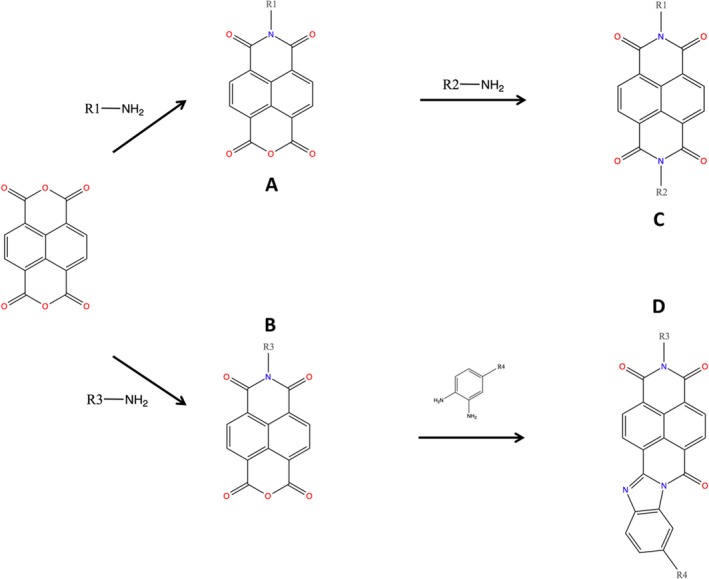
Synthesis of bivalent naphthalimide analog series. Final products (C/D) were screened for NGF inhibitory properties.
Compound sample preparation for SPR analysis
Each compound of interest was weighed on an analytical balance and dissolved in the appropriate amount of DMSO to give a 10 mmol/L solution. The samples were diluted with running buffer (0.01 mol/L HEPES, 0.15 mol/L NaCl, 3 mmol/L EDTA and 0.05% v/v Tween 20; pH 7.5) to yield solutions for the binding experiments with concentrations varying from 200 μmol/L to 0.7 μmol/L and for IC50‐binding experiments with concentrations varying from 3.16 nmol/L to 316 μmol/L.
Analog‐binding affinity to immobilized NGF
Binding affinities of the bivalent naphthalimide analogs to immobilized NGF were determined as previously described, using a Biacore T200 SPR (Kennedy et al. 2016). Prior to the analyte injection, the series S CM5 chip was conditioned with three 30 sec cycles of running buffer (0.01 mol/L HEPES, 0.15 mol/L NaCl, 3 mmol/L EDTA and 0.05% v/v Tween 20; pH 7.5) followed by three start up cycles, allowing the response to stabilize before the analyte injection. Data were collected at a temperature of 25 ᵒC. Individual compound samples were tested from the lowest to the highest concentrations, separated by a 15 sec stabilization period after each sample in each compound series. During each sample cycle, the analyte was injected for 60 sec at a flow rate of 30 μL/min. A dissociation period was monitored for 30 sec after analyte injection before regeneration was induced with 1.0 mol/L NaCl for 120 sec at a flow rate of 30 μL/min to wash any remaining analyte from the sensor chip before running the next sample.
PC12 cell culture
PC12 cells were obtained from the American Type Culture Collection (ATCC; USA). Cell passage 18–21 were used for all experiments. PC12 cells were maintained in Dulbecco's Modified Eagles Medium supplemented with 10% fetal bovine serum (FBS) (Gibco; USA). Media was supplemented with working stock concentration of penicillin (0.1 U/mL) and streptomycin (0.1 U/mL). PC12 cells were seeded at 50% confluence and were passaged at 90% confluence in T25 cm2 tissue culture‐treated flasks (Corning; USA). PC12 cells were incubated at 37°C in 5% CO2.
PC12 toxicity assay
PC12 toxicity assay was performed by plating cells at a density of 200 cells/well on Terasaki plates (Greiner Bio‐One; USA) at 37°C and 5% CO2. Cells were exposed to 100 μmol/L of compound of interest in the Terasaki well for 72 h in the absence of NGF. Cells on the lower horizontal surface of the well were imaged using an EVOS XL Core cell imaging system at 20 × phase observation. The total cells per well were counted and compared to control. Three runs of each experiment were performed, and each condition was tested in quadruplicate.
PC12 NGF‐dependent neurite outgrowth assay
PC12 cells were plated 24 h prior to treatment at a density of 200 cells/well and were grown on Terasaki plates (Greiner Bio‐One; USA) at 37°C and 5% CO2. PC12 cells were exposed to 500 pM of NGF (Life Technologies; Canada) preincubated with varying concentrations of each compound of interest (31.6 nmol/L – 100 μmol/L). Terasaki wells were imaged 72 h after exposure to NGF. Cells on the lower horizontal surface of the well were imaged using, an EVOS XL Core cell imaging system at 20 x phase observation. Differentiation events were manually scored when a cell developed a process with a constant calibre from the origin to the terminal and its length was equal or greater than 1 x cell body diameter. Wells with fewer than 50 cells or greater than 350 cells were excluded from the analysis. Three trials of each experiment were performed, and each condition was tested in quadruplicate.
TrkA phosphorylation and western blotting analysis
TrkA phosphorylation was conducted by the modification of methods previously described (Ross et al. 1998). NGF (50 pM) was incubated with the analog series compound (10 to 1 μmol/L) for 1 h in HKR buffer. 1x106 cells/mL PC12 cells suspended in HKR buffer were treated with the NGF‐analog complex for 15 min at 37°C with occasional agitation. Following the incubation, an immunoprecipitation was performed, using the Pierce Classic IP Kit (Thermo, USA). The cells were lysed and exposed to Protease Inhibitor Cocktail (Pierce, USA). Trk‐antibody (Cell Signaling, USA) was then incubated with the cell lysate at 4°C overnight. Protein A/G coated agarose matrix was used to precipitate the immune complex which was then separated by electrophoresis, using an 8% SDS‐PAGE gel. A western blot was performed to transfer the immune complex to PVDF membrane. The membrane was blocked in 5% skim milk TBS‐T and probed with phosphotyrosine primary antibody 4G10 (Millipore, USA) followed by the secondary HRP‐linked Goat Anti‐Mouse (Santa Cruz Biotechnology, USA) antibody before being visualized by autoradiography.
IC50 determination using SPR spectroscopy
TrkA was immobilized to a CM5 sensor surface as previously described (Forsell et al. 2013). Prior to analyte injection, the Series S CM5 chip was conditioned with three 30 sec cycles of running buffer (0.01 mol/L HEPES, 0.15 mol/L NaCl, 3 mmol/L EDTA and 0.05% v/v Tween 20; pH 7.5) followed by three start up cycles, allowing the response to stabilize before analyte injection. Data were collected at a temperature of 25°C. Individual compound samples were tested from lowest to highest concentrations, separated by a 15 sec stabilization period after each sample in each compound series. During each sample cycle, the analyte was injected for 60 sec at a flow rate of 30 μL/min. A dissociation period was monitored for 120 sec after the analyte injection. Regeneration occurred with a 10 mmol/L Glycine solution at pH 2.0 for 15 sec at a flow rate of 30 μL/min to wash any remaining analyte from the sensor chip before running the next sample.
Statistical analysis
Statistical analysis was performed, using Prism Graphpad 5.0 (San Diego; USA). Error bars are expressed as standard error of the mean with a sample size of n = 3. Inhibition curves were fit using a nonlinear regression model.
Results
Molecular modeling flexible docking
Previous studies have suggested that loop I, II, and IV of each NGF monomer are responsible for binding to TrkA (Barker 2007; Wehrman et al. 2007), whereas loop I and IV are necessary for NGF binding to p75NTR (He and Garcia 2004; Barker 2007) (Fig. 2). Previous drug discovery efforts have been focused on a putative binding domain for small molecule NGF inhibitors in the loop I/IV cleft of the NGF monomer (Colquhoun et al. 2004; Eibl et al. 2013a). Our flexible docking identified a novel‐binding domain located in the loop II/IV cleft of NGF for a series of bivalent naphthalimide compounds which were derived using similar molecular features to previously reported small molecule NGF inhibitors. This novel‐binding domain demonstrated higher binding capabilities than previously suggested docking areas. The pharmacological targeting domain included NGF monomer residues Glu41, Val42, Asn43, Ile44, Asn45, Asn46, Ser47, Val48, Phe49, Gln96, Ala97, Ala98, and Trp99. Fig. 2 demonstrates the protomol which was derived from the described residues of NGF.
Figure 2.
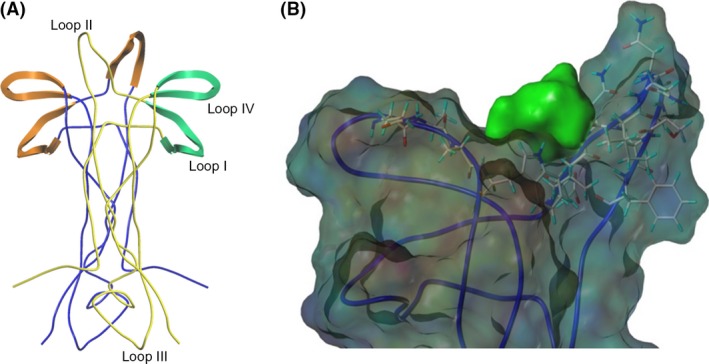
(A)The dimeric structure of NGF (RCSB: 1BET) (McDonald et al. 1991) is presented in ribbon format. NGF monomers are colored blue and yellow. Structural features of the yellow NGF monomer are labeled. Orange highlights represent structural features of the blue NGF monomer which are responsible for TrkA binding. Green structural features highlighted represent areas responsible for p75NTR binding of the yellow NGF monomer. (B) Theoretical docking experiments identified a docking site for bivalent naphthalimide scaffold at loop II/IV domain of NGF. NGF is represented as a ribbon structure (RCSB: 1BET) (McDonald et al. 1991). Resides Glu41, Val42, Asn43, Ile44, Asn45, Asn46, Ser47, Val48, Phe49, Gln96, Ala97, Ala98, and Trp99 were selected to create a protomol binding domain for bivalent naphthalimide analogs represented in green.
The results of the molecular modeling flexible docking score for each bivalent naphthalimide analog was measured. The scoring function predicts the binding affinity of analogs to NGF by counting the number of interactions between entities (hydrophobic, hydrophilic, hydrogen bonds and rotatable bonds in the complex). The scoring output is represented in units of –log(KD). The results of compound BVNP‐0187 were chosen as a representative bivalent naphthalimide scaffold analog following flexible docking at the loop II/IV cleft of NGF (Fig. 3). BVNP‐0187, like previously reported small molecule NGF inhibitors, has a ridged backbone structure comprised of a bivalent naphthalimide motif (IUPAC name: 6,13‐diazatetracyclo[6.6.2.04,16.011,15]hexadeca‐1,3,8,10,15‐pentaene‐5,7,12,14‐tetrone moiety) containing a butanoic acid residue in –R1 and 2‐hydroxy‐5‐benzoic acid residue occupying –R2 (Table 1). Flexible docking determined that BVNP‐0187 had the highest theoretical docking score of all analogs screened. Flexible docking suggested that four hydrogen bonds are formed between BVNP‐0187 and NGF residues Asn43, Asn45, and Trp99 to stabilize within the loop II/IV cleft and measured differences of electrostatic potential were found at the BVNP‐0187 docking domain (Fig. 3).
Figure 3.
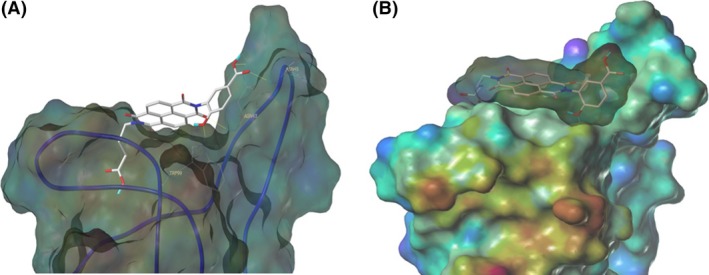
Schematic representation of BVNP‐0187 docked to NGF as determined by molecular modeling. Flexible theoretical docking experiments predict that the bivalent naphthalimide scaffold is suited to bind at the loop II/IV cleft of NGF. (A) Hydrogen bonds are represented at residues Asn43, Asn45, and Trp99 of NGF to stabilize BVNP‐0187 within the loop II/IV cleft. (B) Differences in electrostatic potential at the docking domain. Blue areas represent electron‐poor regions where as a gradient to red represents electron‐rich regions.
Table 1.
Residue description of bivalent naphthalimide analogs
| Compound | ‐R1 | ‐R2 | ‐R3 | ‐R4 |
|---|---|---|---|---|
| BVNP‐0182 | (CH2)3CO2H | (CH2)3CO2H | ||
| BVNP‐0183 |
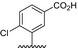
|
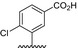
|
||
| BVNP‐0184 |
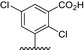
|
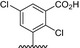
|
||
| BVNP‐0185 |
|
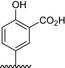
|
||
| BVNP‐0186 |
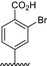
|
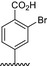
|
||
| BVNP‐0187 |
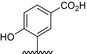
|
(CH2)3CO2H | ||
| BVNP‐0188 |
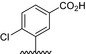
|
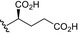
|
||
| BVNP‐0189 |
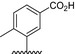
|
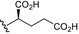
|
||
| BVNP‐0190 |
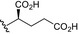
|
CO2H | ||
| BVNP‐0191 |
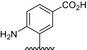
|
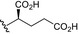
|
||
| BVNP‐0192 |
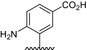
|
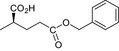
|
||
| BVNP‐0193 |
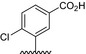
|
CO2H | ||
| BVNP‐0194 | (CH2)3CO2H | CO2H | ||
| BVNP‐0195 |
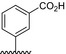
|
CO2H | ||
| BVNP‐0196 |
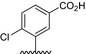
|
Cl | ||
| BVNP‐0197 |
|
NO2 | ||
| BVNP‐0198 |
|
CO2Me |
Analogs were identified as specific binders of NGF using SPR analysis, and were shown to exhibit inhibitory properties in NGF‐TrkA‐mediated signaling. Analog series were synthesized around a bivalent naphthalimide scaffold with varied side chains substitutions at –R1, and –R2 or –R3 and –R4 (Fig. 1).
Affinity of bivalent naphthalimide compounds to immobilized NGF
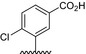
The affinity of bivalent naphthalimide analogs with a total score from flexible docking >3 (~mmol/L binding affinity) was determined using a two‐site steady‐state analysis to immobilized NGF. A concentration series ranging from 0.70 μmol/L to 200 μmol/L was injected over immobilized NGF for a 60 sec contact time followed by a 30 sec dissociation phase. Concentration values were determined based on results from preliminary binding experiments in which bivalent naphthalimide compounds reached near‐saturation levels of the immobilized NGF on the sensor surface. The response obtained from each bivalent naphthalimide sample was plotted against concentration using the BIA evaluation software (Version 2.0) and was evaluated using a two‐site binding model as previously described (Kennedy et al. 2016). 17 bivalent naphthalimide analogs were identified as specific binders of NGF (Fig. 4) with binding affinities ranging from 2.63 μmol/L to 118.10 μmol/L. The 17 analogs showed no significant binding to immobilized TrkA, nor to other neurotrophin family members immobilized on the sensory surface, therefore described specific for NGF (data not shown). Structures of these analogs are summarized in Table 1. Three previously reported compounds, ALE‐0540, PD 90780 and Ro 08‐2750 were also screened to determine the binding affinities (KD) for NGF. The KD of ALE‐0540, PD 90780 and Ro 08‐2750 were determined to be 49.71 μmol/L, 35.83 μmol/L and 32.39 μmol/L, respectively, and none showed binding to immobilized TrkA. The total binding score measured during flexible docking was related to the binding affinity measured with SPR binding (Fig. 5), suggesting binding to immobilized NGF is occurring similar to theoretical docking experiments.
Figure 4.
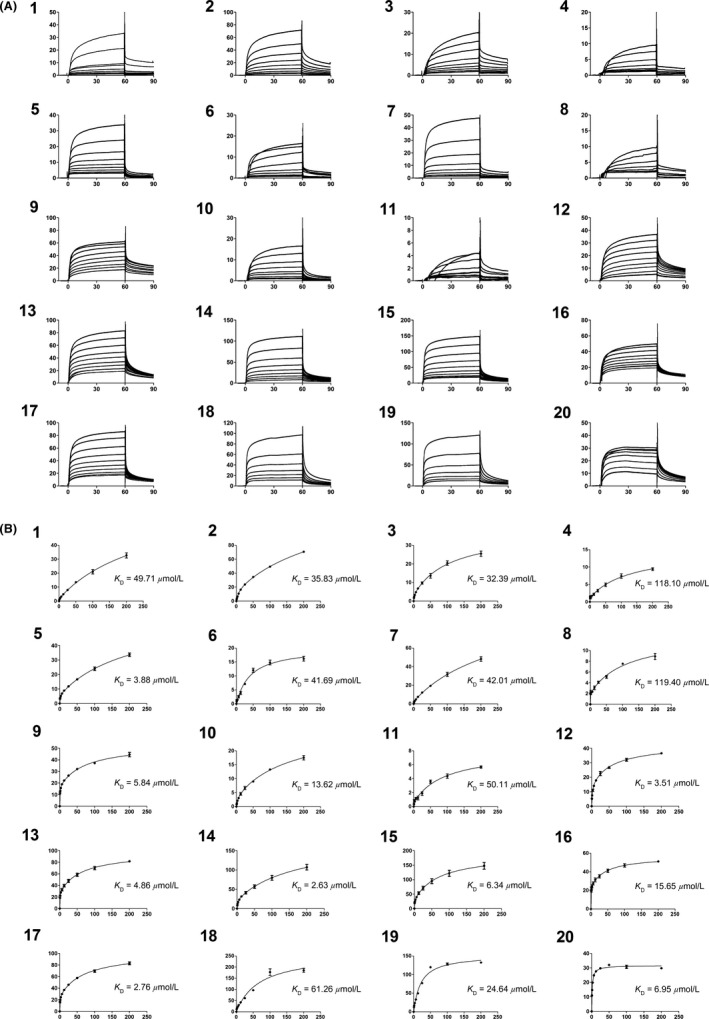
SPR subtracted sensograms and specific binding curves for the 17 bivalent naphthalimide analog found to be specific binders of NGF, as well as three previously reported compounds – ALE‐0540, PD 90780 and Ro 08‐2750 (1–3, respectively). 4–20 represent BVNP‐0182‐BVNP‐0198, respectively. Data points represent the mean of triplicate measure. Error bars represent the standard error of the mean with an n = 3. (A) Blank subtracted sensograms which describe the association and dissociation of each compound. In each sensogram, the x‐axis represents time. 0–60 sec represents the association of the compound to NGF; at 60 sec the analyte flow stops and a dissociation period is represented from 60 to 90 sec prior to chip regeneration at 90 sec. Y axis represents the response of binding in response units (RU). 1 RU = 1 pg/mm2. The lines represent small molecule concentrations, from top to bottom: 200 μmol/L, 100 μmol/L, 50 μmol/L, 25 μmol/L, 12.5 μmol/L, 6.25 μmol/L, 3.13 μmol/L, 1.56 μmol/L and 0.78 μmol/L respectively. (B) Two‐site steady‐state binding models describing the specific binding of each compound to NGF. X axis represents concentration (μmol/L) and Y axis represents subtracted response (response from active flow cell minus response from reference flow cell) (RU). Each data point represents the subtracted response from the saturated association phase (last 10 sec) of the subtracted sensograms from each compound concentration (refer to A). Binding affinity (KD) for each compound was calculated using the BIA evaluation software using a 2:1 binding model.
Figure 5.
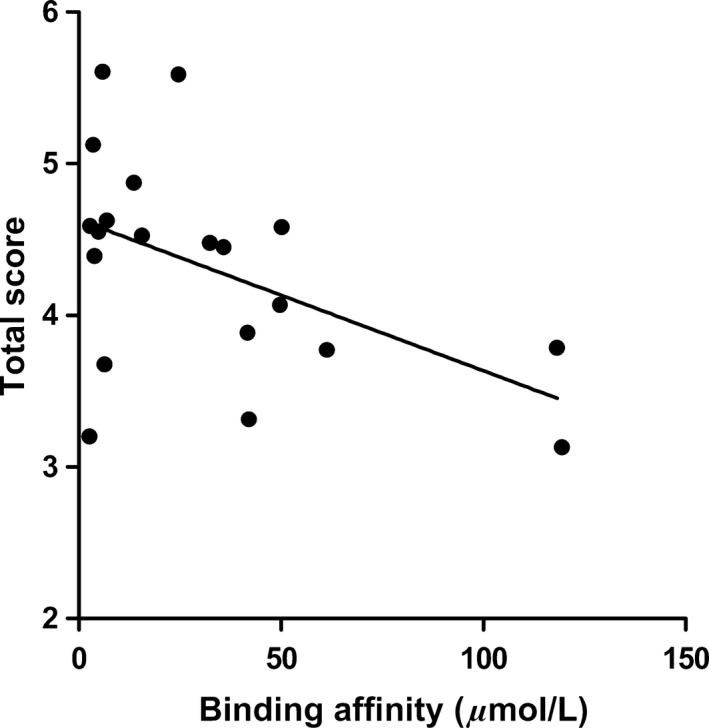
The relationship between total score calculated during molecular modeling flexible docking and the KD measured by SPR spectroscopy for the 17 bivalent naphthalimide analogs and three previously reported compounds (ALE‐0540, PD 90780 and Ro 08‐2750). X‐axis: KD (μmol/L) measured using SPR – Data points represent the mean of triplicate trials. A concentration range (0.70–200 μmol/L) of each analog was injected over immobilized NGF. Y‐axis: total score (−log(KD)) measured during flexible docking. (F(1,18) = 5.807; P = 0.0269; R 2 = 0.2439).
Inhibitory effects of bivalent naphthalimide analogs on NGF‐dependent PC12 cell differentiation
In order to assess the toxicity of the bivalent naphthalimide analogs in vitro, PC12 cells were exposed to 100 μmol/L of each compound for 72 h in the absence of NGF. The cell count did not significantly alter from the control conditions (Fig. 6) and the morphology of PC12 cells was also unaltered following the 72 h exposure.
Figure 6.
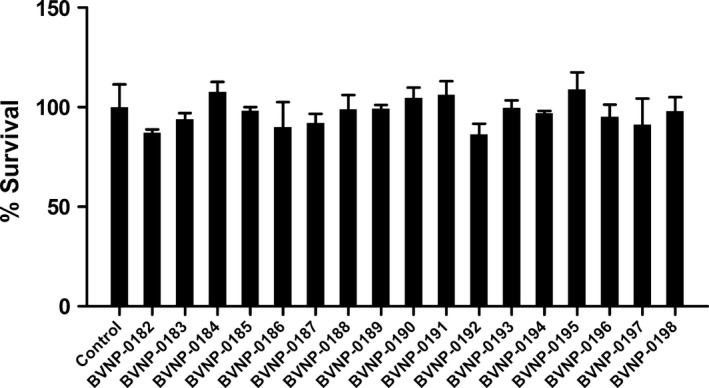
In vitro toxicity of bivalent naphthalimide analog series. PC12 cells were incubated in the absence (control) or presence of 100 μmol/L of each analog for 72 h. The average control cell count per well was considered 100% viability. Each analog‐treated sample is represented by the average percentage of the control cell count. Standard error of the mean count is shown with error bars (n = 3). Treatment of PC12 cells with the analogs shown no significant difference in cell count (P > 0.05) implicating no toxic effect of the analogs in vitro.
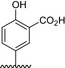
The inhibitory effects of the 17 bivalent naphthalimide analogs which were found to be specific NGF binders were tested in the NGF‐dependent PC12 cell differentiation assay along with ALE‐0540, PD 90780 and Ro 08‐2750 (Table 2). The three previously reported compounds demonstrated IC50 values of 2.44 μmol/L, 33.22 μmol/L, and 11.96 μmol/L, respectively (Table 2). Of the 17 bivalent naphthalimide analogs, three compounds demonstrated inhibitory effects more efficient than previously reported NGF‐inhibitors, which we defined as an IC50 < 2 μmol/L. BVNP‐0188 and BVNP‐0198 demonstrated IC50 values of 1.70 μmol/L and 1.08 μmol/L, respectively. The most efficient analog in NGF‐dependent PC12 cell differentiation inhibition was BVNP‐0197 which demonstrated an IC50 value of 89.5 nmol/L (Fig. 7). BVNP‐0197 has a rigid backbone structure (IPUAC name: 3,10,17‐Triazahexacyclo[13.6.2.02,10.04,9.012,22.019,23]tricosa‐1(21),2,4,6,12,14,19,22‐nonaene‐11,16,18‐trione moiety) and contains a 2‐chloro‐5‐benzoic acid residue in –R3 and nitro group occupying –R4. Flexible docking molecular modeling experiments predicted four hydrogen bonds at NGF residues Asn43, Thr92 and Trp99 to stabilize BVNP‐0197 within the loop II/IV cleft, as well as measured electrostatic potential differences at the docking domain (Fig. 8). A total binding score of 5.5898 was measured for BVNP‐0197 docking to the loop II/IV cleft, estimating a binding affinity of low micromolar range to NGF. Docking of BVNP‐0197 to proNGF measured a binding score of 3.1542, estimating a binding affinity of millimolar range. This change in binding affinity is marked by a decrease in hydrophobic contacts and a reduction in hydrogen bonds between BVNP‐0197 and proNGF as compared to NGF.
Table 2.
Inhibitory effects of novel and previously reported compounds
| Compound | IC50 PC12 neurite outgrowth |
|---|---|
| ALE‐0540 | 2.44 μmol/L |
| PD 90780 | 33.22 μmol/L |
| Ro 08‐2750 | 11.96 μmol/L |
| BVNP‐0182 | >100 μmol/L |
| BVNP‐0183 | 16.86 μmol/L |
| BVNP‐0184 | >100 μmol/L |
| BVNP‐0185 | 11.08 μmol/L |
| BVNP‐0186 | 15.18 μmol/L |
| BVNP‐0187 | 5.45 μmol/L |
| BVNP‐0188 | 1.70 μmol/L |
| BVNP‐0189 | >100 μmol/L |
| BVNP‐0190 | 4.27 μmol/L |
| BVNP‐0191 | 4.33 μmol/L |
| BVNP‐0192 | 2.46 μmol/L |
| BVNP‐0193 | 7.09 μmol/L |
| BVNP‐0194 | 5.35 μmol/L |
| BVNP‐0195 | 6.50 μmol/L |
| BVNP‐0196 | 5.37 μmol/L |
| BVNP‐0197 | 0.089 μmol/L |
| BVNP‐0198 | 1.08 μmol/L |
A PC12 cell‐based neurite outgrowth assay was used to score the relative inhibitory effects of each bivalent naphthalimide analog and three previously reported compounds (ALE‐0540, PD 90780 and Ro 08‐2750) in the presence of 500 pM NGF for 72 h. PC12 cells were treated with a concentration range from 100 μmol/L to 31.6 nmol/L. Each trial condition was completed in quadruplicate wells, replicated with an n = 3. Bold values represent analogues with a higher inhibitory property than previously reported compounds.
Figure 7.
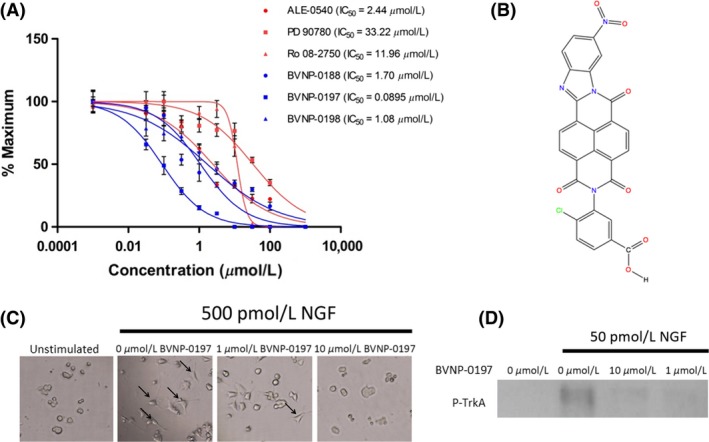
BVNP‐0197 demonstrated inhibitory effects in the nanomolar range in the PC12 differentiation assay. (A) Detailed dose–response experiments were conducted for all 17 bivalent naphthalimide compounds and three previously reported compounds. Previously reported compounds ALE‐0540, PD 90780 and Ro 08‐2750 were determined to have IC 50 values of 2.44 μmol/L, 33.22 μmol/L and 11.96 μmol/L, respectively. Three bivalent naphthalimide compounds were shown to have inhibitory effects more efficient than the previously reported compounds. BVNP‐0188, and BVNP‐0198 were determined to have IC 50 values of 1.70 μmol/L and 1.08 μmol/L, respectively, whereas BVNP‐0197 was found to have an IC 50 value of 89.5 nmol/L. Previously reported compounds are depicted in red where novel compounds are in blue. Data points represent a mean value of quadruplicate results with an n = 3. Error bars are represented as standard error of the mean with an n = 3. (B) The chemical structure of BVNP‐0197. IUPAC name 4‐Chloro‐3‐{6‐nitro‐11,16,18‐trioxo‐3,10,17‐triazahexacyclo[13.6.2.02,10.04,9.012,22.019,23]tricosa‐1(21),2,4,6,8,12,14,19,22‐nonaen‐17‐yl}benzoic acid. (C) BVNP‐0197 functionally inhibits NGF‐mediated PC12 cell process formation. In the presence of 500 pM NGF, PC12 cells were treated with varying concentrations of BVNP‐0197 (100– 0 μmol/L). Images from 0, 1 and 100 μmol/L are shown. In the presence of NGF and the absence of inhibitor, PC12 cells underwent cell process formation (arrows). Process formation significantly decreased with the addition of increasing concentrations of BVNP‐0197. An unstimulated (0 pM NGF) sample was prepared to demonstrate the NGF‐dependence in cell process formation. (D) BVNP‐0197 inhibits TrkA‐mediated phosphorylation in PC12 cells exposed to 50 pM NGF for 1 h at 37°C.
Figure 8.
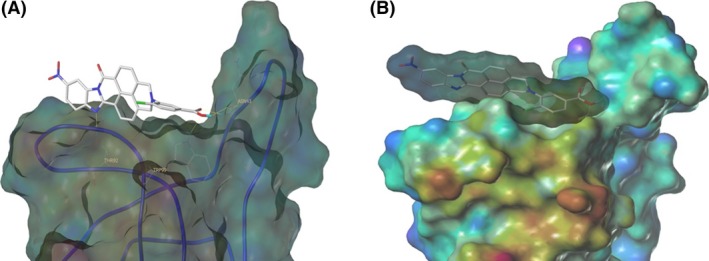
Schematic representation of BVNP‐0197 docked to NGF as determined by molecular modeling. Flexible theoretical docking experiments predict that the bivalent naphthalimide scaffold is suited to bind at the loop II/IV cleft of NGF. (A) Four hydrogen bonds are represented at residues Asn43, Thr92 and Trp99 of NGF to stabilize BVNP‐1097 within the loop II/IV cleft. (B) The differences in electrostatic potential at the docking domain. Blue areas represent electron‐poor regions where a gradient to red represents electron‐rich regions.
To test the inhibitory effect of BVNP‐0197 on NGF‐mediated signaling, the phosphorylation status of TrkA was assessed when PC12 cells were incubated with control media, NGF or NGF/BVNP‐0197. Cells were treated with their respective conditions for 15 min prior to cell lysis. The phosphorylated TrkA signal for NGF‐treated cells was distinctly increased over media control‐treated cells (Fig. 7). This increase in response was diminished by both 10 μmol/L and 1 μmol/L BVNP‐0197 incubated with NGF for 1 h at 37°C (Fig. 7). These results confirm that BVNP‐0197 inhibits NGF‐mediated TrkA signaling in vitro and may be targeted for further therapeutic development.
Inhibitory effects of bivalent naphthalimide analogs
To determine the inhibitory properties of the 17 bivalent naphthalimide analogs using SPR spectroscopy, a dilution series of each analog (3.16 nmol/L to 316.23 μmol/L) was incubated for 1 h with 10 nmol/L NGF prior to a 60 sec injection over immobilized TrkA on the sensor surface. Each injection over immobilized TrkA was followed by a 120 sec dissociation phase prior to regeneration before the next injection. A response for each sample was assessed using SPR spectroscopy and compared to a control sample of 10 nmol/L NGF with no analogpresent. Dose response curves were generated to determine IC50 values using the BIA evaluation software (Version 2.0) and were evaluated, using a one‐site steady‐state‐binding model. The 17 analogs demonstrated IC50 values ranging from 18.5 nmol/L to 81.40 μmol/L, which were related to the IC50 values calculated from neurite outgrowth assays (Fig. 9).
Figure 9.
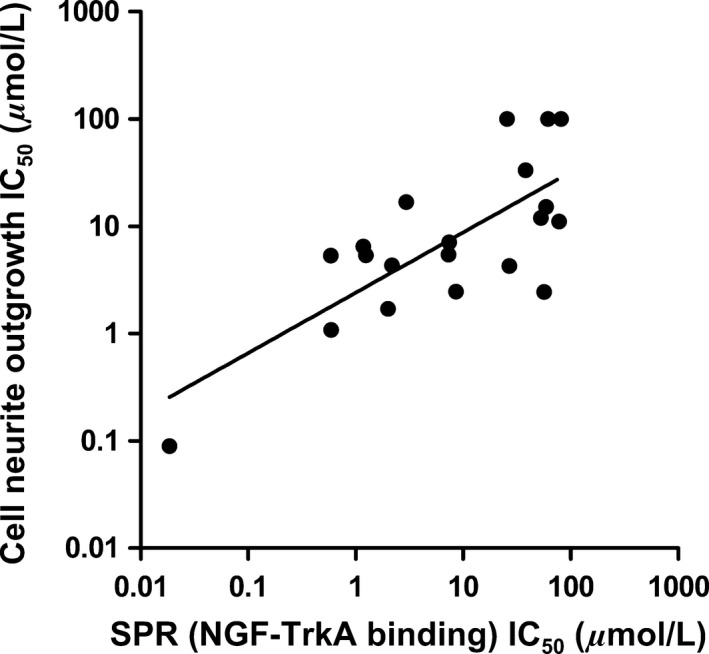
The relationship of the IC 50 measured by SPR spectroscopy for the 17 bivalent naphthalimide analogs and the three previously reported compounds (ALE‐0540, PD 90780 and Ro 08‐2750) and the IC 50 measured using the PC12 cell NGF‐dependent differentiation assay. X‐axis: IC 50 (μmol/L) measured using the SPR–Various concentrations of analog were incubated for 1 h with 10 nmol/L NGF prior to injection over immobilized TrkA. Y‐axis: IC 50 (μmol/L) measured following PC12 cell treatment of increasing analog concentration and 500 pM NGF for 72 h. Each data point represents the mean of triplicate results. (F(1,18) = 22.69; P = 0.0002; R 2 = 0.5576).
Discussion
In the present study, we identified a novel bivalent naphthalimide compound, BVNP‐0197, with a higher NGF‐inhibitory efficiency than previously described compounds utilizing cell‐free SPR technology and cell‐based PC12 neurite outgrowth and TrkA phosphorylation assays. BVNP‐0197 is also predicted to bind to NGF in a novel binding domain found in the loop II/IV cleft.
Historically, most drug discovery efforts in NGF‐mediated pain signaling therapeutics have been directed toward identifying small molecules which inhibit signaling through the receptor TrkA. Small molecules such as K252a have been reported, however, pharmacological specificity has limited the advancement of such compounds into clinical trials (Watson et al. 2008). NGF‐mimetic peptides have also been evaluated, however no peptide‐based strategies has advanced into clinical trials (Eibl et al. 2012). Mimetic strategies have seen success in vivo neuropathic pain models, although their success has been attributed to the beneficial effects of supplementing NGF in maintaining opiate receptors and may see potential in the treatment of peripheral neuropathies (Colangelo et al. 2008).
Success of targeting a protein in a protein‐receptor system was first demonstrated with the approval of anti‐tumor necrosis factor (TNF) antibodies for the therapeutic treatment of rheumatoid arthritis (Elliot et al. 2008) and Crohn's disease (Deželak et al. 2016; Tolentino et al. 2016). Similar strategies have been explored with anti‐NGF antibodies, such as Tanezumab, for the treatment of NGF‐mediated pain. Although seemingly effective, anti‐NGF antibodies are still facing several therapeutic drawbacks with potential serious side effects involving autoimmune responses. Antibody‐mediated pain management strategies are also highly specific and are restricted from crossing the blood brain barrier to the central nervous system (Bannwarth and Kostine 2014).
Similar strategies of NGF inhibition have been introduced using small molecule NGF‐inhibitors. Small molecules such as ALE‐0540 (Owolabi et al. 1999), Ro 08‐2750 (Niederhauser et al. 2000), PD 90780 (Colquhoun et al. 2004), and PQC‐083 (Eibl et al. 2013a) have demonstrated efficiency in binding and inhibiting NGF from interacting with its receptors. Another small molecule Y1036 (Eibl et al. 2010), has been introduced as a multipotent neurotrophin inhibitor by binding to not only NGF, but also BDNF and inhibiting their action on receptors. Compounds such as ALE‐0540, Ro 08‐2750, PD 90780 and PQC‐083 have been shown to bind to the loop I/IV cleft and sufficiently alter the molecular topology in a manner such that NGF can no longer bind efficiently to its receptors (Eibl et al. 2010, 2013a). This loop region of NGF has the highest degree of variance among all the neurotrophins and is responsible for the selectivity in receptor selection among the neurotrophin family members. Alternatively Y1036 has been described to bind at the neurotrophin hydrophobic interface, the region conserved among all neurtrophin family members, and therefore loses the specificity for NGF (Eibl et al. 2013b).
SPR spectroscopy has been used to study biomolecular interactions and has proven effective in determining how a potential drug therapeutic will interact with their target receptor(s). Previously used screening strategies for NGF inhibitors included the use of radioisotopes such as 125I (Owolabi et al. 1999; Colquhoun et al. 2004; Eibl et al. 2013a). SPR is advantageous over labeling strategies because it eliminates the need of fluorescent reporter molecules or radioisotope tags which adds the concern of altering the molecular interactions being examined. Recently, there has been a relationship established between the inhibitory potential of NGF‐inhibiting small molecules described using 125I‐NGF analysis and the high affinity binding measured by SPR techniques (Kennedy et al. 2016). This correlation between inhibitory potential and binding affinity introduced SPR as a viable option for efficient and rapid screening of small molecule entities for identification of novel NGF‐inhibitors.
The present study introduces SPR spectroscopy for screening of a novel bivalent naphthalimide analog series to determine a novel NGF‐inhibitor, BVNP‐0197, with a higher potency than previously reported molecules. Molecular modeling techniques were used to identify lead analogs that targeted an area on NGF specific for TrkA docking, in addition to reporting that NGF binding is of higher affinity than binding to proNGF. These results are of particular importance as it would be expected that the bivalent naphthalimide analogs bind to both proNGF and NGF in vivo. This model, supported by others characterizing small molecule binding to NGF/proNGF (Sheffield et al. 2016) suggests that the lead analogs would bind both proteins, however have a higher affinity for NGF. SPR screening of these lead analogs revealed 17 novel compounds which specifically bind to NGF with binding affinities between 3 μmol/L ‐ 119 μmol/L. This study also provides novel evidence that the bivalent naphthalimide analogs described, as well as the three historic compounds examined (ALE‐0540, Ro 08‐2750, and PD 90780) are true NGF‐inhibitors as they do not bind to TrkA to inhibit NGF‐TrkA‐mediated signaling. The in vitro cell count and morphology of PC12 cells treated with the bivalent naphthalimide analog series, including BVNP‐0197, did not significantly differ from untreated cells after 72 h in culture. The NGF‐dependent cell assay determined that three analogs had higher efficiency in NGF‐ inhibition than previously reported molecules (IC50 < 2 μmol/L). BVNP‐0188, BVNP‐0197 and BVNP‐0198 demonstrated IC50 values calculated from neurite outgrowth assays at 1.70 μmol/L, 89.5 nmol/L and 1.08 μmol/L respectively. IC50 values determined using PC12 neurite outgrowth assays were also related to the IC50 values determined for each analog using SPR binding assays (Fig. 9) demonstrating that SPR could be utilized in future therapeutic screening strategies prior to entering in vitro studies.
It is expected that BVNP‐0197 may have several potential applications in the field of neurotrophin research. For example, BVNP‐0197 may serve as a therapeutic with the ability to inhibit NGF‐TrkA‐mediated pain signaling since the predicted docking domain interferes with TrkA binding. BVNP‐0197 and the other bivalent naphthalimide analogs examined in this study may also serve as evidence for the development of future compounds that inhibit neurotrophin‐mediated pain and other NGF‐related disorders. Due to similar homology, it is expected that BVNP‐0197 would also bind to proNGF and disrupt binding to TrkA. Molecular modeling results suggest that docking of BVNP‐0197 to proNGF would occur at a lower (millimolar) affinity than docking to NGF, however, further investigation with SPR, or using a cell‐based assay would be required to determine if BVNP‐0197 holds any inhibitory action for proNGF‐TrkA binding. SPR analysis of the bivalent naphthalimide analog–NGF interaction may allow for the development of compounds with varied binding kinetics thus leading to more efficient therapeutic options for NGF dysregulation. Further analysis with animal models will be required to validate BVNP‐0197 as a potential therapeutic in clinical use for pathologies associated to NGF dysregulation, such as osteoarthritic pain and neurodegenerative diseases.
Authorship Contributions
Participated in research design: Kennedy, Laamanen, M. Ross, Vohra, Boreham, Scott, G. Ross.Conducted experiments: Kennedy, Laamanen M. Ross.
Performed data analysis: Kennedy, Laamanen.Wrote or contributed to the writing of the manuscript: Kennedy, Laamanen, M. Ross Vohra, Boreham, Scott, G. Ross.
Disclosures
None declared.
Kennedy A. E., Laamanen C. A., Ross M. S., Vohra R., Boreham D. R., Scott J. A., Ross G. M.. Nerve growth factor inhibitor with novel‐binding domain demonstrates nanomolar efficacy in both cell‐based and cell‐free assay systems. Pharma Res Per, 5(5), 2017, e00339 https://doi.org/10.1002/prp2.339
References
- Bannwarth B, Kostine M (2014). Targeting Nerve Growth Factor (NGF) for Pain Management: What Does the Future Hold for NGF Antagonists? Drugs 74: 619–626. [DOI] [PubMed] [Google Scholar]
- Barker PA (2007). High affinity not in the vicinity? Neuron 53: 1–4. [DOI] [PubMed] [Google Scholar]
- Beglova N, Maliartchouk S, Ekiel I, Zaccaro MC, Saragovi HU, Gehring K (2000). Design and solution structure of functional peptide mimetics of nerve growth factor. J Med Chem 43: 3530–3540. [DOI] [PubMed] [Google Scholar]
- Bienenstock J, Tomioka M, Matsuda H, Stead RH, Quinonez G, Simon GT, et al. (1987). The role of mast cells in inflammatory processes: evidence for nerve/mast cell interactions. Int Arch Allergy Appl Immunol 82: 238–243. [DOI] [PubMed] [Google Scholar]
- Bracci‐Laudiero L, Aloe L, Caroleo MC, Buanne P, Costa N, Starace G, et al. (2005). Endogenous NGF regulates CGRP expression in human monocytes, and affects HLA‐DR and CD86 expression and IL‐10 production. Blood 106: 3507–3514. [DOI] [PubMed] [Google Scholar]
- Brodie C (1995). Platelet activating factor induces nerve growth factor production by rat astrocytes. Neurosci Lett 186: 5–8. [DOI] [PubMed] [Google Scholar]
- Cahill CM, Dray A, Coderre TJ (2003). Intrathecal nerve growth factor restores opioid effectiveness in an animal model of neuropathic pain. Neuropharmacology 45: 543–552. [DOI] [PubMed] [Google Scholar]
- Colangelo AM, Bianco MR, Vitagliano L, Cavaliere C, Cirillo G, De Gioia L, et al. (2008). A New Nerve Growth Factor‐Mimetic Peptide Active on Neuropathic Pain in Rats. J Neurosci 28: 2698–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun A, Lawrance GM, Shamovsky IL, Riopelle RJ, Ross GM (2004). Differential activity of the nerve growth factor (NGF) antagonist PD90780 [7‐(benzolylamino)‐4,9‐dihydro‐4‐methyl‐9‐oxo‐pyrazolo[5,1‐b]quinazoline‐2‐carboxylic acid] suggests altered NGF‐p75NTR interactions in the presence of TrkA. J Pharmacol Exp Ther 310: 505–511. [DOI] [PubMed] [Google Scholar]
- Cooper MA (2002). Optical biosensors in drug discovery. Nat Rev Drug Discov 1: 515–528. [DOI] [PubMed] [Google Scholar]
- Deželak M, Repnik K, Koder S, Ferkolj I, Potočnik U (2016). A Prospective Pharmacogenomic Study of Crohn's Disease Patients during Routine Therapy with Anti‐TNF‐α Drug Adalimumab: contribution of ATG5, NFKB1, and CRP Genes to Pharmacodynamic Variability. OMICS 20: 296–309. [DOI] [PubMed] [Google Scholar]
- Dyck PJ, Peroutka S, Rask C, Burton E, Baker MK, Lehman KA, et al. (1997). Intradermal recombinant human nerve growth factor induces pressure allodynia and lowered heat‐pain threshold in humans. Neurology 48: 501–505. [DOI] [PubMed] [Google Scholar]
- Eibl JK, Chapelsky SA, Ross GM (2010). Multipotent neurotrophin antagonist targets brain‐derived neurotrophic factor and nerve growth factor. J Pharmacol Exp Ther 332: 446–454. [DOI] [PubMed] [Google Scholar]
- Eibl JK, Strasser BC, Ross GM (2012). Structural, biological, and pharmacological strategies for the inhibition of nerve growth factor. Neurochem Int 61: 1266–1275. [DOI] [PubMed] [Google Scholar]
- Eibl JK, Abdallah Z, Kennedy AE, Scott JA, Ross GM (2013a) Affinity Crosslinking of Y1036 to Nerve Growth Factor Identifies Pharmacological Targeting Domain for Small Molecule Neurotrophin Antagonists. Neurosci Med 2013:290–298. [Google Scholar]
- Eibl JK, Strasser BC, Ross GM (2013b). Identification of novel pyrazoloquinazolinecarboxilate analogues to inhibit nerve growth factor in vitro. Eur J Pharmacol 708: 30–37. [DOI] [PubMed] [Google Scholar]
- Elliot MJ, Maini RN, Feldmann M, Long‐Fox A, Charles P, Katasikis P, et al. (2008). Treatment of rheumatoid arthritis with chimeric monoclonal antibodies to tumor necrosis factor alpha. Arthritis Rheum 58: S92–S101. [DOI] [PubMed] [Google Scholar]
- Feng D, Kim T, Ozkan E, Light M, Torkin R, Teng KK, et al. (2010). Molecular and structural insight into proNGF engagement of p75NTR and sortilin. J Mol Biol 396: 967–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsell P, Almqvist H, Hillertz P, Akerud T, Otrocka M, Eisele L, et al. (2013). The use of TrkA‐PathHunter assay in high‐throughput screening to identify compounds that affect nerve growth factor signaling. J Biomol Screen 18: 659–669. [DOI] [PubMed] [Google Scholar]
- Fraser S, Cameron M, O'Connor E, Schwickart M, Tanen M, Ware M (2014). Next generation ligand binding assays‐review of emerging real‐time measurement technologies. AAPS J 16: 914–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao J, Ebendal T, Xu X, Wiesenfeld‐Hallin Z, Eriksdotter Jönhagen M (2000). Intracerebroventricular infusion of nerve growth factor induces pain‐like response in rats. Neurosci Lett 286: 208–212. [DOI] [PubMed] [Google Scholar]
- He X‐L, Garcia KC (2004). Structure of nerve growth factor complexed with the shared neurotrophin receptor p75. Science 304: 870–875. [DOI] [PubMed] [Google Scholar]
- Hefti FF, Rosenthal A, Walicke PA, Wyatt S, Vergara G, Shelton DL, et al. (2006). Novel class of pain drugs based on antagonism of NGF. Trends Pharmacol Sci 27: 85–91. [DOI] [PubMed] [Google Scholar]
- Jain AN (1996). Scoring noncovalent protein–ligand interactions: a continuous differentiable function tuned to compute binding affinities. J Comput Aided Mol Des 10: 427–440. [DOI] [PubMed] [Google Scholar]
- Kc R, Li X, Kroin JS, Liu Z, Chen D, Xiao G, et al. (2016). PKCδ null mutations in a mouse model of osteoarthritis alter osteoarthritic pain independently of joint pathology by augmenting NGF/TrkA‐induced axonal outgrowth. Ann Rheum Dis. https://doi.org/10.1136/annrheumdis-2015-208444. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kennedy AE, Sheffield KS, Eibl JK, Murphy MB, Vohra R, Scott JA, et al. (2016). A Surface Plasmon Resonance Spectroscopy Method for Characterizing Small‐Molecule Binding to Nerve Growth Factor. J Biomol Screen 21: 96–100. [DOI] [PubMed] [Google Scholar]
- Lewin GR, Rueff A, Mendell LM (1994). Peripheral and central mechanisms of NGF‐induced hyperalgesia. Eur J Neurosci 6: 1903–1912. [DOI] [PubMed] [Google Scholar]
- Malerba F, Paoletti F, Bruni Ercole B, Materazzi S, Nassini R, Coppi E, et al. (2015). Functional Characterization of Human ProNGF and NGF Mutants: identification of NGF P61SR100E as a “Painless” Lead Investigational Candidate for Therapeutic Applications. PLoS ONE 10: e0136425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald NQ, Lapatto R, Murray‐Rust J, Gunning J, Wlodawer A, Blundell TL (1991). New protein fold revealed by a 2.3‐A resolution crystal structure of nerve growth factor. Nature 354: 411–414. [DOI] [PubMed] [Google Scholar]
- Mir TA, Shinohara H (2013). Two‐dimensional surface plasmon resonance imager: an approach to study neuronal differentiation. Anal Biochem 443: 46–51. [DOI] [PubMed] [Google Scholar]
- Niederhauser O, Mangold M, Schubenel R, Kusznir EA, Schmidt D, Hertel C (2000). NGF ligand alters NGF signaling via p75(NTR) and trkA. J Neurosci Res 61: 263–272. [DOI] [PubMed] [Google Scholar]
- Owolabi JB, Rizkalla G, Tehim A, Ross GM, Riopelle RJ, Kamboj R, et al. (1999). Characterization of antiallodynic actions of ALE‐0540, a novel nerve growth factor receptor antagonist, in the rat. J Pharmacol Exp Ther 289: 1271–1276. [PubMed] [Google Scholar]
- Ross GM, Shamovsky IL, Lawrance G, Solc M, Dostaler SM, Weaver DF, et al. (1998). Reciprocal modulation of TrkA and p75NTR affinity states is mediated by direct receptor interactions. Eur J Neurosci 10: 890–898. [DOI] [PubMed] [Google Scholar]
- Samaranayake H, Wirth T, Schenkwein D, Räty JK, Ylä‐Herttuala S (2009). Challenges in monoclonal antibody‐based therapies. Ann Med 41: 322–331. [DOI] [PubMed] [Google Scholar]
- Schnitzer TJ, Lane NE, Birbara C, Smith MD, Simpson SL, Brown MT (2011). Long‐term open‐label study of tanezumab for moderate to severe osteoarthritic knee pain. Osteoarthritis Cartilage 19: 639–646. [DOI] [PubMed] [Google Scholar]
- Sheffield KSA, Vohra R, Scott JA, Ross GM (2016). Using surface plasmon resonance spectroscopy to characterize the inhibition of NGF‐p75(NTR) and proNGF‐p75(NTR) interactions by small molecule inhibitors. Pharmacol Res 103: 292–299. [DOI] [PubMed] [Google Scholar]
- Skaper SD, Pollock M, Facci L (2001). Mast cells differentially express and release active high molecular weight neurotrophins. Brain Res Mol Brain Res 97: 177–185. [DOI] [PubMed] [Google Scholar]
- Svensson P, Cairns BE, Wang K, Arendt‐Nielsen L (2003). Injection of nerve growth factor into human masseter muscle evokes long‐lasting mechanical allodynia and hyperalgesia. Pain 104: 241–247. [DOI] [PubMed] [Google Scholar]
- Tolentino YFM, Elia PP, Fogaça HS, Carneiro AJV, Zaltman C, Moura‐Neto R, et al. (2016). Common NOD2/CARD15 and TLR4 Polymorphisms Are Associated with Crohn's Disease Phenotypes in Southeastern Brazilians. Dig Dis Sci 61: 2636–2647. [DOI] [PubMed] [Google Scholar]
- Torcia M, Bracci‐Laudiero L, Lucibello M, Nencioni L, Labardi D, Rubartelli A, et al. (1996). Nerve growth factor is an autocrine survival factor for memory B lymphocytes. Cell 85: 345–356. [DOI] [PubMed] [Google Scholar]
- Watson JJ, Allen SJ, Dawbarn D (2008). Targeting nerve growth factor in pain: what is the therapeutic potential? BioDrugs 22: 349–359. [DOI] [PubMed] [Google Scholar]
- Wehrman T, He X, Raab B, Dukipatti A, Blau H, Garcia KC (2007). Structural and mechanistic insights into nerve growth factor interactions with the TrkA and p75 receptors. Neuron 53: 25–38. [DOI] [PubMed] [Google Scholar]
- Wild KD, Bian D, Zhu D, Davis J, Bannon AW, Zhang TJ, et al. (2007). Antibodies to nerve growth factor reverse established tactile allodynia in rodent models of neuropathic pain without tolerance. J Pharmacol Exp Ther 322: 282–287. [DOI] [PubMed] [Google Scholar]
- Zahn PK, Subieta A, Park SS, Brennan TJ (2004). Effect of blockade of nerve growth factor and tumor necrosis factor on pain behaviors after plantar incision. J Pain 5: 157–163. [DOI] [PubMed] [Google Scholar]