

Abstract
One mechanism by which the female sex may protect against elevated coronary vascular tone is inhibition of Ca2+ entry into arterial smooth muscle cells (ASMCs). In vitro findings confirm that high estrogen concentrations directly inhibit voltage‐dependent Cav1.2 channels in coronary ASMCs. For this study, we hypothesized that the nonacute, in vitro exposure of coronary arteries to a low concentration of 17β‐estradiol (17βE) reduces the expression of Cav1.2 channel proteins in coronary ASMCs. Segments of the right coronary artery obtained from sexually mature female pigs were mounted for isometric tension recording. As expected, our results indicate that high concentrations (≥10 μmol/L) of 17βE acutely attenuated Ca2+‐dependent contractions to depolarizing KCl stimuli. Interestingly, culturing coronary arteries for 24 h in a 10,000‐fold lower concentration (1 nmol/L) of 17βE also attenuated KCl‐induced contractions and reduced the contractile response to the Cav1.2 agonist, FPL64176, by 50%. Western blots revealed that 1 nmol/L 17βE decreased protein expression of the pore‐forming α 1C subunit (Cav α) of the Cav1.2 channel by 35%; this response did not depend on an intact endothelium. The 17βE‐induced loss of Cav α protein in coronary arteries was prevented by the estrogen ER α/ER β antagonist, ICI 182,780, whereas the GPER antagonist, G15, did not prevent it. There was no effect of 1 nmol/L 17βE on Cav α transcript expression. We conclude that 17βE reduces Cav1.2 channel abundance in isolated coronary arteries by a posttranscriptional process. This unrecognized effect of estrogen may confer physiological protection against the development of abnormal Ca2+‐dependent coronary vascular tone.
Keywords: Estrogen, sex, coronary artery, calcium channel, α1C subunit, vasodilation
Abbreviations
- 17βE
17β‐estradiol
- Ach
Acetylcholine
- ASMCs
arterial smooth muscle cells
- Cav1.2
L‐type Ca2+ channels
- Cav
voltage‐gated
- Endo
endothelium
- ER
estrogen receptor
- EtOH
ethanol
- FPL64176
2,5‐Dimethyl‐4‐[2‐(phenylmethyl)benzoyl]‐1H‐pyrrole‐3‐carboxylic acid methyl ester
- GPER
G‐protein‐coupled estrogen receptor
- H2O2
hydrogen peroxide
- HRP
horseradish peroxidase
- ICI 182
780, 7α,17β‐[9‐[(4,4,5,5,5‐Pentafluoropentyl)sulfinyl]nonyl]estra‐1,3,5(10)‐triene‐3,17‐diol
- RCA
right coronary artery
- TBST
Tris‐buffered saline containing 0.1% Tween 20
- TSG
thapsigargin
Introduction
The in vivo administration of estrogen to postmenopausal women has a beneficial effect on coronary arterial function by improving blood flow (Gilligan et al. 1994; Puntawangkoon et al. 2010). The improvement in coronary blood flow during estrogen therapy is typically attributed to activation of Ca2+‐activated K+ channels and/or inhibition of voltage‐gated, L‐type Ca2+ (Cav1.2) channels (Bowles et al. 1998a,b). The Cav1.2 channels are the major pathway for voltage‐dependent Ca2+ influx in coronary arterial smooth muscle cells (ASMCs), and as primary mediators of arterial contraction, any imbalance in their activity or expression that favors Ca2+ influx may contribute to elevated vascular tone. For example, the expression of Cav1.2 channels is increased in coronary arteries susceptible to vasospasm (Kuga et al. 2000), and Cav1.2 channels are more abundant in small mesenteric and skeletal muscle arteries of genetically hypertensive rats (Pratt et al. 2002). Accordingly, the latter arteries exhibit accentuated Ca2+‐dependent vascular tone, which is normalized by pharmacological blockers of Cav1.2 channels.
Using porcine coronary arteries, our laboratory (Tummala and Hill 2007; Hill et al. 2010) and others (Han et al. 1995; Teoh et al. 1999) demonstrated that the primary female estrogenic hormone, 17β‐estradiol (17βE), acutely inhibits contractions elicited by depolarizing KCl solutions, suggesting that 17βE reduces Ca2+ influx mediated by voltage‐sensitive Cav1.2 channels. Similarly, Sudhir et al. (1997) demonstrated that vasospasms of porcine coronary arteries are alleviated by the intracoronary infusion of 17βE, implying an inhibitory action of pharmacological concentrations of estrogen on coronary reactivity. Finally, Ullrich et al. (2007) directly confirmed that 17βE (10 μmol/L) inhibits whole‐cell Ca2+ current recorded from Cav1.2 channel‐transfected HEK‐293 cells. In all of these studies, high concentrations (≥1 μmol/L) of 17βE were required to reduce Ca2+ influx or mediate acute relaxation responses. In agreement with these studies (Han et al. 1995; Teoh et al. 1999; Tummala and Hill 2007; Ullrich et al. 2007; Hill et al. 2010), we confirmed that the acute relaxation responses to pharmacological concentrations of 17βE are nongenomic and relate to direct inhibition of Cav1.2 channel function (Hill et al. 2010). However, a recurring concern of these in vitro studies is that the vasodilator effect of high pharmacological 17βE concentrations applied for short exposure times (≤60 min) cannot account for the in vivo effect of estrogen on vascular tone, which apparently results from chronic modulation of vascular contractile mechanisms by a much lower concentration of the circulating hormone. It is likely that 17βE's mechanism(s) of action on vascular tone is related to the duration of exposure (i.e., acute vs. chronic) and needs to be further investigated. For example, the estrogen receptor mediated intracellular signaling events (i.e., kinase phosphorylation) in vascular smooth muscle cells are enhanced with prolonged E2 exposure in culture (Ding et al. 2009).
Several studies have explored whether different in vivo estrogen levels modulate Cav1.2 channel function in ASMCs. Using an ovariectomized rat model, Crews and Khalil (Crews and Khalil 1999) found less Ca2+ influx through Cav1.2 channels in aortae isolated from ovary‐intact compared to ovariectomized females, implying that higher levels of circulating plasma estrogen may reduce the number of functional Cav1.2 channels in ASMCs. Likewise, Bowles (2001) found that coronary arteries of male pigs exhibit a higher expression level of the pore‐forming α 1C subunit (Cav α) of the Cav1.2 channel compared to female animals, and also show more voltage‐dependent Ca2+ current in patch‐clamped coronary ASMCs. Recently, Tharp et al. (2014) elaborated on this study and demonstrated that coronary ASMCs from ovariectomized female pigs have increased Cav1.2 channel current, but not enhanced Cav α protein expression, compared to ovary‐intact female pigs. The authors hypothesized that endogenous female hormones inhibit Cav1.2 channel activity in coronary ASMCs by upregulating the Cav β1 subunit. In this regard, the Cav α pore proteins coassemble in 1:1 stoichiometry with smaller β and α 2 δ subunits to form functional Cav1.2 channels in ASMCs; different β subunits modulate expression and properties of the multiprotein channel (Sonkusare et al. 2006; Kharade et al. 2013; Cox and Fromme 2015). Based on these in vivo studies, the objective of this study was to use an in vitro vessel culture approach to define the effect of 17βE on Cav1.2 channel expression and function. 17β‐Estradiol is the main estrogenic hormone in premenopausal women, and is presumed to be one source of vascular protection (Khalil 2013). We used coronary arteries from sexually mature female pigs as a model because of their close similar to the human coronary vasculature, and similar development of coronary arterial dysfunctions and disease (Jokinen et al. 1985; Tummala and Hill 2007; Sturek 2011).
Materials and Methods
Animals
As previously described (Tummala and Hill 2007), hearts from female Yorkshire pigs (3–4 years of age) were obtained from a local packing plant staffed with a United States Department of Agriculture veterinarian. The distal end of the right coronary artery (RCA) was immediately dissected from the heart in a low Ca2+ Krebs solution (in mmol/L: 138 NaCl, 5 KCl, 0.2 CaCl2, 1 MgCl2, 10 HEPES, 10 glucose, pH 7.4).
Vessel culture
For tension‐recording studies, distal ends of the RCA were carefully sectioned into ring segments (4 mm in length) before placing each ring in the appropriate culture conditions. The RCA designated for real‐time PCR and Western blot analysis were sectioned into longitudinal strips (3 cm in length) prior to vessel culture. To determine the nonacute effect of 17β‐estradiol (17βE; Sigma Chemical Co., St. Louis, MO) on Cav1.2 channel‐mediated coronary reactivity and Cav α expression, arterial segments were incubated at 37°C in a 5% CO2 incubator for 24 h in RPMI 1640 phenol‐free culture media (Sigma). Arteries either were incubated in: (1) control media, (2) media supplemented with 17βE (1 pmol/L to 10 μmol/L), or (3) media supplemented with an equal volume of ethanol solvent (EtOH; 0.1%). In order to implicate specific estrogen receptors (ER), the media in some studies was supplemented either with the ERα/ERβ antagonist, ICI 182,780 (Tocris Bioscience; Ellisville, MO) or the G‐protein‐coupled estrogen receptor (GPER) antagonist, G15 (Tocris Bioscience, Bristol, UK). The latter antagonist reportedly lacks affinity for ERα and ERβ at concentrations up to 10 μmol/L (Dennis et al. 2009; Yu et al. 2011). To evaluate the role of endothelium on the 17βE effects, the endothelium was removed in a subset of studies by rubbing the luminal vessel surface with a wooden toothpick before vessel culture; its removal was later verified using scanning electron microscopy.
Vascular reactivity
Isometric tension recording was performed using freshly isolated arterial rings or rings cultured for 24 h under one of the three conditions described above. Rings were suspended in organ baths (World Precision Instruments, Sarasota, FL) containing an oxygenated (95% O2: 5% CO2) modified Krebs solution (in mmol/L: 138 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 10 glucose, pH 7.4). All rings were initially stretched to their optimal length (Tummala and Hill 2007). To evaluate 17βE‐induced relaxation, the freshly isolated rings were preconstricted with a depolarizing solution containing high (60 mmol/L) KCl (in mmol/L: 83 NaCl, 60 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 10 glucose, pH 7.4) followed by generation of a concentration–response relationship with 17βE (0.1–100 μmol/L) or its EtOH solvent. As reported and discussed earlier (Hill et al. 2010), each 17βE concentration was equilibrated with the ring for 60 min (to achieve a steady‐state relaxation response) before the next sequential concentration was added.
The acute effect of 17βE on depolarization‐induced contractions also was evaluated using freshly isolated rings of RCA. An initial 80 mmol/L KCl‐mediated contraction was induced before incubating each ring in a different 17βE concentration (1 pmol/L to 100 μmol/L) or its ethanol solvent (0.1%) for 60 min. This was followed by the generation of a KCl concentration–response relationship (15–80 mmol/L) in all rings. The composition of the KCl solution was a similar to the 60 mmol/L KCl solution described above except for the equimolar substitution of NaCl for KCl.
The concentration–response relationship with KCl was evaluated in rings cultured for 24 h in EtOH, or in 1 pmol/L, 1 nmol/L, and 10 μmol/L 17βE. In contrast to the freshly used rings above, cultured rings were not exposed to 80 mmol/L KCl before generating the KCl concentration–response relationship; instead an “untreated group” was cultured without 17βE or EtOH. During the generation of the KCl concentration–response relationship, rings were allowed to incubate with each KCl solution for only 10 min before exchanging it for the sequential KCl concentration to minimize depolarization‐induced Cav1.2 channel upregulation (Pesic et al. 2004).
In a subset of studies, the endothelium was denuded from arterial rings using a wooden toothpick prior to vessel culture. After a 24 h culture, the rings were suspended in organ baths and endothelial function was verified by the ability of bradykinin (0.1 μmol/L; Sigma) to relax a 35 mmol/L KCl depolarizing contraction. In studies using the Cav1.2 channel agonist, FPL64176, rings were exposed to acetylcholine (10 μmol/L; Sigma) and thapsigargin (10 μmol/L; Sigma) to release and eliminate the reuptake of Ca2+ back into the sarcoplasmic reticulum (SR), and thereby eliminate a potential effect of FPL64176 on SR Ca2+ release (Wasserstrom et al. 2002). A steady‐state contraction was generated to FPL64176 (3 μmol/L; Tocris Bioscience) followed by application of the Cav1.2 channel antagonist, nifedipine (10 μmol/L; Sigma). Only arterial rings that contracted in response to FPL64176 and relaxed back to baseline in response to nifedipine were used for analysis.
Immunoblots
Cultured strips of RCA were homogenized and membrane lysate prepared (Liu et al. 2001). Equivalent amounts of membrane proteins were run on a 4–15% polyacrylamide gradient gel. After electrophoresis, the proteins were electroblotted to a PVDF membrane. The membrane was subsequently blocked overnight (at 4°C) with 5% non‐fat dry milk dissolved in a Tris‐buffered saline containing 0.1% Tween 20 (TBST). After decanting off the blocking solution, the membrane was incubated in a 1:100 dilution of rabbit polyclonal anti‐α 1C (Alomone Labs, Jerusalem, Israel) for 60 min followed by three washes (10 min each) using TBST. The membrane was then incubated with horseradish peroxidase (HRP) conjugated goat anti‐rabbit IgG (1:1000; Santa Cruz Biotech, Santa Cruz, CA) for 60 min and subsequently washed (3x) with TBST. Chemiluminescence permitted detection of the HRP‐labeled antibody using an AlphaImager Documentation and Analysis System (Alpha Innotech Corp., San Leandro, CA). Likewise, band densitometry was analyzed using AlphaImager software. Smooth muscle β‐actin (1:1000; Abcam, Cambridge, MA) was used as a loading control in Western blots.
Real‐time PCR
The relative abundance of Cav α transcript in arterial strips cultured for 24 h in EtOH or 17βE (1 nmol/L) was determined by real‐time PCR (Tobin et al. 2009). Total RNA was isolated using the RNeasy® protect mini‐kit (Qiagen, Germantown, MD). The resulting RNA was subjected to DNase treatment using DNA‐freeTM kit (Ambion, Austin, TX) to remove any contaminating DNA. Five hundred ng of the RNA isolate was reverse transcribed (iScript cDNA synthesis kit; Biorad, Hercules, CA) to generate cDNA. The cDNA was generated at 25°C for 5 min, followed by 42°C for 30 min and 85°C for 5 min. The cDNA sequences were then amplified by RT‐PCR (iQTM SYBR® Green Supermix Kit; Biorad, Hercules, CA) using Cav α and β‐actin specific primers, and an iCycler iQ® Multicolor Real Time PCR Detection System (Biorad, Hercules, CA). The forward and reverse primers used to amplify cDNA were reported earlier (Hirenallur et al. 2008): Cav α ‐ 5′‐ccgcccactaccaagatcaac‐3′ (forward) and 5′‐gcatctcgggctcctcctc‐3′ (reverse); β‐actin ‐ 5′‐accactggcattgtcatggactct‐3′ (forward) and 5′‐tcttcatgaggtagtcggtcaggt‐3′ (reverse). After initial denaturation at 95°C for 3 min, the following temperature‐cycling profile was used for amplification (40 cycles): 95°C for 10 sec denaturing and 62°C for 1 min for annealing and extension. β‐actin was used as an internal standard. Each amplification reaction contained 5 ng of the cDNA product. Control reactions contained all components except the cDNA template. The relative abundance of Cav α transcript in EtOH and 17βE‐treated arteries was quantified by the ΔΔCt method, and reported as percent of EtOH expression (Livak and Schmittgen 2001).
Scanning electron microscopy
As reported by us earlier (Hill and Muldrew 2014), scanning electron microscopy was used to verify the integrity of the endothelium. Arterial strips were pinned lumen side up on a wax surface in individual glass jars, then fixed with 2% glutaraldehyde (Ted Pella, Inc., Redding, CA) for 60 min, and then postfixed with 4% osmium tetroxide (Ted Pella, Inc.) for an additional 60 min. The specimens were dehydrated using ascending ratios of acetone/ethanol (1:1, 2:1, 4:1) followed by descending ratios of acetone/hexamethyldisilazane (4:1, 2:1, 1:1). Next, the samples were sputter‐coated with gold particles and the luminal surface observed using a PSEM II 2000 scanning electron microscope (FEI, Hillsboro, OR).
Statistics
All data are expressed as the mean ± SE for the number (n) of animals within each group. When comparing two groups, significance was determined using an unpaired t‐test. The KCl concentration–response curves were analyzed using two‐way repeated measures ANOVA with a Holm–Sidak post hoc analysis. Significant effect of multiple 17βE concentrations was evaluated using one‐way ANOVA followed by Bonferroni's post hoc analysis. Potential endothelial effects were analyzed using a two‐way ANOVA (without replication) followed by Tukey HSD post hoc analysis. The EC50 values (‐log of the effective concentration required to generate a 50% maximal response) were calculated and analyzed using GraphPad Prism 4.0 (GraphPad Software Inc., San Diego, CA). Statistical analysis was conducted using JMP 8 Statistical Discovery (SAS Institute Inc., USA) using a level of critical significance of α = 0.05; therefore, significance was defined as P < 0.05.
Results
Vascular reactivity responses
High concentrations of 17βΕ were necessary to relax depolarizing KCl‐induced contractions (Fig. 1), which fully rely on voltage‐dependent Ca2+ influx through Cav1.2 channels. The progressive relaxation mediated by 17βE (0.1–100 μmol/L) significantly attenuated contraction by 17.7 ± 2.4% and 61.3 ± 5.4% at concentrations of 10 μmol/L and 100 μmol/L 17βE, respectively (n = 7). EtOH served as a solvent and time control for the relaxation experiment. Likewise, when arterial rings were incubated with 17βE (1 pM to 100 μmol/L) for 60 min prior to generation of KCl concentration–response curves, only 17βE concentrations of 10 μmol/L and 100 μmol/L significantly attenuated KCl contractions (Fig. 2; n = 5). A 17βE concentration of 1 nmol/L did not acutely relax arteries precontracted by KCl (data not shown) and preincubation for 60 min in 1 nmol/L 17βE did not suppress KCl concentration–response curves as shown in Figure 2.
Figure 1.
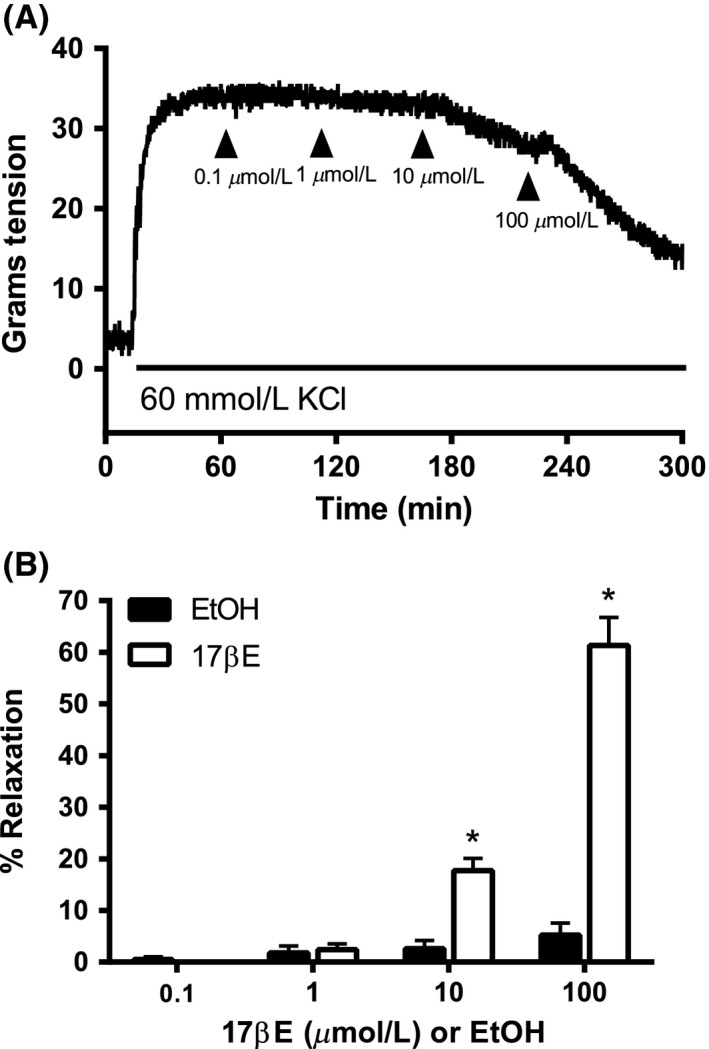
Pharmacological concentrations of 17β‐estradiol (17βE) are required to relax coronary arterial rings precontracted by 60 mmol/L KCl. (A) Typical recording shows concentration‐dependent relaxation by 17βE of a KCl‐contracted arterial ring. Exposure to KCl is indicated by the horizontal line. (B) Average values (mean ± SE) for 17βE‐induced relaxation of KCl precontracted rings. Equal volumes of EtOH served as solvent control. *P < 0.05 using an unpaired t‐test, EtOH versus 17βE groups (n = 7 pigs).
Figure 2.
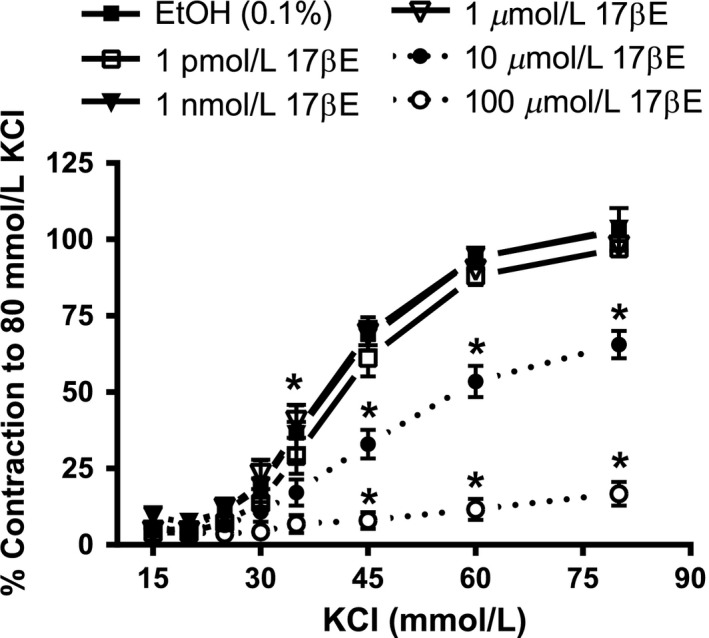
A short‐term (60 min) preexposure to 17βE necessitates the use of high, pharmacological 17βE concentrations to inhibit depolarization‐induced contractions to KCl. Rings were incubated for 60 min in 1 pmol/L, 1 nmol/L, 1 μmol/L, 10 μmol/L, or 100 μmol/L 17βE, or its EtOH solvent, before generating cumulative concentration–response relationships to KCl. The KCl‐induced tension development is expressed as percent (%) of the maximal contractile response to 80 mmol/L KCl before incubating rings in 17βE or EtOH. The data are shown as the mean ± SE and analysis was conducted using two‐way repeated measures ANOVA (Holm–Sidak post hoc). *P < 0.05, 10 μmol/L or 100 μmol/L 17βE versus EtOH at the same KCl concentration (n = 5 pigs).
In contrast, prolonged preexposure of similar arteries to 1 nmol/L 17βE attenuated depolarization‐induced contractions (Fig. 3). Coronary arterial rings cultured for 24 h prior to tension recording in RPMI media containing 1 nmol/L 17βE exhibited an ~25% attenuation of the concentration–response curves to KCl (Fig. 3). This inhibitory effect was accentuated in arterial rings preincubated in 10 μmol/L 17βE for 24 h, whereas 1 pM 17βE had no significant effect on the KCl contraction. The acute (1 h) versus longer term (24 h) effects of 1 nmol/L 17βE on KCl contraction are compared in Table 1.
Figure 3.
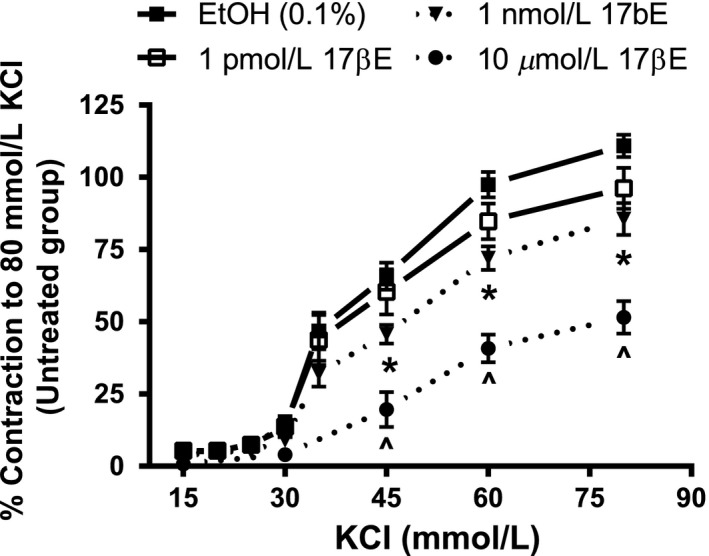
Incubation of coronary arterial rings in a low concentration (1 nmol/L) of 17βΕ for 24 h attenuates the concentration–response curve to KCl. Cumulative concentration–response relationships to KCl were generated after rings were cultured for 24 h in 1pM, 1 nmol/L, or 10 μmol/L 17βΕ or ethanol solvent (EtOH), or left untreated (no addition of 17βΕ or EtOH). The KCl contractions are expressed as percent (%) of the maximal contractile response to 80 mmol/L KCl of the untreated group (n = 5 pigs). The data are shown as the mean ± SE and analysis was conducted using two‐way repeated measures ANOVA (Holm–Sidak post hoc). *P < 0.05, EtOH versus 1 nmol/L 17βΕ at the same KCl concentration. Likewise, ^denotes P < 0.05, EtOH versus 10 μmol/L 17βΕ at the same KCl concentration.
Table 1.
Comparison between effects of acute (1 h) and prolonged (24 h) exposure to 17β‐estradiol (17βE) on EC50 values and maximal tension (Tmax) of KCl concentration–response relationships
| Exposure time | [17βE] or EtOH Solvent | n | EC50 (mmol/L)a | Tmax b |
|---|---|---|---|---|
| 1 h | EtOH (0.1%) | 5 | 39.89 ± 1.01 | 102.81 ± 1.86 |
| 1 pmol/L 17βE | 5 | 41.41 ± 1.01 | 96.96 ± 1.18 | |
| 1 nmol/L 17βE | 5 | 40.87 ± 1.02 | 103.33 ± 7.01 | |
| 24 h | EtOH (0.1%) | 5 | 41.01 ± 1.07 | 110.87 ± 3.83 |
| 1 pM 17βE | 5 | 39.67 ± 1.07 | 96.17 ± 7.15 | |
| 1 nmol/L 17βE | 5 | 44.06 ± 1.08 | 85.59 ± 5.50c |
KCl concentration required to reach one‐half maximal response to 80 mmol/L KCl.
Maximal contraction to 80 mmol/L KCl. Data are expressed as percent of maximal contractile response to the initial addition of 80 mmol/L KCl.
P < 0.05, EtOH versus 1 nmol/L 17βE.
17βE for 24 h results in loss of Cav α protein
Corroborating our isometric tension‐recording findings of reduced Ca2+‐dependent contractions after prolonged exposure to 1 nmol/L 17βE, we also found that a 24 h exposure to 1 nmol/L 17βE decreased the expression of the pore‐forming α 1C subunit of the Cav1.2 channel, Cav α, by 35 ± 0.9% (Fig. 4A and B). The Western blots shown in Figure 4 depict immunoreactive doublet bands corresponding to the short (~200 kD) and long (~240 kD) forms of the Cav α subunit. The EtOH solvent (0.1%) and 1 pmol/L 17βE did not significantly affect Cav α protein expression. Immunodensity of the internal standard, β‐actin, was similar between treatment groups.
Figure 4.
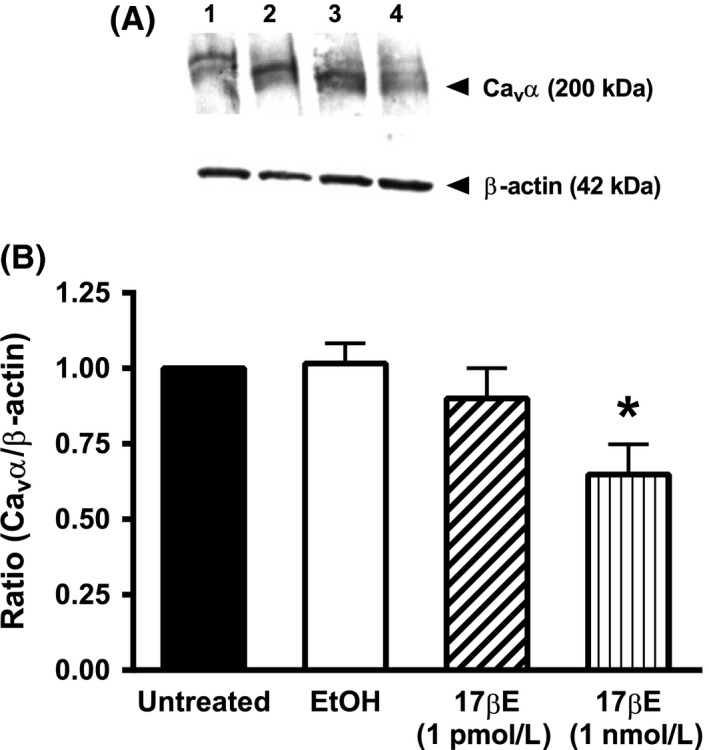
Exposure to 17βΕ for 24 h decreases the expression of the α 1C pore‐forming subunit (Cav α) of the Cav1.2 channel in coronary arterial strips. (A) Diffuse immunoreactive band corresponding to the Cav α protein; the short (~200 kD) and long (~240 kD) forms of Cav α are revealed as a doublet in some blots. β‐Actin (42 kDa) was used as an internal standard. Protein lysates were obtained from cultured arteries exposed for 24 h to the following conditions: lane 1 = untreated; lane 2 = EtOH (0.1%); lane 3 = 1 pmol/L 17βΕ; lane 4 = 1 nmol/L 17βΕ. B, The densitometric ratio corresponding to Cav α in arterial lysates exposed for 24 h to EtOH or 1 nmol/L 17βΕ compared to untreated arteries. Results indicate that 1 nmol/L 17βΕ for 24 h decreases Cav α protein abundance. Data represent mean ± S.E. (n = 5 pigs) and analyzed using one‐way ANOVA followed by Bonferroni's post hoc analysis. There was no difference in β‐actin expression between groups.
Real‐time PCR was used to determine if 1 nmol/L 17βE reduced Cav α subunit abundance by transcriptional regulation. Initially, we standardized the real‐time PCR assay before comparing Cav α subunit mRNA levels. First, the presence of a single peak in the melt curve analysis inferred a single amplification product corresponding to Cav α amplified product. Second, we verified that the real‐time PCR assay was adequately sensitive to detect 50% change in the expression of amplification products corresponding to Cav α mRNA. Standard amplification curves generated for Cav α using twofold serial dilutions of cDNA from 12.5 ng to 1.56 ng per amplification reaction resulted in a one‐cycle (1.0 ± 0.1) increase in threshold cycle as predicted, thereby demonstrating sensitive detection of changes in transcript level in this range. After assay verification, the expression of Cav α transcript was compared between arteries cultured in EtOH solvent or 1 nmol/L 17βE for 24 h (Fig. 5A and B). Representative real‐time PCR curves indicated an average cycle threshold of 26.33 ± 0.52 for the detection of Cav α in EtOH‐treated arteries (n = 6) compared to 26.36 ± 0.59 in 17βE‐treated arteries (n = 6), implying similar abundance of the Cav α subunit transcript as estimated by the ΔΔCt method (Fig. 5C). In the same arterial preparations, as expected, similar cycle thresholds of 16.26 ± 0.35 (EtOH, n = 6) and 16.52 ± 0.39 (17βE, n = 6) also were calculated for the internal standard β‐actin.
Figure 5.
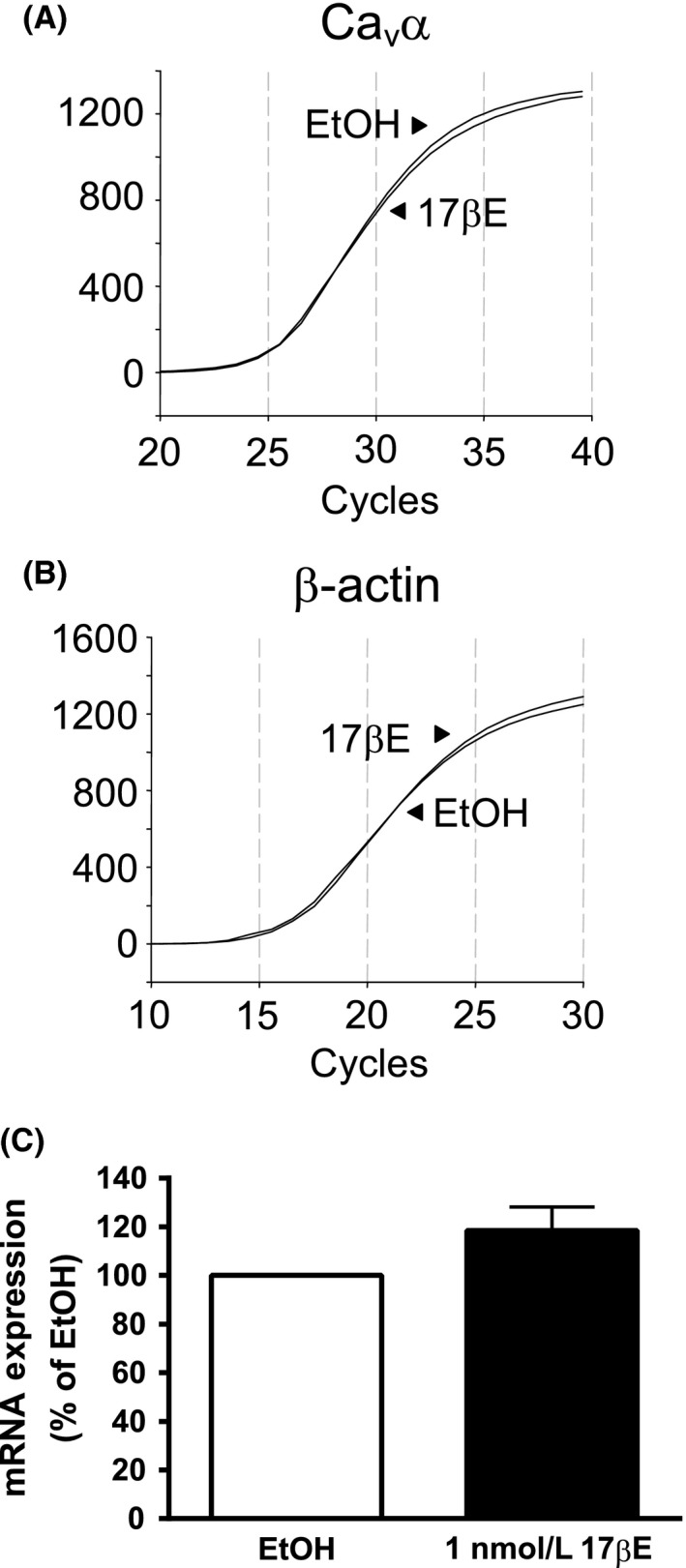
Exposure of arterial segments to 1 nmol/L 17βE for 24 h failed to alter expression of Cav α transcript. (A and B) Real‐time PCR amplifications of Cav α (A) and internal standard β‐actin (B) are similar between arteries cultured for 24 h in RPMI media containing 1 nmol/L 17βΕ or its solvent (EtOH). Relative fluorescence units. (B) Abundance of Cav α transcript is not significantly different between 17βΕ and EtOH‐treated arteries as analyzed by the ΔΔCt method. Data are expressed as percent (%) transcript expression in EtOH (n = 6 pigs).
17βE‐induced inhibition of KCl contractions and Cav α expression is not endothelium dependent
The effect of 24 h exposure to 1 nmol/L 17βE on Cav1.2 channel function also was evaluated by comparing the contractile response to the Cav1.2 channel agonist, FPL64176 (3 μmol/L), between coronary rings exposed to the EtOH solvent or 17βE. Studies were performed in endothelium intact (+Endo) and denuded (‐Endo) arterial rings to evaluate if the inhibitory effect of 1 nmol/L 17βE on Cav1.2 channel‐dependent contraction was mediated by endothelium‐derived factors. In addition to its intended role as a Cav1.2 channel agonist, FPL64176 reportedly can induce Ca2+ release from the sarcoplasmic reticulum (SR) by opening ryanodine receptors (Wasserstrom et al. 2002; Vaithianathan et al. 2010). Therefore, as shown in Figure 6A, arteries were simultaneously exposed to 10 μmol/L acetylcholine (Ach) and 10 μmol/L thapsigargin (TSG) to induce SR Ca2+ release and prevent its reuptake by inhibiting Ca2+‐ATPase, respectively. Arteries incubated in 1 nmol/L 17βE elicited a contractile response to FPL64176 that was 47 ± 0.56% and 60 ± 0.33% less (n = 6) compared to control arteries (EtOH) in +Endo and ‐Endo rings, respectively (Fig. 6B). Overall, the presence or absence of the endothelium had no effect on FPL64176‐induced tension development (P = 0.26).
Figure 6.
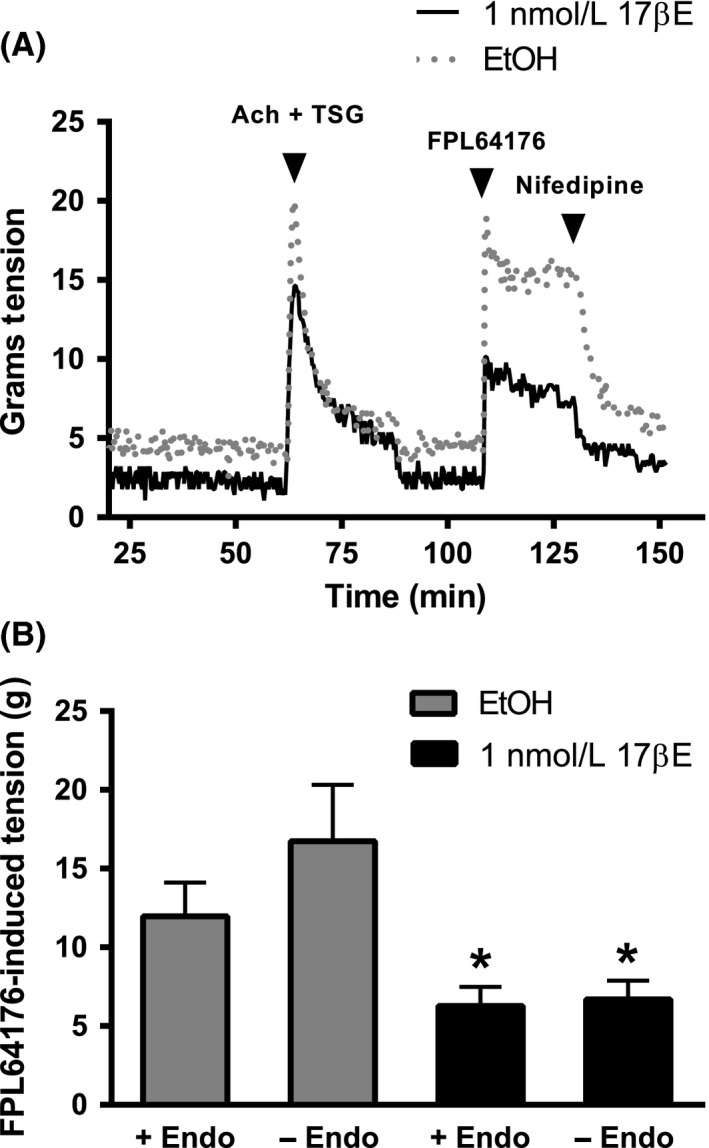
Coronary arterial segments cultured for 24 h in 1 nmol/L 17βΕ show attenuated contractions to Cav1.2 channel activation by 3 μmol/L FPL64176. Endothelium intact (+Endo) and denuded (‐Endo) arteries were cultured in 1 nmol/L 17βΕ or its solvent (EtOH). A, Experimental protocol used to evaluate the contractile response to the Cav1.2 channel agonist, FPL64176 (3 μmol/L). To prevent a potentially confounding influence of FPL‐induced Ca2+ release from sarcoplasmic reticulum (SR), rings initially were exposed to acetylcholine (Ach, 10 μmol/L) and thapsigargin (TSG, 10 μmol/L) before evaluating the contractile response to FPL64176. Complete relaxation of FPL64176‐induced contractions by 10 μmol/L nifedipine confirmed dependence on Cav1.2 channels. (B) Composite data from studies depicted in panel A. Data are expressed as mean ± S.E. and analyzed using a two‐way ANOVA (without replication) followed by Tukey HSD post hoc analysis. * P < 0.05, EtOH ± Endo versus 17βΕ ± Endo; (n = 7 pigs).
As shown by anti‐Cav α Western blots (Fig. 7), coronary arterial strips cultured in 1 nmol/L 17βE for 24 h exhibited a significant decrease in expression of the pore‐forming Cav α subunit of the Cav1.2 channel in +Endo and ‐Endo arterial strips. The +Endo and –Endo arteries exposed to 17βE exhibited a densitometric ratio that was 62 ± 9% and 74 ± 14% of the control +Endo arteries treated with EtOH solvent, respectively. Western blot data are expressed as a ratio of Cav α expression in +Endo arteries exposed to EtOH. In a control set of experiments, the EtOH solvent had no effect on Cav α subunit expression between +Endo and ‐Endo arteries (n = 6, data not shown); the densitometric ratio (Cav α/β‐actin) was 1.01 ± 0.09 and 0.93 ± 0.17 for +Endo and –Endo arteries, respectively.
Figure 7.
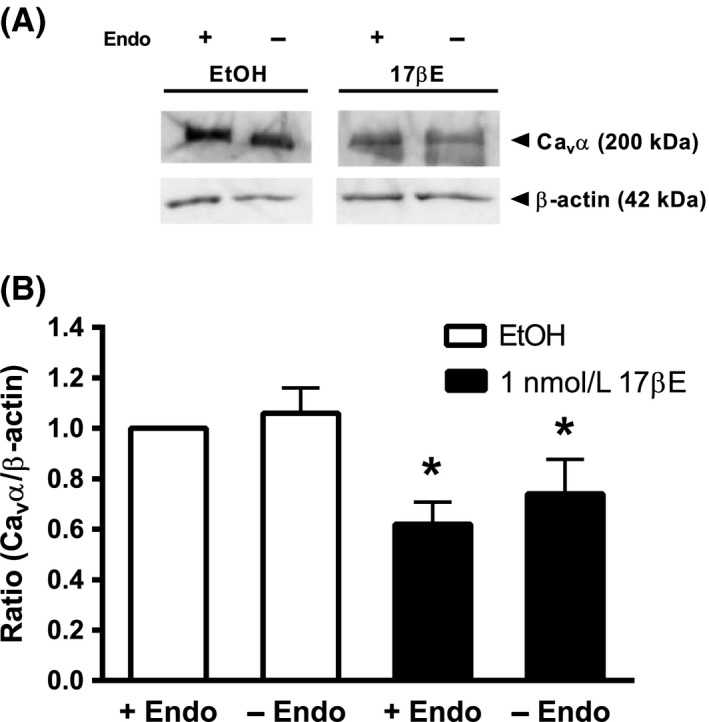
17βΕ decreases expression of the Cav α pore‐forming subunit independently of endothelium. Upper panel shows a representative anti‐Cav α immunoblot from arteries with intact (+Endo) or denuded (‐Endo) endothelium, which were cultured for 24 h in RPMI media containing 1 nmol/L 17βΕ or ethanol (EtOH). 17βΕ decreased the densitometric ratio in intact and denuded arteries. β‐actin served as an internal standard. The images are from different parts of the same gel. The Cav α and β‐actin groupings were imaged separately. Band intensity between groups was normalized to β‐actin and expressed as a densitometric ratio of the EtOH+Endo group. Data represent mean ± SE (n = 7 pigs). *P < 0.05, EtOH ± Endo versus 17βΕ ± Endo using a two‐way ANOVA (without replication) followed by Tukey HSD post hoc analysis.
17βE‐induced loss of Cav α protein requires ERα/ERβ
Previously in Figs. 4 and 7, we demonstrated a significant reduction in the Cav α subunit protein by 1 nmol/L 17βE. Figure 8A shows that the addition of the estrogen receptor (ERα/ERβ) antagonist, ICI 182,780 (10 μmol/L), to the RPMI media significantly prevented the decrease in Cav α protein expression induced by 24 h exposure to 1 nmol/L 17βE (n = 7). To investigate whether the G‐protein‐coupled estrogen receptor (GPER) also may mediate the 17βE‐induced reduction in Cav1.2 channel expression, arterial strips were incubated for 24 h in 1 nmol/L 17βE in the presence of 10 μmol/L G15, a selective GPER antagonist. This intervention failed to prevent the 17βE‐induced decrease in Cav α protein expression (Fig. 8B, n = 4).
Figure 8.
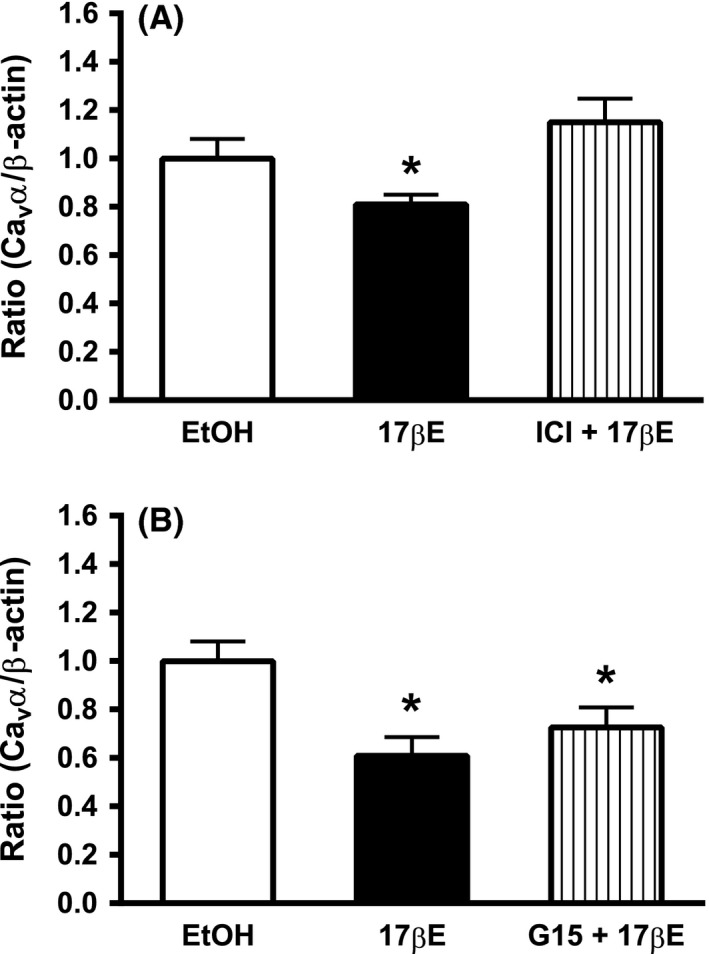
Loss of Cav α in cultured arteries exposed to 1 nmol/L 17βΕ for 24 h is prevented by pharmacological block of ER α/ER β. (A) The ER α/ER β antagonist ICI 182,780 (ICI, 10 μmol/L) prevents 17βΕ‐induced downregulation of the Cav α subunit. Data represent mean ± SE (n = 7 pigs). (B) The G‐protein‐coupled estrogen receptor (GPER) antagonist, G15 (10 μmol/L) failed to prevent a 17βΕ‐induced loss of Cav α. Data represent mean ± SEM. (n = 4 pigs) and are expressed as a densitometric ratio. *P < 0.05 from EtOH using one‐way ANOVA followed by Bonferroni's post hoc analysis.
Discussion
Evidence suggests that circulating estrogen contributes to protection of the female sex against elevated coronary vascular tone by reducing Ca2+ entry into ASMCs (Crews et al. 1999; Orshal and Khalil 2004; Tharp et al. 2014). Indeed, high concentrations of estrogen inhibit Ca2+ influx and attenuate Ca2+‐dependent contractions in isolated coronary arteries (Han et al. 1995; Teoh et al. 1999; Hill et al. 2010). However, a confounding finding is that in vitro inhibition of vascular reactivity is only observed at high concentrations of estrogen, which may exceed physiological concentrations by at least 1000‐fold (Han et al. 1995; Teoh et al. 1999; Salom et al. 2002; Hill et al. 2010; Reslan et al. 2013). This study addresses this concern with two major findings. First, we observed that a prolonged (24 h) rather than acute exposure to 17βE increases its potency as an inhibitor of Ca2+‐dependent contractions, such that 1 nmol/L 17βE significantly depresses contractions induced by depolarizing KCl and the Cav1.2 channel agonist, FPL64176. Second, to our knowledge, we provide the first in vitro findings to directly demonstrate that 17βE results in a posttranscriptional decrease in Cav1.2 channel expression. Our results corroborate in vivo sex studies, which have indirectly suggested that the presence of circulating estrogens in female subjects is associated with lower Ca2+ channel expression in coronary ASMCs, and thus, less Ca2+‐dependent tone developed by the coronary arteries (Bowles 2001; Bowles et al. 2004; Tharp et al. 2014). Because Cav1.2 channels are the predominant pathway for voltage‐dependent Ca2+ influx in ASMCs of the coronary circulation (Bowles et al. 1998a), decreasing their expression in the plasma membrane of ASMCs will increase arterial diameter and promote coronary blood flow.
We used 1 nmol/L 17βE to correlate with previous in vitro studies that used 1 nmol/L 17βE as a near “physiological” E2 concentration to evaluate its effect on the vascular mechanics of arteries (Keung et al. 2005, 2011; Cignarella et al. 2009; Lekontseva et al. 2011). Therefore, we did not attempt to use a definite plasma 17βE concentration from cycling sows or premenopausal women because it is difficult to extrapolate in vitro results (which can differ based upon the experimental model) to the in vivo condition. In premenopausal women, circulating estrogen is primarily composed of 17βE, and its concentration approximates 0.2 nmol/L before ovulation; this value rises to 1 nmol/L at ovulation (Marsh et al. 2011), which is comparable to the 1 nmol/L 17βE concentration used in this study. In mature sows, the plasma estrogen concentration varies from 20 to 840 pmol/L depending on the estrous cycle (Cook et al. 1977; Chatrath et al. 2003). Despite differences in circulating estrogen levels between premenopausal women and our female pig model, there are many advantages to using porcine coronary arteries as a model in this study. The structural similarities of the cardiovascular system between pigs and humans have been well documented (Swindle et al. 2012). In addition, pig coronary arteries similarly express estrogen receptors (ER), ERα and ERβ, as well as the G‐protein‐coupled estrogen receptor, GPER (Traupe et al. 2007; Yu et al. 2011). Overall, circulating plasma estrogens have been well documented to be responsible for differences in coronary vascular reactivity between sexes in swine (Barber and Miller 1997; Jones et al. 1999; Bowles 2001; Heaps and Bowles 2002).
The acute and nongenomic inhibition of Cav1.2 channels in porcine coronary arteries by high concentrations of 17βE has been demonstrated by us and others (Han et al. 1995; Hill et al. 2010). As shown in this study, when coronary arteries are acutely (≤60 min) exposed to 17βE, a 17βE concentration of ≥1 μmol/L is necessary to inhibit contractile responses to depolarizing KCl solutions. Similarly, our study confirms findings by Teoh et al. (2000) that acutely exposing porcine coronary arteries to a lower 17βE concentration of 1 nmol/L fails to inhibit Ca2+‐dependent contractions. It is likely that the apparent lack of in vitro sensitivity to 17βE is partly due to the larger amount of connective tissue and increased diffusion distance of coronary arteries compared to small, resistance arteries. Only after a 24 h exposure to 17βE, were we able to demonstrate that 1 nmol/L 17βE attenuates the depolarization‐induced contractile response to KCl and inhibits contractions elicited by the Cav1.2 channel agonist, FPL64176. Both of these contractile responses are mediated by voltage‐dependent Ca2+ influx through Cav1.2 channels, as verified by their reversal by nifedipine, a selective Cav1.2 channel antagonist (Wang et al. 2011; Owen et al. 2013).
Our findings further suggest that the attenuation of Ca2+‐dependent contractions observed after a 24 h exposure to 1 nmol/L 17βE is mediated by a posttranscriptional downregulation of the pore‐forming Cav α subunit of the Cav1.2 channel. To our knowledge, no studies have reported an effect of near physiological estrogen levels on Cav α expression in ASMCs, although it is well recognized that Cav1.2 channels are dynamically regulated in vivo by circulating factors and disease states. For example, an overabundance of arterial Cav1.2 channels is a shared feature of pig, rat, and mouse models of hypertension, which also exhibit elevated myogenic tone and arterial reactivity (Molero et al. 2002; Pratt et al. 2002; Pesic et al. 2004; Hirenallur et al. 2008; Kharade et al. 2013). Likewise, Wang et al. (2008) reported that angiotensin II increases Cav α subunit expression without changes in Cav α mRNA levels in cultured rat mesenteric arteries, implying involvement of posttranscriptional regulation of Cav1.2. However, Tharp et al. (2014) reported that ASMCs of female pigs subjected to ovariectomy exhibited a twofold increase in Ca2+ current in the absence of altered Cav α transcript or protein expression. Overall, these findings infer that the particular experimental model may determine the mechanism by which the abundance of Cav1.2 channels is regulated in the plasma membrane (Mazzuca et al. 2015).
Importantly, our results suggest that the loss of Cav1.2 channels in coronary arteries in response to 24 h exposure to 1 nmol/L 17βE is prevented by the estrogen receptor (ERα/ERβ) antagonist ICI 182,780, whereas the GPER antagonist G15 has no effect. It remains unlikely that G15 was ineffective as a GPER inhibitor in our studies, because Yu et al. (2011) previously used a similar concentration of G15 (3 μmol/L) to inhibit the GPER‐mediated vasorelaxation induced by its selective agonist, G‐1, in porcine coronary arteries. Instead, our findings imply that 17βE may bind to ERα/ERβ to alter posttranscriptional regulation of Cav1.2 channels, a process that has not been defined in ASMCs. However, in nonvascular cell types and heterologous expression systems, the posttranscriptional mechanisms that regulate Cav1.2 channel expression include ubiquitination and proteosomal degradation of the Cav α subunit (Altier et al. 2011; Rougier et al. 2011). Thus, it is possible that ERα/ERβ signaling disrupts the fine balance between Cav1.2 channel biogenesis and degradation, potentially resulting in a loss of Cav1.2 channels at the plasma membrane.
The relaxation response to high, pharmacological concentrations of estrogen relies on direct inhibition of Ca2+ influx into coronary ASMCs, and does not require the vascular endothelium (Gilligan et al. 1994; Han et al. 1995; Nakajima et al. 1995; Hill et al. 2010; Yang and Reckelhoff 2011). However, the endothelium has been implicated in the regulation of Cav1.2 channel expression in ASMCs (Wang et al. 2008, 2011). Therefore, this study also sought to define the contribution of endothelium to the 17βE‐induced loss of Cav1.2 channels and Ca2+‐dependent contractions. However, we failed to find a role for the endothelium in 17βE actions. Instead, the 17βE‐mediated decrease in Cav1.2 channel numbers appears to be mediated by ERα/ERβ expressed by ASMCs, and we suggest the downregulation of Cav1.2 channels by ERα/ERβ may attenuate the development of abnormal coronary arterial tone. The ERα/ERβ‐mediated, endothelium‐independent dilation and decrease in voltage‐gated Ca2 + influx has been similarly shown using rat mesenteric microvessels (Mazzuca et al. 2015).
Although our porcine coronary artery model offers many experimental advantages, several limitations of this study should be acknowledged. First, as discussed earlier, we did not use a defined “physiological” plasma 17βE concentration. Although our porcine coronary artery model offers many experimental advantages, we cannot ensure our results directly translate to the human condition. Second, the limitations of in vitro vascular studies that remove ASMCs from modulating endogenous influences and place them in artificial vessel culture conditions are appreciated. In earlier studies (Lindqvist et al. 1999; Thorne and Paul 2003), and also here, we used serum‐free RPMI media to maintain arterial contractility and avoid cell proliferation. Third, we attempted to perform whole‐cell patch clamp on ASMCs freshly isolated from our cultured coronary arteries to directly confirm a loss of functional Cav1.2 channels in response to 17βE. However, we were unable to maintain the high resistance (≥10 GΩ) seals required for high‐quality recordings of Ca2+ currents, although we routinely record ion currents from ASMCs freshly isolated from many other arterial preparations (Pesic et al. 2004; Hirenallur et al. 2008; Thakali et al. 2010; Kharade et al. 2013). For this reason, our study relied on two different vascular reactivity assays using depolarizing KCl solution and FPL64176, which directly activate Cav1.2 channels in ASMCs in situ to allow for detection of changes in Cav1.2 channel function.
In conclusion, this study provides initial evidence that a nonacute, low concentration of 17βE for 24 h results in loss of functional Cav1.2 channels in porcine coronary arteries, and this event appears to rely on posttranscriptional modifications enacted by 17βE through ERα/ERβ signaling. This finding is significant because most in vitro studies have used high pharmacological estrogen concentrations to emphasize the antagonistic effects of estrogen on Ca2+ signaling in ASMCs and on arterial contraction. It is possible that this previously unrecognized effect of estrogen contributes to the attenuation of coronary vascular contractions reported by sex studies in females compared to males (Jones et al. 1999; Bowles 2001; Tharp et al. 2014).
Disclosure
None declared.
Author Contributions
Research design: Hill, Dalton, Joseph, and Rusch; Conducted experiments: Hill, Dalton, Joseph, and Thakali; Performed data analysis: Hill, Dalton, Joseph, and Rusch; Contributed to the writing of the manuscript: Hill, Dalton, Joseph, Thakali, and Rusch.
Acknowledgements
We appreciate Odom's Tennessee Pride Sausage, Inc., Little Rock, Arkansas for their donation of the pig hearts for this study. Undergraduates at the University of Central Arkansas involved in this study were Sean Necessary, Quinton Kaufman, Edouard Niyonsaba, and Mohamed Idrissa Moussa. Technical assistance with the SEM was provided by Mr. Jerry Mimms (Dept. of Biology, University of Central Arkansas).
Hill B. J.F., Dalton R. J., Joseph B. K., Thakali K. M., Rusch N. J.. 17β‐estradiol reduces Cav1.2 channel abundance and attenuates Ca2+‐dependent contractions in coronary arteries. Pharma Res Per,5(5), 2017, e00358, https://doi.org/10.1002/prp2.358
References
- Altier C, Garcia‐Caballero A, Simms B, You H, Chen L, Walcher J, et al. (2011). The Cavβ subunit prevents RFP2‐mediated ubiquitination and proteasomal degradation of L‐type channels. Nat Neurosci 14: 173–180. [DOI] [PubMed] [Google Scholar]
- Barber DA, Miller VM (1997). Gender differences in endothelium‐dependent relaxations do not involve NO in porcine coronary arteries. Am J Physiol 273: H2325–H2332. [DOI] [PubMed] [Google Scholar]
- Bowles DK (2001). Gender influences coronary L‐type Ca2+ current and adaptation to exercise training in miniature swine. J Appl Physiol 91: 2503–2510. [DOI] [PubMed] [Google Scholar]
- Bowles DK, Hu Q, Laughlin MH, Sturek M (1998a). Exercise training increases L‐type calcium current density in coronary smooth muscle. Am J Physiol 275: H2159–H2169. [DOI] [PubMed] [Google Scholar]
- Bowles DK, Laughlin MH, Sturek M (1998b). Exercise training increases K+‐channel contribution to regulation of coronary arterial tone. J Appl Physiol 84: 1225–1233. [DOI] [PubMed] [Google Scholar]
- Bowles DK, Maddali KK, Ganjam VK, Rubin LJ, Tharp DL, Turk JR, et al. (2004). Endogenous testosterone increases L‐type Ca2+ channel expression in porcine coronary smooth muscle. Am J Physiol Heart Circ Physiol 287: H2091–H2098. [DOI] [PubMed] [Google Scholar]
- Chatrath R, Ronningen KL, LaBreche P, Severson SR, Jayachandran M, Bracamonte MP, et al. (2003). Effect of puberty on coronary arteries from female pigs. J Appl Physiol 95: 1672–1680. [DOI] [PubMed] [Google Scholar]
- Cignarella A, Bolego C, Pelosi V, Meda C, Krust A, Pinna C, et al. (2009). Distinct roles of estrogen receptor‐alpha and beta in the modulation of vascular inducible nitric‐oxide synthase in diabetes. J Pharmacol Exp Ther 328: 174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook B, Hunter RH, Kelly AS (1977). Steroid‐binding proteins in follicular fluid and peripheral plasma from pigs, cows and sheep. J Reprod Fertil 51: 65–71. [DOI] [PubMed] [Google Scholar]
- Cox RH, Fromme S (2015). Expression of calcium channel subunit variants in small mesenteric arteries of WKY and SHR. Am J Hypertens 28: 1229–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews JK, Khalil RA (1999). Gender‐specific inhibition of Ca2+ entry mechanisms of arterial vasoconstriction by sex hormones. Clin Exp Pharmacol Physiol 26: 707–715. [DOI] [PubMed] [Google Scholar]
- Crews JK, Murphy JG, Khalil RA (1999). Gender differences in Ca2+ entry mechanisms of vasoconstriction in Wistar‐Kyoto and spontaneously hypertensive rats. Hypertension 34: 931–936. [DOI] [PubMed] [Google Scholar]
- Dennis MK, Burai R, Ramesh C, Petrie WK, Alcon SN, Nayak TK, et al. (2009). In vivo effects of a GPR30 antagonist. Nat Chem Biol 5: 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Gros R, Limbird LE, Chorazyczewski J, Feldman RD (2009). Estradiol‐mediated ERK phosphorylation and apoptosis in vascular smooth muscle cells requires GPR 30. Am J Physiol Cell Physiol 297: C1178–C1187. [DOI] [PubMed] [Google Scholar]
- Gilligan DM, Quyyumi AA, Cannon RO III (1994). Effects of physiological levels of estrogen on coronary vasomotor function in postmenopausal women. Circulation 89: 2545–2551. [DOI] [PubMed] [Google Scholar]
- Han SZ, Karaki H, Ouchi Y, Akishita M, Orimo H (1995). 17β‐Estradiol inhibits Ca2+ influx and Ca2+ release induced by thromboxane A2 in porcine coronary artery. Circulation 91: 2619–2626. [DOI] [PubMed] [Google Scholar]
- Heaps CL, Bowles DK (2002). Gender‐specific K+ channel contribution to adenosine‐induced relaxation in coronary arterioles. J Appl Physiol 92: 550–558. [DOI] [PubMed] [Google Scholar]
- Hill BJ, Muldrew E (2014). Oestrogen upregulates the sarcoplasmic reticulum Ca2+ ATPase pump in coronary arteries. Clin Exp Pharm Physiol 41: 430–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill BJ, Gebre S, Schlicker B, Jordan R, Necessary S (2010). Nongenomic inhibition of coronary constriction by 17β‐estradiol, 2‐hydroxyestradiol, and 2‐methoxyestradiol. Can J Physiol Pharmacol 88: 147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirenallur SD, Haworth ST, Leming JT, Chang J, Hernandez G, Gordon JB, et al. (2008). Upregulation of vascular calcium channels in neonatal piglets with hypoxia‐induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 295: L915–L924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokinen MP, Clarkson TB, Prichard RW (1985). Animal models in atherosclerosis research. Exp Mol Pathol 42: 1–28. [DOI] [PubMed] [Google Scholar]
- Jones AW, Rubin LJ, Magliola L (1999). Endothelin‐1 sensitivity of porcine coronary arteries is reduced by exercise training and is gender dependent. J Appl Physiol 87: 1172–1177. [DOI] [PubMed] [Google Scholar]
- Keung W, Vanhoutte PM, Man RY (2005). Acute impairment of contractile responses by 17β estradiol is cAMP and protein kinase G dependent in vascular smooth muscle cells of the porcine coronary arteries. Br J Pharmacol 144: 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keung W, Chan ML, Ho EY, Vanhoutte PM, Man RY (2011). Non‐genomic activation of adenylyl cyclase and protein kinase G by 17β estradiol in vascular smooth muscle of the rat superior mesenteric artery. Pharmacol Res 64: 509–516. [DOI] [PubMed] [Google Scholar]
- Khalil RA (2013). Estrogen, vascular estrogen receptor and hormone therapy in postmenopausal vascular disease. Biochem Pharmacol 86: 1627–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharade SV, Sonkusare SK, Srivastava AK, Thakali KM, Fletcher TW, Rhee SW, et al. (2013). The β3 subunit contributes to vascular calcium channel upregulation and hypertension in angiotensin II‐infused C57BL/6 mice. Hypertension 61: 137–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuga T, Shimokawa H, Hirakawa Y, Kadokami Y, Arai Y, Fukumoto Y, et al. (2000). Increased expression of L‐type calcium channels in vascular smooth muscle cells at spastic site in a porcine model of coronary artery spasm. J Cardiovasc Pharmacol 35: 822–828. [DOI] [PubMed] [Google Scholar]
- Lekontseva O, Chakrabarti S, Jiang Y, Cheung CC, Davidge ST (2011). Role of neuronal nitric‐oxide synthase in estrogen‐induced relaxation in rat resistance arteries. J Pharmacol Exp Ther 339: 367–375. [DOI] [PubMed] [Google Scholar]
- Lindqvist A, Nordstrîm I, Malmqvist U, Nordenfelt P, Hellstrand P (1999). Long‐term effects of Ca2+ on structure and contractility of vascular smooth muscle. Am J Physiol 277: C64–C73. [DOI] [PubMed] [Google Scholar]
- Liu X, Rusch NJ, Striessnig J, Sarna SK (2001). Down‐regulation of L‐type calcium channels in inflamed circular smooth muscle cells of the canine colon. Gastroenterology 120: 480–489. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001). Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐ΔΔ CT) Method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- Marsh EE, Shaw ND, Klingman KM, Tiamfook‐Morgan TO, Yialamas MA, Sluss PM, et al. (2011). Estrogen levels are higher across the menstrual cycle in African‐American women compared with Caucasian women. J Clin Endocrinol Metab 96: 3199–3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzuca MQ, Mata KM, Li W, Rangan SS, Khalil RA (2015). Estrogen receptor subtypes mediate distinct microvascular dilation and reduction in [Ca2+]i in mesenteric microvessels of female rat. J Pharmacol Exp Ther 352: 291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molero MM, Giulumian AD, Reddy VB, Ludwig LM, Pollock JS, Pollock DM, et al. (2002). Decreased endothelin binding and [Ca2+]i signaling in microvessels of DOCA‐salt hypertensive rats. J Hypertens 20: 1799–1805. [DOI] [PubMed] [Google Scholar]
- Nakajima T, Kitazawa T, Hamada E, Hazama H, Omata M, Kurachi Y (1995). 17β‐estradiol inhibits the voltage‐dependent L‐type Ca2+ currents in aortic smooth muscle cells. Eur J Pharmacol 294: 625–635. [DOI] [PubMed] [Google Scholar]
- Orshal JM, Khalil RA (2004). Gender, sex hormones, and vascular tone. Am J Physiol Regul Integr Comp Physiol 286: R233–R249. [DOI] [PubMed] [Google Scholar]
- Owen MK, Witzmann FA, McKenney ML, Lai X, Berwick ZC, Moberly SP, et al. (2013). Perivascular adipose tissue potentiates contraction of coronary vascular smooth muscle: influence of obesity. Circulation 128: 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesic A, Madden JA, Pesic M, Rusch NJ (2004). High blood pressure upregulates arterial L‐type Ca2+ channels: is membrane depolarization the signal? Circ Res 94: e97–e104. [DOI] [PubMed] [Google Scholar]
- Pratt PF, Bonnet S, Ludwig LM, Bonnet P, Rusch NJ (2002). Upregulation of L‐type Ca2+ channels in mesenteric and skeletal arteries of SHR. Hypertension 40: 214–219. [DOI] [PubMed] [Google Scholar]
- Puntawangkoon C, Morgan TM, Herrington DM, Hamilton CA, Hundley WG (2010). Submaximal exercise coronary artery flow increases in postmenopausal women without coronary artery disease after estrogen and atorvastatin. Menopause 17: 114–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reslan OM, Yin Z, do Nascimento GR, Khalil RA (2013). Subtype‐specific estrogen receptor‐mediated vasodilator activity in the cephalic, thoracic, and abdominal vasculature of female rat. J Cardiovasc Pharmacol 62: 26–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rougier JS, Albesa M, Abriel H, Viard P (2011). Neuronal precursor cell‐expressed developmentally down‐regulated 4‐1 (NEDD4‐1) controls the sorting of newly synthesized CaV 1.2 calcium channels. J Biol Chem 286: 8829–8838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salom JB, Burguete MC, Perez‐Asensio FJ, Centeno JM, Torregrosa G, Alborch E (2002). Acute relaxant effects of 17β estradiol through non‐genomic mechanisms in rabbit carotid artery. Steroids 67: 339–346. [DOI] [PubMed] [Google Scholar]
- Sonkusare S, Palade PT, Marsh JD, Telemaque S, Pesic A, Rusch NJ (2006). Vascular calcium channels and high blood pressure: pathophysiology and therapeutic implications. Vascul Pharmacol 44: 131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturek M (2011). Ca2+ regulatory mechanisms of exercise protection against coronary artery disease in metabolic syndrome and diabetes. J Appl Physiol 111: 573–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhir K, Ko E, Zellner C, Wong HE, Hutchison SJ, Chou TM, et al. (1997). Physiological concentrations of estradiol attenuate endothelin 1‐induced coronary vasoconstriction in vivo. Circulation 96: 3626–3632. [DOI] [PubMed] [Google Scholar]
- Swindle MM, Makin A, Herron AJ, Clubb FJ Jr, Frazier KS (2012). Swine as models in biomedical research and toxicology testing. Vet Pathol 49: 344–356. [DOI] [PubMed] [Google Scholar]
- Teoh H, Leung SW, Man RY (1999). Short‐term exposure to physiological levels of 17β‐estradiol enhances endothelium‐independent relaxation in porcine coronary artery. Cardiovasc Res 42: 224–231. [DOI] [PubMed] [Google Scholar]
- Teoh H, Quan A, Leung SW, Man RY (2000). Differential effects of 17β‐estradiol and testosterone on the contractile responses of porcine coronary arteries. Br J Pharm 129: 1301–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakali KM, Kharade SV, Sonkusare SK, Rhee SW, Stimers JR, Rusch NJ (2010). Intracellular Ca2+ silences L‐type Ca2+ channels in mesenteric veins: mechanism of venous smooth muscle resistance to calcium channel blockers. Circ Res 106: 739–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tharp DL, Ivey JR, Shaw RL, Bowles DK (2014). Ovariectomy increases L‐type calcium channel activity in porcine coronary smooth muscle. Menopause 21: 661–668. [DOI] [PubMed] [Google Scholar]
- Thorne GD, Paul RJ (2003). Effects of organ culture on arterial gene expression and hypoxic relaxation: role of the ryanodine receptor. Am J Physiol Cell Physiol 284: C999–C1005. [DOI] [PubMed] [Google Scholar]
- Tobin AA, Joseph BK, Al Kindi HN, Albarwani S, Madden JA, Nemetz LT, et al. (2009). Loss of cerebrovascular Shaker‐type K+ channels: a shared vasodilator defect of genetic and renal hypertensive rats. Am J Physiol Heart Circ Physiol 297: H293–H303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traupe T, Stettler CD, Li H, Haas E, Bhattacharya I, Minotti R, et al. (2007). Distinct roles of estrogen receptors alpha and beta mediating acute vasodilation of epicardial coronary arteries. Hypertension 49: 1364–1370. [DOI] [PubMed] [Google Scholar]
- Tummala S, Hill BJ (2007). The enhanced endothelin‐1‐induced contraction in cultured coronary arteries from mature female pigs is not antagonized by 17β‐estradiol. Vascul Pharmacol 46: 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullrich ND, Koschak A, MacLeod KT (2007). Oestrogen directly inhibits the cardiovascular L‐type Ca2+ channel Cav1.2. Biochem Biophys Res Commun 361: 522–527. [DOI] [PubMed] [Google Scholar]
- Vaithianathan T, Narayanan D, Asuncion‐Chin MT, Jeyakumar LH, Liu J, Fleischer S, et al. (2010). Subtype identification and functional characterization of ryanodine receptors in rat cerebral artery myocytes. Am J Physiol Cell Physiol 299: C264–C278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WZ, Pang L, Palade P (2008). Angiotensin II causes endothelial‐dependent increase in expression of Cav1.2 protein in cultured arteries. Eur J Pharmacol 599: 117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Pang L, Palade P (2011). Angiotensin II upregulates Cav1.2 protein expression in cultured arteries via endothelial H2O2 production. J Vasc Res 48: 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserstrom JA, Wasserstrom LA, Lokuta AJ, Kelly JE, Reddy ST, Frank AJ (2002). Activation of cardiac ryanodine receptors by the calcium channel agonist FPL‐64176. Am J Physiol Heart Circ Physiol 283: H331–H338. [DOI] [PubMed] [Google Scholar]
- Yang XP, Reckelhoff JF (2011). Estrogen, hormonal replacement therapy and cardiovascular disease. Curr Opin Nephrol Hypertens 20: 133–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Ma H, Barman SA, Liu AT, Sellers M, Stallone JN, et al. (2011). Activation of G protein‐coupled estrogen receptor induces endothelium‐independent relaxation of coronary artery smooth muscle. Am J Physiol Endocrinol Metab 301: E882–E888. [DOI] [PMC free article] [PubMed] [Google Scholar]