

Abstract
Metformin is a common co‐medication for many diseases and the victim of clinical drug‐drug interactions (DDIs) perpetrated by cimetidine, trimethoprim and pyrimethamine, resulting in decreased active renal clearance due to inhibition of organic cation transport proteins and increased plasma exposure of metformin. To understand whether area under the plasma concentration–time curve (AUC) increases relate to absorption, in vitro inhibitory potencies of these drugs against metformin transport by human organic cation transporter (OCT) 1, and the apical to basolateral absorptive permeability of metformin across Caco‐2 cells in the presence of therapeutic intestinal concentrations of cimetidine, trimethoprim or pyrimethamine, were determined. Whilst all inhibited OCT1, none enhanced metformin's absorptive permeability (~0.5 × 10−6 cm/sec) suggesting that DDI AUC changes are not related to absorption. Subsequently, to understand whether inhibition of renal transporters are responsible for AUC increases, in vitro inhibitory potencies against metformin transport by human OCT2, multidrug and toxin extrusion (MATE) 1 and MATE2‐K were determined. Ensuing IC 50 values were incorporated into mechanistic static equations, alongside unbound maximal plasma concentration and transporter fraction excreted values, in order to calculate theoretical increases in metformin AUC due to inhibition by cimetidine, trimethoprim or pyrimethamine. Calculated theoretical fold‐increases in metformin exposure confirmed solitary inhibition of renal MATE1 to be the likely mechanism underlying the observed exposure changes in clinical DDIs. Interestingly, clinically observed increases in metformin AUC were predicted more closely when the renal transporter fraction excreted value derived from oral metformin administration, rather than intravenous, was utilized in theoretical calculations, likely reflecting the “flip‐flop” pharmacokinetic profile of the drug.
Keywords: Drug‐drug interaction, MATE1, metformin, OCT2, pharmacokinetics
Abbreviation
- AUC
area under the plasma concentration–time curve
- BCA
bicinchoninic acid
- Cmax
maximum plasma concentration
- DDI
drug‐drug interaction
- ƒe
fraction excreted value
- IC50
inhibitory concentration that produces 50% inhibition
- Ki
absolute inhibition constant
- MATE
multidrug and toxin extrusion
- OCT
organic cation transporter
Introduction
Metformin is a biguanide drug used for the treatment of type 2 diabetes and is becoming a common comedication in patients due to the prevalence of diabetes as a comorbidity in many disease areas. As an organic cation at physiological pH, metformin has minimal passive membrane permeability and therefore requires active transport to cross membranes in order to gain entry into cells. Following absorption, metformin is principally eliminated as unchanged drug via active renal tubular secretion into urine (Graham et al., 2011). From three intravenous human mass balance studies, an overall mean of 88 % of total metformin plasma clearance is attributed to renal elimination (Sirtori et al. 1978; Pentikäinen et al. 1979; Tucker et al. 1981). Additionally, based on metformin renal clearance values reported from eight clinical studies (3 mass balance above and 5 DDI), on average 75 % of metformin renal clearance is due to active transporter processes (deriving a fraction excreted value, ƒe, of 0.66) and 25% due to passive filtration (120 mL/min; GFR × f u = 1) (Somogyi et al. 1987; Wang et al. 2008; Kusuhara et al. 2011; Grün et al. 2013; Müller et al. 2015). The transporters responsible for the active renal elimination of metformin are organic cation transporter (OCT) 2, located on the basolateral membrane of renal proximal tubule epithelial cells, and multidrug and toxin extrusion (MATE) 1/2‐K, located on the apical membrane of tubule epithelial cells, which are responsible for the vectorial uptake of metformin from blood and its efflux into urine, respectively (Song et al. 2008, Tsuda et al. 2009a; Glucophage® label).
Drug‐drug interactions (DDIs) with metformin are of clinical concern as elevated plasma concentrations of metformin are associated with an increased risk of lactic acidosis (Glucophage® label, Stage et al. 2015). DDIs resulting in less than a 2‐fold increase in the plasma exposure (AUC) of metformin have been observed clinically between metformin and cimetidine (Somogyi et al. 1987; Wang et al. 2008), metformin and trimethoprim (Grün et al. 2013; Müller et al. 2015) and metformin and pyrimethamine (Kusuhara et al. 2011), and are all accompanied by a reduction in its active renal elimination attributed to inhibition of renal organic cation transport. Initially, inhibition of OCT2 was deemed responsible for the observed clinical interactions perpetrated by these drugs (Wang et al. 2008). However, the finding that these drugs were more potent inhibitors of MATE transporters compared to OCT2 shifted opinion to inhibition of MATE1 and/or MATE2‐K as the primary cause of the majority of metformin DDIs (Tsuda et al. 2009b; Ito et al. 2012; Hillgren et al. 2013).
Despite this revised knowledge and using mechanistic static equations putting [I]/K i into context with transporter ƒe (Elsby et al. 2012, 2016), it has not so far been possible to reconcile predicted maximum theoretical metformin AUC increases due to renal transporter inhibition with the clinically observed AUC increases, with the former usually over‐predicting the latter using inhibitor K i values reported in the literature. As a consequence of this, and coupled with the observation from AUC profiles of metformin in the presence of interacting drugs that suggest a possible increase in absorption, it is conceivable that an interaction at the level of the intestine may contribute to observed metformin DDIs. Based on studies in polarized Caco‐2 cell monolayers, metformin crosses into enterocytes via transporters, primarily OCT1 located on the apical brush‐border membrane, but once inside the cell has limited basolateral efflux and so is not absorbed transcellularly (Han et al. 2013, 2015). Rather, metformin is hypothesised to undergo absorption via the paracellular route between enterocytes, and as the drug transits along the intestinal tract and luminal concentrations decrease, the transporters such as OCT1 reverse direction and release metformin down its concentration gradient back into the gut lumen for further absorption (Proctor et al. 2008). It is this depot phenomenon that likely gives rise to the slow absorption of metformin resulting in it exhibiting “flip‐flop” pharmacokinetics (Yáñez et al. 2011).
In this study, we sought to investigate the in vitro inhibitory potential of cimetidine, trimethoprim and pyrimethamine against metformin transport mediated by the organic cation transporters OCT1, OCT2, MATE1 and MATE2‐K in order to provide IC50 (K i) values that could be used in mechanistic static predictions of theoretical exposure change due to DDI. Furthermore, we assessed whether these same perpetrator compounds could enhance the absorptive permeability of metformin in Caco‐2 cell monolayers, using substrate and inhibitor concentrations relevant to expected clinical intestinal concentrations, in order to investigate whether an intestinal interaction may contribute towards observed DDIs.
Materials and Methods
Materials
Metformin, cimetidine, trimethoprim, pyrimethamine, ammonium chloride, sodium butyrate, non‐essential amino acids, MES, human serum albumin (HSA), lucifer yellow and HEPES were purchased from Sigma‐Aldrich (Poole, Dorset, UK). [14C]‐Metformin was purchased from American Radiolabeled Chemicals (St Louis, MO) and Optiphase Supermix liquid scintillation cocktail, 24‐well liquid scintillation counting visiplates and 96‐well isoplates were purchased from PerkinElmer Life and Analytical Sciences (Buckinghamshire, UK). Hanks balanced salt solution (Gibco™ HBSS; containing CaCl2 and MgCl2), Dulbecco's modified eagle medium (Gibco™ DMEM; high glucose with GlutaMax and pyruvate), fetal bovine serum (Gibco™; heat inactivated) and mammalian protein extraction reagent (M‐PER) were purchased from Fisher Scientific (Loughborough, UK). All other chemicals, solvents, and reagents were purchased from Fisher Scientific.
Biocoat™ Poly‐D‐lysine 24‐well multiwell plates, human organic cation transporter (OCT) 1 (SLC22A1)‐, OCT2 (SLC22A2)‐, multidrug and toxin extrusion protein (MATE) 1 (SLC47A1)‐, and MATE2‐K (SLC47A2)‐expressing TransportoCells™ and vector control cells were supplied by Corning BV Life Sciences (Amsterdam, The Netherlands). Fasted simulated intestinal fluid (FaSSIF) powder was purchased from Biorelevant (London, UK). Millicell‐96 multiwell cell culture insert plates (with polycarbonate membranes; 0.4 μm pore size, 0.12 cm2 surface area) and Millicell 96‐well transport companion plates were purchased from Millipore (Watford, Hertfordshire, UK). 96‐Well deep well Abgene polypropylene or shallow round‐bottomed polypropylene plates were supplied by Fisher Scientific. Caco‐2 cells (HTB37, supplied at passage number 17) were obtained from American Type Culture Collection.
Time linearity and kinetic determination of the transporter‐mediated uptake of metformin by OCT1, OCT2, MATE1, and MATE2‐K
OCT1, OCT2, MATE1, MATE2‐K and control cell lines were seeded in cell culture medium (consisting of DMEM supplemented with 10% (w/v) fetal bovine serum and 1% (v/v) nonessential amino acids) at 3–4 × 105 cells per well in 24‐well poly‐D‐lysine coated plates to achieve a preassay confluence of typically 80–95%. The media was changed 3–4 h postseeding and the cells were cultured at 37°C, 8% CO2 for 24 h (media for MATE cells, and corresponding control cells, contained 2 mmol/L sodium butyrate). Prior to the assay, cells were washed twice with prewarmed uptake buffer (HBSS containing 10 mmol/L HEPES, pH 7.4) then left to preincubate in warm uptake buffer for 10 min (MATE cells and corresponding control cells were preincubated in warm uptake buffer solution containing 40 mmol/L ammonium chloride for 20 min). After the preincubation period, uptake buffer was removed and the appropriate incubation solutions were added to the wells. Incubation solutions contained ≤1.2% (v/v) DMSO. For time linearity experiments performed in transporter‐expressing cells and corresponding control cells (triplicate wells per condition), [14C]‐metformin (100 μmol/L; 37 kBq/mL) was incubated at 37°C for 1, 2, 3, 5, 10 and 15 min with OCT1, MATE1 or MATE2‐K cells, and 2, 5, 10, 15, 30, and 45 min with OCT2 cells. At the end of the incubation, active transport processes were terminated by removing (via aspiration) the incubation solutions, immediately washing the cells twice with ice cold uptake buffer and then placing plates on ice. Following the wash steps, M‐PER (400 μL) was added to each well and cells were lysed for at least 5 min at 250 rpm on an orbital shaker. An aliquot (300 μL) of cell lysate was added to a white walled, clear bottomed 24‐well visiplate, liquid scintillation cocktail (2 mL) was added, and samples were counted on a Microbeta2 scintillation counter (PerkinElmer) in order to determine the total radioactivity (disintegrations per minute; dpm) taken up in cells. Separately, the protein content of cell lysates (25 μL) was determined using a Bicinchoninic acid (BCA) protein assay kit according to the manufacturer's instructions.
Using the optimum linear incubation time determined above (OCT1 = 10 min; OCT2 = 5 min, MATE1/2‐K = 1.5 min), the uptake of [14C]‐metformin into transporter‐expressing cells and vector control cells was determined at 37°C over a range of concentrations (10, 30, 100, 300, 1000, 3000 and 10,000 μmol/L, OCT1 and MATE1/2‐K; 1, 10, 100, 300, 1000, 3000 and 10,000 μmol/L, OCT2) in order to determine an apparent K m.
In vitro SLC transporter inhibition assessment
Incubations were performed as described above using the optimum linear incubation times stated for the Km determinations and a metformin probe substrate concentration that was at least 10× lower than the apparent Km determined for each respective transporter.
For OCT1 assessment, uptake of the probe substrate [14C]‐metformin (100 μmol/L) was determined (in triplicate wells per condition, over three separate occasions) at 37°C in OCT1‐expressing cells and vector control cells, in the absence and presence of cimetidine (1‐3000 μmol/L), trimethoprim (1‐300 μmol/L) or pyrimethamine (0.3‐100 μmol/L). For OCT2 assessment, uptake of the probe substrate [14C]‐metformin (100 μmol/L) was determined (in triplicate wells per condition, over three separate occasions) at 37°C in OCT2‐expressing cells and vector control cells, in the absence and presence of cimetidine (3‐3000 μmol/L), trimethoprim (1‐1000 μmol/L) or pyrimethamine (0.3‐100 μmol/L). For MATE1 assessment, uptake of the probe substrate [14C]‐metformin (100 μmol/L) was determined (in triplicate wells per condition, over three separate occasions) at 37°C in MATE1‐expressing cells and vector control cells, in the absence and presence of cimetidine (0.1‐100 μmol/L), trimethoprim (0.1‐100 μmol/L) or pyrimethamine (0.003‐1 μmol/L). For MATE2‐K assessment, uptake of the probe substrate [14C]‐metformin (100 μmol/L) was determined (in triplicate wells per condition, over three separate occasions) at 37°C in MATE2‐K‐expressing cells and vector control cells, in the absence and presence of cimetidine (0.1‐100 μmol/L) or trimethoprim (0.03‐30 μmol/L).
Data analysis
The determined total uptake of probe substrate [14C]‐metformin into cells (pmol) was normalised to the protein (mg) content of each well to calculate the uptake activity (pmol/mg). For K m determination experiments the calculated uptake activity (pmol/mg) was also normalized for incubation time (min) to give uptake rate (pmol/min/mg). Uptake rate of probe substrate into transporter‐expressing cells was corrected for that determined into vector control cells to calculate the transporter‐mediated (corrected) uptake rate. For K m determinations, corrected uptake rate (pmol/min/mg) was plotted against nominal substrate concentration and fitted using SigmaPlot 12.5 (Michaelis–Menten equation). For IC50 determinations, corrected uptake activity (pmol/mg) was converted to percentage (vehicle) control activity, which was subsequently plotted against nominal inhibitor concentration. Curves were fitted using SigmaPlot 12.5 (four‐or five parameter logistic equation) to determine the concentration that produces half‐maximal inhibition of probe substrate transport (IC50).
Assessment of metformin apparent permeability across polarised Caco‐2 cell monolayers in the absence and presence of cimetidine, trimethoprim, and pyrimethamine
Caco‐2 cells between passage numbers 50–60 were used for experiments. Cells were seeded in cell culture medium (consisting of DMEM supplemented with 10% (w/v) fetal bovine serum, 2 mmol/L l‐glutamine, 1% (v/v) nonessential amino acids, 50 U/mL penicillin and 50 μg/mL streptomycin) onto Millicell‐96 multiwell insert plates at 1 × 105 cells/cm2 and cultured at 37°C in an atmosphere of 5% CO2 with a relative humidity of 95 %. Media was changed every two or three days and plates were used for transport studies on day 20 postseeding. Cell monolayers were washed twice with prewarmed (37°C) transport buffer (HBSS containing 25 mmol/L HEPES and 4.45 mmol/L glucose, pH 7.4) for basolateral compartments and prewarmed MES‐transport buffer (HBSS containing 10 mmol/L MES and 4.45 mmol/L glucose, pH 6.5) for apical compartments. Cells were then preincubated with transport buffer (basolateral compartments) or MES‐transport buffer (apical compartments) for 40 min at 37°C prior to the addition of donor and receiver solutions.
Following this preincubation period, donor solutions of transport buffer, or MES‐transport buffer, containing [14C]‐metformin with or without test inhibitor were added to basolateral (total volume = 210 μL) or apical compartments (total volume = 90 μL), respectively. Donor compartments also contained the cell monolayer integrity marker lucifer yellow (100 μmol/L). Transport buffer or MES‐transport buffer containing DMSO or test inhibitor was added to the corresponding receiver compartments. Donor and receiver solutions contained a final DMSO concentration of ≤1 % (v/v). Following a 90‐min incubation at 37°C, receiver compartments were sampled (50 μL) into 96‐well isoplates (followed by the addition of 200 μL liquid scintillation cocktail) and the amount of [14C]‐metformin was quantified by liquid scintillation counting on a Microbeta2 counter, and used to calculate apparent permeability (Papp), as described previously (Elsby et al. 2008). Mass balance (percent recovery) of metformin was calculated as described previously (Elsby et al. 2008). The amount of lucifer yellow appearing in the receiver compartments was quantified on a fluorescence plate reader and used to calculate permeation across the cell monolayer. Cell monolayer integrity was deemed acceptable for a well if the determined P app for lucifer yellow was ≤1.0 cm/sec (× 10−6).
The bidirectional [apical‐to‐basolateral (A–B) and basolateral‐to‐apical (B–A)] apparent permeability of [14C]‐metformin across Caco‐2 cell monolayers (buffer pH 6.5/7.4; apical/basolateral) was assessed in triplicate wells per incubation condition at metformin concentrations of 10, 100, 1000 and 10,000 μmol/L. Further assessment in buffer (pH 6.5/7.4) was conducted at a single [14C]‐metformin concentration of 10,000 μmol/L in the absence and presence of cimetidine (1000 μmol/L), trimethoprim (500 μmol/L) or pyrimethamine (200 μmol/L). Subsequently, this latter experiment was repeated in buffer containing FaSSIF (pH 6.5) and buffer containing 1 % (w/v) HSA (pH 7.4), present in the apical and basolateral compartments, respectively.
Mechanistic static predictions of AUC changes for known clinical DDIs with metformin based upon determined in vitro OCT2 and MATE1 inhibitory data
The OCT2 and MATE1 IC50 (equating to K i) values obtained for cimetidine, trimethoprim, and pyrimethamine were incorporated into the adapted Rowland‐Matin mechanistic static equation below, as described previously by Elsby et al. (2012, 2016), to predict the change in metformin AUC based upon inhibition of a fraction excreted (ƒe) value of 0.66 for renal OCT2/MATE1:
where K i = absolute inhibition constant (equating to IC50 if the probe [S] ≪≪ K m in the inhibition assay and assuming competitive inhibition, based on the Cheng‐Prusoff equation; Cheng and Prusoff (1973)) and [I] = unbound maximum plasma concentration (C max).
Additionally, the predicted change in metformin AUC based upon inhibition of OCT2/MATE1 with a lower fraction excreted value of 0.39 was also determined. This lower ƒe value (0.39) is derived from the renal clearance determined in human mass balance studies following oral administration of metformin, whereas the higher ƒe value (0.66) is derived from the more usual determination of renal clearance contribution following intravenous administration.
Results
Time linearity and kinetic determination of the transporter‐mediated uptake of metformin by OCT1, OCT2, MATE1, and MATE2‐K
Uptake of the probe substrate [14C]‐metformin (100 μmol/L) into transporter‐expressing cells was linear from 5 to 15 min for OCT1, up to 5 min for OCT2, up to 2 min for MATE1 and up to 2 min for MATE2‐K (Fig. 1), giving acceptable minimum uptake ratios over control cells of 5, 18, 47, and 9, respectively. Concentration‐dependent increases in the transporter‐mediated (corrected) uptake of [14C]‐metformin were observed with all transporters, giving apparent K m values ranging from 1608 μmol/L for OCT2 to 5422 μmol/L for OCT1 (Table 1 and Fig. 1).
Figure 1.
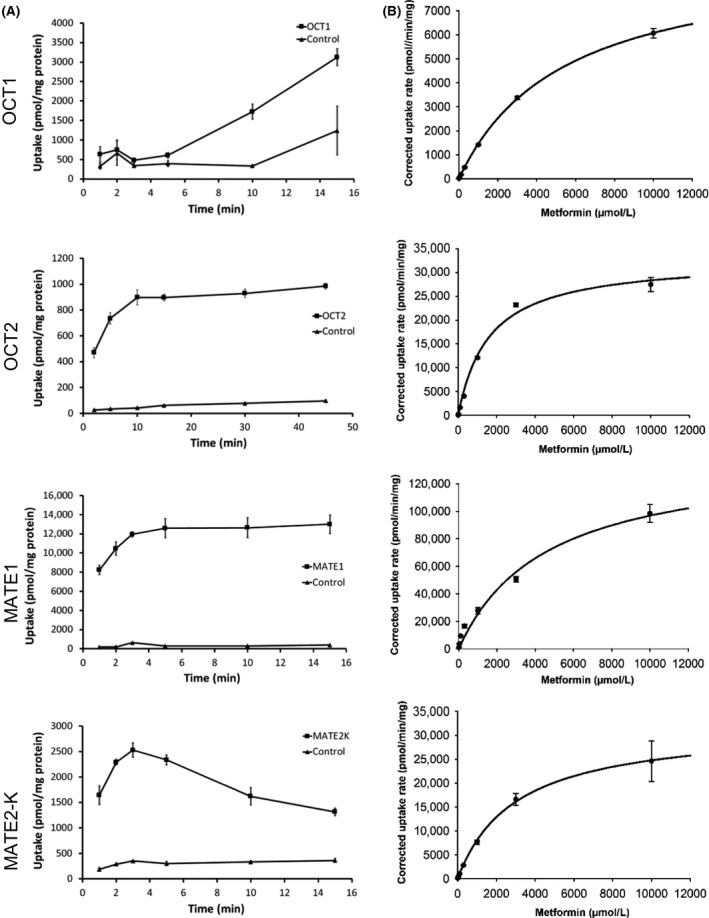
Time linearity (A) and Michaelis–Menten kinetic analysis (B) of metformin transport mediated by OCT1, OCT2, MATE1, and MATE2‐K. Data are expressed as mean (±SD) of triplicate wells per condition.
Table 1.
K m and V max kinetic parameters for transporter‐mediated uptake of metformin
| Transporter | Kinetic parameters (±SE) | |
|---|---|---|
| K m (μmol/L) | V max (pmol/min/mg protein) | |
| OCT1 | 5422 ± 369 | 9370 ± 297 |
| OCT2 | 1608 ± 175 | 32918 ± 1165 |
| MATE1 | 4565 ± 903 | 141000 ± 12283 |
| MATE2‐K | 2986 ± 817 | 32147 ± 3401 |
K m, Michaelis–Menten constant; SE, standard error from fitting; V max maximal velocity of uptake.
Assessment of cimetidine, trimethoprim, and pyrimethamine as inhibitors of organic cation transporters in vitro
Cimetidine, trimethoprim, and pyrimethamine demonstrated concentration‐dependent inhibition of OCT1‐, OCT2‐, MATE1‐ and MATE2‐K‐mediated transport of [14C]‐metformin (100 μmol/L) giving the mean IC50 curves shown in Figure 2. The determined mean IC50 values for each inhibitor from three separate experiments are shown in Table 2. The inhibitory potential (IC50) of cimetidine towards MATE1 was approximately 225‐fold and 170‐fold more potent than towards OCT1 and OCT2, respectively. A similar trend was observed for trimethoprim which produced a MATE1 IC50 that was approximately 10‐fold and 52‐fold more potent compared to OCT1 and OCT2. Of all the inhibitors tested, pyrimethamine gave the lowest IC50 versus MATE1 (0.131 μmol/L), which again was more potent (35‐fold) compared with OCT1 and OCT2 transporters.
Figure 2.
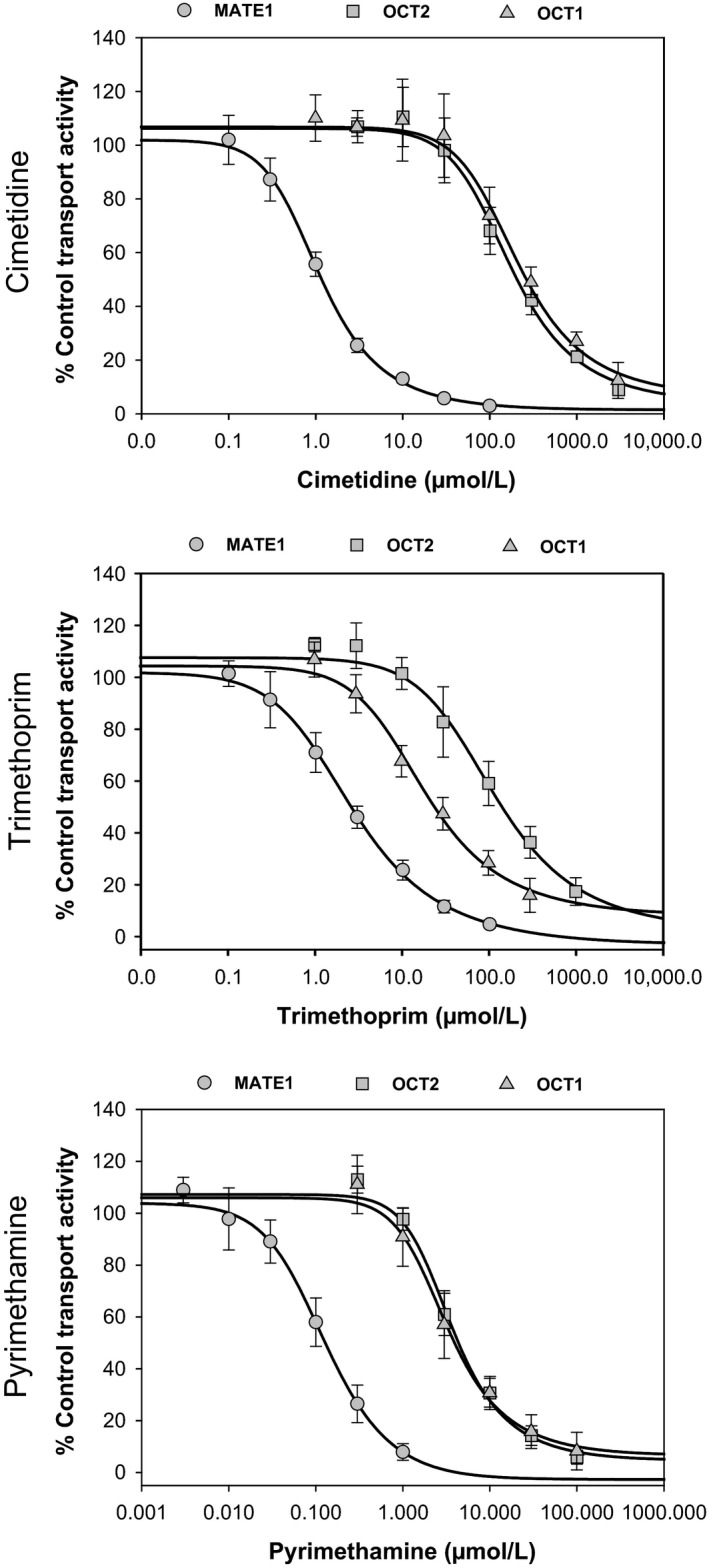
Mean concentration‐dependent inhibition of OCT1‐, OCT2‐, and MATE1‐mediated transport of [14C]‐metformin (100 μmol/L) by cimetidine, trimethoprim, and pyrimethamine. Data are expressed as mean (±SD) from a minimum of triplicate wells over three experimental occasions per inhibitor.
Table 2.
IC50 values for inhibition of transporter‐mediated uptake of metformin by cimetidine, trimethoprim, and pyrimethamine
| Inhibitor | IC50 (≅K i) values (μmol/L) (Mean ± SD) | |||
|---|---|---|---|---|
| OCT1 | OCT2 | MATE1 | MATE2‐K | |
| Cimetidine | 275 ± 32.2 | 207 ± 19.6 | 1.22 ± 0.0870 | 3.34 ± 1.02 |
| Trimethoprim | 27.7 ± 3.80 | 137 ± 58.0 | 2.64 ± 0.271 | 0.353 ± 0.0584 |
| Pyrimethamine | 4.46 ± 0.770 | 4.55 ± 1.12 | 0.131 ± 0.0401 | N.D. |
IC50,, half maximal inhibitory concentration; K i,, absolute inhibition constant (equates to IC50 in these assays as probe substrate concentration utilized is ≪≪K m); ND, not determined.
Assessment of DDI potential versus intestinal OCT1 inhibition using FDA static equation approaches gave theoretical intestinal luminal concentration [I2]/K i ratios of 23, 99, and 180 for cimetidine (400 mg dose; [I 2] = 6341 μmol/L), trimethoprim (200 mg dose; [I 2] = 2756 μmol/L) and pyrimethamine (50 mg dose; [I 2] = 804 μmol/L), respectively. Determined C max free/K i ratios for the inhibition potential of cimetidine, trimethoprim, and pyrimethamine toward OCT2 and MATE1 are shown in Tables 3 and 4, respectively, and for all inhibitors were less than 0.1 for OCT2, or greater than 2 (>0.1) for MATE1.
Table 3.
Predicted versus observed AUC increases of metformin with various coadministered drugs based upon inhibition of renal OCT2
| Perpetrator drug | Dose (mg) | f u | [C max] (μmol/L) | [C max free] (μmol/L) | OCT2 K i (μmol/L) | [C max free]/K i ratio | Predicted AUC fold‐increase (ƒe = 0.66) | Predicted AUC fold‐increase (ƒe = 0.39) | Observed AUC fold‐increase (metformin dose mg) |
|---|---|---|---|---|---|---|---|---|---|
| Cimetidine | 400 | 0.80a |
9.6 5.1 |
7.68 4.08 |
207 |
0.037 0.020 |
1.02 1.01 |
1.01 1.01 |
1.46 (250 mg)b
1.54 (500 mg)c |
| Trimethoprim | 200 | 0.56 |
14d
7.6 |
7.84 4.26 |
137 |
0.057 0.031 |
1.04 1.02 |
1.02 1.01 |
1.37 (500 mg)e
1.30 (850 mg)f |
| Pyrimethamine | 50 | 0.13 | 2.29 | 0.298 | 4.55 | 0.065 | 1.04 | 1.02 | 1.39 (250 mg)g |
f u, fraction unbound (taken from the Bactrim and Daraprim drug labels accessed via Drugs@FDA database; https://www.accessdata.fda.gov/scripts/cder/daf); C max Mean steady‐state maximum plasma concentration for total (bound plus unbound) drug measured in the clinical interaction study with metformin; Ki absolute inhibition constant (assuming competitive inhibition; equates to IC50 in these assay as probe substrate concentration utilised is ≪≪K m).
Somogyi and Gugler (1983).
Somogyi et al. (1987).
Wang et al. (2008).
Hruska et al. (2005).
Grün et al. (2013).
Müller et al. (2015).
Kusuhara et al. (2011).
Table 4.
Predicted versus observed AUC increases of metformin with various coadministered drugs based upon inhibition of renal MATE1
| Perpetrator drug | Dose (mg) | f u | [C max] (μmol/L) | [C max free] (μmol/L) | MATE1 K i (μmol/L) | [C max free]/K i ratio | Predicted AUC fold‐increase (ƒe = 0.66) | Predicted AUC fold‐increase (ƒe = 0.39) | Observed AUC fold‐increase (metformin dose mg) |
|---|---|---|---|---|---|---|---|---|---|
| Cimetidine | 400 | 0.80a |
9.6 5.1 |
7.68 4.08 |
1.22 |
6.30 3.34 |
2.32 2.03 |
1.51 1.43 |
1.46 (250 mg)b
1.54 (500 mg)c |
| Trimethoprim | 200 | 0.56 |
14d
7.6 |
7.84 4.26 |
2.64 |
2.97 1.61 |
1.98 1.69 |
1.41 1.32 |
1.37 (500 mg)e
1.30 (850 mg)f |
| Pyrimethamine | 50 | 0.13 | 2.29 | 0.298 | 0.131 | 2.27 | 1.85 | 1.37 | 1.39 (250 mg)g |
f u, fraction unbound (taken from the Bactrim and Daraprim drug labels accessed via Drugs@FDA database; https://www.accessdata.fda.gov/scripts/cder/daf))
C max, Mean steady‐state maximum plasma concentration for total (bound plus unbound) drug measured in the clinical interaction study with metformin; K i, absolute inhibition constant (assuming competitive inhibition; equates to IC50 in these assay as probe substrate concentration utilized is ≪≪K m)
Somogyi and Gugler (1983)
Somogyi et al. (1987)
Wang et al. (2008)
Hruska et al. (2005)
Grün et al. (2013)
Müller et al. (2015)
Kusuhara et al. (2011)
Effect of cimetidine, trimethoprim, and pyrimethamine on the apparent permeability of metformin across polarised Caco‐2 cell monolayers
The determined bidirectional Papp (cm/sec × 10−6) of a range of concentrations of metformin in buffer (pH6.5/7.4) is shown in Figure 3 and was approximately 0.5 cm/sec (× 10−6) in both the A‐B and B‐A directions across all metformin concentrations (10–10,000 μmol/L). Furthermore, there was no change to the determined A‐B or B‐A Papp values of metformin (10,000 μmol/L) in the presence of cimetidine, trimethoprim or pyrimethamine when incubations were conducted either in buffer alone (pH6.5/7.4), or in FaSSIF containing buffer (pH6.5) apically with 1 % HSA (w/v) containing buffer (pH 7.4) basolaterally (Fig. 3).
Figure 3.
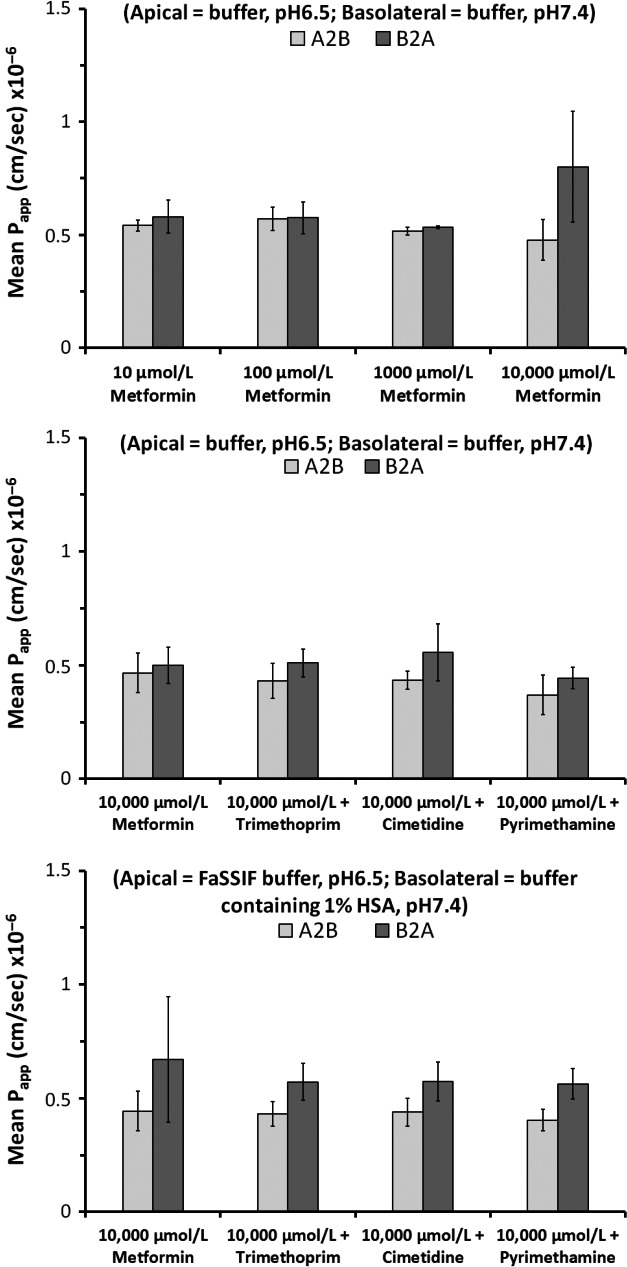
Mean bidirectional apparent permeability of a range of concentrations of metformin (10‐10,000 μmol/L), and of a single concentration of metformin (10,000 μmol/L) in the absence and presence of cimetidine (1000 μmol/L), trimethoprim (500 μmol/L) or pyrimethamine (200 μmol/L), across polarized Caco‐2 cell monolayers at pH6.5/7.4. Incubations with inhibitors were conducted both in buffer alone or buffer containing FaSSIF and 1 % (w/v) human serum albumin. Data are expressed as mean (±SD) of n =3–8 wells per condition.
Predicted versus observed AUC changes for known clinical DDIs with metformin based upon determined in vitro OCT2 and MATE1 inhibitory data
Using the drug inhibitory affinities determined above for cimetidine, trimethoprim, and pyrimethamine versus OCT2 and their pharmacokinetic parameters given in Table 3, calculations were performed with mechanistic static equations in order to predict the theoretical fold‐increase in metformin AUC that would occur as a consequence of these drugs' inhibition of renal OCT2. As anticipated, the calculated theoretical fold increases in exposure due to inhibition of OCT2 (with ƒe = 0.66 or 0.39) gave AUC increases that were below bioequivalence (<1.25; ranging from 1.01–1.04) indicating no DDI potential via this mechanism.
In contrast, the calculated theoretical fold increases in AUC due to inhibition of renal MATE1 by cimetidine, trimethoprim, and pyrimethamine were 2.32/2.03, 1.98/1.69, and 1.85 using an ƒe value of 0.66, or 1.51/1.43, 1.41/1.32, and 1.37 using an ƒe value of 0.39, respectively. Predicted AUC increases were within 39‐86 % or 2‐11 % of clinically observed AUC increases when using an ƒe value of 0.66, or an ƒe value of 0.39, respectively.
Discussion
Metformin is widely prescribed to patients due to the prevalence of diabetes as a comorbidity in many diseases (Bloomer et al. 2013). As a consequence of being a common comedication, and because of concern that elevated plasma concentrations are associated with an increased risk of lactic acidosis (Glucophage® label, DeFronzo et al. 2016), there is a requirement to assess the potential for drugs to perpetrate DDIs with metformin during development (Giacomini et al. 2010; Hillgren et al. 2013). Such assessment involves initial in vitro evaluation of the drug as an inhibitor of transporters, followed (if warranted) by a clinical interaction study (US FDA draft DDI guidance 2012). Pivotal to assessment is the prior understanding of the mechanism(s) underlying clinically observed DDIs in order to identify the critical target metformin disposition pathway(s).
Cimetidine, trimethoprim, and pyrimethamine perpetrate clinical DDIs with metformin by reducing its renal clearance through inhibition of renal organic cation transporters thereby increasing the C max and AUC of metformin (Somogyi et al. 1987; Wang et al. 2008; Kusuhara et al. 2011; Grün et al. 2013; Müller et al. 2015). In order to confirm the likely mechanism underlying these DDIs, the in vitro inhibitory properties of cimetidine, trimethoprim, and pyrimethamine against OCT2‐, MATE1‐ and MATE2‐K‐mediated metformin transport were assessed in transfected cells to generate IC50 values that could be incorporated into mechanistic equations for computing AUC increases due to transporter inhibition. Inhibition experiments were conducted over a linear incubation time using a metformin probe substrate concentration that was at least 10‐fold lower than its determined K m for transporter‐mediated uptake (Table 1), such that calculated IC50 values equate to K i (assuming competitive inhibition; Cheng and Prusoff 1973). This is approach is valid as cimetidine, trimethoprim, and pyrimethamine are reported to be competitive inhibitors of MATE1 (Kusuhara et al. 2011; Ito et al. 2012; Müller et al. 2015). Determined IC50 (K i) values (Table 2) versus OCT2, MATE1 and MATE2‐K were in reasonable concordance with literature values and were much lower (35‐ to 170‐fold; Fig. 1) for MATEs versus OCT2 (Kusuhara et al. 2011; Ito et al. 2012; Müller et al. 2015; Shen et al., 2016). However, only inhibition constants for OCT2 and MATE1 were subsequently incorporated into mechanistic equations for predicting metformin AUC increases as the study of Prasad et al. (2016) determined that these, and not MATE2‐K, are the major cationic transporter proteins expressed in human renal proximal tubules; representing 26% and 18% of renal transporter protein. The reported minimal detectable expression of MATE2‐K protein suggests MATE2‐K may have limited functional activity in human kidney. This notion is further supported clinically by the finding that the MATE2‐K selective inhibitor nizatidine did not increase metformin AUC, nor did it reduce renal clearance in an interaction study, despite its unbound C max/K i ratio being 1.05 indicating the potential for a DDI in vivo through inhibition of MATE2‐K (Morrissey et al. 2016). Collectively, these suggest that MATE2‐K is not involved in renal disposition of metformin in humans and that inhibition of MATE2‐K is not important in manifesting metformin clinical DDIs.
Unbound C max/Ki ratios for inhibition of OCT2 by cimetidine, trimethoprim or pyrimethamine were less than 0.1 resulting in calculated fold‐increases in metformin AUC of ≤1.04 (using ƒe = 0.66), confirming that DDI via this mechanism is unlikely in vivo (Table 3). In contrast, unbound C max/K i ratios for inhibition of MATE1 were 3.34‐6.3, 1.61‐2.97 and 2.27 for cimetidine, trimethoprim and pyrimethamine, respectively, confirming inhibition of this pathway as being clinically relevant to observed DDIs (Table 4) and agreeing with published literature (Tsuda et al. 2009b; Ito et al. 2012). Unbound C max is the correct concentration to put into context with K i as intracellular concentrations of cimetidine and trimethoprim in renal proximal tubule cells are not anticipated to accumulate above, but rather equilibrate with, plasma levels since they are transported substrates of passive facilitative OCT2 (Bactrim™ label, Severance et al. 2017). However, incorporating these ratios and an ƒe = 0.66 resulted in over‐predicted calculated maximum theoretical fold‐increases in metformin AUC due to MATE1 inhibition compared with clinically observed changes (Table 4).
It is difficult to reconcile this disconnect, however, examinations of metformin AUC profiles from DDI studies with cimetidine, trimethoprim, and pyrimethamine show an increase in the initial rising phase of the curve with parallel terminal phases, suggesting that elevated C max and AUC could result from increased metformin absorption. This was investigated using polarized Caco‐2 cell monolayers that, like enterocytes, have been demonstrated to express OCT1 functionally on the apical membrane of cells (Han et al. 2013). OCT1 is the major transporter involved in intestinal uptake of metformin, creating a depot for absorption (Han et al. 2015), and would be expected to be inhibited by cimetidine, trimethoprim, and pyrimethamine in vivo based on calculated [I 2]/K i ratios (>10), using IC50 values from this study (Table 2) which agreed with literature values (Ito et al. 2012; Müller et al. 2015). The determined absorptive (apical to basolateral) permeability (~0.5 cm/sec × 10−6) of 10, 100, 1000 and 10,000 μmol/L metformin in Caco‐2 cells (Fig. 3) was consistent with values reported previously by Song et al. (2016) and Proctor et al. (2008). A concentration of 10,000 μmol/L was chosen to investigate the effect of inhibitors on permeability as this reflects metformin's maximum aqueous solubility; lying between the theoretical intestinal concentration (dose in mol/250 mL) of 250 mg and 500 mg doses of metformin. Additionally, inhibitor concentrations chosen ensured (1) that they were greater than theoretical enterocyte concentrations ([I gut max]; Agarwal et al. 2013) for clinical doses of cimetidine (528 μmol/L), trimethoprim (230 μmol/L) or pyrimethamine (67 μmol/L) and, (2) that they were >3 times determined OCT1 IC50 values to give reasonable or complete inhibition of OCT1. Inhibition of OCT1 would act to reduce the apical uptake of metformin making more available for paracellular absorption (Han et al. 2015), yet the observed absence of an effect of cimetidine, trimethoprim or pyrimethamine on enhancing metformin absorptive permeability across Caco‐2 cells in this study suggests that DDI AUC profile changes are not related to increased metformin absorption.
Interestingly, in the clinical scenario where there is increased metformin renal clearance due to pregnancy, the resulting reduction in C max and AUC of metformin is accompanied by a lowering of the initial rising phase of the AUC profile without a change in the terminal phase (Eyal et al. 2010; Fig 4). This is consistent with the fact that metformin exhibits “flip‐flop” pharmacokinetics (Tucker et al. 1981), where the rate of absorption of a drug is slower than its rate of elimination, thereby the terminal phase of the AUC profile reflects the absorption rate and the initial rising phase represents the elimination of drug (Yáñez et al. 2011). Consequently, the increase in the initial rising phase of the metformin AUC curve observed in DDIs, giving rise to increased exposure, must result from reduction in renal elimination due to inhibition of MATE1.
Figure 4.
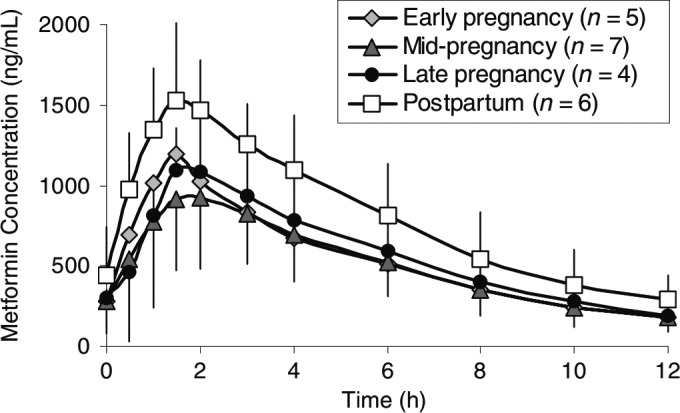
Figure reproduced from Eyal et al. (2010) to illustrate how a documented decrease in metformin (500 mg) renal clearance in pregnancy changes (lowers) the initial rising phase of the AUC profile of a drug (metformin) that exhibits “flip‐flop” pharmacokinetics.
This is feasible as inhibition of renal MATE1 would cause intracellular concentrations of metformin to increase, which in turn would act to reduce the blood‐to‐cell concentration gradient of metformin as the driving force for passive facilitative OCT2 transport activity. Consequently, metformin uptake into renal proximal tubule cells would be reduced leading to elevated metformin plasma concentrations as elimination is decreased. This hypothesis was also proposed by Burt et al. (2016) as part of development of a PBPK model towards understanding metformin‐cimetidine DDI. Furthermore, it is supported in vivo by the observation that, following intravenous administration, the 4.2‐fold increase in metformin AUC (giving ƒe = 0.76) in Mate1 (−/−) null mice compared with wildtype mice (Tsuda et al. 2009a) is identical in effect to the 4.5‐fold increase in metformin AUC (giving ƒe = 0.77) observed in Oct1/2 (−/−) null mice (Higgins et al. 2012), confirming a complete reduction in Oct1/2 activity towards dual substrates in the absence of functional Mate1.
Having established that increases in metformin AUC observed clinically with cimetidine, trimethoprim and pyrimethamine are due to a reduction in renal clearance via MATE1 inhibition, the over‐prediction of theoretical AUC change needs to be addressed. This is likely attributed to deriving an incorrect ƒe value (0.66) for MATE1. For conventional oral drugs (whose elimination rate <<<absorption rate) ƒe values are derived from total plasma clearance obtained in human intravenous balance studies (Elsby et al. 2012). However, with metformin, due to “flip‐flop” pharmacokinetics, it is being renally eliminated from plasma faster than it can be absorbed. Since absorption is still occurring in parallel to active renal elimination, the extent of renal elimination is reduced (compared to intravenous determination) and would therefore derive a lower ƒe value. Indeed, taking oral metformin human mass balance into account (Pentikäinen et al. 1979; Tucker et al. 1981; Kusuhara et al. 2011), on average 52% of the dose is eliminated in urine, deriving an ƒe = 0.39. Furthermore, using this revised ƒe value better predicted (within 11% of clinical values) the clinically observed increases in metformin AUC based on inhibition of MATE1 by cimetidine, trimethoprim, and pyrimethamine. For future studies, it would be interesting to see if the model of Burt et al. (2016) still required the 8 to 18‐fold decrease in cimetidine Ki to capture the change in metformin kinetics if this revised renal ƒe value was taken into account.
Such a differential for renal transporter ƒe has also been observed based on metformin AUC increases of 2.9‐fold and 4.2‐fold in Oct1/2 (−/−)mice, deriving values of 0.66 and 0.76, following oral and intravenous administration, respectively. If such a differential did not exist then the fold increase in metformin AUC in null mice (due to the absence of Oct1/2 protein) compared to wild type mice would be identical regardless of metformin route of administration. Interestingly, oral AUC profiles of metformin in mice showed the same protracted absorption profile (Higgins et al. 2012) as observed in humans. Moreover, the changed oral AUC profile in null mice compared to wildtype mirrored clinical metformin DDI profile changes due to transporter inhibition, that is, an increase in the initial rising phase.
In conclusion, in vitro DDI studies, coupled with mechanistic static approaches for predicting maximal theoretical increases in exposure, indicate solitary inhibition of renal MATE1 as the likely mechanism underlying the metformin AUC increases in observed clinical interactions perpetrated by cimetidine, trimethoprim, and pyrimethamine.
Authorship Contributions
Participated in research design: Elsby; Conducted experiments: Chidlaw, Outteridge, Pickering, Radcliffe, and Sullivan; Performed data analysis: Chidlaw, Elsby, Outteridge, Pickering, Radcliffe, and Sullivan; Wrote or contributed to the writing of the manuscript: Butler, Chidlaw, Elsby, Jones, Outteridge, Pickering, and Radcliffe.
Disclosure
None declared.
Acknowledgements
This research was funded by Cyprotex Discovery Limited.
Elsby R., Chidlaw S., Outteridge S., Pickering S., Radcliffe A., Sullivan R., Jones H., Butler P.. Mechanistic in vitro studies confirm that inhibition of the renal apical efflux transporter multidrug and toxin extrusion (MATE) 1, and not altered absorption, underlies the increased metformin exposure observed in clinical interactions with cimetidine, trimethoprim or pyrimethamine. Pharma Res Per, 5(5), 2017, e00357, https://doi.org/10.1002/prp2.357
References
- Agarwal S, Arya V, Zhang L (2013). Review of P‐gp inhibition data in recently approved new drug applications: utility of the proposed [I1]/IC50 and [I2]/IC50 criteria in the P‐gp decision tree. J Clin Pharmacol 53: 228–233. [DOI] [PubMed] [Google Scholar]
- Bactrim™ . (sulfamethoxazole and trimethoprim) injection US FDA drug label, revised March 2017; Sun Pharmaceutical Industries, Inc. Accessed via FDA approved drug products database (www.accessdata.fda.gov/scripts/cder/daf/) on 01 Aug 2017.
- Bloomer J, Derimanov G, Dumont E, Ellens H, Matheny C (2013). Optimizing the in vitro and clinical assessment of drug interaction risk by understanding co‐medications in patient populations. Expert Opin Drug Metab Toxicol 9: 737–751. [DOI] [PubMed] [Google Scholar]
- Burt HJ, Neuhoff S, Almond L, Gaohua L, Harwood MD, Jamei M, et al. (2016). Metformin and cimetidine: physiologically based pharmacokinetic modelling to investigate transporter mediated drug‐drug interactions. Eur J Pharm Sci 88: 70–82. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH (1973). Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 22: 3099–3108. [DOI] [PubMed] [Google Scholar]
- Daraprim® (pyrimethamine) . US FDA drug label, March 2003; GlaxoSmithKline. Accessed via FDA approved drug products database (www.accessdata.fda.gov/scripts/cder/daf/) on 10 May 2017.
- DeFronzo R, Fleming GA, Chen K, Bicsak TA (2016). Metformin‐associated lactic acidosis: current perspectives on causes and risk. Metabolism 65: 20–29. [DOI] [PubMed] [Google Scholar]
- Elsby R, Surry DD, Smith VN, Gray AJ (2008). Validation and application of Caco‐2 assays for the in vitro evaluation of development candidate drugs as substrates or inhibitors of P‐glycoprotein to support regulatory submissions. Xenobiotica 38: 1140–1164. [DOI] [PubMed] [Google Scholar]
- Elsby R, Hilgendorf C, Fenner K (2012). Understanding the critical disposition pathways of statins to assess drug‐drug interaction risk during drug development: it's not just about OATP1B1. Clin Pharmacol Ther 92: 584–598. [DOI] [PubMed] [Google Scholar]
- Elsby R, Martin P, Surry D, Sharma P, Fenner K (2016). Solitary inhibition of the breast cancer resistance protein efflux transporter results in a clinically significant drug‐drug interaction with rosuvastatin by causing up to a 2‐fold increase in statin exposure. Drug Metab Dispos 44: 398–408. [DOI] [PubMed] [Google Scholar]
- Eyal S, Easterling TR, Carr D, Umans JG, Miodovnik M, Hankins GD, et al. (2010). Pharmacokinetics of metformin during pregnancy. Drug Metab Dispos 38: 833–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomini KM, Huang S‐M, Tweedie DJ, Benet LZ, Brouwer KLR, Chu X, et al. (2010). Membrane transporters in drug development. Nat Rev Drug Discov 9: 215–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glucophage® (metformin hydrochloride) . US FDA drug label, revised April 2017; Bristol‐Myers Squibb Company. Accessed via FDA approved drug products database (www.accessdata.fda.gov/scripts/cder/daf/) on 26 May 2017.
- Graham GG, Punt J, Arora M, Day RO, Doogue MP, Duong JK, et al. (2011). Clinical pharmacokinetics of metformin. Clin Pharmacokinet 50: 81–98. [DOI] [PubMed] [Google Scholar]
- Grün B, Kiessling MK, Burhenne J, Riedel K‐D, Weiss J, Rauch G, et al. (2013). Trimethoprim–metformin interaction and its genetic modulation by OCT2 and MATE1 transporters. Br J Clin Pharmacol 76: 787–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han TK, Everett RS, Proctor WR, Ng CM, Costales CL, Brouwer KL, et al. (2013). Organic cation transporter 1 (OCT1/mOct1) is localized in the apical membrane of Caco‐2 cell monolayers and enterocytes. Mol Pharmacol 84: 182–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han T, Proctor WR, Costales CL, Cai H, Everett RS, Thakker DR (2015). Four cation‐selective transporters contribute to apical uptake and accumulation of metformin in Caco‐2 cell monolayers. J Pharmacol Exp Ther 352: 519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins JW, Bedwell DW, Zamek‐Gliszczynski MJ (2012). Ablation of both organic cation transporter (OCT)1 and OCT2 alters metformin pharmacokinetics but has no effect on tissue drug exposure and pharmacodynamics. Drug Metab Dispos 40: 1170–1177. [DOI] [PubMed] [Google Scholar]
- Hillgren KM, Keppler D, Zur AA, Giacomini KM, Stieger B, Cass CE, et al. (on behalf of the International Transporter Consortium) (2013). Emerging transporters of clinical importance: an update from the International Transporter Consortium. Clin Pharmacol Ther 94: 52–63. [DOI] [PubMed] [Google Scholar]
- Hruska MW, Amico JA, Langaee TY, Ferrell RE, Fitzgerald SM, Frye RF (2005). The effect of trimethoprim on CYP2C8 mediated rosiglitazone metabolism in human liver microsomes and healthy subjects. Br J Clin Pharmacol 59: 70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Kusuhara H, Yokochi M, Toyoshima J, Inoue K, Yuasa H, et al. (2012). Competitive inhibition of the luminal efflux by multidrug and toxin extrusions, but not basolateral uptake by organic cation transporter 2, is the likely mechanism underlying the pharmacokinetic drug‐drug interactions caused by cimetidine in the kidney. J Pharmacol Exp Ther 340: 393–403. [DOI] [PubMed] [Google Scholar]
- Kusuhara H, Ito S, Kumagai Y, Jiang M, Shiroshita T, Moriyama Y, et al. (2011). Effects of a MATE protein inhibitor, pyrimethamine, on the renal elimination of metformin at oral microdose and at therapeutic dose in healthy subjects. Clin Pharmacol Ther 89: 837–844. [DOI] [PubMed] [Google Scholar]
- Morrissey KM, Stocker SL, Chen EC, Castro RA, Brett CM, Giacomini KM (2016). The effect of nizatidine, a MATE2K selective inhibitor, on the pharmacokinetics and pharmacodynamics of metformin in healthy volunteers. Clin Pharmacokinet 55: 495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller F, Pontones CA, Renner B, Mieth M, Hoier E, Auge D, et al. (2015). N(1)‐methylnicotinamide as an endogenous probe for drug interactions by renal cation transporters: studies on the metformin‐trimethoprim interaction. Eur J Clin Pharmacol 71: 85–94. [DOI] [PubMed] [Google Scholar]
- Prasad B, Johnson K, Billington S, Lee C, Chung GW, Brown CD, et al. (2016). Abundance of drug transporters in the human kidney cortex as quantified by quantitative targeted proteomics. Drug Metab Dispos 44: 1920‐1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pentikäinen PJ, Neuvonen PJ, Penttilä A (1979). Pharmacokinetics of metformin after intravenous and oral administration to man. Eur J Clin Pharmacol 16: 195–202. [DOI] [PubMed] [Google Scholar]
- Proctor WR, Bourdet DL, Thakker DR (2008). Mechanisms underlying saturable intestinal absorption of metformin. Drug Metab Dispos 36: 1650–1658. [DOI] [PubMed] [Google Scholar]
- Severance AC, Sandoval PJ, Wright SH (2017). Correlation between apparent substrate affinity and OCT2 transporter turnover. J Pharmacol Exp Ther 362: 405–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Liu T, Jiang H, Titsch C, Taylor K, Kandoussi H, et al. (2016). Cynomolgus monkey as a clinically relevant model to study transport involving renal organic cation transporters: in vitro and in vivo evaluation. Drug Metab Dispos 44: 238‐249. [DOI] [PubMed] [Google Scholar]
- Sirtori CR, Franceschini G, Galli‐Kienle M, Cighetti G, Galli G, Bondioli A, et al. (1978). Disposition of metformin (N, N‐dimethylbiguanide) in man. Clin Pharmacol Ther 24: 683–693. [DOI] [PubMed] [Google Scholar]
- Somogyi A, Gugler R (1983). Clinical pharmacokinetics of cimetidine. Clin Pharmacokinet 8: 463–495. [DOI] [PubMed] [Google Scholar]
- Somogyi A, Stockley C, Keal J, Rolan P, Bochner F (1987). Reduction of metformin renal tubular secretion by cimetidine in man. Br J Clin Pharmacol 23: 545–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song IS, Shin HJ, Shim EJ, Jung IS, Kim WY, Shon JH, et al. (2008). Genetic variants of the organic cation transporter 2 influence the disposition of metformin. Clin Pharmacol Ther 84: 559–562. [DOI] [PubMed] [Google Scholar]
- Song IH, Zong J, Borland J, Jerva F, Wynne B, Zamek‐Gliszczynski MJ, et al. (2016). The Effect of dolutegravir on the pharmacokinetics of metformin in healthy subjects. J Acquir Immune Defic Syndr 72: 400–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stage TB, Brøsen K, Christensen MMH (2015). A comprehensive review of drug‐drug interactions with metformin. Clin Pharmacokinet 54: 811–824. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Terada T, Mizuno T, Katsura T, Shimakura J, Inui K‐I (2009a). Targeted disruption of the multidrug and toxin extrusion 1 (Mate1) gene in mice reduces renal secretion of metformin. Mol Pharmacol 75: 1280–1286. [DOI] [PubMed] [Google Scholar]
- Tsuda M, Terada T, Ueba M, Sato T, Masuda S, Katsura T, et al. (2009b). Involvement of human multidrug and toxin extrusion 1 in the drug interaction between cimetidine and metformin in renal epithelial cells. J Pharmacol Exp Ther 329: 185–191. [DOI] [PubMed] [Google Scholar]
- Tucker GT, Casey C, Phillips PJ, Conner H, Ward JD, Woods HF (1981). Metformin kinetics in healthy subjects and in patients with diabetes mellitus. Br J Clin Pharmacol 12: 235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- US Food and Drug Administration (2012). Draft guidance for industry: Drug interaction studies – study design, data analysis, implications for dosing, and labelling recommendations (available at http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf; accessed 01 December 2014).
- Wang ZJ, Yin OQ, Tomlinson B, Chow MS (2008). OCT2 polymorphisms and in‐vivo renal functional consequence: studies with metformin and cimetidine. Pharmacogenet Genomics 18: 637–645. [DOI] [PubMed] [Google Scholar]
- Yáñez JA, Remsberg CM, Sayre CL, Forrest ML, Davies NM (2011). Flip‐flop pharmacokinetics‐delivering a reversal of disposition: challenges and opportunities during drug development. Ther Deliv 2: 643–672. [DOI] [PMC free article] [PubMed] [Google Scholar]