

Ventricular myocytes isolated from the myosin-binding protein C knockout hypertrophic cardiomyopathy mouse model demonstrate decreased repolarizing K+ currents and action potential and QT interval prolongation, linking cellular repolarization abnormalities with arrhythmia susceptibility and the risk for sudden cardiac death in hypertrophic cardiomyopathy.
Keywords: myosin-binding protein C, cardiomyopathy, arrhythmia, repolarization, K+ channels
Abstract
Hypertrophic cardiomyopathy (HCM) is one of the most common genetic cardiac diseases and among the leading causes of sudden cardiac death (SCD) in the young. The cellular mechanisms leading to SCD in HCM are not well known. Prolongation of the action potential (AP) duration (APD) is a common feature predisposing hypertrophied hearts to SCD. Previous studies have explored the roles of inward Na+ and Ca2+ in the development of HCM, but the role of repolarizing K+ currents has not been defined. The objective of this study was to characterize the arrhythmogenic phenotype and cellular electrophysiological properties of mice with HCM, induced by myosin-binding protein C (MyBPC) knockout (KO), and to test the hypothesis that remodeling of repolarizing K+ currents causes APD prolongation in MyBPC KO myocytes. We demonstrated that MyBPC KO mice developed severe hypertrophy and cardiac dysfunction compared with wild-type (WT) control mice. Telemetric electrocardiographic recordings of awake mice revealed prolongation of the corrected QT interval in the KO compared with WT control mice, with overt ventricular arrhythmias. Whole cell current- and voltage-clamp experiments comparing KO with WT mice demonstrated ventricular myocyte hypertrophy, AP prolongation, and decreased repolarizing K+ currents. Quantitative RT-PCR analysis revealed decreased mRNA levels of several key K+ channel subunits. In conclusion, decrease in repolarizing K+ currents in MyBPC KO ventricular myocytes contributes to AP and corrected QT interval prolongation and could account for the arrhythmia susceptibility.
NEW & NOTEWORTHY Ventricular myocytes isolated from the myosin-binding protein C knockout hypertrophic cardiomyopathy mouse model demonstrate decreased repolarizing K+ currents and action potential and QT interval prolongation, linking cellular repolarization abnormalities with arrhythmia susceptibility and the risk for sudden cardiac death in hypertrophic cardiomyopathy.
hypertrophic cardiomyopathy (HCM) is one of the most common genetic cardiac diseases (1:500 in the general population) (28, 42) and among the leading causes of sudden cardiac death (SCD) in the young (1, 29). HCM is caused by mutations in sarcomere proteins. The altered sarcomere structure and/or function leads to pathological hypertrophy, abnormal heart function, and, importantly, arrhythmias and sudden death (14).
The pathophysiological link between abnormal sarcomere structure and arrhythmia is not clearly defined, likely varies with mutation type, and has been the subject of extensive research (11, 12). As in any other cardiac pathology leading to arrhythmia, two major components exist: initiation (triggering) of the abnormal action potential (AP) and propagation of the AP throughout the myocardium. Arrhythmias can result from a variety of mechanisms including reentry, triggered activity, and abnormal automaticity. Propagation of the arrhythmia is dependent on tissue and structural abnormalities leading to reentry circuits (39). HCM is usually characterized by cardiac fibrosis and cellular disarray, which have been shown to be important factors in reentry circuit formation and arrhythmia propagation (12) as well as a significant clinical risk factor for SCD (38).
There are also findings that support a role for abnormal cellular electrophysiological alterations in arrhythmia initiation in HCM; however, myocyte electrophysiological remodeling has not been clearly defined (11). A major electrocardiographic finding in HCM is prolongation of the QT interval, resulting from ventricular myocyte AP prolongation, in humans and animal models (8, 15, 20, 46). QT prolongation in HCM emphasizes the important arrhythmogenic role of abnormal cellular excitability in HCM.
Abnormal intracellular Ca2+ handling has been shown to play a key role in the pathophysiology of HCM (4, 12). Alterations in ion channel expression and function have also been established. Most previous studies concentrated on the arrhythmogenic role of Ca2+ and Na+ currents in HCM (9, 41, 44). Repolarizing K+ currents have not been investigated in detail in HCM and are the focus of the present work.
Myosin-binding protein C (MyBPC) mutations are commonly involved in HCM. It is estimated that ~40% of identified mutations in individuals with HCM occur in MyBPC (17). MyBPC knockout (KO) mice have been developed to study the bases of MyBPC mutation-induced HCM and functional abnormalities (16), with the majority of the work focused on structural and mechanical dysfunction (22, 26, 43).
The aim of the present study was to explore and define the cellular substrates for arrhythmias in MyBPC KO mice, focusing specifically on repolarizing K+ channels. We tested the hypothesis that K+ currents are decreased in MyBPC KO mice and that these changes contribute to AP prolongation.
The experiments performed establish that MyBPC KO mice demonstrate diverse ventricular arrhythmias, QT and AP prolongation, and decreased repolarizing K+ currents and K+ channel subunit mRNA transcript levels. Our results suggest that remodeling of repolarizing K+ currents contributes to abnormal cellular excitability and arrhythmia susceptibility in HCM.
METHODS
Experimental animals.
Homozygous cardiac MyBPC KO mice were kindly provided by the laboratory of Dr. Richard Moss (University of Wisconsin). These mice were generated as previously described and were maintained on an SV/129 background (16). Control wild-type (WT) SV/129 mice were obtained from Taconic Farms (Germantown, NY). Six- to nine-month-old, age-matched, male mice were used according to protocols approved by the Institutional Animal Care and Use Committee of the Lewis Katz School of Medicine at Temple University and the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health.
Telemetry and electrocardiography.
Implanted ETA-F20 telemetric transmitters (DSI, St. Paul, MN) were used to obtain continuous lead II electrocardiography (ECG) recordings with an AD Instruments PowerLab (Colorado Springs, CO) system on awake mice. Animals were housed in a room with 12:12-h light-dark cycle for 7 days to allow recovery from the surgery. Telemetry recordings were performed over a total of 24 h (equivalent to standard recording time of a Holter monitor in humans) net recording time (30 min of recording per each hour, over 48 h spanning over 2 consecutive days). The tracings were analyzed, looking for ventricular arrhythmia, by scrolling manually throughout the recordings. The data were stored and subsequently analyzed with the ECG module in the LabChart 8 software by two separate examiners who were blinded to the genetic information of the animals. To obtain ECG intervals, 2-min samples [as previously described (48)] from the telemetry tracings were analyzed for each mouse at noontime. Identical analyses were performed for each mouse at midnight to assess for daytime and nighttime variability. In each recording, the QT interval was determined as the time interval between the initiation of the QRS complex and the end of the T wave, defined as the time the T wave returned to the baseline. The measurement is shown in Fig. 3A. QT intervals were corrected for heart rate using the following formula: corrected QT (QTc) = QT/(√RR/100) (30).
Fig. 3.

Telemetric electrocardiographic recordings. A and B: representative ECG waveforms from awake WT (A) and KO (B) animals marked with the different intervals that were measured. C: corrected QT (QTc) interval analyzed from telemetric recordings of WT (n = 4) and KO (n = 4) animals at noontime and midnight. Mean ± SE values are plotted. **P < 0.01.
Echocardiography.
Anesthetized (0.5–1.5% isoflurane) mice underwent transthoracic echocardiography using a Vevo2100 ultrasound system (VisualSonics, Toronto, ON, Canada) with a 30-MHz MS-400 transducer.
Body temperature was maintained using a heated platform. Respiratory rates and ECGs were monitored throughout the study. For consistency, heart rate was maintained between 400 and 450 beats/min. Images were acquired in the short-axis B-mode and M-mode at the level of the left ventricular (LV) midpapillary muscles as well as the parasternal long axis for analysis of cardiac function and dimensions.
Tissue processing and histology.
Hearts were rinsed with PBS, weighed, and then perfusion fixed with 10% formalin and paraffin embedded for histology following a previously published protocol (10). Tissue blocks were sent to AML Laboratories (St. Augustine, FL) for sectioning and staining for Masson’s trichrome (Sigma-Aldrich, St. Louis, MO) to assess for tissue histology and collagen deposition. Myocyte cross-sectional area was measured in tissue sections stained with wheat germ agglutinin (ThermoFisher Scientific) and quantified with National Institutes of Health ImageJ software (http://rsbweb.nih.gov/ij/). At least 100 myocytes from 3 sections of the heart were analyzed per animal to assess myocyte cross-sectional area.
Myocyte isolation and cellular electrophysiology.
Adult mouse LV cardiomyocytes were isolated as previously described (27, 50), maintained at room temperature, and used within 12 h of isolation. For cellular electrophysiology experiments, myocytes were placed in a chamber mounted on an inverted Nikon microscope and perfused with normal physiological solution (Tyrode) containing 1 mM Ca2+. All experiments were done in a heated chamber at a temperature of 31°C as previously described (50). To measure Ca2+-insensitive K+ currents, myocytes were bathed in normal Tyrode solution containing 0.2 mM CdCl2 to block Ca2+ currents. Cells were dialyzed for 10 min after patch rupture with pipette solution containing (in mM) 120 K aspartate, 20 KCl, 5 Na2ATP, 1 MgCl2, 10 HEPES, 1 CaCl2, and 10 EGTA (pH with KOH to 7.2). EGTA was included in the pipette solution to minimize Ca2+-sensitive K+ currents and Na+/Ca2+ exchange current. The myocyte was ramped to −50 mV from a holding potential (Vhold) of −80 mV to inactivate Na+ channels and depolarized to different test potentials (−120 to 70 mV) in a 10-mV increment for 4,000 ms to record K+ currents. The bath solution was then switched to normal Tyrode solution containing 0.2 mM CdCl2 and 2 mM 4-aminopyridine (4-AP). Once a stable effect of 4-AP was observed, the depolarization steps were applied again. The end of this 4-AP-insensitive K+ current was considered as the sustained K+ current (Isus) and inwardly rectifying current (IK1). The current sensitive to 4-AP contained mainly the transient outward K+ current (Ito) and slowly decaying K+ current (IK,slow).
APs were measured with ruptured whole cell configuration with an Axoclamp 2B amplifier in the Bridge mode. The cell was dialyzed with the following pipette solution (in mM) for 5 min: 120 K aspartate, 20 KCl, 5 Na2ATP, 1 MgCl2, and 10 HEPES (pH with KOH to 7.2). The bath solution was normal Tyrode solution. APs were elicited by 2-ms depolarizations (5–15 mV) at 0.5 Hz for 5 min.
Analysis was performed using Clampfit 10.5 software offline.
RNA isolation and quantitative RT-PCR.
Total RNA was isolated from WT and KO mouse hearts using the mirVana Isolation Kit (Ambion) followed by DNase treatment to eliminate contaminating genomic DNA (Qiagen). Reverse transcription reaction was performed using the RT2 First Strand Kit (Qiagen) according to the manufacturer’s instructions. Real-time quantitative PCR was performed using RT2 SYBR Green ROX qPCR Mastermix (Qiagen). Data generated from the mouse heart samples were normalized to hypoxanthine guanine phosphoribosyl transferase (HPRT) expression. Fold changes of the transcript level in KO mice compared with WT mice were calculated with the method, where Ct is cycle threshold. The primer sets that were used in the experiments are shown in Table 1.
Table 1.
Sequence-specific primers used in SYBR green RT-PCR
| Subunit | Gene | Forward Primer | Reverse |
|---|---|---|---|
| TASK1 | KCNK3 | 5′-CCTTCTACTTCGCCATCACC-3′ | 5′-AACATGCAGAACACCTTGC-3′ |
| Kvβ1 | KCNAB1 | 5′-GACCTCTCTCCAATCGCTGA-3′ | 5′-CTGAATGGCACCAAGGTTTT-3′ |
| HPRT | 5′-AGCCCCAAAATGGTTAAGGT-3′ | 5′-CAAGGGCATATCCAACAACA-3′ |
Commercial primers (Qiagen) were also used for the following targets: KCND2, KCND3, KCNIP2, KCNA4, KCNJ2, KCNJ12, KCNH2, KCNQ1, KCNE1, KCNA5, KCNB1, KCNK2, and KCNK5. Their sequences are proprietary protected. HPRT, hypoxanthine guanine phosphoribosyl transferase.
Data analysis.
Data are presented as means ± SE. Unpaired t-tests were performed to detect significance using Excel and GraphPad Prism 6 software. P < 0.05 was considered significant (*P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 in the figures).
RESULTS
MyBPC KO mice demonstrate severe hypertrophy, tissue fibrosis, and contractile dysfunction.
MyBPC KO mice developed severe cardiac hypertrophy (Fig. 1A) with an elevated heart weight-to-body weight ratio (WT: 5.2 ± 0.3 mg/g, n = 10; KO: 7.5 ± 0.4 mg/g, n = 9; P < 0.001; Fig. 1B). Masson’s trichrome-stained tissue sections at the mid-LV level showed gross chamber remodeling and overt interstitial fibrosis in KO mice, as evidenced by patchy blue staining (Fig. 1, A and C). Higher magnification revealed significant differences in cardiomyocyte morphology, with cellular hypertrophy and myofibrillar disarray in KO mice (Fig. 1C). Wheat germ agglutinin staining also revealed increased extracellular matrix consistent with tissue fibrosis and significantly increased myocyte cross-sectional area (WT: 233 ± 8.1 μm2, N = 3 animals, n > 100 myocytes/animal; KO: 537.8 ± 46.6 μm2, N = 4 animals, n > 100 myocytes/animal; P < 0.01; Fig. 1, D and E).
Fig. 1.

Heart size, morphology, histology, and myocyte cross-sectional area. A: representative cross sections of wild-type (WT) and knockout (KO) hearts at the mid left ventricular (LV) level stained with Masson’s trichrome. The viable myocardium is pink, and the fibrotic area is blue. B: heart weight-to-body weight (HW/BW) ratio in WT (n = 10) and KO (n = 10) animals. Mean ± SE values are plotted. ***P < 0.001. C: representative magnified cross sections at the mid-LV level of WT and KO hearts stained with Masson’s trichrome. Cardiomyocytes are pink, and their nuclei are dark blue. The fibrotic area is light blue. D: representative magnified cross sections at the mid-LV level of WT and KO hearts stained with wheat germ agglutinin (WGA) for the extracellular matrix and tissue fibrosis. E: myocyte cross-sectional area in WT (N = 3 animals, n > 100 myocytes/animal) and KO (N = 4 animals, n > 100 myocytes/animal) animals as analyzed from WGA-stained sections. Mean ± SE values are plotted. **P < 0.01.
Consistent with a previous report (16), echocardiography demonstrated severe hypertrophy (Fig. 2, A and B) with increased anterior wall diameter (WT: 1.06 ± 0.07 mm, n = 5; KO: 1.55 ± 0.16 mm, n = 5; P < 0.05; Fig. 2C) and posterior wall diameter (WT: 0.73 ± 0.03 mm, n = 5; KO: 1.26 ± 0.12 mm, n = 5; P < 0.01; Fig. 2D). Ejection fraction (WT: 64.4 ± 4.9%, n = 5; KO: 39.5 ± 2.8%, n = 5; P < 0.01; Fig. 2E) and fractional shortening (WT: 35.3 ± 3.6%, n = 5; KO: 19.2 ± 1.5%, n = 5; P < 0.01; Fig. 2F) were significantly reduced in KO animals. Chamber dilation was also noted with increased LV diastolic diameter (WT: 4.06 ± 0.16 mm, n = 5; KO: 4.77 ± 0.28 mm, n = 5; P = 0.06; Fig. 2G) and systolic diameter (WT: 2.65 ± 0.24 mm, n = 5; KO: 3.86 ± 0.28 mm, n = 5; P < 0.05; Fig. 2H) in KO mice.
Fig. 2.

Cardiac structure and function measured by echocardiography. A and B: representative parasternal short-axis M-mode (A) and parasternal long-axis images (B) demonstrating severe hypertrophy with increased wall thickness in KO hearts. C–H: LV anterior wall diastolic diameter (LVAWDd; C), posterior wall diastolic diameter (LVPWDd; D), ejection fraction (EF; E), fractional shortening (FS; F), LV diastolic diameter (LVDd; G), and LV systolic diameter (LVSd; H) in WT (n = 5) and KO (n = 5) animals. Mean ± SE values are plotted. *P < 0.05; **P < 0.01.
KO mice exhibit prolonged QTc intervals and overt ventricular arrhythmias.
Telemetric electrocardiographic (lead II ECG) recordings were performed on awake mice, as detailed in methods. Representative ECG waveforms are shown in Fig. 3, A and B. KO mice demonstrated significant prolongation of the QTc interval at noontime (WT: 36.7 ± 3.3 ms, n = 4; KO: 60.9 ± 3, n = 4; P < 0.01) and midnight (WT: 37.4 ± 2.7 ms, n = 4; KO: 60.8 ± 4.1, n = 4; P < 0.01) without significant daytime variability (Fig. 3C). Averaging noon and midnight values for all ECG intervals revealed that there were no significant differences in the durations of the RR, PR, and QRS intervals in KO mice compared with WT mice (Table 2).
Table 2.
ECG parameters observed in WT and KO mice
| WT | KO | P Value | |
|---|---|---|---|
| RR interval, ms | 103.2 ± 5.7 | 106.9 ± 2.5 | 0.6 |
| PR interval, ms | 33.6 ± 1.2 | 30.8 ± 2.3 | 0.37 |
| QRS duration, ms | 14.3 ± 1 | 18.3 ± 1.3 | 0.63 |
| QT interval, ms | 37.5 ± 2 | 62.9 ± 2.5 | 0.002* |
| Corrected QT interval, ms | 37 ± 2 | 60.8 ± 2.3 | 0.001* |
Values are means ± SE. P values of RR interval, PR interval, QRS duraction, QT interval, and corrected QT interval in awake wild-type (WT; n = 4) and knockout (KO; n = 4) animals, as analyzed from telemetric electrocardiographic recordings, are shown.
P < 0.01.
Telemetry recordings revealed that three of four WT animals demonstrated premature ventricular contractions (PVCs), with a total of eight for all WT animals, over the total net time of 24 h (Fig. 4, A and C, and representative trace in Fig. 4D). None of the four WT animals demonstrated high-grade ventricular ectopy. All 4 KO animals demonstrated ventricular ectopy with a total of 61 events over total net time of 24 h (Fig. 4A). PVCs were noted in all KO animals. Comparison of the average ventricular ectopy events between WT and KO animals did not reach significance (WT: 2 ± 0.8, n = 4; KO: 15 ± 12, n = 4; P = 0.32; Fig. 4B), since it was prominent in only one of the KO animals. This KO animal demonstrated diverse ventricular ectopy including PVCs, couplets, and nonsustained ventricular tachycardia (NSVT; 3–5 beats in a row; Fig. 4C). Representative tracings are shown in Fig. 4, E–I.
Fig. 4.

Arrhythmias in WT and KO animals. A: total ventricular ectopy events over the total net time of 24 h in WT (n = 4) and KO (n = 4) animals. B: average ventricular ectopy events over the total net time of 24 h in WT (n = 4) and KO (n = 4) animals. Mean ± SE values are plotted. NS, not significant. C: ventricular ectopy categories noted over the total net time of 24 h in WT (n = 4) and KO (n = 4) animals. D–I: representative tracings of ventricular arrhythmias noted in WT and KO animals, including premature ventricular contractions [PVCs; WT (D) and KO (E)], couplets (KO; F), three-beat nonsustained ventricular tachycardia (NSVT; KO; G), four-beat NSVT (KO; H), and five-beat NSVT (KO; I).
APs are prolonged and K+ currents are decreased in KO cardiomyocytes.
Whole cell current- and voltage-clamp experiments demonstrated that cardiomyocyte membrane capacitance was significantly increased, representing myocyte hypertrophy (WT: 234.4 ± 15.6 pF, N = 3 animals, n = 15 cells; KO: 469 ± 44.2 pF, N = 3 animals, n = 18 cells; P < 0.0001; Fig. 5A).
Fig. 5.
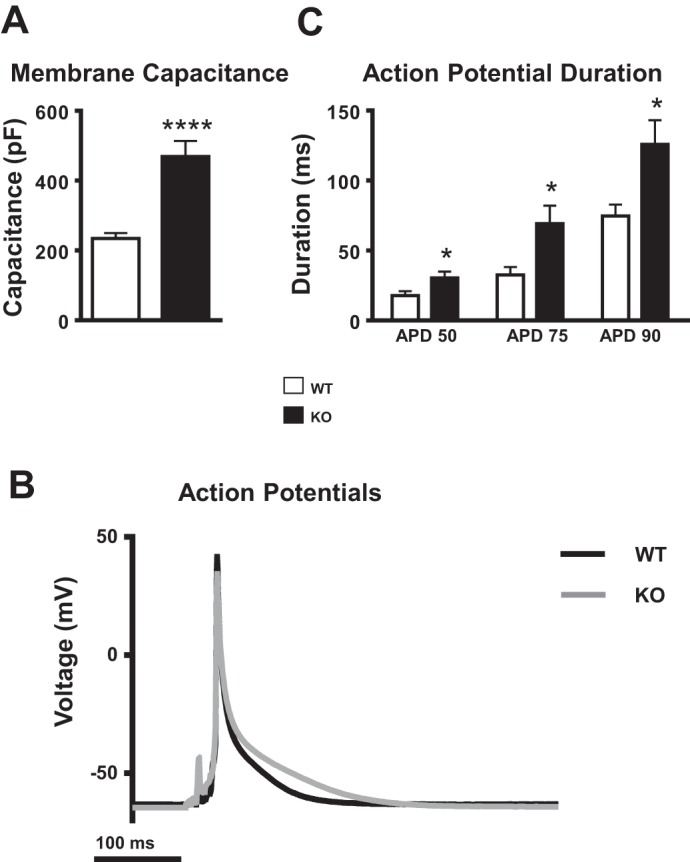
Myocyte membrane capacitance and current-clamp recordings of action potentials (APs). A: membrane capacitance in WT (N = 3 animals, n = 15 cells) and KO (N = 3 animals, n = 18 cells) myocytes. Mean ± SE values are plotted. ****P < 0.0001. B: representative AP waveforms in WT and KO myocytes. C: AP duration (APD) at 50% repolarization (APD50), 75% repolarization (APD75), and 90% repolarization (APD90) in WT (n = 12) and KO (n = 11) myocytes. Mean ± SE values are plotted. *P < 0.05.
APs induced at 0.5 Hz were significantly prolonged in KO myocytes at all durations examined: AP duration (APD) at 50% repolarization (APD50; WT: 17.7 ± 3.2 ms, n = 12; KO: 30.4 ± 4.5 ms, n = 11; P < 0.05), 75% repolarization (APD75; WT: 32.6 ± 5.6 ms; KO: 69.2 ± 12.8 ms, n = 11; P < 0.05), and 90% repolarization (APD90; WT: 74.7 ± 8.1 ms; KO: 125.7 ± 17.3 ms; P < 0.05; Fig. 5, B and C).
Repolarizing K+ current densities (normalized to cell capacitance) were decreased in KO myocytes. Representative current tracings are shown in Fig. 6, A and B. Currents were evoked in response to 4-s voltage steps to test potentials between −120 and 100 mV from a Vhold of −70 mV. Peak outward K+ currents (IK peak) measured in WT (n = 15) and KO (n = 14) myocytes demonstrated a 33–39% decrease in KO myocytes (Fig. 6C).
Fig. 6.

Voltage-clamp recordings of repolarizing K+ currents. A and B: representative whole cell K+ currents (normalized to membrane capacitance) recorded from WT (A) and KO (B) animals. Currents were evoked in response to (4-s) voltage steps to test potentials between −100 and 60 mV from a holding potential (Vhold) of −70 mV. C: peak outward K+ currents (normalized to membrane capacitance) recorded from WT (n = 15) and KO (n = 14) myocytes. Mean ± SE values are plotted. *P < 0.05; **P < 0.01; ***P < 0.001. D and E: representative 4-aminopyridine (4-AP)-insensitive K+ currents (normalized to membrane capacitance) recorded from WT (D) and KO (E) animals. Currents were evoked in response to (4-s) voltage steps to test potentials between −100 and 60 mV from a Vhold of −70 mV. F: peak 4-AP-insensitive K+ currents (normalized to membrane capacitance) recorded from WT (n = 13) and KO (n = 13) myocytes. Mean ± SE values are plotted. *P < 0.05; **P < 0.01. G and H: representative 4-AP-sensitive K+ currents (normalized to membrane capacitance) recorded from of WT (G) and KO (H) animals. Currents were evoked in response to (4-s) voltage steps to test potentials between −20 and 60 mV from a Vhold of −70 mV. I: peak 4-AP-sensitive K+ currents (normalized to membrane capacitance) recorded from WT (n = 9) and KO (n = 10) myocytes. Mean ± SE values are plotted. *P < 0.05; **P < 0.01.
Myocytes were then exposed to 4-AP. There was a significant reduction of the 4-AP-insensitive sustained outward plateau current in KO myocytes. Representative current tracings are shown in Fig. 6, D and E. These currents, evoked in response to voltage steps to test potentials between 20 and 100 mV from a Vhold of −70 mV, in WT (n = 13) and KO (n = 13) myocytes, demonstrated a 21–23% reduction (Fig. 6F). 4-AP-insensitive IK1 was also decreased by 16–30%, (voltage steps to test potentials between −120 and 100 mV; Fig. 6F).
The most significant differences in KO myocytes were in the 4-AP-sensitive currents, which include Ito and IK,slow1. Representative current tracings are shown in Fig. 6, G and H. These currents, measured in response to voltage steps to test potentials between 0 and 60 mV from a Vhold of −70 mV in WT (n = 9) and KO (n = 10) myocytes, were significantly reduced (46–61%) in KO myocytes (Fig. 6I).
K+ channel mRNA levels are decreased in KO hearts.
Quantitative RT-PCR analysis revealed decreased mRNA levels of several K+ channel transcripts in KO mice compared with WT mice. Transcripts of Ito, the fast and slow components of Ito (Ito,fast and Ito,slow, respectively), demonstrated the most significant reduction (Fig. 7A): KCND2, coding for the Kv4.2 subunit, was down by 3.1-fold to a relative level of 0.32 ± 0.05 (P < 0.001); KCND3, coding for the Kv4.3 subunit, was down by 1.7-fold to a relative level to 0.58 ± 0.07 (P < 0.01); KCNIP2, coding for the regulatory subunit KChIP2, was down by 1.6-fold to a relative level of 0.62 ± 0.14 (P < 0.05); and KCNA4, coding for Kv1.4 subunit, was down by 2.4-fold to a relative level of 0.41 ± 0.14 (P < 0.01). The IK1 transcript KCNJ2, coding for Kir2.1, was down by 2.4-fold to a relative level of 0.41 ± 0.05 (P < 0.05; Fig. 7B). Slow delayed rectifier K+ current (IKs) transcript KCNE1, coding for the minK regulatory subunit, was also significantly reduced by 2.3-fold to a relative level of to 0.43 ± 0.12 (P < 0.05; Fig. 7C). Other transcripts coding for additional subunits of Ito (Fig. 7A), inward rectifier (Fig. 7B), delayed rectifier (Fig. 7C), and two-pore domain (K2P) currents (Fig. 7D) were tested, and although some demonstrated a trend for decreased levels, the differences did not reach statistical significance.
Fig. 7.
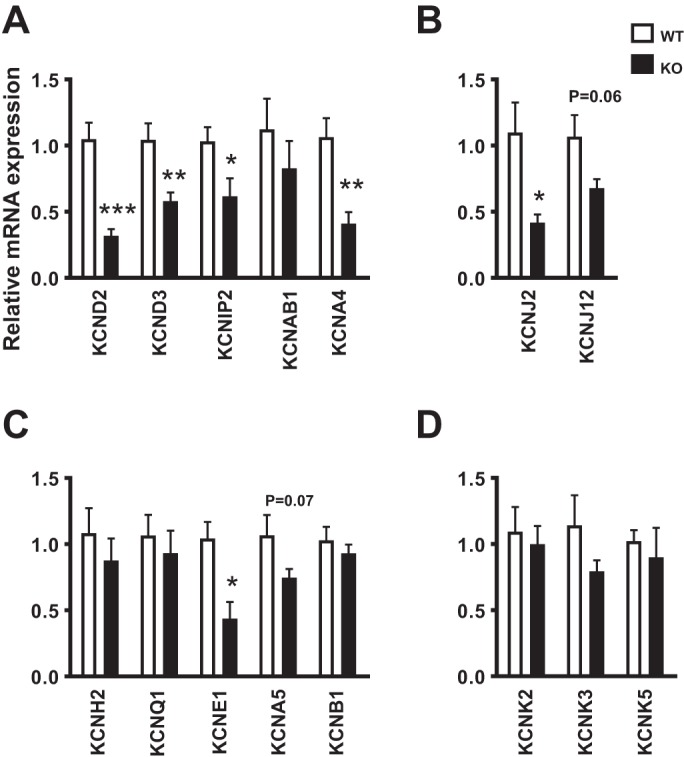
Transcript expression of K+ channel subunits: mRNA relative transcript levels as measured by quantitative RT-PCR in WT (n = 7) and KO (n = 7) animals. Mean ± SE values are plotted. *P < 0.05; **P < 0.01; ***P < 0.001. A: transient outward voltage-gated K+ currents [Ito,fast: KCND2 (Kv4.2), KCND3 (Kv4.3), KCNIP2 (KChIP2), and KCNAB1 (Kvβ1); Ito,slow: KCNA4 (Kv1.4)]. B: inward rectifier current [IK1: KCNJ2 (Kir2.1) and KCNJ12 (Kir2.1)]. C: delayed rectifier voltage-gated K+ currents [IKr: KCNH2 (hERG); IKs: KCNQ1 (KvLQT1) and KCNE1 (minK); IK,slow1: KCNA5 (Kv1.5); IK,slow2: KCNB1 (Kv2.1)]. D: two-pore domain current [Iss: KCNK2 (TREK1), KCNK3 (TASK1), and KCNK5 (TASK2)].
DISCUSSION
Why HCM patients die suddenly was the question that was the driving force for this study. This question has been the center of extensive investigation. Clinical and basic studies have supported several mechanisms: tissue fibrosis and cellular disarray causing microreentry circuits, coronary bridging and vascular instability leading to ischemia, remodeling of Ca2+ currents, abnormal intracellular Ca2+ handling, and remodeling of late Na+ currents (4, 9, 11, 12, 23, 44). The idea that repolarizing K+ currents might also be involved in electrical remodeling in HCM has not been well studied and is the topic of the present study.
Remodeling of repolarization in HCM.
The main finding of our study is the significant remodeling of outward repolarizing currents in MyBPC KO ventricular myocytes, an HCM model system (Fig. 6). To the best of our knowledge, this is the first HCM animal model in general, and MyBPC specifically, in which such a change has been described.
Supporting our findings, remodeling of K+ currents has been reported in an in vitro study of human cardiomyocytes isolated from HCM patients (9). Although mouse cardiac electrophysiology is inherently different from human, an animal model enables the controlled and dynamic exploration of in vivo pathophysiology, diagnostic studies, and potential therapeutic intervention (11).
The basis for this study was the clinical observation of QT prolongation and repolarization abnormalities in HCM patients (15, 20). This phenotype was evident in the animal model (Fig. 3), which also recapitulated the structural, histological (Fig. 1), echocardiographic (Fig. 2), and arrhythmogenic substrate (Fig. 4). Of note, QT prolongation was noted in HCM patients with mutations in several other HCM genes (myosin heavy chain 7, thin filament, Z disk) in addition to MyBPC (20) as well as in a different animal model [myosin heavy chain and troponin-I double mutation HCM mouse model (46)], suggesting that our findings are likely not gene/model specific.
Interestingly, heterozygous animals of the same MyBPC model that demonstrate only mild contractile dysfunction without ventricular hypertrophy exhibited prolonged QTc interval on surface ECG, with intermediate values between WT and our homozygous KO animals (8). Although most human patients with HCM carry heterozygous mutations, there are patients who harbor homozygous, compound heterozygous (2 different mutations in the same gene), and double (such as MyBPC together with MYH7) sarcomere gene mutations and usually are characterized with a severe phenotype (3, 31). This underscores the relevance of exploring our homozygous animals with the pronounced phenotype.
Arrhythmia susceptibility in KO mice.
SCD in HCM is mediated primarily by ventricular fibrillation, usually preceded by PVCs and ventricular tachycardia (37). In our study, one of the four KO mice exhibited diverse ventricular ectopy including frequent NSVT. Untriggered arrhythmias at rest are not common in HCM. The proportion that was noted in our study (25%) is similar to the proportion described in other animal/human studies (2, 6, 19, 32). The ventricular arrhythmias that have been previously described in several other HCM mouse models were mainly attributed to the structural arrhythmogenic substrate, remodeling of Ca2+ currents, abnormal Ca2+ handling, and hypertrophy per se (5, 6, 13, 18, 24, 40, 47). Although electrocardiographic repolarization abnormalities and QT prolongation were also noted in these studies, K+ currents were not explored. We suggest that reductions in repolarizing K+ currents contribute to APD and QT prolongation and the associated ventricular arrhythmias.
Molecular components underlying the downregulation of ionic currents.
Correlating with the cellular electrophysiological results, we found that various mRNA transcripts of K+ channel subunits were decreased (Fig. 7). This reduction was noted in each of the K+ current components but was most prominent in Ito (Fig. 7A). Ito is the dominant repolarizing current in the mouse affecting APD and shape (33, 35).
It is well established that reduced K+ currents in general, and Ito specifically, are commonly observed in mouse models with cardiac hypertrophy and failure regardless of the underlying cause (25, 49). However, physiological cardiac hypertrophy associated with training resulted in the opposite outcome, with upregulation of repolarizing currents (48). These results indicate that changes in repolarizing K+ currents are not specific to the hypertrophy per se but are related to the physiological or pathological processes causing hypertrophy.
Implications.
In addition to sarcomeric cardiomyopathies, these results may also be relevant to other forms of genetic malformation syndromes, neuromuscular disorders, and metabolic cardiomyopathies. These cardiomyopathies usually present in early childhood, harbor poor prognosis, and are characterized by severe hypertrophy, electrocardiographic and repolarization abnormalities, arrhythmias, and sudden death. There is a paucity of investigation in this field, and further research into the pathophysiology of these orphan diseases is clearly needed.
There is a growing interest in investigating the interaction between membrane ion channels in general, and K+ channels in particular, and the intracellular cytoskeleton and sarcomere (35, 36). It has been suggested that sarcomeric cardiomyopathies share a final common pathway involving the cell membrane and the intracellular components (7, 45). Our intriguing results suggest a link between both ends, leading to an abnormal excitation-contraction coupling, contractile dysfunction, and arrhythmias.
Study limitations.
As in any other studies exploring cellular electrophysiology in mice, one should be aware of the limitation originating from the major differences in the repolarizing currents between mice and larger mammals, with dominance of Ito effecting different APD and shape in mice. The significance of our findings should be considered in this context. Although Ito has an important role in larger mammals, including humans, controlling the initial phase of AP repolarization and determining the driving force for the Ca2+ entry through L-type Ca2+ channels (36), it is important to mention that larger mammals have longer APDs and that APD is less dependent on changes in Ito. This limitation, however, is inherent to the rodent experimental system and does not limit the capability to study disease mechanisms in highly controlled in vivo models (21, 34).
Conclusions.
In summary, MyBPC KO HCM mice demonstrate an overt phenotype with severe cardiac hypertrophy, contractile dysfunction, and arrhythmia susceptibility. Electrocardiographic recordings revealed prolongation of the QTc interval consistent with repolarization abnormality and diverse ventricular arrhythmias. Cellular electrophysiological recordings demonstrated reduced K+ currents and AP prolongation. Furthermore, quantitative RT-PCR experiments established reduced K+ channel transcript levels. These results suggest that remodeling of repolarizing K+ currents may contribute to the arrhythmia susceptibility and could be the target for novel therapies for the prevention of SCD in HCM.
GRANTS
A. Toib received a research grant from the Philadelphia Health and Education Corporation Fund. X. Chen received National Heart, Lung, and Blood Institute (NHLBI) Grant HL-88243. S. Mohsin received a Scientist Development Grant from the American Heart Association. S. R. Houser received NHLBI Grants HL-89312, HL-91799, and HL-33921.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.T. and S.R.H. conceived and designed research; A.T., C.Z., G.B., X.Z., M.W., H.K., E.F., R.M.B., D.M.T., S.H., and X.C. performed experiments; A.T., G.B., M.W., Y.Y., C.D.T., H.K., T.E.S., N.Z., D.M.T., S.H., and P.G. analyzed data; A.T., C.Z., G.B., X.Z., M.W., Y.Y., C.D.T., H.K., N.Z., D.M.T., S.H., P.G., X.C., S.M., and S.R.H. interpreted results of experiments; A.T., C.D.T., and T.E.S. prepared figures; A.T. and S.M. drafted manuscript; A.T., G.B., M.W., C.D.T., H.K., E.F., R.M.B., D.M.T., S.H., P.G., X.C., S.M., and S.R.H. edited and revised manuscript; A.T., C.Z., G.B., X.Z., M.W., Y.Y., C.D.T., H.K., T.E.S., E.F., R.M.B., N.Z., D.M.T., S.H., P.G., X.C., S.M., and S.R.H. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Richard Moss (University of Wisconsin) for providing the MyBPC KO mouse line and Dr. Kai-Chien Yang (National Taiwan University) for his critical review and research advice.
REFERENCES
- 1.Ackerman M, Atkins DL, Triedman JK. Sudden cardiac death in the young. Circulation 133: 1006–1026, 2016. doi: 10.1161/CIRCULATIONAHA.115.020254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adabag AS, Casey SA, Kuskowski MA, Zenovich AG, Maron BJ. Spectrum and prognostic significance of arrhythmias on ambulatory Holter electrocardiogram in hypertrophic cardiomyopathy. J Am Coll Cardiol 45: 697–704, 2005. doi: 10.1016/j.jacc.2004.11.043. [DOI] [PubMed] [Google Scholar]
- 3.Alcalai R, Seidman JG, Seidman CE. Genetic basis of hypertrophic cardiomyopathy: from bench to the clinics. J Cardiovasc Electrophysiol 19: 104–110, 2008. doi: 10.1111/j.1540-8167.2007.00965.x. [DOI] [PubMed] [Google Scholar]
- 4.Ashrafian H, McKenna WJ, Watkins H. Disease pathways and novel therapeutic targets in hypertrophic cardiomyopathy. Circ Res 109: 86–96, 2011. doi: 10.1161/CIRCRESAHA.111.242974. [DOI] [PubMed] [Google Scholar]
- 5.Baudenbacher F, Schober T, Pinto JR, Sidorov VY, Hilliard F, Solaro RJ, Potter JD, Knollmann BC. Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J Clin Invest 118: 3893–3903, 2008. doi: 10.1172/JCI36642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berul CI, McConnell BK, Wakimoto H, Moskowitz IP, Maguire CT, Semsarian C, Vargas MM, Gehrmann J, Seidman CE, Seidman JG. Ventricular arrhythmia vulnerability in cardiomyopathic mice with homozygous mutant Myosin-binding protein C gene. Circulation 104: 2734–2739, 2001. doi: 10.1161/hc4701.099582. [DOI] [PubMed] [Google Scholar]
- 7.Bowles NE, Bowles KR, Towbin JA. The “final common pathway” hypothesis and inherited cardiovascular disease. The role of cytoskeletal proteins in dilated cardiomyopathy. Herz 25: 168–175, 2000. doi: 10.1007/s000590050003. [DOI] [PubMed] [Google Scholar]
- 8.Cheng Y, Wan X, McElfresh TA, Chen X, Gresham KS, Rosenbaum DS, Chandler MP, Stelzer JE. Impaired contractile function due to decreased cardiac myosin binding protein C content in the sarcomere. Am J Physiol Heart Circ Physiol 305: H52–H65, 2013. doi: 10.1152/ajpheart.00929.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coppini R, Ferrantini C, Yao L, Fan P, Del Lungo M, Stillitano F, Sartiani L, Tosi B, Suffredini S, Tesi C, Yacoub M, Olivotto I, Belardinelli L, Poggesi C, Cerbai E, Mugelli A. Late sodium current inhibition reverses electromechanical dysfunction in human hypertrophic cardiomyopathy. Circulation 127: 575–584, 2013. doi: 10.1161/CIRCULATIONAHA.112.134932. [DOI] [PubMed] [Google Scholar]
- 10.Duran JM, Makarewich CA, Sharp TE, Starosta T, Zhu F, Hoffman NE, Chiba Y, Madesh M, Berretta RM, Kubo H, Houser SR. Bone-derived stem cells repair the heart after myocardial infarction through transdifferentiation and paracrine signaling mechanisms. Circ Res 113: 539–552, 2013. doi: 10.1161/CIRCRESAHA.113.301202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Force T, Bonow RO, Houser SR, Solaro RJ, Hershberger RE, Adhikari B, Anderson ME, Boineau R, Byrne BJ, Cappola TP, Kalluri R, LeWinter MM, Maron MS, Molkentin JD, Ommen SR, Regnier M, Tang WH, Tian R, Konstam MA, Maron BJ, Seidman CE. Research priorities in hypertrophic cardiomyopathy: report of a Working Group of the National Heart, Lung, and Blood Institute. Circulation 122: 1130–1133, 2010. doi: 10.1161/CIRCULATIONAHA.110.950089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frey N, Luedde M, Katus HA. Mechanisms of disease: hypertrophic cardiomyopathy. Nat Rev Cardiol 9: 91–100, 2011. doi: 10.1038/nrcardio.2011.159. [DOI] [PubMed] [Google Scholar]
- 13.Gehrmann J, Berul CI. Cardiac electrophysiology in genetically engineered mice. J Cardiovasc Electrophysiol 11: 354–368, 2000. doi: 10.1111/j.1540-8167.2000.tb01806.x. [DOI] [PubMed] [Google Scholar]
- 14.Gersh BJ, Maron BJ, Bonow RO, Dearani JA, Fifer MA, Link MS, Naidu SS, Nishimura RA, Ommen SR, Rakowski H, Seidman CE, Towbin JA, Udelson JE, Yancy CW; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines; American Association for Thoracic Surgery; American Society of Echocardiography; American Society of Nuclear Cardiology; Heart Failure Society of America; Heart Rhythm Society; Society for Cardiovascular Angiography and Interventions; Society of Thoracic Surgeons . 2011 ACCF/AHA guideline for the diagnosis and treatment of hypertrophic cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 124: e783–e831, 2011. doi: 10.1161/CIR.0b013e318223e2bd. [DOI] [PubMed] [Google Scholar]
- 15.Gray B, Ingles J, Medi C, Semsarian C. Prolongation of the QTc interval predicts appropriate implantable cardioverter-defibrillator therapies in hypertrophic cardiomyopathy. JACC Heart Fail 1: 149–155, 2013. doi: 10.1016/j.jchf.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 16.Harris SP, Bartley CR, Hacker TA, McDonald KS, Douglas PS, Greaser ML, Powers PA, Moss RL. Hypertrophic cardiomyopathy in cardiac myosin binding protein-C knockout mice. Circ Res 90: 594–601, 2002. doi: 10.1161/01.RES.0000012222.70819.64. [DOI] [PubMed] [Google Scholar]
- 17.Ho CY, Charron P, Richard P, Girolami F, Van Spaendonck-Zwarts KY, Pinto Y. Genetic advances in sarcomeric cardiomyopathies: state of the art. Cardiovasc Res 105: 397–408, 2015. doi: 10.1093/cvr/cvv025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huke S, Venkataraman R, Faggioni M, Bennuri S, Hwang HS, Baudenbacher F, Knollmann BC. Focal energy deprivation underlies arrhythmia susceptibility in mice with calcium-sensitized myofilaments. Circ Res 112: 1334–1344, 2013. doi: 10.1161/CIRCRESAHA.113.301055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jansson K, Dahlström U, Karlsson E, Nylander E, Walfridsson H, Sonnhag C. The value of exercise test, Holter monitoring, and programmed electrical stimulation in detection of ventricular arrhythmias in patients with hypertrophic cardiomyopathy. Pacing Clin Electrophysiol 13: 1261–1267, 1990. doi: 10.1111/j.1540-8159.1990.tb02025.x. [DOI] [PubMed] [Google Scholar]
- 20.Johnson JN, Grifoni C, Bos JM, Saber-Ayad M, Ommen SR, Nistri S, Cecchi F, Olivotto I, Ackerman MJ. Prevalence and clinical correlates of QT prolongation in patients with hypertrophic cardiomyopathy. Eur Heart J 32: 1114–1120, 2011. doi: 10.1093/eurheartj/ehr021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaese S, Verheule S. Cardiac electrophysiology in mice: a matter of size. Front Physiol 3: 345, 2012. doi: 10.3389/fphys.2012.00345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kensler RW, Harris SP. The structure of isolated cardiac myosin thick filaments from cardiac Myosin binding protein-C knockout mice. Biophys J 94: 1707–1718, 2008. doi: 10.1529/biophysj.107.115899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kessler EL, Boulaksil M, van Rijen HV, Vos MA, van Veen TA. Passive ventricular remodeling in cardiac disease: focus on heterogeneity. Front Physiol 5: 482, 2014. doi: 10.3389/fphys.2014.00482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Knollmann BC, Kirchhof P, Sirenko SG, Degen H, Greene AE, Schober T, Mackow JC, Fabritz L, Potter JD, Morad M. Familial hypertrophic cardiomyopathy-linked mutant troponin T causes stress-induced ventricular tachycardia and Ca2+-dependent action potential remodeling. Circ Res 92: 428–436, 2003. doi: 10.1161/01.RES.0000059562.91384.1A. [DOI] [PubMed] [Google Scholar]
- 25.Knollmann BC, Knollmann-Ritschel BE, Weissman NJ, Jones LR, Morad M. Remodelling of ionic currents in hypertrophied and failing hearts of transgenic mice overexpressing calsequestrin. J Physiol 525: 483–498, 2000. doi: 10.1111/j.1469-7793.2000.t01-1-00483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Korte FS, McDonald KS, Harris SP, Moss RL. Loaded shortening, power output, and rate of force redevelopment are increased with knockout of cardiac myosin binding protein-C. Circ Res 93: 752–758, 2003. doi: 10.1161/01.RES.0000096363.85588.9A. [DOI] [PubMed] [Google Scholar]
- 27.Makarewich CA, Zhang H, Davis J, Correll RN, Trappanese DM, Hoffman NE, Troupes CD, Berretta RM, Kubo H, Madesh M, Chen X, Gao E, Molkentin JD, Houser SR. Transient receptor potential channels contribute to pathological structural and functional remodeling after myocardial infarction. Circ Res 115: 567–580, 2014. doi: 10.1161/CIRCRESAHA.115.303831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation 92: 785–789, 1995. doi: 10.1161/01.CIR.92.4.785. [DOI] [PubMed] [Google Scholar]
- 29.Maron BJ, Haas TS, Murphy CJ, Ahluwalia A, Rutten-Ramos S. Incidence and causes of sudden death in U.S. college athletes. J Am Coll Cardiol 63: 1636–1643, 2014. doi: 10.1016/j.jacc.2014.01.041. [DOI] [PubMed] [Google Scholar]
- 30.Mitchell GF, Jeron A, Koren G. Measurement of heart rate and Q-T interval in the conscious mouse. Am J Physiol Heart Circ Physiol 274: H747–H751, 1998. [DOI] [PubMed] [Google Scholar]
- 31.Mohiddin SA, Begley DA, McLam E, Cardoso JP, Winkler JB, Sellers JR, Fananapazir L. Utility of genetic screening in hypertrophic cardiomyopathy: prevalence and significance of novel and double (homozygous and heterozygous) beta-myosin mutations. Genet Test 7: 21–27, 2003. doi: 10.1089/109065703321560895. [DOI] [PubMed] [Google Scholar]
- 32.Monserrat L, Elliott PM, Gimeno JR, Sharma S, Penas-Lado M, McKenna WJ. Non-sustained ventricular tachycardia in hypertrophic cardiomyopathy: an independent marker of sudden death risk in young patients. J Am Coll Cardiol 42: 873–879, 2003. doi: 10.1016/S0735-1097(03)00827-1. [DOI] [PubMed] [Google Scholar]
- 33.Nerbonne JM. Molecular basis of functional myocardial potassium channel diversity. Card Electrophysiol Clin 8: 257–273, 2016. doi: 10.1016/j.ccep.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nerbonne JM. Studying cardiac arrhythmias in the mouse: a reasonable model for probing mechanisms? Trends Cardiovasc Med 14: 83–93, 2004. doi: 10.1016/j.tcm.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 35.Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol Rev 85: 1205–1253, 2005. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- 36.Niwa N, Nerbonne JM. Molecular determinants of cardiac transient outward potassium current (Ito) expression and regulation. J Mol Cell Cardiol 48: 12–25, 2010. doi: 10.1016/j.yjmcc.2009.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Mahony C, Elliott P, McKenna W. Sudden cardiac death in hypertrophic cardiomyopathy. Circ Arrhythm Electrophysiol 6: 443–451, 2013. doi: 10.1161/CIRCEP.111.962043. [DOI] [PubMed] [Google Scholar]
- 38.Prinz C, Schwarz M, Ilic I, Laser KT, Lehmann R, Prinz EM, Bitter T, Vogt J, van Buuren F, Bogunovic N, Horstkotte D, Faber L. Myocardial fibrosis severity on cardiac magnetic resonance imaging predicts sustained arrhythmic events in hypertrophic cardiomyopathy. Can J Cardiol 29: 358–363, 2013. doi: 10.1016/j.cjca.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 39.Rubart M, Zipes DP. Mechanisms of sudden cardiac death. J Clin Invest 115: 2305–2315, 2005. doi: 10.1172/JCI26381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schober T, Huke S, Venkataraman R, Gryshchenko O, Kryshtal D, Hwang HS, Baudenbacher FJ, Knollmann BC. Myofilament Ca sensitization increases cytosolic Ca binding affinity, alters intracellular Ca homeostasis, and causes pause-dependent Ca-triggered arrhythmia. Circ Res 111: 170–179, 2012. doi: 10.1161/CIRCRESAHA.112.270041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Semsarian C, Ahmad I, Giewat M, Georgakopoulos D, Schmitt JP, McConnell BK, Reiken S, Mende U, Marks AR, Kass DA, Seidman CE, Seidman JG. The L-type calcium channel inhibitor diltiazem prevents cardiomyopathy in a mouse model. J Clin Invest 109: 1013–1020, 2002. doi: 10.1172/JCI200214677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol 65: 1249–1254, 2015. doi: 10.1016/j.jacc.2015.01.019. [DOI] [PubMed] [Google Scholar]
- 43.Stelzer JE, Fitzsimons DP, Moss RL. Ablation of myosin-binding protein-C accelerates force development in mouse myocardium. Biophys J 90: 4119–4127, 2006. doi: 10.1529/biophysj.105.078147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tardiff JC, Carrier L, Bers DM, Poggesi C, Ferrantini C, Coppini R, Maier LS, Ashrafian H, Huke S, van der Velden J. Targets for therapy in sarcomeric cardiomyopathies. Cardiovasc Res 105: 457–470, 2015. doi: 10.1093/cvr/cvv023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Towbin JA. Inherited cardiomyopathies. Circ J 78: 2347–2356, 2014. doi: 10.1253/circj.CJ-14-0893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsoutsman T, Kelly M, Ng DC, Tan JE, Tu E, Lam L, Bogoyevitch MA, Seidman CE, Seidman JG, Semsarian C. Severe heart failure and early mortality in a double-mutation mouse model of familial hypertrophic cardiomyopathy. Circulation 117: 1820–1831, 2008. doi: 10.1161/CIRCULATIONAHA.107.755777. [DOI] [PubMed] [Google Scholar]
- 47.Wolf CM, Moskowitz IP, Arno S, Branco DM, Semsarian C, Bernstein SA, Peterson M, Maida M, Morley GE, Fishman G, Berul CI, Seidman CE, Seidman JG. Somatic events modify hypertrophic cardiomyopathy pathology and link hypertrophy to arrhythmia. Proc Natl Acad Sci USA 102: 18123–18128, 2005. doi: 10.1073/pnas.0509145102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang KC, Foeger NC, Marionneau C, Jay PY, McMullen JR, Nerbonne JM. Homeostatic regulation of electrical excitability in physiological cardiac hypertrophy. J Physiol 588: 5015–5032, 2010. doi: 10.1113/jphysiol.2010.197418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang KC, Nerbonne JM. Mechanisms contributing to myocardial potassium channel diversity, regulation and remodeling. Trends Cardiovasc Med 26: 209–218, 2016. doi: 10.1016/j.tcm.2015.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang X, Ai X, Nakayama H, Chen B, Harris DM, Tang M, Xie Y, Szeto C, Li Y, Li Y, Zhang H, Eckhart AD, Koch WJ, Molkentin JD, Chen X. Persistent increases in Ca(2+) influx through Cav1.2 shortens action potential and causes Ca(2+) overload-induced afterdepolarizations and arrhythmias. Basic Res Cardiol 111: 4, 2016. doi: 10.1007/s00395-015-0523-4. [DOI] [PMC free article] [PubMed] [Google Scholar]