

Congenital heart block is a rare disease of the fetal heart caused by anti-Ro autoantibodies in the mother that result in death and lifelong heart pacing of survivors. This study provides in vivo and in vitro transcriptome support that injury may be mediated by an effect of interferon on the fetal fibroblasts.
Keywords: autoimmune congenital heart block, cardiac fibroblast, RNASeq
Abstract
The signature lesion of SSA/Ro autoantibody-associated congenital heart block (CHB) is fibrosis and a macrophage infiltrate, supporting an experimental focus on cues influencing the fibroblast component. The transcriptomes of human fetal cardiac fibroblasts were analyzed using two complementary approaches. Cardiac injury conditions were simulated in vitro by incubating human fetal cardiac fibroblasts with supernatants from macrophages transfected with the SSA/Ro-associated noncoding Y ssRNA. The top 10 upregulated transcripts in the stimulated fibroblasts reflected a type I interferon (IFN) response [e.g., IFN-induced protein 44-like (IFI44L), of MX dynamin-like GTPase (MX)1, MX2, and radical S-adenosyl methionine domain containing 2 (Rsad2)]. Within the fibrotic pathway, transcript levels of endothelin-1 (EDN1), phosphodiesterase (PDE)4D, chemokine (C-X-C motif) ligand (CXCL)2, and CXCL3 were upregulated, while others, including adenomedullin, RAP guanine nucleotide exchange factor 3 (RAPGEF3), tissue inhibitor of metalloproteinase (TIMP)1, TIMP3, and dual specificity phosphatase 1, were downregulated. Agnostic Database for Annotation, Visualization and Integrated Discovery analysis revealed a significant increase in inflammatory genes, including complement C3A receptor 1 (C3AR1), F2R-like thrombin/trypsin receptor 3, and neutrophil cytosolic factor 2. In addition, stimulated fibroblasts expressed high levels of phospho-MADS box transcription enhancer factor 2 [a substrate of MAPK5 (ERK5)], which was inhibited by BIX-02189, a specific inhibitor of ERK5. Translation to human disease leveraged an unprecedented opportunity to interrogate the transcriptome of fibroblasts freshly isolated and cell sorted without stimulation from a fetal heart with CHB and a matched healthy heart. Consistent with the in vitro data, five IFN response genes were among the top 10 most highly expressed transcripts in CHB fibroblasts. In addition, the expression of matrix-related genes reflected fibrosis. These data support the novel finding that cardiac injury in CHB may occur secondary to abnormal remodeling due in part to upregulation of type 1 IFN response genes.
NEW & NOTEWORTHY Congenital heart block is a rare disease of the fetal heart associated with maternal anti-Ro autoantibodies which can result in death and for survivors, lifelong pacing. This study provides in vivo and in vitro transcriptome-support that injury may be mediated by an effect of Type I Interferon on fetal fibroblasts.
absent structural abnormalities, the development of congenital heart block (CHB) in the second trimester of pregnancy is almost universally associated with fetal exposure to transplacentally acquired maternal autoantibodies reactive with SSA/Ro ribonucleoproteins (11). However, penetrance of disease despite autoantibody exposure is low, suggesting that antibodies are necessary but insufficient to cause disease and the final pathway to permanent injury is variable: kept totally in check in most exposed fetuses (normal sinus rhythm), subclinical in others (first-degree block), and fully executed in very few (advanced block) (10). Remarkably, the clinical progression from normal sinus rhythm to advanced block can occur in less than a week, as demonstrated by serial fetal echocardiography, or within 24 h, as detected by ambulatory heart rhythm surveillance (17). Cardiac injury is most often identified in utero between 18 and 24 wk of gestation (9) and associated with significant mortality (17.5%) and morbidity (70% require pacing) (23, 30). In the presence of disease beyond the conduction system, the case fatality rate approaches 50% (23, 30).
Histological correlates from fetuses and neonates electively terminated or dying with CHB consistently display extensive fibrosis and calcification of the atrioventricular (AV) node. Protein expression of transforming growth factor (TGF)-β has been noted at septal regions, extracellularly in the fibrous matrix, and intracellularly in macrophage infiltrates (2, 12–15). In vivo consequences of TGF-β activation are reflected by the presence of fibroblast transdifferentiation into myofibroblasts. Additional pathological findings include endocardial fibroelastosis and papillary muscle fibrosis. Particularly in affected hearts obtained close to the time of initial detection in the second trimester, macrophages and multinucleated giant cells are present in septal and thickened fibrous subendocardial regions in close proximity to IgG and apoptotic cells (15, 37). Accordingly, critical insights into the pathogenesis of disease are likely to be gained by focusing on fetal cardiac fibroblast reactivity to antibody-mediated inflammation.
Using an in vitro approach as well as a translational approach, we evaluated fibroblast transcriptomes in the context of anti-SSA/Ro-mediated inflammation and injury. The in vitro approach used cultured cardiac fibroblasts isolated via a Langendorff preparation from otherwise healthy, electively terminated second-trimester fetuses. Incubation with supernatants from macrophages transfected with SSA/Ro-associated noncoding Y ssRNA (hY3) (2, 13) was used as a proxy to simulate the cascade to scar in CHB as supported by the histological evidence. Translation to human disease leveraged an unprecedented opportunity to interrogate the transcriptome of fibroblasts from an electively terminated fetal heart with AV nodal block progressing from second to third degree in 2 days. Fibroblasts were similarly isolated by a Langendorff preparation but immediately flow sorted and evaluated in the absence of any stimuli or coculturing and compared with flow “healthy” fetal cardiac fibroblasts obtained under otherwise identical conditions.
MATERIALS AND METHODS
Specimens.
Affected and control hearts were obtained following written informed consent from the New York University (NYU) Institutional Review Board as part of the Research Registry for Neonatal Lupus. Several hearts were obtained from elective terminations of pregnancy at 20–22 wk of gestation from fetuses in which there was no known cardiac disease. These were used for the generation of cultured fibroblasts. However, one was used specifically for sorting by flow cytometry to match the one CHB heart. The anti-SSA/Ro-positive mother of the CHB fetus was a 35-yr-old African American woman with two previous children, one child with CHB and the other child healthy. In this pregnancy, she was treated with 400 mg hydroxychloroquine daily and intravenous immunoglobulin at 1 g/kg every 3 wk beginning at gestational week 12. At 19 wk, the fetal echocardiogram revealed a heart rate of 140 beats/min with rare intermittent episodes of slowing at 100 beats/min. It was unclear if this was sinus bradycardia, blocked atrial premature contractions or second-degree block. The mother was treated with 4 mg dexamethasone. Two days later, the fetus was in complete block and was electively terminated.
Isolation, preparation, and treatment of cardiac fibroblasts.
Aortas from healthy fetal hearts were cannulated using continuous perfusion of the coronary arteries by a Langendorff preparation with the addition of proteolytic enzymes to yield a single cell suspension of primary human fetal cardiac cells for the culturing experiments as previously described (13). The same Langendorff preparation was used to isolate cell suspensions from one healthy heart and the CHB heart. Through the use of a cell sorting flow cytometer, isolates yielded DAPI-negative cells, which were podoplanin positive, CD45 negative, and CD31 negative.
Treatment of cardiac fibroblasts.
For the in vitro approach, human fetal fibroblasts were cultured in fibroblast basal media (Lonza) plus 10% FCS. Fibroblasts were plated at a density of 5 × 105 cells/well (6-well plate). After being serum starved, fibroblasts were incubated for 16 h with the following: 1) supernatants generated from macrophages transfected with hY3, 2) no treatment, 3) BIX-02189 (10 μM) alone, and 4) BIX-02189 as a cotreatment with condition 1. The protocol to generate hY3 macrophage supernatants used human macrophages derived from healthy control monocytes in accordance with a previously published protocol (12). Macrophages were treated briefly with or without 2.5 μg/ml hY3. After 18 h, supernatants from control macrophages and activated “hY3-transfected” macrophages (an in vitro model of heart block), as previously described (2), were harvested. Type I interferon (IFN) activity was detected using a reporter cell assay (38). Supernatants from macrophages transfected with hY3 were evaluated using a well-established approach that measures IFN-responsive transcripts in a WISH cell line.
Immunofluorescence.
Fibroblasts were cultured on glass coverslips at equal density per well, treated and subsequently washed, fixed with 4% paraformaldehyde, and permeabilized with acetone. To assess phospho-MADS box transcription enhancer factor 2 (MEF2C), the protocol involved the addition of primary antibody (1:100) and species-specific TRITC-conjugated secondary antibody (1:200) along with DNA dye (Hoechst 33342). For a field at ×20, images were recorded with a digital camera.
RNASeq.
In each condition, fibroblasts were lysed in QIAzol solution (Qiagen) for RNA extraction using the Direct-zol RNA Miniprep kit (ZymoResearch) in accordance with the manufacturer’s instructions. Fibroblast RNA was then quantified using a Nanodrop ND-2000 spectrophotometer. Downstream assays were performed in the NYU Genome Technology Center. To assess protein-coding expression profiles, RNASeq libraries were made using the SMARTer Ultra Low RNA kit by Illumina using 10 ng total RNA and used to prepare cDNA libraries. Each condition was barcoded. Libraries were run on an Illumina 2500 by using a paired-end 50-bp protocol. Sequencing reads were then demultiplexed and converted to FASTQ format with Illumina FASTQ software.
Single-read raw sequencing data were received in FASTQ format. Quality control metrics were measured on the FASTQ files using the FASTQC 0.11.4 tool. Read mapping was then performed using Tophat 2.0.9 against the hg19 human reference genome. The resulting BAM alignment files were processed using the HTSeq 0.6.1 python framework and respective hg19 GTF gene annotation, obtained from the University of California-Santa Cruz database. The Bioconductor package DESeq2 (3.2) was then used to identify differentially expressed genes. RNA transcriptomes in FASTQ format are available on request from the corresponding author.
Genes were defined as upregulated if their expression increased by twofold (log2-transformed ratio > 1) in the presence of hY3 macrophage supernatants relative to the untreated condition and downregulated if their expression decreased by 40% (log2-transformed ratio < −0.724). The Database for Annotation, Visualization and Integrated Discovery (DAVID) was used in conjunction with a statistical routine (19) to assign differentially regulated genes to functional clusters.
Quantitative PCR.
Total RNAs were used to prepare cDNA libraries. For real-time quantitative (q)PCR analysis of MX dynamin-like GTPase (MX)1, the primers were 5′-GTTTCCGAAGTGGACATCGCA-3′ (forward) and 5′-CTGCACAGGTTGTTCTCAGC-3′ (reverse). Other targets included endothelin-1 (EDN1) and RAP guanine nucleotide exchange factor 3 (RAPGEF3), with gene expression and quantification using END1 primers [5′-AGAGTGTGTCTACTTCTGCCA-3′ (forward) and 5′-CTTCCAAGTCCATACGGAACAA-3′ (reverse)] along with RAPGEF3 primers [5′-GACCGGAAGTACCACCTTAGG-3′ (forward) and 5′-AGATTCCCACAACTTGGCTCC-3′ (reverse)]. Levels of expression were normalized by parallel amplification and quantification of the GAPDH mRNA level with primers 5′-ACCACAGTCCATGCCATCAC-3′ (forward) and 5′-TCCACCACCCTGTTGCTGTA-3′ (reverse). Brilliant SYBR green RT-PCR (Invitrogen) was used as the qPCR mix.
Statistics.
The Mann-Whitney test was used to compare the relative gene expression of EDN1 and RAPGEF3 in the fibroblasts of the different groups and to evaluate the targeted transcripts of WISH cells. Values of P ≤ 0.05 were considered significant.
RESULTS
Characterization of fetal fibroblasts stimulated with supernatants from macrophages transfected with ssRNA.
In vitro experiments were performed in accordance with previous laboratory work, in which a model of anti-SSA/Ro-associated injury exploits the cross talk between macrophages stimulated with the ssRNA component of the SSA/Ro immune complex and the effects of the generated supernatants on cultured cardiac fibroblasts (13). The initial approach compared the transcriptome of cultured untreated healthy primary human fetal fibroblasts with the transcriptome of fibroblasts stimulated with supernatants of hY3 (SSA/Ro 60-associated ssRNA)-transfected macrophages. Treatment with hY3 macrophage supernatants resulted in increased expression of 2,659 genes and decreased expression of 4,954 genes. An inspection of transcripts ranked by fold change in expression yielded genes reflecting a type I IFN response (Fig. 1A).
Fig. 1.

Differential expression of targeted and agnostically determined candidate genes of fibroblasts treated with supernatants from human SSA/Ro-associated noncoding Y ssRNA [hY3; a component of SSA/Ro 60-associated ssRNA]-transfected macrophages. Interferon (IFN) genes (A), inflammatory genes (B), fibrotic genes (C), and antifibrotic genes (D) are displayed as a heat map of fibroblasts treated with hY3 macrophage supernatant (MΦ; n = 1) or untreated (mean, n = 2) and a bar graph showing the log2-transformed ratio (y-axis) versus targeted gene (x-axis). An inspection of transcripts ranked by fold change in expression yielded genes reflecting a type I IFN response. By agnostic Database for Annotation, Visualization and Integrated Discovery (DAVID) analysis, inflammatory genes were also highly expressed. For targeted candidates, fibroblasts treated with hY3 macrophage supernatants expressed a pattern of fibrotic genes that is consistent with a path toward fibrosis.
Given the enrichment of type I IFN-responsive genes in the fibroblast transcriptome, a WISH cell line (41) was selected to evaluate supernatants from macrophages transfected with hY3 because of the line’s capacity to elicit IFN-responsive transcripts. As shown in Fig. 2, IFN-induced protein with tetratricopeptide repeats 1 (IFIT1) transcripts were significantly increased in WISH cells treated with hY3 macrophage supernatants but not macrophage supernatants alone (n = 7, P = 0.02).
Fig. 2.
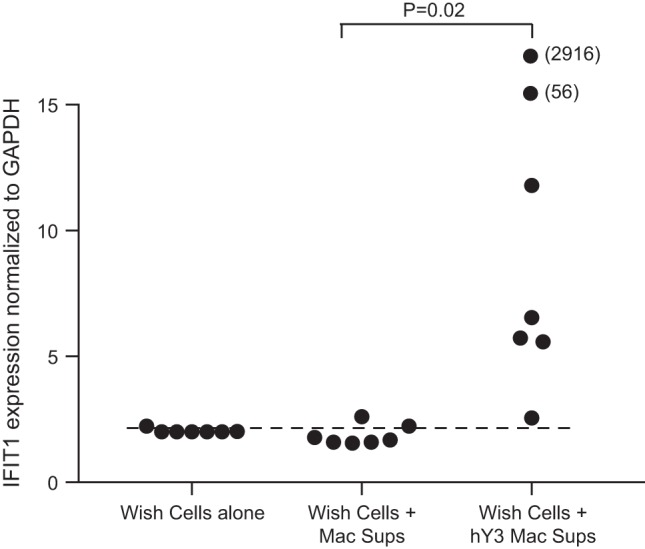
Assessment of activity of the IFN-α pathway in hY3 macrophage supernatants (Mac Sups). To authenticate the presence of IFN-α in hY3 macrophage supernatants, WISH cells were incubated without treatment, with macrophage supernatants, and with hY3-treated macrophage supernatants overnight. RNA was isolated from WISH cells, and an evaluation of the transcripts IFN-induced protein with tetratricopeptide repeats 1 (IFIT1) was performed. Values on the y-axis are the log2-transformed values of (where CT is threshold cycle); the x-axis shows the treatment of WISH cells. Values appearing above the dashed line represent IFIT1 levels greater than means + 2SD. The results showed that IFIT1 was highly abundant within the transcriptome reported by WISH cells given hY3 macrophage supernatants but not untreated macrophage supernatants (P = 0.02, Fisher’s exact test).
DAVID annotation was used to draw biological salience from the large number of differentially regulated genes. Genes residing in the top category (defense response category) of the number one cluster included complement C3A receptor (C3AR)1, chemokine (C-X-C motif) ligand (CXCL)10, F2R-like thrombin/trypsin receptor 3 (F2RL3), and neutrophil cytosolic factor 2 (NCF2) (Table 1 and Fig. 1B).
Table 1.
Top four DAVID GO categories of fibroblasts treated with hY3 macrophage supernatants versus untreated fibroblasts
| Category | Category P Value | False Discovery Rate |
|---|---|---|
| Defense response | 1.79 × 10−10 | 3.23 × 10−7 |
| Response to wounding | 3.14 × 10−8 | 5.66 × 10−5 |
| Inflammatory response | 3.93 × 10−8 | 7.09 × 10−5 |
| Innate immune response | 1.83 × 10−8 | 3.29 × 10−5 |
DAVID, Database for Annotation, Visualization and Integrated Discovery; GO, Gene Ontology; hY3, human SSA/Ro-associated noncoding Y ssRNA.
Targeted approaches to filtering of the transcriptome used candidates consistent with a profibrotic environment. Healthy fetal fibroblasts exposed to supernatants from hY3-transfected macrophages showed increased expression of profibrotic transcripts and decreased expression of antifibrotic transcripts (Fig. 1, C and D). Among genes that promote fibrosis, EDN1 and phosphodiesterase (PDE)4D, which reduce cAMP, as well as CXCL2 and CXCL3 were increased in cultured fibroblasts treated with hY3 macrophage supernatants compared with untreated fibroblasts. In contrast, genes that resist fibrosis, such as RAPGEF3, adenomedullin (ADM), dual specificity phosphatase 1 (DUSP1), tissue inhibitor of metallopeptidase (TIMP)1, and TIMP3, were downregulated.
ERK5 promotes fibrotic differentiation of fetal cardiac fibroblasts treated with supernatants from activated macrophages.
Based on a targeted approach, the next set of experiments explored pathways that might be operative in generating the profibrotic phenotype with expression of EDN1, PDE4D, and others (Fig. 1C) of the stimulated cardiac fibroblasts. Treatment with hY3 macrophage supernatants resulted in high levels of phospho-MEF2C [a substrate of MAPK5 (ERK5) (5)] as measured by immunofluorescence, a finding consistent with previously established associations between ERK5, phospho-MEF2C, and fibrosis (33). Cotreatment using BIX-02189, a specific inhibitor of ERK5, abrogated the accumulation of phospho-MEF2C (Fig. 3A). There was no effect of treatment with BIX-02189 alone.
Fig. 3.

Evidence of ERK5 activation and its inhibition on the expression of IFN, inflammatory, fibrotic, and antifibrotic genes in stimulated fibroblasts. Human fetal fibroblasts at equal cell numbers were evaluated for the expression of phospho-MADS box transcription enhancer factor 2 (MEF2C), a proxy of ERK5 activation. It was noted that phospho-MEF2C was detected by fibroblasts treated with hY3 macrophage supernatants (A) (results are representative of 3 experimentsm magnification: ×40). Inflammatory, fibrotic, and antifibrotic genes (B) and IFN genes (C) are displayed as log2-transformed ratios (y-axis) versus the targeted gene (x-axis) in stimulated fibroblasts in the presence and absence of BIX-02189, an ERK5 inhibitor. The robust expression of IFN genes in stimulated fibroblasts was not inhibited by BIX-02189. However, cotreatment of fibroblasts with BIX-02189 yielded a profile of attenuated fibrotic genes and increased antifibrotic genes.
The addition of BIX-02189 restored to baseline levels 632 of the 2,659 genes (23%) that were induced by hY3 macrophage supernatants and 1,369 of the 4,954 genes (27%) that were decreased. For example, PDE4D, EDN1, and TGF-β were downregulated in the presence of BIX-02189 (Fig. 3B), whereas DUSP1, ADM, and RAPGEF3 were upregulated. In contrast, the upregulation of IFN response genes by stimulated fibroblasts was unaffected by cotreatment with BIX-02189 (Fig. 3C). qPCR confirmed the upregulation of MX1 transcripts after treatment, but there was no effect by cotreatment with BIX-02189 (Fig. 4, top). For EDN1, qPCR confirmed the upregulation of transcript after treatment with hY3 macrophage supernatants and subsequent attenuation by BIX-02189 (Fig. 4, middle). Specifically, upregulation of the EDN1 transcript was attenuated by BIX-02189 (37.3 ± 8.8 vs. 4.9 ± 1.6, n = 3, P < 0.02). In contrast, downregulation of RAPGEF3 was reversed by BIX-02189 (0.79 ± 0.01 vs. 1.37 ± 0.1, n = 3, P = 0.006).
Fig. 4.
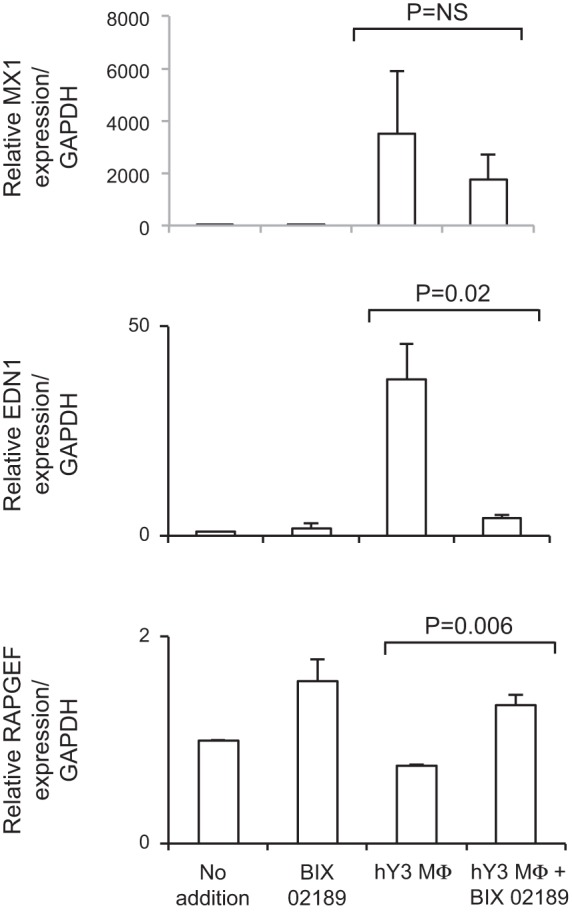
Effect of ERK5 inhibition on the expression of MX dynamin-like GTPase 1 (MX1), endothelin-1 (EDN1), and RAP guanine nucleotide exchange factor 3 (RAPGEF3) in stimulated fibroblasts. The upregulation of the EDN1 transcript in response to hY3 macrophage supernatants for 18 h was attenuated, whereas the downregulation of RAPGEF3 was reversed by BIX-02189. In contrast, the expression of MX1 was unaffected by cotreatment with BIX-02189. Shown on the y-axis are (transcript normalized to GAPDH) values, and shown on the x-axis are conditions including untreated, BIX-02189, hY3 macrophage supernatants, and hY3 macrophage supernatants + BIX-02189. Results are representative of three experiments. Bars represent means ± SE.
Mining the transcriptome of human fetal fibroblasts isolated from a 19-wk-old heart with CHB.
To translate the in vitro findings to human disease, the fibroblast transcriptomes in a fetal heart dying with CHB and a gestational age-approximated fetal heart from an elective termination were compared. A single cell suspension was prepared after tissue processing of each fetal heart, and both were applied to cell sorting. The protocol yielded cell populations that were enriched in leukocytes, endothelial cells, and fibroblasts (Fig. 5, A–D). The resultant RNA was used to generate cDNA libraries for downstream RNASeq analysis. The enriched populations expressed the appropriate transcripts (Fig. 5E).
Fig. 5.

Authentication of enriched leukocytes, endothelial cells, and fibroblasts based on flow sorting and transcriptome analysis. CHB heart (A and B) and control heart (C and D) were stained following the protocols outlined in materials and methods. After the selection of viable (DAPI negative) cells (not shown), anti-CD45 and anti-CD31 were used to separate leukocytes and endothelial cells, respectively (A and C). Note within the same panels the utility of a portion of the unstained cells (gate, black) to further separate isolated cells using anti-podoplanin (B and D). Flow panels show the gating, as indicated on the labels of y- and x-axes of A–D, to isolate CD45, CD31, and podoplanin fractions that are enriched populations of leukocytes, endothelial cells, and fibroblasts (E). As shown in E, RNA abundance of varied isolated cells after flow sorting (which was derived from RNASeq) was consistent with the lineage assignment.
Consistent with the in vitro data, fibroblasts of the CHB heart showed a significant increase in expression of IFN-α-responsive genes. Inspection of transcripts ranked by fold change in expression revealed five expressing a type 1 IFN response gene, including IFIT1B, OAS2, CXCL10, IFN-induced protein 44-like (IFI44L), and BCL6B (Table 2 and Fig. 6A).
Table 2.
Upregulated genes for CHB versus healthy flow-sorted fibroblasts
| Rank | Gene Symbol | Gene Name | Comment | Fold Increase* |
|---|---|---|---|---|
| 1 | ELTD1 | Adhesion G protein-coupled receptor L4 | Angiogenesis | 100 |
| 2 | IFIT1B | IFN-induced protein with tetratricopeptide repeats 1B | IFN pathway | 67 |
| 3 | GSTM1 | Glutathione-S-transferase μ1 | Cell metabolism | 57 |
| 4 | OAS2 | 2′-5′-Oligoadenylate synthetase 2 | IFN pathway | 47 |
| 5 | TM4SF18 | Transmembrane 4 L six family member 18 | Cell adhesion | 47 |
| 6 | CXCL10 | Chemokine (C-X-C motif) ligand 10 | IFN pathway | 43 |
| 7 | IFI44L | IFN-induced protein 44 like | IFN pathway | 38 |
| 8 | ACSL6 | Acyl-CoA synthetase long-chain family member 6 | Cell metabolism | 36 |
| 9 | BCL6B | B cell CLL/lymphoma 6B | IFN pathway | 36 |
| 10 | LCP1 | Lymphocyte cytosolic protein 1 | Cell adhesion | 30 |
CHB, SSA/Ro autoantibody-associated congenital heart block; IFN, interferon.
Ratio of transcripts per million (TPM) of CHB/healthy flow fibroblasts.
Fig. 6.

Comparison of expression of targeted and agnostically determined candidate genes of flow-sorted (fl) fibroblasts derived from CHB and healthy (control) human fetal hearts. IFN genes (A), inflammatory genes (B), fibrotic genes (C), and antifibrotic genes (D) are displayed as heat maps and bar graphs showing log2-transformed ratios (y-axis) versus the targeted gene (x-axis). Flow CHB fibroblasts expressed high levels of IFN, inflammatory, and fibrotic genes and low levels of antifibrotic genes compared with flow healthy fibroblasts.
In considering the impact of type 1 IFN on the fibroblast transcriptome, IFN regulatory factor (IRF)1 and IRF7, previously associated with corticosteroid resistance and alteration of metabolic pathways, respectively (4, 47), were also upregulated in the CHB heart. Glutamate receptor interacting protein 1(GRIP1; NCOA2), which binds to both IRF1 and the glucocorticoid receptor, was also expressed at higher levels in the affected heart compared with the control heart. In contrast, IFN-sensitive transcripts [MMP-19, collagen (COL)6A2, CXCL14, EGF6, and integrin subunit-α11 (ITGA11)], whose encoded proteins act on the cell matrix, were expressed at lower levels in the CHB heart. There was a 2.7-fold increase in the expression of peroxisome proliferator-activated receptor-γ coactivator-1α (PPARGC1A), a critical transcriptional coactivator of metabolic gene expression, along with high levels of metabolic transcripts (SLC25A19, ATP1B1, and ATP1A3) previously linked to PPARGC1A (39).
Using an analytic strategy of the transcriptome that paralleled the data sets generated from the in vitro cocultures, flow CHB fibroblasts were found to express the same high levels of proinflammatory genes as fetal cardiac fibroblasts treated with hY3 macrophage supernatants (e.g., C3AR1, CXCL10, F2RL3, and NCF2; Fig. 6B). With regard to fibrosis, again reflecting the in vitro studies, profibrotic genes were upregulated (e.g., EDN1, PDE4D, CXCL2, CXCL3, 1, and CPEB4; Fig. 6C) and antifibrotic candidates were downregulated [e.g., DUSP1, TIMP3, mesoderm-specific transcript (MEST), ADM, TIMP1, Clusterin (CLU), and RAPGEF3].
Three of the top DAVID Gene Ontology categories show overlap between the flow-sorted CHB fibroblasts and in vitro fetal cardiac fibroblasts treated with hY3 macrophage supernatants, including defense response, response to wounding, and the inflammatory response (Table 3).
Table 3.
Top four DAVID GO categories from analysis of upregulated genes of CHB versus healthy human fetal fibroblasts
| Category | Category P Value | False Discovery Rate | Category Shared With hY3 Macrophage Supernatants Versus Untreated |
|---|---|---|---|
| Defense response | 2.11 × 10−26 | 3.75 × 10−23 | Yes |
| Intrinsic to membrane | 4.90 × 10−16 | 6.11 × 10−13 | No |
| Inflammatory Response | 9.74 × 10−18 | 1.73 × 10−14 | Yes |
| Response to wounding | 4.26 × 10−15 | 7.49 × 10−12 | Yes |
The category intrinsic to the cell membrane that includes pathways with components of the receptor tyrosine kinase (ERBB) network [e.g., neuregulin-1 (NRG1), HB-EGF, and epiregulin (EREG)] was increased in flow-sorted CHB cardiac fibroblasts but not in vitro stimulated fibroblasts. Similarly, components of a negative feedback loop [e.g., mitogen-inducible gene 6 (MIG6)/receptor-associated late transducer (RALT) and SPRY] were downregulated in CHB cardiac fibroblasts but not in cultured fibroblasts (Table 4).
Table 4.
Targeted transcripts of flow-sorted CHB fibroblasts and flow-sorted healthy fibroblasts for IFN, the ERBB network, and cardiac function
| Candidate | CHB | Healthy |
|---|---|---|
| Impact of type 1 IFN on the transcriptome | ||
| PPARGC1A | 11.3* | 9.9 |
| IRF1 | 9.6 | 8.3 |
| NCOA2 | 8.8 | 7.8 |
| IRF7 | 8.9 | 8.0 |
| ATP1B1 | 11.9 | 11.1 |
| SLC25A19 | 6.9 | 6.4 |
| ATP1A3 | 11.2 | 10.8 |
| CXCL14 | 1.1 | 2.7 |
| ITGA11 | 6.7 | 8.3 |
| COL6A2 | 12.4 | 14.3 |
| MMP19 | 5.5 | 7.8 |
| Intrinsic to the membrane/related to the ERBB network | ||
| EREG | 7.1 | 6.1 |
| NRG1 | 5.4 | 1.7 |
| HBEGF | 9.6 | 8.0 |
| ERRFI1, MIG6/RALT | 7.1 | 8.1 |
| SPRY1 | 8.7 | 9.2 |
| Cardiac function/cardiac remodeling | ||
| NPPB | 13.6 | 11.1 |
| MME | 2.7 | 7.8 |
| TTN | 17.1 | 15.0 |
| XIRP2 | 11.5 | 8.4 |
| ELN | 12.1 | 14.3 |
ERBB, ErbB receptor tyrosine kinase. *Units of log2(TPM).
With a focus on cardiac homeostasis, several injury-related gene products were upregulated, such as NT-pro-B-type natriuretic peptide (NPPB), xin actin binding repeat containing 2 (XIRP2), and titin (TTN), while genes affording protection were downregulated [membrane metalloendopeptidase (MME) and elastin (ELN)].
DISCUSSION
On the basis of the transcriptomes of fibroblasts analyzed using two complementary approaches, interferogenic, profibrotic, and inflammatory genes contribute to the pathogenesis of CHB. The in vitro model used hY3 macrophage supernatants as a proxy of Fcγ receptor-mediated delivery of hY3 ssRNA, influencing the macrophage phenotype (16). The interferogenic signature included an upregulation of transcripts encoding antiviral effectors IFI44L, MX1, MX2, and radical S-adenosyl methionine domain containing 2 (Rsad2) (48). Within profibrotic pathways, a panel of matrix transcripts revealed that levels of EDN1, PDE4D, CXCL2, and CXCL3 were twofold higher in hY3-stimulated fibroblasts compared with untreated fibroblasts. In addition, levels of genes that resist matrix accumulation, including ADM, RAPGEF3, TIMP1, TIMP3, and DUSP1, were downregulated. Agnostic DAVID analysis revealed inflammatory genes including C3AR1, F2RL3, and NCF2. Supporting clinical translation, these results were reflected in the transcriptome of fibroblasts from a 19-wk-old fetal heart dying with CHB.
The highly reactive fibroblast transcriptome reflects an IFN-enriched environment. This observation extends earlier work by our group, which revealed that macrophages are a source of IFN-α (16). Seemingly paradoxical in the pathogenesis of CHB, IFNs have been shown to exert antifibrotic effects. In studies of the fibrotic comorbidities of chronic hepatitis C, initial evidence suggested that type 1 IFNs resisted fibrosis (1, 28). Exposure of fibroblasts to IFN-α resulted in inhibition of proliferation (54) along with inhibition of collagen synthesis (22) mediated by IRF7. IFN-α also inhibited bleomycin-induced pulmonary fibrosis (3), and pretreatment with IFN-α strongly suppressed ConA-induced fibrosis and hepatocellular injury (55). However, data are emerging that the potential contribution of IFN-α to fibrosis is more complex. IFN treatment resulted in a loss of genes encoding secreted components of the extracellular matrix, including several secreted proteinases that possibly stabilized their targets (39). In addition, it has been shown that type I IFN promotes the expression of PPARGC1A (53), which encodes PGC-1α. This protein is a key component controlling metabolic gene expression, which, when upregulated, may influence injury, especially at early hypertrophic responses to pressure overload (34).
The relationship between IFN-α and profibrotic consequences has been previously addressed in scleroderma. Indeed, Kim et al. (32) demonstrated that IFN-α production was significantly higher in patients with diffuse compared with limited scleroderma and was particularly associated with lung fibrosis. The authors postulated that IFN-α might contribute to disease as a result of a vasculopathic effect, an exuberant immune response, or some as yet-unknown effect on profibrotic factors. In a separate cohort, Eloranta et al. (24) also reported that serum IFN-α levels were associated with lung fibrosis. In contrast to the results discussed above on hepatitis C, IFN-α was ineffective in patients with scleroderma (6). Moreover, IFN-α is known to have an antiangiogenic effect, and it retards wound healing (27, 52). Although the antiangiogenic effects have previously been explained by IFN-α-mediated antiproliferative activity or induction of apoptosis of endothelial cells, a more recent study (44) has shown that prolonged exposure to IFN-α induces replicative senescence of endothelial cells.
There is also an association of type 1 IFN and corticosteroid resistance (47), which may provide an insight into the observation that dexamethasone does not prevent heart block or consistently halt its advance to complete block (8, 25, 30). An anti-inflammatory effect of glucocorticoids (GC) involves the recruitment of a GR corepressor complex, which includes glucocorticoid receptor (GCR) and GRIP1. GRIP1 is attenuated by IRF1 (47), and it has been recently shown that increased levels of IRF1 attenuated a GR reporter activity in GRIP1-transfected cells (4). Perhaps IFN confers a relative GC resistance by increasing IRF1, which might then saturate the available pool of GRIP1 and essentially out compete the GC/GCR complex.
TGF-β has been an attractive candidate in considering the fibrosing component in CHB. A central role of TGF-β is assigned to its receptor ALK5, which phosphorylates mothers against DDP homolog 2 (SMAD2) and SMAD3 (canonical) and thus initiates their translocation to the nucleus (43). The spectrum of TGF-β signaling also includes ancillary pathways such as an activation of MAPKs, the phosphoinositide 3-kinase Akt cascade, and focal adhesion kinase (20). MAPKs have also been shown to regulate the activity of MEK5/ERK5, which recruit the transcription factor MEF2C, which is involved in fibrosis (31). Fibroblast phospho-MEF2C was induced by the hY3-stimulated macrophage supernatant. Specific inhibition of ERK5 with the small molecule BIX-02189 improved survival rate and inhibited pulmonary fibrosis in a bleomycin-induced lung fibrosis model (33). Phospho-MEF2C has been shown to exert a pathological reaction to stress during heart failure (21). Transgenic mice expressing a dominant negative MEF2C profile demonstrated attenuated postnatal myocardial growth in calcineurin-induced hypertrophy (35), and, in a parallel finding, the antisense depletion of MEF2C decreased injury and a pathological contribution by PGC-1α (40, 45).
The agnostic approach provided an opportunity to confirm previously reported biomarkers. For example, from our gene interrogations, NT-proBNP (NPPB) was detected at higher levels in the CHB condition, while neprilysin (MME; which neutralizes NPPB) was detected at lower levels. These results are consistent with the observation of Saxena et al. (51) that the levels of NPPB were significantly elevated in the cord blood of CHB neonates compared with anti-SSA/Ro-exposed neonates without cardiac disease. The data also support the assertion that heart dysfunction occurs secondary to abnormal remodeling. For example, the cardiomyopathy marker XIRP2 was upregulated, and there was a respective loss and gain of vascular stiffness resisting genes, such as ELN, and vascular stiffness promoting genes, such as TTN.
This fetus developed recurrent CHB despite the prophylactic use of intravenous immunoglobulin and hydroxychloroquine. With regard to the former, this is particularly disconcerting since intravenous immunoglobulin was given at an inflammatory dose of 1 g/kg based on two prospective studies demonstrating that lower replacement doses of 400 mg/kg do not prevent recurrent CHB (26, 46). However, it is acknowledged that no therapy is always efficacious. Although the biology of the fetus did clearly support fibroblast reactivity likely in response to macrophage activation, there may have been environmental and genetic influences not overcome by simply increasing Fcγ inhibiting receptors, an expected effect of intravenous immunoglobulin (42, 50), or dampening Toll-like receptor signaling, an expected effect of hydroxychloroquine (16).
Several limitations of this study are acknowledged. The primary focus was to evaluate the influence of persistently stimulated macrophages on a fibroblast phenotype. However, the macrophage-fibroblast system may not fully recapitulate the pathological process. For example, it has been reported that anti-SSA/Ro antibodies directly bind living cultured cardiomyocytes and perturb Ca2+ electrogenesis (7, 49). Although this may be one mechanism in addition to binding of apoptotic cardiocytes and the formation of immune complexes, both likely result in macrophage infiltration [supported by autopsy studies (14, 15, 18, 37)] influencing the fibroblast contribution to scar. In addition, the macrophage-fibroblast axis may not fully explain the predilection for the conduction system. However, extranodal diseases such as endocardial fibroelastosis, dilated cardiomyopathy, and valvular abnormalities are seen in nearly 20% of cardiac neonatal lupus cases (29). The use of hY3 macrophage supernatants containing a varied and complex amalgam of stimuli is a potential limitation. In addition, the in vitro CHB injury model does not incorporate relevant biomechanical stimuli that are likely contributing to the phenotype of the CHB fibroblasts in the injured tissue. For in vivo assessments, the pool of flow-sorted fibroblasts may contain contaminating cells of nonfibroblast origin. While it can be argued that fibroblasts are isolated from the whole diseased heart and not the specific area of injury, fibrosis can extend beyond the AV node (18). Although the analysis of only one affected fetus precludes generalizability to all affected cases, the data are consistent with our experimental model, and another opportunity to harvest cells in short temporal proximity to an electively terminated CHB fetus is unlikely.
In summary, data generated from both an in vitro model and fibroblasts isolated from a fetus with rapid progression of clinical disease from incomplete to complete block within a few days support the hypothesis that interferogenic, profibrotic, and inflammatory genes contribute to the pathogenesis of CHB (Fig. 7). The novel finding was the detection of IFN-responsive transcripts with the potential to amplify the burden of disease through recruitment of mononuclear cells and biological properties related to energy metabolism and matrix regulation. Further implicating IFN-responsive transcripts in pathogenesis, Lisney et al. (36) very recently reported that anti-SSA/Ro-positive mothers of children with CHB express IFN-α at higher levels, although the children per se were not evaluated. Thus, a reasonable prediction is that IFNs may contribute to both susceptibility and regulatory factors, each of which could influence the fate of anti-SSA/Ro exposure and whether the proposed pathologic cascade eventuates in irreversible third-degree heart block.
Fig. 7.
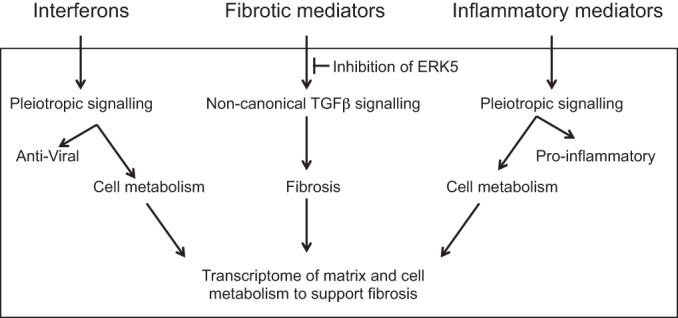
Interferogenic, profibrotic and inflammatory genes and their impact on the phenotype of fibroblast toward fibrosis pathways of autoimmune CHB. Upstream pathological events result in the release of IFNs, fibrotic, and inflammatory mediators by human macrophages treated with immune complexes of Ro60-hY3 and anti-Ro60. The highly reactive fibroblast transcriptome reflects an hY3 macrophage supernatant-enriched environment. For IFNs and inflammatory mediators in addition to well-documented antiviral and pro-inflammatory actions, it has been shown that type I IFN and inflammatory C3A promotes the expression of key component-controlling metabolic gene expression, which, when upregulated, may contribute to the cellular metabolism that is needed to support fibrosis. While “canonical pathways” represent common properties related to the SMAD3 pathway, noncanonical pathways, such as MAPKs, inclusive of MEK5/ERK5, are those that deviated from the canonical paradigm. In our model, for the association of phenotype and fibroblasts treated with hY3 macrophage supernatants, transforming growth factor (TGF)-β signaling by noncanonical is supportive. Specific inhibition of ERK5 with the small molecule BIX-02189 may forestall fibrosis.
GRANTS
This work was supported by National Institutes of Health (NIH) Grants R37-AR-042455, 3-R37-AR-042455-21S1, and 3-R37-AR-042455-21S2 (to J. Buyon); the Research Registry for Neonatal Lupus (NIH Grant N01-AR-4-2220, to J. Buyon); a Lupus Foundation of America Lifeline grant (to J. Buyon); and NIH Grants R03-HD-069986 and 1-R01-HD-079951-01A1 (to J. Buyon). For the genomics facility, the access of service of RNASeq relied on a shared resource, which is partially supported by a Cancer Center Support Grant (NIH Grant P30-CA-016087).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
R.M.C., A.J.M., and J.P.B. conceived and designed research; R.M.C., M.B., and J.P.B. analyzed data; R.M.C. and J.P.B. interpreted results of experiments; R.M.C. prepared figures; R.M.C. and J.P.B. drafted manuscript; R.M.C. and J.P.B. edited and revised manuscript; R.M.C. and J.P.B. approved final version of manuscript; A.J.M., T.J., and S.E.R. performed experiments.
ACKNOWLEDGMENTS
The authors are grateful to Ben Wainwright for excellent editorial assistance.
REFERENCES
- 1.Alric L, Duffaut M, Selves J, Sandre K, Mularczyck M, Izopet J, Desmorat H, Bureau C, Chaouche N, Dalbergue B, Vinel JP. Maintenance therapy with gradual reduction of the interferon dose over one year improves histological response in patients with chronic hepatitis C with biochemical response: results of a randomized trial. J Hepatol 35: 272–278, 2001. doi: 10.1016/S0168-8278(01)00110-6. [DOI] [PubMed] [Google Scholar]
- 2.Alvarez D, Briassouli P, Clancy RM, Zavadil J, Reed JH, Abellar RG, Halushka M, Fox-Talbot K, Barrat FJ, Buyon JP. A novel role of endothelin-1 in linking Toll-like receptor 7-mediated inflammation to fibrosis in congenital heart block. J Biol Chem 286: 30444–30454, 2011. doi: 10.1074/jbc.M111.263657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Azuma A, Li YJ, Abe S, Usuki J, Matsuda K, Henmi S, Miyauchi Y, Ueda K, Izawa A, Sone S, Hashimoto S, Kudoh S. Interferon-β inhibits bleomycin-induced lung fibrosis by decreasing transforming growth factor-β and thrombospondin. Am J Respir Cell Mol Biol 32: 93–98, 2005. doi: 10.1165/rcmb.2003-0374OC. [DOI] [PubMed] [Google Scholar]
- 4.Bhandare R, Damera G, Banerjee A, Flammer JR, Keslacy S, Rogatsky I, Panettieri RA, Amrani Y, Tliba O. Glucocorticoid receptor interacting protein-1 restores glucocorticoid responsiveness in steroid-resistant airway structural cells. Am J Respir Cell Mol Biol 42: 9–15, 2010. doi: 10.1165/rcmb.2009-0239RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Black BL, Olson EN. Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu Rev Cell Dev Biol 14: 167–196, 1998. doi: 10.1146/annurev.cellbio.14.1.167. [DOI] [PubMed] [Google Scholar]
- 6.Black CM, Silman AJ, Herrick AI, Denton CP, Wilson H, Newman J, Pompon L, Shi-Wen X. Interferon-α does not improve outcome at one year in patients with diffuse cutaneous scleroderma: results of a randomized, double-blind, placebo-controlled trial. Arthritis Rheum 42: 299–305, 1999. doi:. [DOI] [PubMed] [Google Scholar]
- 7.Boutjdir M, Chen L, Zhang ZH, Tseng CE, DiDonato F, Rashbaum W, Morris A, el-Sherif N, Buyon JP. Arrhythmogenicity of IgG and anti-52-kD SSA/Ro affinity-purified antibodies from mothers of children with congenital heart block. Circ Res 80: 354–362, 1997. doi: 10.1161/01.RES.80.3.354. [DOI] [PubMed] [Google Scholar]
- 8.Breur JM, Visser GH, Kruize AA, Stoutenbeek P, Meijboom EJ. Treatment of fetal heart block with maternal steroid therapy: case report and review of the literature. Ultrasound Obstet Gynecol 24: 467–472, 2004. doi: 10.1002/uog.1713. [DOI] [PubMed] [Google Scholar]
- 9.Brito-Zerón P, Izmirly PM, Ramos-Casals M, Buyon JP, Khamashta MA. The clinical spectrum of autoimmune congenital heart block. Nat Rev Rheumatol 11: 301–312, 2015. doi: 10.1038/nrrheum.2015.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buyon JP, Clancy RM, Friedman DM. Autoimmune associated congenital heart block: integration of clinical and research clues in the management of the maternal/foetal dyad at risk. J Intern Med 265: 653–662, 2009. doi: 10.1111/j.1365-2796.2009.02100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buyon JP, Hiebert R, Copel J, Craft J, Friedman D, Katholi M, Lee LA, Provost TT, Reichlin M, Rider L, Rupel A, Saleeb S, Weston WL, Skovron ML. Autoimmune-associated congenital heart block: demographics, mortality, morbidity and recurrence rates obtained from a national neonatal lupus registry. J Am Coll Cardiol 31: 1658–1666, 1998. doi: 10.1016/S0735-1097(98)00161-2. [DOI] [PubMed] [Google Scholar]
- 12.Clancy RM, Alvarez D, Komissarova E, Barrat FJ, Swartz J, Buyon JP. Ro60-associated single-stranded RNA links inflammation with fetal cardiac fibrosis via ligation of TLRs: a novel pathway to autoimmune-associated heart block. J Immunol 184: 2148–2155, 2010. doi: 10.4049/jimmunol.0902248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clancy RM, Askanase AD, Kapur RP, Chiopelas E, Azar N, Miranda-Carus ME, Buyon JP. Transdifferentiation of cardiac fibroblasts, a fetal factor in anti-SSA/Ro-SSB/La antibody-mediated congenital heart block. J Immunol 169: 2156–2163, 2002. doi: 10.4049/jimmunol.169.4.2156. [DOI] [PubMed] [Google Scholar]
- 14.Clancy RM, Backer CB, Yin X, Kapur RP, Molad Y, Buyon JP. Cytokine polymorphisms and histologic expression in autopsy studies: contribution of TNF-α and TGF-β 1 to the pathogenesis of autoimmune-associated congenital heart block. J Immunol 171: 3253–3261, 2003. doi: 10.4049/jimmunol.171.6.3253. [DOI] [PubMed] [Google Scholar]
- 15.Clancy RM, Kapur RP, Molad Y, Askanase AD, Buyon JP. Immunohistologic evidence supports apoptosis, IgG deposition, and novel macrophage/fibroblast crosstalk in the pathologic cascade leading to congenital heart block. Arthritis Rheum 50: 173–182, 2004. doi: 10.1002/art.11430. [DOI] [PubMed] [Google Scholar]
- 16.Clancy RM, Markham AJ, Reed JH, Blumenberg M, Halushka MK, Buyon JP. Targeting downstream transcription factors and epigenetic modifications following Toll-like receptor 7/8 ligation to forestall tissue injury in anti-Ro60 associated heart block. J Autoimmun 67: 36–45, 2016. doi: 10.1016/j.jaut.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cuneo BF, Ambrose SE, Tworetzky W. Detection and successful treatment of emergent anti-SSA mediated fetal atrioventricular block. Am J Obstet Gynecol 215: 527−528, 2016. doi: 10.1016/j.ajog.2016.07.002. [DOI] [PubMed] [Google Scholar]
- 18.Cuneo BF, Fruitman D, Benson DW, Ngan BY, Liske MR, Wahren-Herlineus M, Ho SY, Jaeggi E. Spontaneous rupture of atrioventricular valve tensor apparatus as late manifestation of anti-Ro/SSA antibody-mediated cardiac disease. Am J Cardiol 107: 761–766, 2011. doi: 10.1016/j.amjcard.2010.10.059. [DOI] [PubMed] [Google Scholar]
- 19.Dennis G Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 4: 3, 2003. doi: 10.1186/gb-2003-4-5-p3. [DOI] [PubMed] [Google Scholar]
- 20.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 425: 577–584, 2003. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 21.Dirkx E, da Costa Martins PA, De Windt LJ. Regulation of fetal gene expression in heart failure. Biochim Biophys Acta 1832: 2414–2424, 2013. doi: 10.1016/j.bbadis.2013.07.023. [DOI] [PubMed] [Google Scholar]
- 22.Duncan MR, Berman B. Gamma interferon is the lymphokine and beta interferon the monokine responsible for inhibition of fibroblast collagen production and late but not early fibroblast proliferation. J Exp Med 162: 516–527, 1985. doi: 10.1084/jem.162.2.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eliasson H, Sonesson SE, Sharland G, Granath F, Simpson JM, Carvalho JS, Jicinska H, Tomek V, Dangel J, Zielinsky P, Respondek-Liberska M, Freund MW, Mellander M, Bartrons J, Gardiner HM; Fetal Working Group of the European Association of Pediatric Cardiology . Isolated atrioventricular block in the fetus: a retrospective, multinational, multicenter study of 175 patients. Circulation 124: 1919–1926, 2011. doi: 10.1161/CIRCULATIONAHA.111.041970. [DOI] [PubMed] [Google Scholar]
- 24.Eloranta ML, Franck-Larsson K, Lövgren T, Kalamajski S, Rönnblom A, Rubin K, Alm GV, Rönnblom L. Type I interferon system activation and association with disease manifestations in systemic sclerosis. Ann Rheum Dis 69: 1396–1402, 2010. doi: 10.1136/ard.2009.121400. [DOI] [PubMed] [Google Scholar]
- 25.Friedman DM, Kim MY, Copel JA, Llanos C, Davis C, Buyon JP. Prospective evaluation of fetuses with autoimmune-associated congenital heart block followed in the PR Interval and Dexamethasone Evaluation (PRIDE) Study. Am J Cardiol 103: 1102–1106, 2009. doi: 10.1016/j.amjcard.2008.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Friedman DM, Llanos C, Izmirly PM, Brock B, Byron J, Copel J, Cummiskey K, Dooley MA, Foley J, Graves C, Hendershott C, Kates R, Komissarova EV, Miller M, Paré E, Phoon CK, Prosen T, Reisner D, Ruderman E, Samuels P, Yu JK, Kim MY, Buyon JP. Evaluation of fetuses in a study of intravenous immunoglobulin as preventive therapy for congenital heart block: results of a multicenter, prospective, open-label clinical trial. Arthritis Rheum 62: 1138–1146, 2010. doi: 10.1002/art.27308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garmendía G, Miranda N, Borroso S, Longchong M, Martínez E, Ferrero J, Porrero P, López-Saura P. Regression of infancy hemangiomas with recombinant IFN-α2b. J Interferon Cytokine Res 21: 31–38, 2001. doi: 10.1089/107999001459132. [DOI] [PubMed] [Google Scholar]
- 28.Hiramatsu N, Hayashi N, Kasahara A, Hagiwara H, Takehara T, Haruna Y, Naito M, Fusamoto H, Kamada T. Improvement of liver fibrosis in chronic hepatitis C patients treated with natural interferon alpha. J Hepatol 22: 135–142, 1995. doi: 10.1016/0168-8278(95)80420-X. [DOI] [PubMed] [Google Scholar]
- 29.Izmirly PM, Saxena A, Kim MY, Wang D, Sahl SK, Llanos C, Friedman D, Buyon JP. Maternal and fetal factors associated with mortality and morbidity in a multi-racial/ethnic registry of anti-SSA/Ro-associated cardiac neonatal lupus. Circulation 124: 1927–1935, 2011. doi: 10.1161/CIRCULATIONAHA.111.033894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Izmirly PM, Saxena A, Sahl SK, Shah U, Friedman DM, Kim MY, Buyon JP. Assessment of fluorinated steroids to avert progression and mortality in anti-SSA/Ro-associated cardiac injury limited to the fetal conduction system. Ann Rheum Dis 75: 1161–1165, 2016. doi: 10.1136/annrheumdis-2015-208311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kato Y, Zhao M, Morikawa A, Sugiyama T, Chakravortty D, Koide N, Yoshida T, Tapping RI, Yang Y, Yokochi T, Lee JD. Big mitogen-activated kinase regulates multiple members of the MEF2 protein family. J Biol Chem 275: 18534–18540, 2000. doi: 10.1074/jbc.M001573200. [DOI] [PubMed] [Google Scholar]
- 32.Kim D, Peck A, Santer D, Patole P, Schwartz SM, Molitor JA, Arnett FC, Elkon KB. Induction of interferon-α by scleroderma sera containing autoantibodies to topoisomerase I: Association of higher interferon-α activity with lung fibrosis. Arthritis Rheum 58: 2163–2173, 2008. doi: 10.1002/art.23486. [DOI] [PubMed] [Google Scholar]
- 33.Kim S, Lim JH, Woo CH. ERK5 inhibition ameliorates pulmonary fibrosis via regulating Smad3 acetylation. Am J Pathol 183: 1758–1768, 2013. doi: 10.1016/j.ajpath.2013.08.014. [DOI] [PubMed] [Google Scholar]
- 34.Lehman JJ, Kelly DP. Transcriptional activation of energy metabolic switches in the developing and hypertrophied heart. Clin Exp Pharmacol Physiol 29: 339–345, 2002. doi: 10.1046/j.1440-1681.2002.03655.x. [DOI] [PubMed] [Google Scholar]
- 35.Lin Q, Schwarz J, Bucana C, Olson EN. Control of mouse cardiac morphogenesis and myogenesis by transcription factor MEF2C. Science 276: 1404–1407, 1997. doi: 10.1126/science.276.5317.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lisney AR, Szelinski F, Reiter K, Burmester GR, Rose T, Dörner T. High maternal expression of SIGLEC1 on monocytes as a surrogate marker of a type I interferon signature is a risk factor for the development of autoimmune congenital heart block. Ann Rheum Dis 76: 1476–1480, 2017. doi: 10.1136/annrheumdis-2016-210927. [DOI] [PubMed] [Google Scholar]
- 37.Llanos C, Friedman DM, Saxena A, Izmirly PM, Tseng CE, Dische R, Abellar RG, Halushka M, Clancy RM, Buyon JP. Anatomical and pathological findings in hearts from fetuses and infants with cardiac manifestations of neonatal lupus. Rheumatology (Oxford) 51: 1086–1092, 2012. doi: 10.1093/rheumatology/ker515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mavragani CP, Niewold TB, Moutsopoulos NM, Pillemer SR, Wahl SM, Crow MK. Augmented interferon-α pathway activation in patients with Sjögren’s syndrome treated with etanercept. Arthritis Rheum 56: 3995–4004, 2007. doi: 10.1002/art.23062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Minsky N, Roeder RG. Control of secreted protein gene expression and the mammalian secretome by the metabolic regulator PGC-1α. J Biol Chem 292: 43−50, 2017. doi: 10.1074/jbc.C116.761049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Naya FJ, Black BL, Wu H, Bassel-Duby R, Richardson JA, Hill JA, Olson EN. Mitochondrial deficiency and cardiac sudden death in mice lacking the MEF2A transcription factor. Nat Med 8: 1303–1309, 2002. doi: 10.1038/nm789. [DOI] [PubMed] [Google Scholar]
- 41.Niewold TB, Rivera TL, Buyon JP, Crow MK. Serum type I interferon activity is dependent on maternal diagnosis in anti-SSA/Ro-positive mothers of children with neonatal lupus. Arthritis Rheum 58: 541–546, 2008. doi: 10.1002/art.23191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nimmerjahn F, Ravetch JV. The antiinflammatory activity of IgG: the intravenous IgG paradox. J Exp Med 204: 11–15, 2007. doi: 10.1084/jem.20061788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oh SP, Seki T, Goss KA, Imamura T, Yi Y, Donahoe PK, Li L, Miyazono K, ten Dijke P, Kim S, Li E. Activin receptor-like kinase 1 modulates transforming growth factor-β 1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci USA 97: 2626–2631, 2000. doi: 10.1073/pnas.97.6.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pammer J, Reinisch C, Birner P, Pogoda K, Sturzl M, Tschachler E. Interferon-α prevents apoptosis of endothelial cells after short-term exposure but induces replicative senescence after continuous stimulation. Lab Invest 86: 997–1007, 2006. doi: 10.1038/labinvest.3700461. [DOI] [PubMed] [Google Scholar]
- 45.Pereira AH, Clemente CF, Cardoso AC, Theizen TH, Rocco SA, Judice CC, Guido MC, Pascoal VD, Lopes-Cendes I, Souza JR, Franchini KG. MEF2C silencing attenuates load-induced left ventricular hypertrophy by modulating mTOR/S6K pathway in mice. PLoS One 4: e8472, 2009. doi: 10.1371/journal.pone.0008472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pisoni CN, Brucato A, Ruffatti A, Espinosa G, Cervera R, Belmonte-Serrano M, Sánchez-Román J, García-Hernández FG, Tincani A, Bertero MT, Doria A, Hughes GR, Khamashta MA. Failure of intravenous immunoglobulin to prevent congenital heart block: findings of a multicenter, prospective, observational study. Arthritis Rheum 62: 1147–1152, 2010. doi: 10.1002/art.27350. [DOI] [PubMed] [Google Scholar]
- 47.Rogatsky I, Luecke HF, Leitman DC, Yamamoto KR. Alternate surfaces of transcriptional coregulator GRIP1 function in different glucocorticoid receptor activation and repression contexts. Proc Natl Acad Sci USA 99: 16701–16706, 2002. doi: 10.1073/pnas.262671599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nat Rev Immunol 8: 559–568, 2008. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salomonsson S, Sonesson SE, Ottosson L, Muhallab S, Olsson T, Sunnerhagen M, Kuchroo VK, Thorén P, Herlenius E, Wahren-Herlenius M. Ro/SSA autoantibodies directly bind cardiomyocytes, disturb calcium homeostasis, and mediate congenital heart block. J Exp Med 201: 11–17, 2005. doi: 10.1084/jem.20041859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Samuelsson A, Towers TL, Ravetch JV. Anti-inflammatory activity of IVIG mediated through the inhibitory Fc receptor. Science 291: 484–486, 2001. doi: 10.1126/science.291.5503.484. [DOI] [PubMed] [Google Scholar]
- 51.Saxena A, Izmirly PM, Han SW, Briassouli P, Rivera TL, Zhong H, Friedman DM, Clancy RM, Buyon JP. Serum biomarkers of inflammation, fibrosis, and cardiac function in facilitating diagnosis, prognosis, and treatment of anti-SSA/Ro-associated cardiac neonatal lupus. J Am Coll Cardiol 66: 930–939, 2015. doi: 10.1016/j.jacc.2015.06.1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stout AJ, Gresser I, Thompson WD. Inhibition of wound healing in mice by local interferon alpha/beta injection. Int J Exp Pathol 74: 79–85, 1993. [PMC free article] [PubMed] [Google Scholar]
- 53.Sweeney TE, Suliman HB, Hollingsworth JW, Welty-Wolf KE, Piantadosi CA. A toll-like receptor 2 pathway regulates the Ppargc1a/b metabolic co-activators in mice with Staphylococcal aureus sepsis. PLoS One 6: e25249, 2011. doi: 10.1371/journal.pone.0025249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tamm I, Kikuchi T, Wang E, Pfeffer LM. Growth rate of control and beta-interferon-treated human fibroblast populations over the course of their in vitro life span. Cancer Res 44: 2291–2296, 1984. [PubMed] [Google Scholar]
- 55.Tanabe J, Izawa A, Takemi N, Miyauchi Y, Torii Y, Tsuchiyama H, Suzuki T, Sone S, Ando K. Interferon-β reduces the mouse liver fibrosis induced by repeated administration of concanavalin A via the direct and indirect effects. Immunology 122: 562–570, 2007. doi: 10.1111/j.1365-2567.2007.02672.x. [DOI] [PMC free article] [PubMed] [Google Scholar]