

Abstract
Diallyl sulfide (DAS), a selective inhibitor of CYP2E1, has shown protective effects against alcohol‐ and acetaminophen‐induced hepatotoxicity in many studies. However, DAS is also a CYP2E1 substrate that on metabolism produces toxic metabolites and causes cytotoxicity. The objective of this study was to find a potent DAS analog as a CYP2E1 inhibitor and has the characteristic of producing less toxic metabolites. We selected seven commercially available compounds that are similar to DAS (DAS analogs). First, we performed ligand‐CYP2E1 docking study to determine the binding mode and binding energy. The analysis suggested a relative potential for these DAS analogs as CYP2E1 inhibitor. We then performed a comprehensive inhibition kinetics of DAS analogs and determined the relative IC50, K i, and types of inhibition compared to that of DAS. The results showed that compared to DAS, diallyl ether and allyl methyl sulfide have lower K i values (3.1 and 4.4 μmol/L, respectively, vs. 6.3 μmol/L for DAS) and IC50 values (6.3 and 11.4 μmol/L, respectively, vs. 17.3 μmol/L for DAS). However, allyl methyl sulfide and thiophene showed similar inhibitory capacities to that of DAS, and four other DAS analogs showed lower potency than DAS. In conclusion, we have found relatively more potent inhibitors of CYP2E1, which have lower toxicity than DAS. These compounds can replace DAS not only as a tool for in vitro and in vivo studies that involve CYP2E1 inhibition, but also can lead the way for their use in preventing CYP2E1‐mediated hepatic toxicity of alcohol and acetaminophen.
Keywords: CYP2E1, cytochrome P450 2E1, diallyl sulfide, enzyme inhibition, molecular modeling
Abbreviations
- 5,1 HA
5‐hexen‐1‐amine
- AES
allyl ethyl sulfide
- AMS
allyl methyl sulfide
- CYP
Cytochrome P450
- DAS
diallyl sulfide
- DE
diallyl ether
- PEA
2‐(prop‐2‐en‐1‐yloxy) ethan‐1‐amine
- PEXA
2‐prop‐2‐enoxyacetamide
- ROS
reactive oxygen species
- TP
thiophene
Introduction
Diallyl sulfide (DAS), a thioether present in garlic extract, has garnered significant attention from scientific communities since the early 1990s. Researchers have explored the use of DAS for its multifarious applications that includes anticancer, antioxidant and anti‐inflammatory properties (Rao et al. 2015). DAS has been used most frequently in the prevention against toxicities induced by xenobiotics such as alcohol and acetaminophen by selectively inhibiting cytochrome P450 2E1 (CYP2E1) (Rao et al. 2015). CYP2E1 is involved in the metabolism of more than 85 xenobiotics (Trafalis et al. 2010). CYP2E1‐mediated metabolism produces reactive oxygen species (ROS) and toxic metabolites, which causes cytotoxicity. For example, CYP2E1 metabolism of acetaminophen (APAP) causes production of a toxic metabolite N‐acetyl‐p‐benzoquinone imine, and APAP‐induced liver injury accounts for more than half of overdose‐related liver failure in the United States (Yoon et al. 2016). In addition, ethanol is known to induce CYP2E1, which subsequently metabolizes ethanol and causes ROS and acetaldehyde mediated cellular cytotoxicity and cancer of hepatic and nonhepatic organs/systems (Lu and Cederbaum 2008; Jin et al. 2013). DAS, through CYP2E1 inhibition, has shown protective effects against ethanol‐ and APAP‐mediated hepatoxicities in several studies (Swaminathan et al. 2013; Zhang et al. 2013; Raza and John 2015). DAS has also been extensively used both in vitro and in vivo study as a tool to inhibit CYP2E1 in hepatic as well as a number of extrahepatic cells (Jin et al. 2012).
However, despite DAS being a selective inhibitor of CYP2E1, it has failed to achieve clinical relevance as a drug, as well as, its application in chronic in vitro/vivo study as a tool due to its own potential toxicity. At a relatively high concentration and chronic exposure, DAS is toxic to hepatic and extrahepatic cells (Rao et al. 2015). In addition, although DAS is being used as a tool to inhibit CYP2E1, it is poorly characterized with regard to CYP2E1 inhibition. Our major goal is to find a superior alternative to DAS, which overcomes the limitations of DAS, and can be used as a better research tool to inhibit CYP2E1 in various pathological conditions. Furthermore, novel DAS analogs have the potential to be used as adjuvant therapy in various pathological conditions. In this study, we selected seven commercially available compounds, which resemble DAS structure. These compounds were characterized for binding affinity using ligand‐docking analysis followed by CYP2E1 inhibition study.
Materials and Methods
Chemicals
Diallyl sulfide (DAS), allyl methyl sulfide (AMS), allyl ethyl sulfide (AES), diallyl ether (DE), thiophene (TP), 2‐(prop‐2‐en‐1‐yloxy) ethan‐1‐amine (PEA), and 5‐hexen‐1‐amine (5,1 HA) were purchased from Sigma‐Aldrich (St. Louis, MO). 2‐prop‐2‐enoxyacetamide (PEXA) was purchased from Aldlab Chemicals (Woburn, MA). The stock solutions for these compounds were prepared in acetonitrile that was bought from Sigma.
Molecular docking study
Molecular modeling studies were performed using the Schrodinger Molecular Modeling Suite 2015 (Schrödinger, LLC, NY) as described (Chen et al. 2012; Lu et al. 2014). The initial models of human CYP2E1 for docking were taken from the Protein Data Bank. Among the six X‐ray crystal structures available for human CYP2E1, we chose the 3KOH for docking DAS and all the analogs based on completeness, resolution, and ligand binding pose (Porubsky et al. 2010). Protein and grid preparations were performed using the Protein Preparation Wizard with default protocol, and the grid of the CYP2E1 active site containing heme iron was defined. All compounds were prepared using the Ligprep module, before they were docked into the active site with metal constraints using the Glide module in the software.
CYP2E1 inhibition assay
The inhibition of CYP2E1 activity by DAS or its analogs was measured using standard para‐nitrophenol (p‐NP) hydroxylation assay as described before (Cederbaum 2014). Briefly, the final reaction mixture contained 50 mmol/L potassium phosphate buffer (pH 7.4), 2 pmol/μL of CYP2E1 baculosomes (Thermo Fisher Scientific, Waltham, MA), and varying concentration of p‐NP (5, 15, 30, 50, and 100 μmol/L) and inhibitors (5, 10, 25, 50, 100, and 200 μmol/L). On pre‐incubation at 37°C for 5 min, the reaction was initiated by adding 26 mmol/L NADPH and carried out for an hour at 37°C. On terminating the reaction by 20% (w/v) trichloroacetic acid, the reaction was neutralized by 2 mol/L NaOH and absorbance was measured at 535 nm using a microplate reader (Cytation™ 5 Cell Imaging Multi‐Mode Reader, BioTek, VT). The absorbance without an inhibitor was normalized to 100% activity of CYP2E1.
CYP2E1 kinetic data analysis
The CYP2E1 inhibition kinetics were analyzed using GraphPad Prism 5 software (GraphPad Software Inc., San Diego, CA). The half maximal inhibitory concentration (IC50) was determined by fitting curve to data by nonlinear regression. The Michaelis–Menten constant (K m) and maximal reaction velocity (V max) were obtained using the Michaelis–Menten nonlinear regression equation. The experimental data were re‐analyzed with the appropriate equation to determine the inhibition type and inhibition constant (K i). Goodness‐of‐fit criteria were used to define the inhibition pattern.
Results and Discussion
Docking study of the analogs with CYP2E1 active site
To find a superior DAS alternative, we performed a structure activity study. The central sulfur atom in DAS structure interacts with the heme of CYP2E1 active site, thereby acting as an inhibitor. However, DAS is also metabolized at the same position resulting in production of toxic sulfur metabolites such as diallyl sulfoxide and diallyl sulfone (Rao et al. 2015). We analyzed seven compounds (AMS, AES, DE, TP, PEA, 5,1 HA, and PEXA) that resemble DAS, but have slightly different chemical moieties (Fig. 1A). We chose analogs that altered DAS at the central hetero atom by replacing sulfur with oxygen (DE, PEA, and PEXA), or by completely removing sulfur (5,1 HA). These alterations should result in DAS analogs retaining their ability to interact with the heme of CYP2E1 active site, but losing their ability to be oxidized into toxic metabolites. We also chose DAS analogs that had stronger nucleophiles at the carbon atom adjacent to the hetero atom (PEA, PEXA, and 5,1 HA) to increase the binding strength of the analogs to the active site. Furthermore, we chose compounds with smaller side chain (AES and AMS), which is likely to improve the binding affinity as well (Rao et al. 2015). Low molecular monocyclic aromatic compounds are also a viable option as the binding site of CYP2E1 (Fig. 1B) exists within a narrow channel, unlike other CYP active sites (Collom et al. 2008). Therefore, we chose a cyclic analog of DAS (TP), which is likely to strengthen the binding as an inhibitor.
Figure 1.
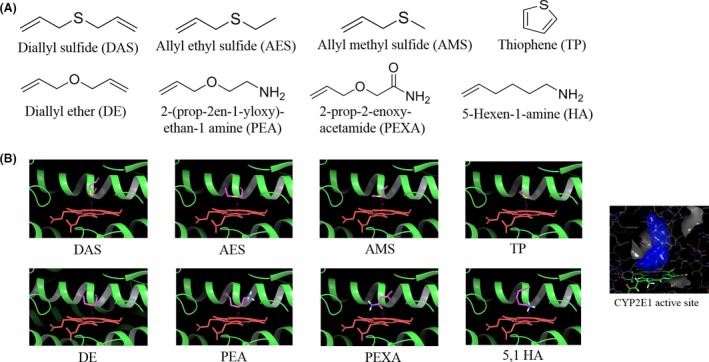
(A) The structures of the seven commercially available DAS analogs that were purchased and used for this study. (B) Docking of DAS and analogs with human CYP2E1 crystal structures using Schrödinger Molecular Modeling Suite 2015. All the compounds were prepared using the Ligprep module and then docked into the active site with metal constraints. The results showed similar binding mode for all DAS analogs.
To determine the potential binding mode and molecular interactions between the analogs and CYP2E1 at the active site, we performed a docking study. All the compounds generally showed similar binding mode, in that they occupied approximately the same space surrounded by a group of hydrophobic residues and the “necklace” of phenylalanine side chains including Phe298, Phe116, and Phe207, above the heme (Fig. 1B). In each structure, the heteroatom (oxygen or sulfur) interacts with the heme iron with distances of 3.15–3.58 Å, whereas the alkyl chain or aromatic ring extends toward the helix I. Among all the compounds, TP favors a π‐π interaction with the phenylalanine side and shows the best docking score of −4.86 (Table 1). However, unlike the thiophene ring, the alkyl chains in the other compounds are flexible and lack a π‐π interaction with the surrounding residues. Therefore, the study predicts that introducing aromatic moieties would increase interactions with CYP2E1. We also observed a higher docking score for AMS (−3.24) than DAS (−1.56). These results are consistent with our hypothesis.
Table 1.
Summary of the CYP2E1 inhibition kinetics of the analogs
| Analogs | Inhibition type | K i (μmol/L) | IC50 (μmol/L) | Maximal inhibition (%) | Docking score |
|---|---|---|---|---|---|
| Diallyl sulfide (DAS) | Competitive | 6.3 ± 1.0 | 17.3 ± 1.1 | 97 | −1.54 |
| Allyl methyl sulfide (AMS) | Competitive | 4.4 ± 0.9 | 11.4 ± 1.1a | 100 | −3.25 |
| Thiophene (TP) | Competitive | 7.7 ± 1.2 | 19.4 ± 1.1 | 98 | −4.86 |
| Diallyl ether (DE) | Competitive | 3.1 ± 0.2a | 6.3 ± 1.2a | 100 | −0.17 |
| 5‐hexen‐1‐amine (5, 1 HA) | Uncompetitive | ~7.3 (ambiguous) | 74.7 ± 1.2a | 75 | −1.88 |
| Noncompetitive | ~10.5 (ambiguous) | ||||
| 2‐(prop‐2‐en‐1‐yloxy) ethan‐1‐amine (PEA) | Uncompetitive | 19.3 ± 2.3a | 43.7 ± 1.4a | 71 | −1.27 |
| Noncompetitive | 34.1 ± 3.3a | ||||
| 2‐prop‐2‐enoxyacetamide (PEXA) | Uncompetitive | 246 ± 47a | 91.4 ± 1.6a | 20 | −1.73 |
| Noncompetitive | 363 ± 64a |
The inhibition kinetic results (IC50 and K i) are presented as mean ± SE from four experimental replicates. The type of inhibition was also determined as described in Materials and Methods. The docking scores are also presented in the table.
P < 0.05 when compared to DAS.
Docking score does not always depict ligand–receptor interactions accurately. In fact, the flaw of the scoring system is considered to be a major limiting factor of computational docking (Ramirez and Caballero 2016), especially for small ligands for which the number of interactions between a ligand and its receptor is too small to make reliable differentiations among different ligands. The docking programs make simplified assumptions in the assessment of modeled complexes without accounting for physical phenomena that play a role in influencing molecular recognition, such as entropic effects (Kitchen et al. 2004). For example, replacing the sulfur atom of the DAS structure with oxygen (DE) should have resulted in similar or increased binding affinity. However, interestingly, we observed lowest docking score for DE, when we would have expected this compound to be at the least equally potent to DAS in its activity. Moreover, we expected increased binding for the compounds in which modification have been made at the carbon atom adjacent to the heteroatom by integrating stronger nucleophiles (e.g., PEXA). However, we did not observe much increase in docking score for such analogs. Nevertheless, this preliminary docking study provided some rough comparisons among the chosen ligands.
Inhibition of CYP2E1 activity by DAS and its analogs
There is very little information about the precise CYP2E1 inhibition capacity of DAS. In a few studies, DAS has been shown to be a competitive inhibitor of CYP2E1 using human and rat liver microsomes (Brady et al. 1991; Morris et al. 2004). However, these studies were performed either with insufficient or very low concentrations of DAS. In these studies, the reported K i values of DAS therefore differ by a large margin, perhaps due to the lack of sufficiently varying degree of concentrations of both the inhibitor as well as the substrate. The study by Morris et al. used a couple of substrate and inhibitor concentrations and provided a better inhibition kinetic profile. Moreover, at first, we characterized the DAS inhibition kinetics with CYP2E1 using multiple concentrations of both p‐NP and DAS (Fig. 2, Table 1).
Figure 2.
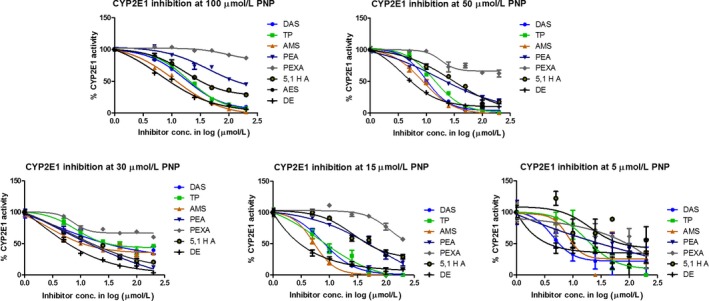
Inhibition of CYP2E1 activity at varying p‐NP substrate and inhibitor concentrations. The absorbance obtained without any inhibitor (vehicle only) was considered as 100% CYP2E1 activity for each substrate concentration. The data were analyzed using nonlinear regression curve fitting. The IC50 was determined and presented in Table 1. The data were also used to plot Michaelis–Menten kinetics curve, and Ki and type of inhibition were determined as described in Materials and Methods.
We used CYP2E1 baculosomes for our study as they have the unique property of expressing only one type of CYP isozyme. This gives an advantage over traditional microsome‐based assays in that there will be no interference/interactive effects from other CYPs in determining the inhibition kinetics. First, we established that CYP2E1 activity is linear at 100 μmol/L substrate concentration for 1 h of reaction (Fig. S1). We then conducted the inhibition assay at five different p‐NP concentrations (5, 15, 30, 50, 100 μmol/L) and six different concentrations of inhibitors (5, 10, 25, 50, 100, and 200 μmol/L). Based on the reported K m value, 100 μmol/L may not be the saturating p‐NP concentration. However, CYP2E1 shows substrate inhibition at >100 μmol/L p‐NP (Collom et al. 2008). Therefore, first we performed the inhibition assay at 100 μmol/L to obtain the IC50 values (Table 1). Subsequently, we used lower concentrations of the substrate to determine the IC50 at each substrate concentration as well as the K i values. Our results showed that DAS has an IC50 of 17.3 ± 1.1 μmol/L and a K i of 6.3 ± 1.0 μmol/L, and it follows competitive inhibition kinetics. Brady et al. used acetone‐induced rat microsome for their study and only one concentration of DAS (125 μmol/L), and they observed that inhibition of CYP2E1 by DAS followed competitive inhibition, which is consistent with our findings (Brady et al. 1991). However, the reported K i value (188 μmol/L) from their study varies by a large margin from our results as well as from the study conducted by another group using human and rat microsome. Morris et al. reported an IC50 value of 3 μmol/L for DAS. However, they used only two lower concentrations (2 and 8 μmol/L) (Morris et al. 2004). The reported Ki values (0.3~1.9 μmol/L) were also determined using limited concentrations of both DAS (0.25 and 1.0 μmol/L for human CYP2E1; 2 and 6 μmol/L for rat CYP2E1) and the substrate. Our results are likely to provide more accurate and reliable IC50 and Ki than the reported values, because we used multiple substrate and DAS concentrations and used CYP2E1‐induced baculosomes.
Furthermore, we performed a complete inhibition kinetics of seven DAS analogs as described above (Fig. 2, Table 1). Of the seven compounds, although AES caused marked inhibition, it did not fit with any inhibition kinetics. Therefore, in subsequent studies, we used only six compounds for complete inhibition kinetic analysis. Results show that, contrary to the docking analysis, DE has significantly lower IC50 and K i values (6.3 ± 1.2*μmol/L and 3.1 ± 0.2*μmol/L, respectively) than DAS (17.3 ± 1.1 μmol/L and 6.3 ± 1.0 μmol/L, respectively). The analog with the highest docking score, TP, showed the similar inhibitory capacity as compared to DAS (IC50 = 19.4 ± 1.1 μmol/L and K i = 7.7 ± 1.2 μmol/L). AMS, which exhibited relatively higher docking score, showed an IC50 of 11.42 ± 1.1*μmol/L and K i of 4.4 ± 0.9 μmol/L, which are lower than DAS. Similar to DAS, these analogs showed competitive inhibition of the CYP2E1 activity. Thus, AMS, TP, and DE showed complete inhibition at saturating substrate concentration (Table 1), suggesting that their nature of inhibition for CYP2E1 is similar to DAS. Other analogs such as AES, 5,1 HA, PEA, and PEXA, despite having similar binding conformation and score, did not exhibit desirable inhibitory activity and mostly followed uncompetitive and/or noncompetitive inhibition kinetics.
As expected, the replacement of oxygen and/or changing the size of side chain of DAS improved the binding of oxygen or sulfur with the heme of CYP2E1, and showed better inhibition profiles with DE and AES than DAS. This mode of binding is expected to produce metabolite by CYP2E1, which may be less toxic to the metabolites produced by the oxidation of sulfur. It is interesting to note that compounds that showed higher than or similar to DAS binding affinity (IC50 or K i) also showed similar inhibition kinetics to DAS. However, the compounds that showed lower binding affinity than DAS showed very different inhibition kinetics. These compounds appear to bind the CYP2E1 active site in two different modes; one through oxygen and the other through nitrogen. The two binding modes appeared to have weaken the overall binding affinity. We had initially expected that nitrogen moieties on PEA, PEXA, and 5,1 HA is likely to strengthen the binding of the heme of CYP2E1.
These results are significant in terms of finding a superior alternative to DAS. Despite having substantial pharmacological activities and being used in numerous research studies for many years, DAS has failed to consolidate its credibility to be a potential drug candidate (Rao et al. 2015). Due to its own toxicity, it is difficult to design any long‐term/chronic treatment schedule with DAS. The metabolites, diallyl sulfoxide and diallyl sulfone, by undergoing further metabolic activation to their respective epoxides can cause considerable toxicity. Moreover, these metabolites can cause autocatalytic destruction of the CYP2E1 enzyme (Rao et al. 2015). Zhai et al. has reported that DAS aggravated the isoniazid induced toxic effect by potentiating increased ROS production in primary rat hepatocytes (Zhai et al. 2008). Our own group have found that despite rescuing ethanol‐mediated cell death to a certain extent, DAS itself causes ~15% cell death in SVGA astrocytic cell lines in 24 h (Jin et al. 2013). The preliminary results (unpublished observations) with DAS analogs showed that, even at as high as 200 μmol/L concentrations, TP and DE did not show any significant toxicities for 6 and 4 days of treatment, respectively. However, as expected, DAS caused more than 40% cell death at 200 μmol/L after 2 days of treatment.
In conclusion, this is the first report on a thorough analysis of CYP2E1 inhibition kinetics of DAS and its seven structural analogs. This study provided two DAS analogs that have better inhibition characteristics than DAS, which also appear to show relatively lower toxicity than DAS in early studies. Our extensive toxicity profile study of these compounds, and whether these compounds can rescue alcohol‐ and acetaminophen‐mediated toxicity in hepatic and extra‐hepatic cells, is underway. Overall, the results are very encouraging in the search for a superior alternative to the widely used DAS as a selective CYP2E1 inhibitor as a research tool and/or as a possible combination/preventive therapy in alcohol‐ and other xenobiotic‐mediated toxicity.
Author Contributions
Participated in research design: Mohammad A. Rahman, Wei Li, and Santosh Kumar; Conducted experiments: Mohammad A. Rahman, Narasimha M. Midde, and Xiaoxin Wu; Performed data analysis: Mohammad A. Rahman, Narasimha M. Midde, Wei Li, and Santosh Kumar; Wrote the manuscript: Mohammad A. Rahman and Santosh Kumar.
Disclosure
None declared.
Supporting information
Figure S1. Time kinetics of para nitrophenol hydroxylation.
Acknowledgements
The authors thank the National Institutes of Health for financial support to Dr. Kumar (AA022063).
Rahman M. A., Midde N. M., Wu X., Li W., Kumar S.. Kinetic characterizations of diallyl sulfide analogs for their novel role as CYP2E1 enzyme inhibitors. Pharma Res Per, 5(5), 2017, e00362, https://doi.org/10.1002/prp2.362
References
- Brady JF, Ishizaki H, Fukuto JM, Lin MC, Fadel A, Gapac JM, et al. (1991). Inhibition of cytochrome P‐450 2E1 by diallyl sulfide and its metabolites. Chem Res Toxicol 4: 642–647. [DOI] [PubMed] [Google Scholar]
- Cederbaum AI (2014). Methodology to assay CYP2E1 mixed function oxidase catalytic activity and its induction. Redox Biol 2: 1048–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Ahn S, Wang J, Lu Y, Dalton JT, Miller DD, et al. (2012). Discovery of novel 2‐aryl‐4‐benzoyl‐imidazole (ABI‐III) analogues targeting tubulin polymerization as antiproliferative agents. J Med Chem 55: 7285–7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collom SL, Laddusaw RM, Burch AM, Kuzmic P, Perry MD, Miller GP (2008). CYP2E1 Substrate Inhibition: mechanistic interpretation through an effector site for monocyclic compounds. J Biol Chem 283: 3487–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M, Kumar A, Kumar S (2012). Ethanol‐mediated regulation of cytochrome P450 2A6 expression in monocytes: role of oxidative stress‐mediated PKC/MEK/Nrf2 pathway. PLoS ONE 7: e35505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M, Ande A, Kumar A, Kumar S (2013). Regulation of cytochrome P450 2e1 expression by ethanol: role of oxidative stress‐mediated pkc/jnk/sp1 pathway. Cell Death Dis 4: e554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitchen DB, Decornez H, Furr JR, Bajorath J (2004). Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discovery 3: 935–949. [DOI] [PubMed] [Google Scholar]
- Lu Y, Cederbaum AI (2008). CYP2E1 and Oxidative Liver Injury by Alcohol. Free Radic Biol Med 44: 723–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Chen JJ, Wang J, Li CM, Ahn S, Barrett CM, et al. (2014). Design, Synthesis, and Biological Evaluation of Stable Colchicine Binding Site Tubulin Inhibitors as Potential Anticancer Agents. J Med Chem 57: 7355–7366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris CR, Chen SC, Zhou L, Schopfer LM, Ding X, Mirvish SS (2004). Inhibition by allyl sulfides and phenethyl isothiocyanate of methyl‐n‐pentylnitrosamine depentylation by rat esophageal microsomes, human and rat CYP2E1, and Rat CYP2A3. Nutr Cancer 48: 54–63. [DOI] [PubMed] [Google Scholar]
- Porubsky PR, Battaile KP, Scott EE (2010). Human cytochrome P450 2E1 structures with fatty acid analogs reveal a previously unobserved binding mode. J Biol Chem 285: 22282–22290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez D, Caballero J (2016). Is It Reliable to Use Common Molecular Docking Methods for Comparing the Binding Affinities of Enantiomer Pairs for Their Protein Target? Int J Mol Sci 17(4): 525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao PS, Midde NM, Miller DD, Chauhan S, Kumar A, Kumar S (2015). Diallyl sulfide: potential use in novel therapeutic interventions in alcohol, drugs, and disease mediated cellular toxicity by targeting cytochrome P450 2E1. Curr Drug Metab 16: 486–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raza H, John A (2015). Differential cytotoxicity of acetaminophen in mouse macrophage J774.2 and human hepatoma HepG2 cells: protection by diallyl sulfide. PLoS ONE 10: e0145965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan K, Clemens DL, Dey A (2013). Inhibition of CYP2E1 leads to decreased malondialdehyde‐acetaldehyde adduct formation in VL‐17A cells under chronic alcohol exposure. Life Sci 92: 325–336. [DOI] [PubMed] [Google Scholar]
- Trafalis DT, Panteli ES, Grivas A, Tsigris C, Karamanakos PN (2010). CYP2E1 and risk of chemically mediated cancers. Expert Opin Drug Metab Toxicol 6: 307–319. [DOI] [PubMed] [Google Scholar]
- Yoon E, Babar A, Choudhary M, Kutner M, Pyrsopoulos N (2016). Acetaminophen‐induced hepatotoxicity: a comprehensive update. J Clin Transl Hepatol 4: 131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai Q, Lu SR, Lin Y, Yang QL, Yu B (2008). Oxidative stress potentiated by diallylsulfide, a selective CYP2E1 inhibitor, in isoniazid toxic effect on rat primary hepatocytes. Toxicol Lett 183: 95–98. [DOI] [PubMed] [Google Scholar]
- Zhang RH, Gao JY, Guo HT, Scott GI, Eason AR, Wang XM, et al. (2013). Inhibition of CYP2E1 attenuates chronic alcohol intake‐induced myocardial contractile dysfunction and apoptosis. Biochem Biophys Acta 1832: 128–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Time kinetics of para nitrophenol hydroxylation.