

Abstract
Protein synthesis inhibitors are commonly used for measuring protein degradation rates, but may cause cytotoxicity via direct or indirect mechanisms. This study aimed to identify concentrations providing optimal inhibition in the absence of overt cytotoxicity. Actinomycin D, cycloheximide, emetine, and puromycin were assessed individually, and in two‐, three‐, and four‐drug combinations for protein synthesis inhibition (IC 50) and cytotoxicity (CC 50) over 72 h. Experiments were conducted in HepG2 cells and primary rat hepatocytes (PRH). IC 50 for actinomycin D, cycloheximide, emetine, and puromycin were 39 ± 7.4, 6600 ± 2500, 2200 ± 1400, and 1600 ± 1200 nmol/L; with corresponding CC 50 values of 6.2 ± 7.3, 570 ± 510, 81 ± 9, and 1300 ± 64 nmol/L, respectively, in HepG2 cells. The IC 50 were 1.7 ± 1.8, 290 ± 90, 620 ± 920, and 2000 ± 2000 nmol/L, with corresponding CC 50 values of 0.98 ± 1.8, 680 ± 1300, 180 ± 700, and 1600 ± 1000 (SD) nmol/L, respectively, in PRH. CC 50 were also lower than the IC 50 for all drug combinations in HepG2 cells. These data indicate that using pharmacological interference is inappropriate for measuring protein degradation over a protracted period, because inhibitory effects cannot be extricated from cytotoxicity.
Keywords: cytotoxicity, protein degradation rate, protein synthesis inhibitor
Abbreviations
- CC10
cytoxicity concentration at 10% of maximum (90% cell viability)
- CC50
cytoxicity concentration at 50% of maximum (50% cell viability)
- DDIs
drug–drug interactions
- FICs
fractional inhibitory concentrations
- GST
glutathione S‐transferase
- HBSS
Hank's balanced salt solution
- HepG2
hepatocellular carcinoma cell line
- kdeg
degradation rate constant
- MTT
3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide
- PBPK
physiologically based pharmacokinetic
- PRH
primary rat hepatocyte
Introduction
Protein abundance in a cellular system is a balance between the rate of synthesis and degradation. The ability of the cell to remove and replenish proteins in a dynamic state of constant turnover is paramount to maintaining essential cellular functions. While rates of protein synthesis are readily measurable by time‐course experiments utilising radioisotopes and protein quantification, the rate of degradation (k deg) is often more difficult to determine especially in vivo (Millward et al. 1981; Pratt et al. 2002). This is due to the complex interplay between different protein degradation mechanisms and paucity in understanding the causal signalling mechanisms initiating specific protein degradation. Protein degradation is commonly quantified as half‐life, the time taken for protein to decrease by half (Zhou 2004; Belle et al. 2006; Zhang et al. 2007). This variable is interchangeable with k deg by the following equations assuming first‐order decay kinetics (Belle et al. 2006), where N is the protein intensity, k is the decay rate constant (and –k represents k deg), and t 1/2 is the half‐life:
| (1) |
| (2) |
| (3) |
Physiologically based pharmacokinetic (PBPK) modelling can be used to predict the magnitude and dynamics of drug–drug interactions (DDIs), allowing the investigation of optimal timings for washout periods or switching of drug regimens in clinical practice. Such approaches require robust drug and system parameters (Jamei et al. 2009; Rostami‐Hodjegan 2012). Clearly, k deg is a critical system parameter for the simulation of time‐dependent DDIs, such as those mediated by mechanism‐based inhibition or induction (Venkatakrishnan and Obach 2007; Almond et al. 2009).
Several sources have highlighted the lack of accurate k deg data for metabolising enzymes and transporter proteins as important sources of error in DDI prediction (Obach et al. 2007; Wang 2010). Despite its well‐established impact, there is large disparity in the literature for the k deg of specific proteins and different values are used for the same enzyme across different studies, resulting in inconsistent predictions (Ghanbari et al. 2006; Yang et al. 2008; Wang 2010; Yeo et al. 2011). Proteins have widely varied half‐lives, ranging from minutes to several days, and protein turnover is tightly regulated through multiple molecular mechanisms. Apart from the importance in PBPK, further characterisation of k deg for specific proteins is required for better understanding of cell signalling processes involved in both normal and dysfunctional diseased cell states, thus studies of protein turnover are used in many different areas of cellular and molecular biology.
Traditional methods of protein degradation measurement and derivation of k deg, fall into two experimental designs: (1) quantifying the amount of a specific protein before and after a cell perturbation then measuring the difference in protein abundance and time between the initial and new steady‐state; or (2) quantifying changes in protein abundance by kinetic, time‐course experiments (Alvarez‐Castelao et al. 2012). The kinetic approach is based on an initial cell treatment with protein synthesis inhibitors followed by the quantification of changes in protein content over time by immunoblotting (Dai et al. 2013). Traditional methods of measuring protein degradation generally utilise low level incorporation of radiolabelled amino acids in the form of pulse‐chase analysis, often involving the use of protein synthesis inhibitors to eliminate reincorporation (Zhou 2004; Doherty et al. 2009). The more recent approaches focus on simultaneously measuring the rates of a large number of proteins. For example, stable isotope labelling by amino acids (SILAC) in cell culture followed by mass‐spectrometry (MS) as a common proteomics‐based method for measuring protein turnover rates (Mann 2006; Doherty et al. 2009; Fierro‐Monti et al. 2013; Takahashi et al. 2017) and isobaric tag for relative and absolute quantification (iTRAQ) are also used (Jayapal et al. 2010). The focus of this study was on the more traditional methods of measuring protein degradation utilising protein synthesis inhibitors for pharmacological interference.
The aim of this study was to find a suitable protein synthesis inhibitor or drug combination that provided maximum protein synthesis inhibition with minimum cytotoxicity for subsequent use in measuring protein degradation rates. The four selected inhibitors actinomycin D, cycloheximide, emetine, and puromycin were assessed alone and in combination to determine their suitability for protein degradation studies. Leucine incorporation assays and standard 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assays were employed to determine the level of protein synthesis inhibition and cytotoxicity, respectively, across a range of drug concentrations in immortalised hepatic cell line and primary hepatocytes. Two‐drug combinations were tested for synergy by the modified fixed‐ratio isobologram method. Combinations of three and four inhibitors were assessed at subcytotoxic concentrations of each inhibitor.
Materials and Methods
Materials
Dulbecco's modified eagle medium (DMEM), fetal bovine serum (FBS), trypsin‐EDTA solution, Hank's balanced salt solution (HBSS), thiazolyl blue tetrazolium (TBT), and protein synthesis inhibitors (actinomycin D (A4262), emetine dihydrochloride hydrate (E2375), and puromycin dihydrochloride (P7255)) were purchased from Sigma‐Aldrich (Dorset, UK). HepG2 cells were purchased from American Tissue Culture Collections (ATCC, Virginia). Cryopreserved primary rat hepatocytes, William's E media, plating cocktail, maintenance cocktail, Geltrex® matrix, and collagen I coated plates were purchased from Invitrogen Ltd (Paisley, UK). Cycloheximide (ab120093) was purchased from Abcam (Cambridge, UK). l‐Leucine [4,5‐3H] (MT‐672E) was obtained from Moravek (California). The CellTiter‐Glo cell viability assay and the GSH‐Glo glutathione assay were purchased from Promega (Southampton, UK).
Cell line culture
HepG2 cells were grown in DMEM medium supplemented with 10% FBS solution and were discarded beyond passage 20. The media was changed every 48 h and cells were cultured until 80–90% confluence in a 37°C 5% CO2 humidified incubator. Cell counts were carried out by a Nucleocounter (Chemometec, Denmark).
Primary rat hepatocyte culture
Primary rat hepatocytes (PRH) were purchased from Invitrogen (Paisley, UK), isolated from male Sprague–Dawley rats at 9 weeks old (Lot. RS745). Cryopreserved PRH were thawed in a 37°C water bath for approximately 2 min until contents were around 90% thawed. Once thawed, the hepatocytes were added to 50 mL of prewarmed plating media (William's E media without phenol red supplemented with 5% FBS, 1 μmol/L dexamethasone, 1% solution of penicillin/streptomycin, 4 μg/mL bovine insulin, 2 mmol/L GlutaMAX™, and 15 mmol/L HEPES (CHRM® supplement A), and centrifuged for 3 min at 55g at 18°C and the supernatant fraction discarded. The hepatocytes were then resuspended in plating media at 1 × 106 cells per ml density.
The cell viability of primary human hepatocytes was calculated using the Chemometec NucleoCounter® NC‐100™ according to the manufacturer's protocol. Cells were seeded in collagen coated plates and were incubated for 5 h at 37°C with 5% CO2 and 95% humidity. After 5 h incubation, plating media was discarded and replaced with 0.022 mg/mL of Geltrex® Matrix in maintenance media (William's E media supplemented with 0.1 μmol/Ldexamethasone, 0.5% penicillin/streptomycin 6.25 μg/mL human recombinant insulin, 6.25 μg/mL human transferrin, 6.25 ng/mL selenous acid, 1.25 mg/mL BSA, 5.35 μg/mL linoleic acid, 2 nmol/L GlutaMAX™, and 15 mM HEPES). After incubation overnight, media containing Geltrex® was removed and replaced with varying drug concentrations and controls in maintenance media.
Measuring protein synthesis inhibition by [3H]‐leucine incorporation
HepG2 cells were seeded at 2 × 105 cells per well in DMEM supplemented with 10% FBS and the plates were incubated overnight at 37°C to allow cells to adhere. PRH cells were seeded in collagen coated 24‐well plates at a density of 2 × 105 cells per well. Old media was removed and replaced with 0–100 μmol/L of protein synthesis inhibitors dissolved in DMEM with 10% FBS for HepG2 cells or maintenance media for PRH and incubated for 72 h in a 37°C humidified incubator. In the last 2 h of incubation, cells were pulsed with 2 μCi of [3H]‐leucine without removing the inhibitor. After 2 h, the media containing [3H]‐leucine was removed by aspiration and the cells were washed with HBSS before removal from well by trypsinisation. HepG2 cells were then harvested onto a filtermat using a TomTec cell harvester. The filtermat was sealed in a sample bag with melt‐on scint and the level of protein synthesis was determined by the level of [3H]‐leucine incorporation measured using a MicroBeta detector (Perkin‐Elmer, Cambridge, UK). PRH cells were transferred to scintillation vials and radioactivity was determined using QuantaSmart™ software on a Tri‐Carb scintillation counter (Perkin‐Elmer).
Measuring cell viability by standard MTT Assays
Standard MTT assays were performed on HepG2 and PRH cells to measure cell viability. 2 × 104 cells per well of HepG2 were seeded into 96‐well plates in DMEM with 10% FBS and left overnight in a 37°C humidified incubator to allow cells to adhere to the plate. PRH were seeded in collagen coated 96‐well plates at a density of 2 × 104 cells per well. Old media was removed and replaced with 0–300 μmol/L of protein synthesis inhibitors and incubated for 72 h. A vehicle control and control with no drug was included. A quantity of 20 μL of 5 mg/mL TBT in HBSS was added to each well and incubated for 2 h. A quantity of 100 μL lysis buffer (50% v/v dimethylformahyde and 20% v/v sodium dodecyl sulphate) was added to each well and the plate was incubated overnight at 37°C. The absorbance was quantified at 570 nm by a Tecan GENios micoplate reader (Germany).
Single protein synthesis inhibitor analysis
The protein synthesis inhibitors actinomycin D, cycloheximide, emetine, and puromycin were analysed individually in HepG2 and PRH cells. Actinomycin D was incubated 0–10 μmol/L and 0–0.039 μmol/L and puromycin at 0–20 μmol/L and 0–5 μmol/L for leucine incorporation assays and MTT cytotoxicity assays, respectively. Cycloheximide was incubated at 0–300 μmol/L and emetine at 0–30 μmol/L for both leucine incorporation and MTT assays.
Two‐drug combination fixed‐ratio isobologram analysis
The effects of two‐drug combinations on HepG2 cells were assessed by the modified fixed‐ratio isobologram protocol, which detects synergy, additivity, or antagonism between a pair of drugs (Fivelman et al. 2004). Stock solutions of the drugs were prepared at 10 mmol/L in sterile distilled water. Concentration–response assays were carried out to obtain the IC50 and CC50 of the individual drugs by leucine incorporation and standard MTT assays, respectively. For the six two‐drug combinations, the drug dilutions were made to allow the IC50 or CC50 to fall at about the fourth threefold serial dilution. The dilutions of each of the two drugs in each combination were prepared in seven fixed ratios 6:0, 5:1, 4:2, 3:3, 2:4, 1:5, and 0:6. These mixtures were then serially diluted threefold in quadruplicates to generate a range of eight concentrations for each condition. Protein synthesis inhibition and cell viability assays were conducted as described above to generate a concentration–response curve to calculate the IC50 and CC50 for drugs A and B in each mixture. The fractional inhibitory concentrations (FICs) were calculated using Equation (4), (5), and 6 (Gorka et al. 2013):
| (4) |
| (5) |
| (6) |
Isobologram curves were generated by plotting FICA versus FICB. FICindex = 1 was taken as indicative of an additive effect between drugs A and B, FICindex<1 indicative of synergy and FICindex>1 indicative of antagonism.
Three‐ and four‐drug combination analysis
Three‐drug combinations: actinomycin D, cycloheximide, and emetine; actinomycin D, puromycin, and emetine; actinomycin D, puromycin, and cycloheximide; and puromycin, cycloheximide and emetine, and four‐drug combination: actinomycin D, puromycin, cycloheximide, and emetine were assessed at subcytotoxic concentrations of each drug (determined from the single drug incubation experiments) in HepG2 cells. The three‐ and four‐drug combinations were made up at the CC10 concentrations and measured for level of protein synthesis inhibition by [3H]‐leucine incorporation and assessed for cytotoxicity by several different toxicity assays.
Standard MTT assay
Standard MTT assays were performed on the three‐ and four‐drug combinations using methods described above in HepG2 cells. Further toxicity assays (CellTiter‐Glo®, GSH‐Glo™ glutathione, and trypan blue exclusion) were performed on these combinations to confirm the robustness of MTT assays as a measure of cell viability.
CellTiter‐Glo® luminescent cell viability assay
A CellTiter‐Glo® luminescent cell viability assay was performed on the above drug combinations following 72 h incubation in HepG2 as described in the manufacturer's protocol. Cells were seeded at 2 × 104 cells per well in DMEM with 10% FBS. The assay measures the amount of ATP present that indicates the presence of metabolically active viable cells.
GSH‐Glo™ glutathione assay
GSH‐Glo™ glutathione assays were performed on the above drug combinations following 72 h incubation in HepG2 cells according to the manufacturer's protocol. Cells were seeded at 1 × 104 cells per well in DMEM with 10% FBS. The assay measures the conversion of a luciferin derivative into luciferin in the presence of glutathione and glutathione S‐transferase (GST) as an indication of oxidative stress.
Trypan blue exclusion
HepG2 cells were seeded at 5 × 104 cells per well in DMEM +10% FBS and incubated with the three‐ and four‐drug combinations for 72 h. Following incubation, the cells were washed with HBSS solution and trypsinised for 5 min before being transferred in suspension to Eppendorf tubes. A quantity of 10 μL of cell suspension was added to 10 μL of trypan blue solution and placed on a Countess™ slide. Cell viability was calculated using a Countess™ automated cell counter (LifeTechnologies, UK).
Data analysis
The IC50 (concentration causing 50% protein synthesis inhibition), CC50 (concentration causing 50% cell viability), and CC10 (concentration causing 90% cell viability) were calculated by nonlinear regression of drug concentration versus leucine incorporation and MTT concentration–response graphs, respectively, using Graphpad Prism 3 software. The IC50 and CC50 values derived from the single inhibitor analyses were used for subsequent fixed‐ratio isobologram two‐drug combination analyses.
Results
Single protein synthesis inhibitor
The mean ± SD CC50 for the four protein synthesis inhibitors actinomycin D, cycloheximide, emetine, and puromycin were found at 6.2 ± 7.3, 570 ± 510, 81 ± 9, and 1300 ± 64 nmol/L, respectively, in HepG2 cells and 0.98 ± 1.8, 680 ± 1300, 180 ± 700, and 1600 ± 1000 nmol/L, respectively, in PRH. The IC50 were 39 ± 7.4, 6600 ± 2500, 2200 ± 1400, and 1600 ± 1200 μmol/L, respectively, in HepG2 and 1.7 ± 1.8, 290 ± 90, 620 ± 920, and 2000 ± 2000 nmol/L, respectively, in primary rat hepatocytes. The IC50 and CC50 concentrations were calculated from concentration–response graphs as shown in Figure 1. The CC50 concentrations were lower compared to corresponding IC50 values for all four inhibitor drugs except cycloheximide in PRH; this indicates that the inhibitors were more effective in generating cell death than protein synthesis inhibition and thus unsuitable for further protein degradation studies.
Figure 1.
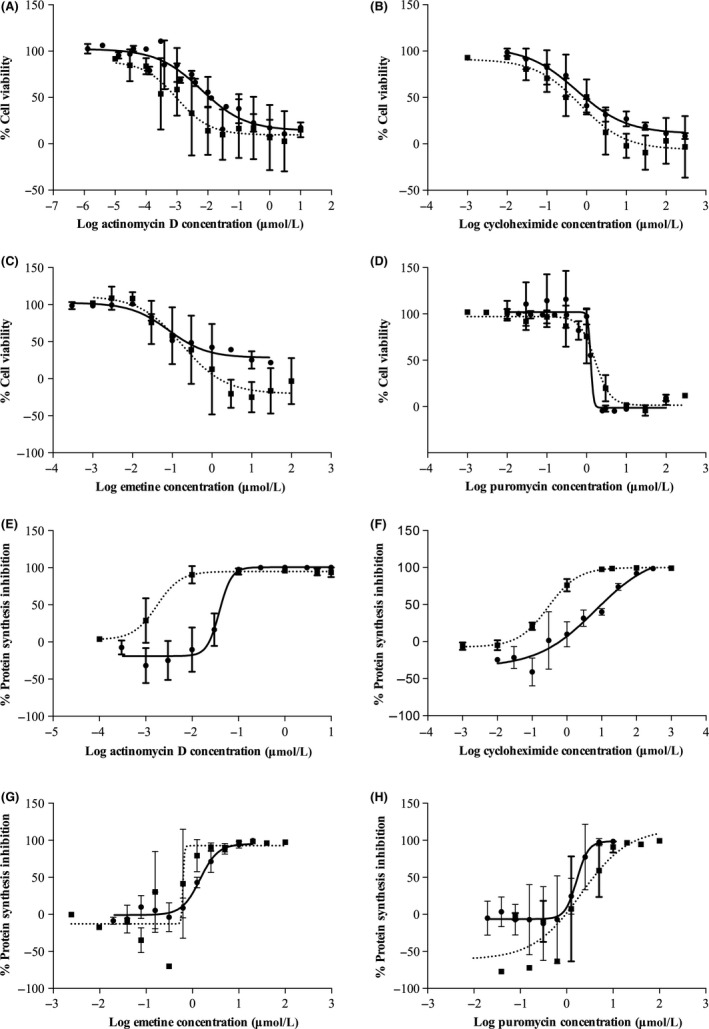
IC 50 and CC 50 of the four individual protein synthesis inhibitors in HepG2 and primary rat hepatocytes (PRH). (A–D) Cell viability was measured by standard MTT assay and expressed as viability as a percentage of untreated control. (E–H) Protein synthesis inhibition across different concentrations of inhibitors was measured by [3H]‐Leucine incorporation assay and shown as percentage of inhibition of control. Dotted line shows PRH and solid line for HepG2 cells. Dose–response curves were produced by Prism software and IC 50 and CC 50 values were calculated from linear regression models. Data are shown as mean ± SD from n = 3 independent experiments.
Figure 2 shows linear regression between the IC50 and CC50 values derived from HepG2 and cryopreserved PRH cells. Figures 2A–C show linear relationships between the IC50 and CC50 between HepG2 and PRH cells for actinomycin D, emetine, and puromycin. Cycloheximide fit in the linear relationship for cytotoxicity but not for protein synthesis inhibition.
Figure 2.
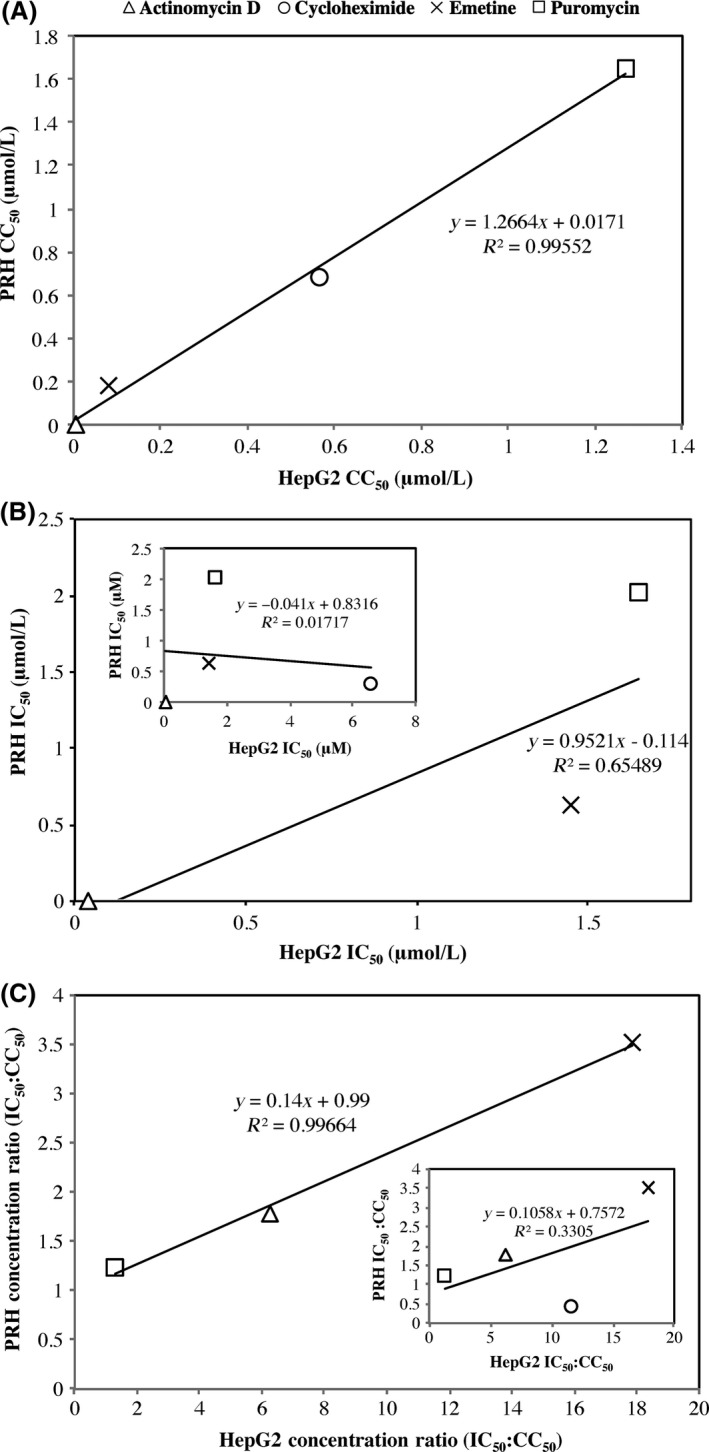
Linear regression analysis of IC 50 and CC 50 between HepG2 and PRH cell types. (A) shows linear regression between CC 50 values of the four protein synthesis inhibitor drugs for the different cell types. (B) shows linear regression between IC 50 values with cycloheximide omitted but shown in inset graph, of the two cell types. (C) shows IC 50:CC 50 ratio of HepG2 and PRH cell types omitting cycloheximide. Cycloheximide is included in the inset graph.
Two‐drug protein synthesis inhibitor combinations
The fixed‐ratio isobologram method was employed to assess additivity, synergy, or antagonism in both protein synthesis inhibition and cytotoxicity between drug pairs. Six combinations of drug pairs for the four inhibitors were analysed. The combinations cycloheximide and emetine, cycloheximide and puromycin, and emetine and puromycin showed antagonism for protein synthesis inhibition at all ratios (as shown in Fig. 3A–C) and were therefore deemed to be unsuitable for protein degradation studies. As such, isobolograms to assess cytotoxicity were not carried out for these combinations. Actinomycin D and emetine showed additivity (no interaction) between the drugs for protein synthesis inhibition and synergy for cytotoxicity, indicating that actinomycin D and emetine did not increase protein synthesis inhibition in combination but did display higher cytoxicity. As such, this combination was also deemed unsuitable for measuring protein degradation rates. Actinomycin D and cycloheximide, and actinomycin D and puromycin did show synergy for protein synthesis inhibition at some ratios. This combination also displayed strong synergy for cytotoxicity at most ratios. Interestingly, at ratios of 5:1 and 4:2 for actinomycin D: cycloheximide and actinomycin D: puromycin, these combinations were synergistic for protein synthesis inhibition and antagonistic for cytotoxicity as seen in Figure 3D and F, respectively. However, despite the synergy for protein synthesis inhibition and antagonism for cytotoxicity at these ratios, the CC50 values for these drug pairs alone and in combination were still lower than the IC50 values and thus cytotoxicity was observed at lower concentrations than those required to inhibit protein synthesis. The CC50 concentrations for actinomycin D in combination with cycloheximide at 5:1 and 4:2 ratios were 12 and 14 nmol/L and the corresponding IC50 concentrations were 28 and 35 nmol/L, respectively. The CC50 values for cycloheximide in combination with actinomycin D at 5:1 and 4:2 ratios were 26 and 12 nmol/L and the corresponding IC50 concentrations were 2500 and 1300 nmol/L respectively. For the combination actinomycin D and puromycin, the CC50 concentrations for actinomycin D at 5:1 and 4:2 ratios were 9.8 and 8.1 nmol/L and the corresponding IC50 concentrations were 16 and 21 nmol/L respectively. As for puromycin, the CC50 values at 5:1 and 4:2 ratios were 60 and 20 nmol/L and the corresponding IC50 concentrations were 690 and 360 nmol/L respectively.
Figure 3.
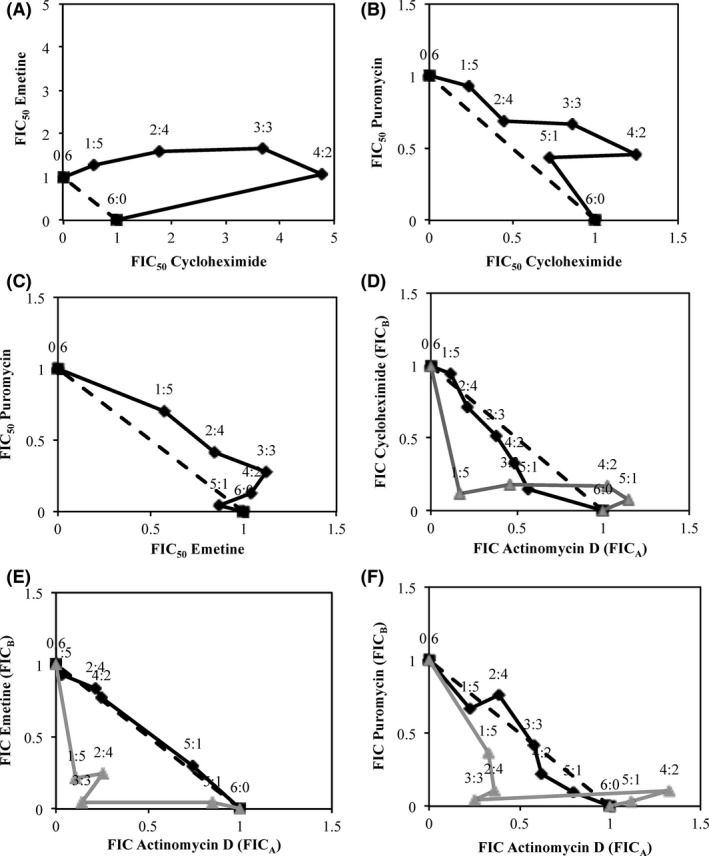
Isobolograms generated based on CC 50 and IC 50 values showing the interaction between protein synthesis inhibitor pairs. Six combinations of inhibitor pairs are shown in (A–F) The dotted line corresponds to the predicted curve if drug pairs showed an additive effect. The black line corresponds to drug pair interactions for protein synthesis inhibition. The grey line shows drug pair interactions for cytotoxicity. FICA and FICB correspond to the fractional inhibitory concentrations of the first and second drugs in each drug pair listed. Cytotoxicity analysis was not performed for cycloheximide–emetine, cycloheximide–puromycin, and emetine–puromycin drug pairs (A–C) as these showed strong antagonism for protein synthesis inhibition. N = 4 independent experiments were carried out in HepG2 cells.
Three‐ and four‐drug combination analysis
The four inhibitors individually and the two‐drug combinations displayed high cell death. Three‐ and four‐drug combinations at subtoxic concentrations (CC10 of each when incubated alone) were, therefore, assessed to investigate whether protein synthesis inhibition could be achieved at concentrations lower or equal to those causing cytotoxicity. The CC10 (90% cell viability concentration) were calculated for each drug to be 0.17, 24, 7.0, and 110 nmol/L for actinomycin D, cycloheximide, emetine, and puromycin, respectively, in HepG2 cells. As mentioned previously, the inhibitors alone displayed a lower concentration for CC50 than IC50 indicating that they were more effective in generating cell death than inhibiting protein synthesis. The four‐drug combination showed a high 76% protein synthesis incorporation (thus low inhibition) and high cytotoxicity across all cytotoxicity assays as seen in Figures 4 and 5. Three‐drug combinations: actinomycin D, cycloheximide, and emetine; actinomycin D, cycloheximide, and puromycin; and actinomycin D, puromycin and emetine also demonstrated low protein synthesis inhibition with high cytotoxicity, also seen in Figures 4 and 5. Although puromycin, cycloheximide, and emetine gave low cell death across the assays, it was also ineffective at inhibiting protein synthesis inhibition, as shown in Figure 4, where level of leucine incorporation is higher than control. Overall, three‐ and four‐drug combinations of these protein synthesis inhibitors were deemed to be unsuitable for further protein degradation studies even at low concentrations.
Figure 4.
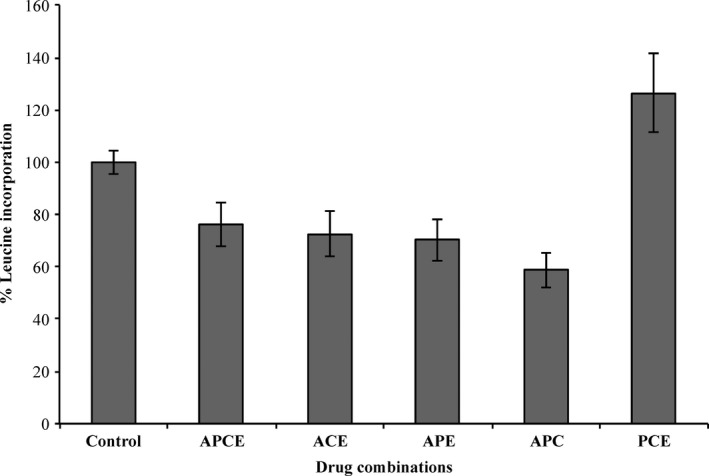
The level of [3H]Leucine incorporation for three‐ and four‐inhibitor combinations at subcytotoxic concentrations (CC 10). Leucine incorporation assays were carried out in HepG2 cells and the percentage of incorporation compared to control was calculated. Combination APCE corresponds to actinomycin D, puromycin, cycloheximide, and emetine; ACE to actinomycin D, cycloheximide, and emetine; APE to actinomycin D, puromycin, and emetine; APC to actinomycin D, puromycin, and cycloheximide; and PCE to puromycin, cycloheximide, and emetine. Data are shown as mean ± S.D from n = 3 independent experiments.
Figure 5.
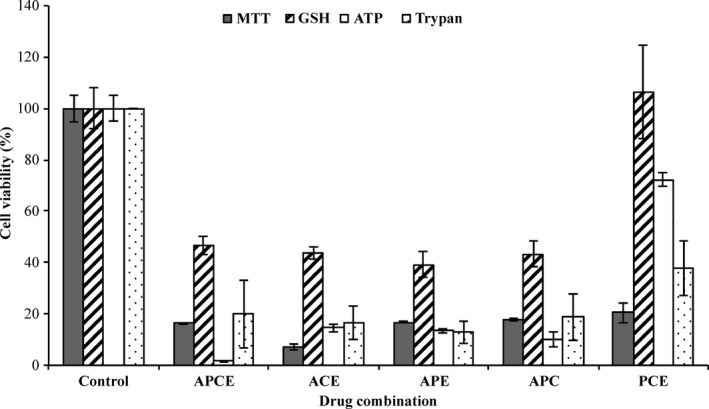
Measuring cytotoxicity for the three‐ and four‐inhibitor combinations at subcytotoxic concentrations. Three‐ and four‐inhibitor combinations were prepared at CC 10 concentrations. A range of cytotoxicity assays including standard MTT, GSH, ATP, and trypan blue exclusion assays were conducted on HepG2 cells. APCE corresponds to actinomycin D, puromycin, cycloheximide, and emetine; ACE to actinomycin D, cycloheximide, and emetine; APE to actinomycin D, puromycin, and emetine; APC to actinomycin D, puromycin, and cycloheximide; and PCE to puromycin, cycloheximide; and emetine. Data are shown as mean ± SD from n = 3 independent experiments.
Discussion
The use of protein synthesis inhibitors is the most common method for measuring protein degradation rates and has been documented by many sources over four decades (Goldberg and Dice 1974; Curfman et al. 1980; Princiotta et al. 2003; Zhou 2004; Belle et al. 2006; Delgado‐Vega et al. 2012; Chistyakov et al. 2014). The more recent approaches focus on simultaneously measuring the rates of a large number of proteins. For example, (SILAC) in cell culture followed by (MS) as a common proteomics‐based method for measuring protein turnover rates (Mann 2006; Doherty et al. 2009; Fierro‐Monti et al. 2013; Takahashi et al. 2017) and (iTRAQ) are also used (Jayapal et al. 2010). However, the wide application of these proteonomic approaches are limited by cost and complexity. The focus of this study was on the more simple traditional methods of measuring protein degradation utilising protein synthesis inhibitors for pharmacological interference. The aim of this study was to define inhibitor concentrations (single or combinations) that provide maximum protein synthesis inhibition with minimum cytotoxicity that could then be used in subsequent experiments to accurately estimate endogenous degradation rates.
For this study, four protein synthesis inhibitors actinomycin D, cycloheximide, emetine, and puromycin were selected based on their different mechanisms of action and previous use in biomedical research. Actinomycin D (Sobell 1985) intercalates DNA forming a stable complex with deoxyguanosine residues, thus blocking movement of RNA polymerase and subsequently transcription. Cycloheximide binds the 60S ribosomal subunit blocking the translocational step in amino acid elongation, thus inhibiting protein synthesis (Schneider‐Poetsch et al. 2010). Emetine inhibits protein synthesis by binding onto the 40S subunit of ribosomes and inhibiting translocation of proteins (Akinboye and Bakare 2011). Puromycin acts as an analogue of the 3′‐terminal end of aminoacyl‐tRNA, which results in premature amino acid chain termination during translation of proteins (Azzam and Algranati 1973).
This study supports reported concerns over the inhibitors being too disruptive to normal cellular function to use to measure natural rates of protein turnover (Yewdell et al. 2011; Geva‐Zatorsky et al. 2012). In all cases, the CC50 concentration for the drugs in combination was lower than the corresponding IC50, suggesting that even in combination protein synthesis inhibition could not be studied in the absence of an effect on other cellular functions. These data suggest that inhibiting mechanisms of protein synthesis by pharmacological interference (even with lower concentration combinations) is not a physiologically appropriate method of measuring k deg because all protein systems, including those involved in protein degradation pathways, are likely to be affected. In support of this, Dai et al. reported that cycloheximide could affect protein degradation by activating the AKT (protein kinase B) leading to downstream effects on the normal functioning of the ubiquitin proteasome degradation (UPD) pathway (Dai et al. 2013). In addition to the drugs disrupting protein degradation machinery, there have been reports of protein synthesis inhibitors actively inducing a range of protein mRNA production that also impact accuracies for calculating protein degradation rates downstream (Hattori and Gross 1995; Schuetz et al. 1995; Stordeur et al. 1995). It should be noted that the incubation time with the protein synthesis inhibitor drugs was for 72 h in the current study and as such, measurement of degradation for proteins with medium or long (over 72 h) half‐lives are likely to be particularly problematic. Further optimisations with shorter incubation periods may be possible for proteins with shorter t 1/2 but robust optimisation will be required.
Protein synthesis inhibitors are commonly used for measuring protein degradation yet in previous studies, there has been little consideration for their cytotoxic effects and virtually none have optimised a specific concentration to use. Several studies have used cycloheximide at millimolar concentrations, which was much higher than the nontoxic concentration range found here (Pan and Haines 1999; Princiotta et al. 2003; Jeong et al. 2005; Xie et al. 2010; Majumder et al. 2012). MTT assays were used as the main method of measuring CC50 and the level of cytotoxicity across the four protein synthesis inhibitor drugs and their combinations. Since MTT assays specifically assess the formazan production pathway as a measure of cellular mitochondrial damage, other forms of cytotoxicity assays including GSH, ATP, and trypan blue exclusion, which assess other mechanisms of cytotoxicity, were carried out to validate the findings. Good agreement across assays and drug combinations was observed with the exception of puromycin, cycloheximide, and emetine in which higher cellular toxicity was detected in MTT than other assays. Despite GSH assays showing higher cell viability across the different drug combinations, it should be noted that GSH assays alone could not be used to predict the CC50 in this study because the results were in disagreement with the other cytotoxicity assays employed. A potential limitation is that protein binding was not assessed in this study. However, it should be recognised that protein binding would be expected to impact both cytotoxicity and protein synthesis inhibition by impacting free‐drug concentration. Thus, the ratio would not be expected to be different.
Earlier studies with actinomycin D and puromycin reported toxicity in HeLa cells at concentrations within the range investigated here. Studies by Sawicki and Godman (1971) showed that at 0.08 μmol/L actinomycin D was sufficient to cause cell toxicity in HeLa cells, which is in agreement with the present findings. Dudani et al. (1988) proposed that puromycin caused cytotoxicity at 0.9 μmol/L in human cell lines, including HeLa cells, which also agreed with the presented results. Dudani et al. also reported a 79.6% protein synthesis inhibition at 0.9 mmol/L in HeLa cells which further supports our findings that puromycin is cytotoxic at concentrations lower than those required for protein synthesis inhibition. Conversely, Yin Low et al. (2009) conducted cytotoxicity assays on emetine in Huh‐7 cells and reported over 90% cell viability at 10 μmol/L which is much higher concentrations than those used here. Although the reason for this disparity is not apparent, cytotoxicity of these inhibitors may vary between different cell types. The single drug analyses were carried out in HepG2 and primary rat hepatocytes with reasonable agreement in protein synthesis inhibition and cytotoxicity for actinomycin D, emetine, and puromycin as shown in the linear relationship displayed in Figure 2. This study was carried out in readily available HepG2 cells and primary rat hepatocytes with the aim of transferring the optimised conditions onto primary human hepatocytes to validate a more physiologically accurate k deg prediction (Wilkening et al. 2003). However, due to the presented findings, an alternative approach to k deg determination is now being explored.
Despite the wide‐ranging importance of protein degradation, there has been no single recognised method for its measurement. However, these data indicate that the use of protein synthesis inhibitors should be avoided. The more recent methods of measuring rates of degradation focus on high‐throughput approaches aiming to quantify many different proteins in parallel; these involve metabolic labelling of proteins of interest followed by MS analysis (Doherty and Beynon 2006). Newly developed quantitative proteonomic methods provide an important alternative to chemical inhibition, however, reproducibility across different experiments and the impact of protein labelling on endogenous protein degradation warrants full investigation.
Author Contributions
C.C wrote the manuscript. A.O, M.S, L.A, and C.C participated in experimental design. C.C conducted experiments with recommendations from P.M and N.J.L. Data was analysed by C.C, and A.O. L.A, P.M, N.J.L, M.S, and A.O contributed to the editing of the manuscript.
Disclosure
The authors report no declaration of interest.
Acknowledgements
We thank Iain Gardner for his contribution to useful scientific discussions. This work was supported by the Medical Research Council [Grant: JXG10848] and Simcyp Limited (a Certara company) [Grant: JXR10935].
Chan C., Martin P., Liptrott N. J., Siccardi M., Almond L., Owen A.. Incompatibility of chemical protein synthesis inhibitors with accurate measurement of extended protein degradation rates. Pharma Res Per, 5(5), 2017, e00359, https://doi.org/10.1002/prp2.359
References
- Akinboye ES, Bakare O (2011). Biological activities of emetine. Open Nat Prod J 4: 8–15. [Google Scholar]
- Almond LM, Yang J, Jamei M, Tucker GT, Rostami‐Hodjegan A (2009). Towards a quantitative framework for the prediction of DDIs arising from cytochrome P450 induction. Curr Drug Metab 10: 420–432. [DOI] [PubMed] [Google Scholar]
- Alvarez‐Castelao B, Ruiz‐Rivas C, Castano JG (2012). A critical appraisal of quantitative studies of protein degradation in the framework of cellular proteostasis. Biochem Res Int 2012: 823597 https://doi.org/10.1155/2012/823597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzam ME, Algranati ID (1973). Mechanism of puromycin action: fate of ribosomes after release of nascent protein chains from polysomes. Proc Natl Acad Sci USA 70: 3866–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belle A, Tanay A, Bitincka L, Shamir R, O'Shea EK (2006). Quantification of protein half‐lives in the budding yeast proteome. Proc Natl Acad Sci USA 103: 13004–13009. https://doi.org/10.1073/pnas.0605420103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chistyakov DV, Aleshin S, Sergeeva MG, Reiser G (2014). Regulation of peroxisome proliferator‐activated receptor beta/delta expression and activity levels by toll‐like receptor agonists and MAP kinase inhibitors in rat astrocytes. J Neurochem 130: 563–574. https://doi.org/10.1111/jnc.12757. [DOI] [PubMed] [Google Scholar]
- Curfman GD, O'Hara DS, Hopkins BE, Smith TW (1980). Suppression of myocardial protein degradation in the rat during fasting. Effects of insulin, glucose, and leucine. Circ Res 46: 581–589. [DOI] [PubMed] [Google Scholar]
- Dai CL, Shi J, Chen Y, Iqbal K, Liu F, Gong CX (2013). Inhibition of protein synthesis alters protein degradation through activation of protein kinase B (AKT). J Biol Chem 288: 23875–23883. https://doi.org/10.1074/jbc.M112.445148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado‐Vega AM, Dozmorov MG, Quiros MB, Wu YY, Martinez‐Garcia B, Kozyrev SV, et al. (2012). Fine mapping and conditional analysis identify a new mutation in the autoimmunity susceptibility gene BLK that leads to reduced half‐life of the BLK protein. Ann Rheum Dis 71: 1219–1226. https://doi.org/10.1136/annrheumdis-2011-200987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty MK, Beynon RJ (2006). Protein turnover on the scale of the proteome. Expert Rev Proteomics 3: 97–110. https://doi.org/10.1586/14789450.3.1.97. [DOI] [PubMed] [Google Scholar]
- Doherty MK, Hammond DE, Clague MJ, Gaskell SJ, Beynon RJ (2009). Turnover of the human proteome: determination of protein intracellular stability by dynamic SILAC. J Proteome Res 8: 104–112. https://doi.org/10.1021/pr800641v. [DOI] [PubMed] [Google Scholar]
- Dudani AK, Gupta RS, Gupta R (1988). Species‐specific differences in the toxicity of puromycin towards cultured human and Chinese hamster cells. FEBS Lett 234: 141–144. [DOI] [PubMed] [Google Scholar]
- Fierro‐Monti I, Racle J, Hernandez C, Waridel P, Hatzimanikatis V, Quadroni M (2013). A novel pulse‐chase SILAC strategy measures changes in protein decay and synthesis rates induced by perturbation of proteostasis with an Hsp90 inhibitor. PLoS ONE 8: e80423 https://doi.org/10.1371/journal.pone.0080423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fivelman QL, Adagu IS, Warhurst DC (2004). Modified fixed‐ratio isobologram method for studying in vitro interactions between atovaquone and proguanil or dihydroartemisinin against drug‐resistant strains of Plasmodium falciparum. Antimicrob Agents Chemother 48: 4097–4102. https://doi.org/10.1128/AAC.48.11.4097-4102.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geva‐Zatorsky N, Issaeva I, Mayo A, Cohen A, Dekel E, Danon T, et al. (2012). Using bleach‐chase to measure protein half‐lives in living cells. Nat Protoc 7: 801–811. https://doi.org/10.1038/nprot.2012.028. [DOI] [PubMed] [Google Scholar]
- Ghanbari F, Rowland‐Yeo K, Bloomer JC, Clarke SE, Lennard MS, Tucker GT, et al. (2006). A critical evaluation of the experimental design of studies of mechanism based enzyme inhibition, with implications for in vitro‐in vivo extrapolation. Curr Drug Metab 7: 315–334. [DOI] [PubMed] [Google Scholar]
- Goldberg AL, Dice JF (1974). Intracellular protein degradation in mammalian and bacterial cells. Annu Rev Biochem 43: 835–869. https://doi.org/10.1146/annurev.bi.43.070174.004155. [DOI] [PubMed] [Google Scholar]
- Gorka AP, Jacobs LM, Roepe PD (2013). Cytostatic versus cytocidal profiling of quinoline drug combinations via modified fixed‐ratio isobologram analysis. Malar J 12: 332 https://doi.org/10.1186/1475-2875-12-332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori Y, Gross SS (1995). Cycloheximide induces nitric oxide synthase mRNA in vascular smooth muscle cells by prolonging mRNA lifetime. Biochem Mol Biol Int 37: 439–445. [PubMed] [Google Scholar]
- Jamei M, Dickinson GL, Rostami‐Hodjegan A (2009). A framework for assessing inter‐individual variability in pharmacokinetics using virtual human populations and integrating general knowledge of physical chemistry, biology, anatomy, physiology and genetics: A tale of ‘bottom‐up’ vs ‘top‐down’ recognition of covariates. Drug Metab Pharmacokinet 24: 53–75. [DOI] [PubMed] [Google Scholar]
- Jayapal KP, Sui S, Philp RJ, Kok YJ, Yap MG, Griffin TJ, et al. (2010). Multitagging proteomic strategy to estimate protein turnover rates in dynamic systems. J Proteome Res 9: 2087–2097. https://doi.org/10.1021/pr9007738. [DOI] [PubMed] [Google Scholar]
- Jeong WS, Keum YS, Chen C, Jain MR, Shen G, Kim JH, et al. (2005). Differential expression and stability of endogenous nuclear factor E2‐related factor 2 (Nrf2) by natural chemopreventive compounds in HepG2 human hepatoma cells. J Biochem Mol Biol 38: 167–176. [DOI] [PubMed] [Google Scholar]
- Majumder P, Chen YT, Bose JK, Wu CC, Cheng WC, Cheng SJ, et al. (2012). TDP‐43 regulates the mammalian spinogenesis through translational repression of Rac1. Acta Neuropathol 124: 231–245. https://doi.org/10.1007/s00401-012-1006-4. [DOI] [PubMed] [Google Scholar]
- Mann M (2006). Functional and quantitative proteomics using SILAC. Nat Rev Mol Cell Biol 7: 952–958. https://doi.org/10.1038/nrm2067. [DOI] [PubMed] [Google Scholar]
- Millward DJ, Bates PC, Rosochacki S (1981). The extent and nature of protein degradation in the tissues during development. Reprod Nutr Dev 21: 265–277. [DOI] [PubMed] [Google Scholar]
- Obach RS, Walsky RL, Venkatakrishnan K (2007). Mechanism‐based inactivation of human cytochrome p450 enzymes and the prediction of drug‐drug interactions. Drug Metab Dispos 35: 246–255. https://doi.org/10.1124/dmd.106.012633. [DOI] [PubMed] [Google Scholar]
- Pan Y, Haines DS (1999). The pathway regulating MDM2 protein degradation can be altered in human leukemic cells. Cancer Res 59: 2064–2067. [PubMed] [Google Scholar]
- Pratt JM, Petty J, Riba‐Garcia I, Robertson DH, Gaskell SJ, Oliver SG, et al. (2002). Dynamics of protein turnover, a missing dimension in proteomics. Mol Cell Proteomics 1: 579–591. [DOI] [PubMed] [Google Scholar]
- Princiotta MF, Finzi D, Qian SB, Gibbs J, Schuchmann S, Buttgereit F, et al. (2003). Quantitating protein synthesis, degradation, and endogenous antigen processing. Immunity 18: 343–354. [DOI] [PubMed] [Google Scholar]
- Rostami‐Hodjegan A (2012). Physiologically based pharmacokinetics joined with in vitro‐in vivo extrapolation of ADME: a marriage under the arch of systems pharmacology. Clin Pharmacol Ther 92: 50–61. https://doi.org/10.1038/clpt.2012.65. [DOI] [PubMed] [Google Scholar]
- Sawicki SG, Godman GC (1971). On the differential cytotoxicity of actinomycin D. J Cell Biol 50: 746–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider‐Poetsch T, Ju J, Eyler DE, Dang Y, Bhat S, Merrick WC, et al. (2010). Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol 6: 209–217. https://doi.org/10.1038/nchembio.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuetz JD, Strom SC, Schuetz EG (1995). Induction of P‐glycoprotein mRNA by protein synthesis inhibition is not controlled by a transcriptional repressor protein in rat and human liver cells. J Cell Physiol 165: 261–272. https://doi.org/10.1002/jcp.1041650207. [DOI] [PubMed] [Google Scholar]
- Sobell HM (1985). Actinomycin and DNA transcription. Proc Natl Acad Sci USA 82: 5328–5331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stordeur P, Schandene L, Durez P, Gerard C, Goldman M, Velu T (1995). Spontaneous and cycloheximide‐induced interleukin‐10 mRNA expression in human mononuclear cells. Mol Immunol 32: 233–239. [DOI] [PubMed] [Google Scholar]
- Takahashi R, Shahidi‐Latham S, Wong S, Chang J (2017). Applying stable isotope labelled amino acids in micropatterned hepatocyte coculture to directly determine the degradation rate constant for CYP3A4. Drug Metab Dispos 45: 581–585. [DOI] [PubMed] [Google Scholar]
- Venkatakrishnan K, Obach RS (2007). Drug‐drug interactions via mechanism‐based cytochrome P450 inactivation: points to consider for risk assessment from in vitro data and clinical pharmacologic evaluation. Curr Drug Metab 8: 449–462. [DOI] [PubMed] [Google Scholar]
- Wang YH (2010). Confidence assessment of the Simcyp time‐based approach and a static mathematical model in predicting clinical drug‐drug interactions for mechanism‐based CYP3A inhibitors. Drug Metab Dispos 38: 1094–1104. https://doi.org/10.1124/dmd.110.032177. [DOI] [PubMed] [Google Scholar]
- Wilkening S, Stahl F, Bader A (2003). Comparison of primary human hepatocytes and hepatoma cell line Hepg2 with regard to their biotransformation properties. Drug Metab Dispos 31: 1035–1042. https://doi.org/10.1124/dmd.31.8.1035. [DOI] [PubMed] [Google Scholar]
- Xie Y, Burcu M, Linn DE, Qiu Y, Baer MR (2010). Pim‐1 kinase protects P‐glycoprotein from degradation and enables its glycosylation and cell surface expression. Mol Pharmacol 78: 310–318. https://doi.org/10.1124/mol.109.061713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Liao M, Shou M, Jamei M, Yeo KR, Tucker GT, et al. (2008). Cytochrome p450 turnover: regulation of synthesis and degradation, methods for determining rates, and implications for the prediction of drug interactions. Curr Drug Metab 9: 384–394. [DOI] [PubMed] [Google Scholar]
- Yeo KR, Walsky RL, Jamei M, Rostami‐Hodjegan A, Tucker GT (2011). Prediction of time‐dependent CYP3A4 drug‐drug interactions by physiologically‐based pharmacokinetic modelling: impact of inactivation parameters and enzyme turnover. Eur J Pharm Sci 43: 160–173. [DOI] [PubMed] [Google Scholar]
- Yewdell JW, Lacsina JR, Rechsteiner MC, Nicchitta CV (2011). Out with the old, in with the new? Comparing methods for measuring protein degradation. Cell Biol Int 35: 457–462. https://doi.org/10.1042/CBI20110055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Low JS, Chen KC, Wu KX, Mah‐LeeNm HCJJ (2009). Antiviral activity of emetine dihydrochloride against dengue virus infection. J Antivir Antiretrovir 1: 062000. [Google Scholar]
- Zhang L, Gurskaya NG, Merzlyak EM, Staroverov DB, Mudrik NN, Samarkina ON, et al. (2007). Method for real‐time monitoring of protein degradation at the single cell level. Biotechniques 42: 446–450. [DOI] [PubMed] [Google Scholar]
- Zhou P (2004). Determining protein half‐lives. Methods Mol Biol 284: 67–77. https://doi.org/10.1385/1-59259-816-1:067. [DOI] [PubMed] [Google Scholar]