

Abstract
The intracellular signaling mechanisms through which TGF-β regulates pulmonary development are incompletely understood. Canonical TGF-β signaling involves Smad2/3 phosphorylation, Smad2/3·Smad4 complex formation and nuclear localization, and gene regulation. Here, we show that physiologically relevant TGF-β1 levels also stimulate Smad1/5 phosphorylation, which is typically a mediator of bone morphogenetic protein (BMP) signaling, in mouse pup pulmonary artery smooth muscle cells (mPASMC) and lung fibroblasts and other interstitial lung cell lines. This cross-talk mechanism likely has in vivo relevance because mixed Smad1/5/8·Smad2/3 complexes, which are indicative of TGF-β-stimulated Smad1/5 activation, were detected in the developing mouse lung using a proximity ligation assay. Although mixed Smad complexes have been shown not to transduce nuclear signaling, we determined that TGF-β stimulates nuclear localization of phosphorylated Smad1/5 and induces the expression of prototypical BMP-regulated genes in the mPASMC. Small-molecule kinase inhibitor studies suggested that TGF-β-regulated Smad1/5 phosphorylation in these cells is mediated by TGF-β-type I receptors, not BMP-type I receptors, but possibly the accessory activin-like kinase (ALK1) receptor. Although work by others suggested that ALK1 is expressed exclusively in endothelial cells in the vasculature, we detected ALK1 mRNA and protein expression in mPASMC in vitro and in mouse pup lungs. Moreover, using an antimurine ALK1 antibody and mPASMC, we determined that ALK1 regulates Smad1/5 phosphorylation by TGF-β. Together, these studies characterize an accessory TGF-β-stimulated BMP R-Smad signaling mechanism in interstitial cells of the developing lung. They also indicate the importance of considering alternate Smad pathways in studies directed at determining how TGF-β regulates newborn lung development.
Keywords: TGF-β signaling, lung development, pulmonary vascular smooth muscle cells
the prototypical member of the TGF-β superfamily of pleiotropic, secreted cytokines, TGF-β plays an important role in regulating cell differentiation, migration, and function and, thereby, controls the development of many organs. Several studies indicate that TGF-β regulates early pulmonary development (reviewed in Refs. 4, 41, and 54). In the embryonic lung of rodents, for example, TGF-β controls mesodermal progenitor cell differentiation (32) and the branching morphogenesis of structures required for the formation of the conducting airways (18, 33, 53, 67, 74–76). However, less is known about how TGF-β regulates the late stages of pulmonary development, when the acinar–gas exchange portions of the lung form. TGF-β and its intracellular signaling elements are dynamically regulated in the late developing lung (1, 2, 42, 46, 47). Moreover, studies suggest that TGF-β overactivation during this stage of pulmonary development disrupts lung microvascular and alveolar development, causing newborn lung disease. For example, adenovirus-mediated active TGF-β overexpression in the newborn rat pup lung was found to cause patchy interstitial fibrosis and decrease pulmonary septation (18). Also, TGF-β overexpression in 1-wk-old transgenic mouse pups was noted to inhibit lung parenchymal cell differentiation, microvascular development, and peripheral airway septation (65). This inhibitory effect of TGF-β on late lung development is mediated through its receptor system (56). Several studies now show that TGF-β is activated during newborn lung injury (1, 40, 42) and directly disrupts pulmonary development. This is because TGF-β inhibition with neutralizing antibodies during newborn lung injury has been demonstrated to protect pulmonary microvascular and alveolar development (40, 42) and improve extracellular matrix formation (29, 68).
The pathways through which TGF-β regulates intracellular signaling in pulmonary cells during lung development remain poorly characterized. It is difficult to extrapolate information from other cell types, because TGF-β signaling depends on the type, origin, and microenvironment of the cell and exhibits differential regulation during development. The three TGF-β isoforms—TGF-β1, TGF-β2, and TGF-β3—are products of different genes. But, in most cases, they signal through the same receptor-mediated system. The TGF-β isoforms are secreted as inactive molecules that bind, through adaptor proteins, to the extracellular matrix. Unlike most cytokines, TGF-β requires extracellular activation before it can exert biological effects within cells. Once activated, TGF-β binds to its type II receptor (TGFβR2) and promotes the recruitment, phosphorylation, and activation, of the TGF-β type I receptors (TGFβR1), activin-like kinase (ALK)4, and ALK5, in a heteromeric receptor complex (21, 36). In some cases, accessory type III receptors, including betaglycan and endoglin, assist in the formation of the TGF-β receptor complex. Once activated, the TGFβR1 phosphorylates serine (S) residues in a COOH-terminal SSXS motif in the TGF-β type I receptor-regulated (R)-Smad proteins, Smad2, and Smad3 (Smad2/3; Ref. 14). After recruiting Smad4, a co-Smad, the TGF-β R-Smads migrate as a complex into the nucleus and regulate gene expression. In a parallel but similar signaling paradigm, bone morphogenetic protein (BMP) engages its type II and I receptors and stimulates the phosphorylation of the BMP R-Smad proteins, Smad1/5/8. After complexing with Smad4, the activated BMP R-Smads enter the nucleus and regulate gene expression. The TGF-β-Smad2/3 and BMP-Smad1/5 signaling pathways coincide in aortic smooth muscle cells (58) and pulmonary artery smooth muscle cells (PASMC) derived from adult humans (50, 57, 60, 71, 72). Moreover, TGF-β and BMP signaling proteins are regulated during late pulmonary development (2), and TGF-β stimulates Smad2/3 phosphorylation in PASMC and other interstitial cells in the developing lung (42, 43).
Accumulating evidence, however, indicates that TGF-β can also cross-stimulate BMP R-Smad signaling in select cells. For example, TGF-β increases Smad1 phosphorylation in human breast cancer cells (34) and other primary cells and cell lines (8, 12, 69). Studies suggest that this TGF-β-stimulated BMP R-Smad cross talk induces the formation of unique mixed TGF-β and BMP R-Smad complexes that do not include Smad4 or stimulate BMP-regulated gene expression (12). However, the TGF-β-mediated BMP R-Smad phosphorylation appears to be variable; it has not been detected in many cell types (8, 22). For example, although TGF-β-stimulated Smad1/5 phosphorylation in aortic, corneal, microvascular, and embryonic endothelial cells (19, 20), it did not in human umbilical vein endothelial cells (64). Moreover, whereas TGF-β increased Smad1/5 phosphorylation in fibroblast cell lines (NIH/3T3), it did not appear to do so in embryonic mouse fibroblast cells (22). TGF-β stimulates Smad1/5/8 phosphorylation in adult human aortic (58) and PASMC (13, 50, 60, 73) and in adult mouse PASMC (22). However, TGF-β has also been observed not to stimulate BMP R-Smad signaling in some of these smooth muscle cells (SMC) (e.g., 61), and the mechanisms through which it stimulates Smad1/5 phosphorylation in the other cases are unknown.
In the current studies, we tested whether TGF-β stimulates BMP R-Smad signaling in interstitial cells from the developing lung. Here, we demonstrate that physiological levels of TGF-β increased Smad1/5 phosphorylation in primary mouse pup PASMC and lung fibroblasts and in PASMC and lung fibroblast cell lines. Moreover, we detected mixed TGF-β and BMP R-Smad complexes in the mouse pup lung, which are indicative of this cross-talk mechanism. Although previous studies showed no TGF-β-mediated nuclear signaling in cells exhibiting TGF-β-stimulated BMP R-Smad phosphorylation (12, 13), in primary mouse PASMC, we detected TGF-β-induced nuclear translocation of phosphorylated (p)Smad1/5 and stimulated the expression of prototypical BMP-regulated genes. Moreover, using a neutralizing antibody, we determined that ALK1 plays a role in regulating TGF-β phosphorylation of Smad1/5 in those cells. Together, these results detail a TGF-β BMP R-Smad cross-talk mechanism not previously reported in PASMC and lung fibroblasts from the developing lung. They also support the importance of considering differential R-Smad signaling pathways in determining how TGF-β regulates the development of the newborn lung.
MATERIALS AND METHODS
Antibodies and reagents.
Smad activation was detected using rabbit anti-pSmad1/5/8 [pS463/465-no. Vli31, 1:1,000; Main Medical Center Research Institute), anti-Smad1 (LS-C7585, 1:500; LifeSpan BioSciences), anti-pSmad2 (pS465/467-no. 3101, 1:500; Cell Signaling Technologies (CST)], and anti-Smad2 (no. 3103, 1:500; CST). These pSmad-reactive antibodies recognize the phosphorylation of the COOH-terminal SSXS motifs of the proteins. ALK1 immunoreactivity was detected in lung tissue using a polyclonal rabbit anti-ALK1 antibody (SAB4503630, 1:1,000; Sigma) and was neutralized in cells using a monoclonal rat anti-mouse (m)ALK1 (MAB770, concentration described below; R&D Systems). An anti-ID1 antibody (ab192303, 1:500; Abcam) was also used in experiments. To characterize the smooth muscle cells, anti-smoothelin antibody (sc-28562, 1:250; Santa Cruz Biotechnology) was used. To assess reference gene expression, an anti-GAPDH antibody (G8795; 1:2,000; Sigma) was employed. Isotype antibodies were obtained from a commercial source (Abcam). Enzyme-conjugated secondary antibodies were purchased from Jackson ImmunoResearch, and Alexa Fluor 546-conjugated ones were obtained from Thermo Fisher Scientific. Recombinant human TGF-β1 and BMP4, mouse (m)ALK1-Fc (all obtained from R&D Systems), and the kinase inhibitors SB505124 (S4696; Sigma) dorsomorphin (ab144821; Abcam) and LDN212854 (SML0965; Sigma) were also used. The cytokines were reconstituted using 4 mM HCl in 1 mg/ml BSA; the stock inhibitor solutions were dissolved in DMSO, according to manufacturer instructions.
Cell isolation and culture.
The Subcommittee for Research Animal Studies at the Massachusetts General Hospital approved the investigations described here. Mouse pup pulmonary artery smooth muscle cells (mPASMC) were isolated from postnatal day (P)10 FVB/NCrl (Charles River Laboratories) using methods described by others (31), using a suspension of iron oxide in agar (25). The mouse pup pulmonary (m)Fibroblasts were obtained from the peripheral lung using previously described methods (6). The cells were identified by their characteristic morphology; the mPASMC were further characterized using an anti-smoothelin antibody and indirect immunofluorescence. CS54, an adult rat PASMC line (also known as PAC1 cells), was generated by A. Rothman (49) and was characterized as before (6); RLF-6 cells, a rat fetal lung cell line, was purchased from American Type Culture Collection. The PASMC and mFibroblast cells were maintained in DMEM containing 4.5 g/l glucose (hDMEM, no. 11995; Life Technologies); the RFL-6 cells were cultured in RPMI 1640 (no. 2633; Life Technologies). Complete media were formulated with 10% (vol/vol) heat-inactivated FBS (SH3008803; Hyclone), 0.29 mg/ml glutamine, 100 U/ml penicillin, and 100 µg/ml streptomycin. The cells were maintained in a humidified 37°C incubator chamber containing 5% CO2 and passaged before becoming confluent using EDTA-trypsin. The mPASMC were used before passage 10.
Experimental protocols.
TGF-β- and BMP-stimulated Smad phosphorylation was determined in the following manner. After reaching 90% confluence, cells residing on 9.5-mm2 dishes were serum restricted using hDMEM containing 0.2% FBS for 24 h. Subsequently, the cells were exposed to TGF-β1, BMP4, or 0.2% DMSO, the latter of which corresponds with the amount of the solvent used in the kinase inhibitor solutions, in the same media for 1 h. In some studies, the cells were pretreated with SB505124, dorsomorphin, or LDN212854 for 1 h before the addition of the ligands or DMSO to the media. Subsequently, the cells were lysed, and protein or RNA was isolated, as described below.
Nuclear pSmad1/5 localization was tested using mPASMC seeded onto 1.7-cm2 chamber slides. After achieving 70% confluence, the cells were serum restricted as above and then treated with TGF-β1, BMP4, or DMSO in the same media for 1 h. Subsequently, pSmad1/5 immunoreactivity in the nuclei was determined in the cells as detailed below.
To detect ALK1 on the surface of mPASMC, the cells were exposed to 15 µg/ml rat anti-mALK1 antibody, with or without 10 µg/ml mALK1-Fc (~1:2.5 molar ratio) preadsorption, or 15 µg/ml rat IgG2b in the serum restriction media in chamber slides overnight. Subsequently, the cells were washed with PBS, fixed with 4% formaldehyde, and then reacted with a fluoroprobe-conjugated anti-rat IgG antibody. To examine the role of ALK1 in mediating TGF-β-stimulated pSmad1/5 nuclear localization, mPASMC on chamber slides were treated with the antibodies described above in the serum-restriction media overnight. Subsequently, the media were removed, and the cells were treated with or without TGF-β1 in the serum restriction media for 1 h. Afterward, nuclear pSmad1/5 immunoreactivity was determined as described below.
Immunoblotting.
For the Smad protein analysis, cells were snap-frozen on dry ice, placed on wet ice, and then scraped into ice-cold lysis buffer containing glycerol, as described by others (60). For the ID1 protein studies, cells were scraped into lysis buffer containing 50 mM Tris·HCl (pH 7.4), 1 mM EDTA, 1 mM dithiothreitol, and protease inhibitors (P8340; Sigma). The lysates were then sonicated, centrifuged to remove insoluble components, and then placed on ice. For the tissue ALK1 protein level analysis, mice were killed using 200 mg/kg pentobarbital sodium intraperitoneal injection, and then whole lung and liver tissues were obtained, blotted to remove excess blood, and then quickly frozen using liquid nitrogen. Subsequently, the tissues were pulverized, and the proteins were solubilized using the ice-cold lysis buffer defined above. The protein concentrations in the tissue and cell lysates were determined using bicinchoninic acid protein assay reagent (23227; ThermoFisher), and equal amounts of protein were resolved using SDS-PAGE and electroblotted onto polyvinylidene difluoride membranes. For the Smad and ID protein analysis, anti-GAPDH reactivity was tested to confirm the protein transfer to the blots. For the ALK1 protein analysis using mixed tissues and cell type samples, 0.1% Ponceau S and 5% acetic acid (wt/vol) blot staining was used for this purpose. In the latter case, the stain was removed from the blot using 0.1 M NaOH and water rinsing before the blocking and antibody reaction. The membranes were blocked using 5% nonfat dry milk in TBS containing 0.05% Tween 20, and then exposed to primary antibodies. Subsequently, immunocomplexes were detected using peroxidase-conjugated secondary antibodies. Enhanced chemiluminescence signals were acquired using a cooled charged-coupled device (CCD) camera system (ChemiDoc XRS; Bio-Rad).
RNA detection using RT-PCR.
mRNA expression was determined using end-point RT-PCR, with 18S RNA expression used as an internal standard, as previously described (6). Briefly, RNA was obtained from tissues and cells using phenol and guanidine isothiocyanate reagent (TRIzol; ThermoFisher), dissolved in RNase-free water, and quantified using an RNA-binding fluoroprobe (RiboGreen; ThermoFisher) and fluorescence spectroscopy. Subsequently, cDNAs were generated using 500 ng RNA, 250 ng of random primer, 1 mM dNTP, 40 units of RNasin, and 200 units of Moloney murine leukemia virus reverse transcriptase. Duplex PCR was performed using 0.05 volume of the cDNA, 500 nM gene-specific primers, 18S RNA primer-competimer mix (Ambion), 5 mM dNTP, 1.1 mM MgCl2, and thermostable DNA polymerase (REDTaq; Sigma). The oligonucleotides used to detail ID1-3 mRNA expression were defined by Alejandre-Alcazar et al. (1). The sense and anti-sense oligonucleotides used to detect the A isoform of ALK1 (NCBI: NM_009612) were 5′-cgg gag acg gag atc tac aa-3′ and 5′-ctc ggg tgc cat gta tct tt-3′. Single amplicons of the expected length were detected in the RT-PCR products using agarose gel electrophoresis and ethidium bromide staining. Epifluorescent-densitometric images of the gels were collected with a CCD camera system (ChemiDoc; Bio-Rad).
Immunofluorescence studies.
Cells were fixed with 4% formaldehyde in TBS and then permeabilized with 0.1% Triton X-100 in TBS. Subsequently, the cells were blocked with 1% goat serum in TBS. The cells were then reacted with the anti-pSmad1/5/8 or smoothelin antibodies, or isotype control ones, in TBS containing 0.1% BSA overnight and then with Alexa Fluor 546-conjugated secondary antibodies and 4′-6- diamidino-2-phenylindole (DAPI), which identifies nuclear DNA. Subsequently, wide-field epifluorescent images were collected employing an integrated microscope (Ti-E; Nikon) and CCD camera system (Clara-E; Andor). For the quantification of nuclear pSmad1/5 localization, random, nonoverlapping images were collected using the microscope system. The mean fluorescence signal was then determined in a region of interest corresponding to the DAPI-stained nuclear DNA using an image processing program (NIS-Elements; Nikon).
Immunohistochemistry.
Mixed R-Smad complexes in mouse pup lungs were detected using a bright-field proximity ligation assay (PLA; Ref. 55), employing previously described methods (15). The pups were killed on P10, and then the lungs were fixed in situ for 10 h by instilling 3% formaldehyde in PBS in the airways at 22 cm H2O water pressure (42). Subsequently, the lungs were removed from the thorax, and a 2–3-mm-thick transverse segment of the left lung was dehydrated, equilibrated in Clear Rite 3 (Richard Allen Scientific), and then embedded in paraffin. Subsequently, 6-µm-thick lung sections were cleared of paraffin and rehydrated before antigen retrieval was performed using 1 mM EDTA, pH 8.0, for 10 min at 95°C. Custom, high-affinity anti-Smad1/5/8 and Smad2/3 antibodies, which were raised against conserved Smad isoform sequences and characterized elsewhere (15), were conjugated to oligonucleotide proximity probes using a Duolink II Probemaker kit (Sigma). After blocking endogenous peroxidase and nonspecific protein binding, the lung sections were incubated overnight at 4°C with both the anti-Smad1/5/8 and Smad2/3 antibodies, to detect mixed Smad complexes, or with the anti-Smad2/3 antibody alone, to control for the detection system. Subsequently, antibody complexes were detected using a Duolink II in situ bright-field kit (Sigma), as directed by the manufacturer. Briefly, the lung sections were extensively washed and then reacted with a proximity probe mix. This solution contains denatured, circularized DNA probes that anneal only to proximate oligonucleotide-conjugated antibodies (17). After washing, the complexed DNA probes are amplified using single-stranded rolling circle amplification and then detected with a peroxidase-labeled hybridization probe and a colorimetric substrate (NovaRed; Vector Laboratories). After the tissues were counterstained with hematoxylin, they were dehydrated and mounted with a coverslip. To identify the Smad complexes in the lung sections, 7.5-µm-thick 0.25-µm-step z-stack images were acquired using a microscope with a motorized stage (Ti-E; Nikon) and integrated CCD camera system (DS-Ri1; Nikon), and then extended focus images were constructed (16).
ALK1 expression in mouse pup lung sections was detected using the following method. Lungs sections underwent antigen retrieval using methods described above and were blocked with 5% goat serum in PBS. Subsequently, they were reacted with a polyclonal rabbit anti-ALK1 or isotype control antibody overnight. After extensive washing, the sections were exposed to an alkaline phosphatase-conjugated secondary antibody, and immune complexes were detected with a substrate (Vector Red; Vector Laboratories). Following a counterstain with hematoxylin, the sections were dehydrated, and a coverslip was mounted. Bright-field images were then acquired and focused, as described above.
Data analysis and statistical methods.
Unless otherwise indicated, experiments were repeated at least three times, and representative images and data are shown. The relative nuclear pSmad1/5 intensity levels were determined by dividing the mean intensity levels corresponding to the nuclear ROI obtained during epifluorescence microscopy by the average signal detected in the control-treated cell group. During the initial study, we arbitrarily assessed TGF-β-stimulated nuclear pSmad1/5 localization in ~100 cells per treatment group. A power analysis using these data indicated that 45 cells per group would be necessary to detect a difference in nuclear Smad1/5 signaling due to anti-ALK1 treatment with 45 cells per group at a P < 0.05 and power of 0.8. The relative gene amplicon densitometric intensity levels were obtained by dividing the amplicon intensity signal by that associated with 18S and then normalizing it to the averaged signal detected in the control-treated cells. The relative pSmad1/5 protein level was determined by dividing the uncalibrated pSmad1/5 chemiluminescent signal level by that associated with GAPDH and then normalizing it to the average signal detected in the TGF-β1-treated control group. The data were analyzed using R (23). Unless otherwise indicated, significance for the tests was determined at P < 0.05.
RESULTS
TGF-β induces BMP R-Smad phosphorylation in mouse pup PASMC and lung fibroblasts, and in pulmonary interstitial cell lines.
To determine whether TGF-β cross-stimulates BMP R-Smad signaling pathways in the developing lung, we assessed the effects of TGF-β on Smad1/5/8 phosphorylation in mouse pup pulmonary artery smooth muscle cells (mPASMC). These cells were isolated from the pulmonary arteries of P10 mouse pups, which are undergoing the alveolar phase of lung development (3). The cells exhibited an SMC phenotype and expressed smoothelin, an SMC-specific gene (63) (data not shown). The cells were treated with 0–2.5 ng/ml TGF-β1. These doses are at the lower range of TGF-β1 levels that are detected in human and mouse tissues (11, 26, 28). For example, 2.5–5 ng/ml of active TGF-β1 has been detected in the bronchoalveolar lavage of human babies during alveolar development (28).
As shown in Fig. 1, immunoblotting revealed that treatment with as small a dose as 0.02 ng/ml TGF-β1 increased pSmad1/5 levels in mPASMC. Two bands with pSmad1/5 immunoreactivity were detected in the mPASMC and other cells used in our studies. Work by others (12) determined that the upper band comprises pSmad1 and pSmad5, while the lower one consists of pSmad5 alone. A ~53-kDa band consistent with phosphorylated Smad8 (also known as Smad9) was not detected during our studies with TGF-β and BMP. Therefore, we will refer to our detection of pSmad1/5 in the results detailed below. As expected, TGF-β1 was found to stimulate Smad2 phosphorylation, and BMP4 increased Smad1/5 phosphorylation. Smad1 and Smad2 levels were not changed by the cytokine treatment. Moreover, we found that TGF-β did not change the expression level of Smad5 in the mPASMC (data not shown). To determine whether TGF-β increases BMP R-Smad phosphorylation in other pulmonary SMC, we also assessed Smad1/5 phosphorylation following TGF-β treatment in CS54 cells, a cloned rat PASMC line (49). TGF-β was found to increase Smad1/5 phosphorylation in these cells as well. However, higher doses of TGF-β1 were required to phosphorylate the Smads in the PASMC line than in the primary PASMC.
Fig. 1.
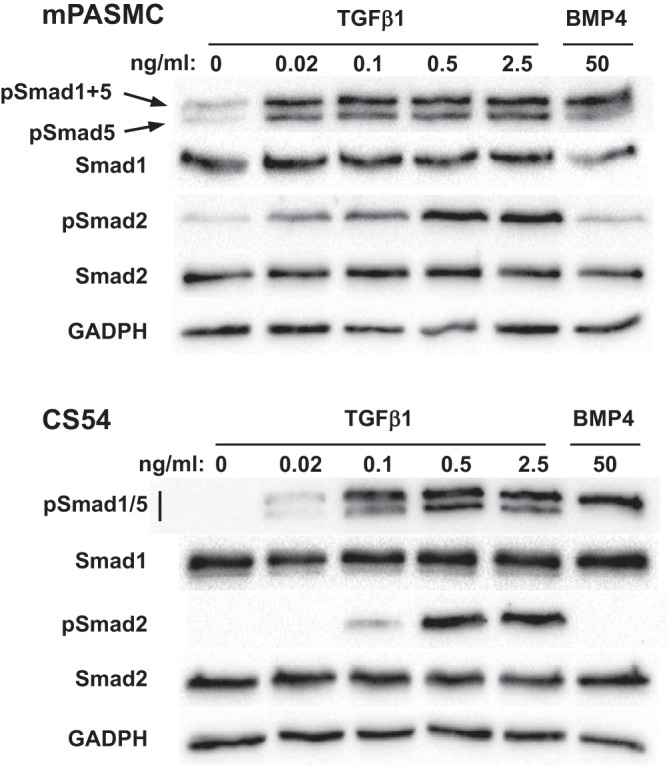
TGFβ stimulates Smad1/5 phosphorylation in primary mouse pup (m)PASMC and in an adult rat PASMC line (CS54). Cells were serum-starved for 24 h and then treated with the indicated amounts of TGFβ1 or BMP4 for 1 h. Cell lysates were then collected, and the protein expression level of the indicated phospho- and total Smads and GAPDH were detected using immunoblotting.
Recently, Schwartz et al. (50) demonstrated that TGF-β1 (2 ng/ml) increases Smad1/5 phosphorylation in NIH/3T3 cells, a fibroblast cell line derived from embryonic mice (59). However, others did not detect phosphorylation of these Smad proteins by TGF-β in mouse fibroblasts (20, 22). Given this variability of TGF-β function in the fibroblasts and the importance of these cells in regulating pulmonary development (9), we next tested whether TGF-β induces Smad1/5 phosphorylation in mouse pup lung fibroblasts (mFibroblasts). The mFibroblasts were isolated from the periphery of P10 mouse pup lungs using a previously described method (6). As shown in Fig. 2, as low a dose as 0.2 ng/ml TGF-β1 stimulated Smad1/5 and Smad2 phosphorylation in these cells. We also found that TGF-β1 stimulated Smad1/5 phosphorylation in a fibroblast cell line derived from embryonic rat lungs (RFL-6 cells). However, similar to the CS54 cells, higher dosages of TGF-β1 were required to induce Smad1/5 phosphorylation in the fibroblast cell line in comparison with the primary newborn lung fibroblasts.
Fig. 2.
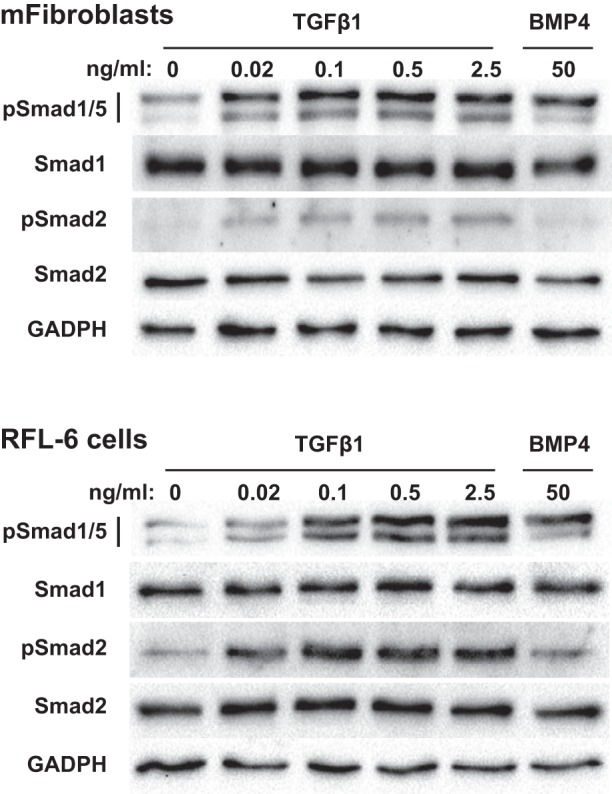
TGFβ stimulates Smad1/5 phosphorylation in mouse pup lung fibroblasts (mFibroblasts) and in a fetal lung fibroblast cell line (RFL-6 cells). The cells were serum-starved for 24 h, treated with the indicated amounts of TGFβ1 or BMP4 for 1 h, and the protein expression levels of TGFβ and BMP R-Smads and GAPDH in soluble cell lysates were determined using immunoblotting.
TGF-β-mediated BMP R-Smad signaling is detected in the developing lung.
To assess whether TGF-β-mediated BMP R-Smad stimulation occurs in vivo, we tested whether Smad1/5/8 and Smad2/3 complexes could be detected in mouse pup lungs. For this work, we used custom, high-affinity Smad protein antibodies and a recently validated PLA (15). This assay detects complexes of mixed TGF-β and BMP R-Smad in tissues at cellular resolution with high sensitivity and specificity. The principles of this assay are shown in Fig. 3A. Fixed and permeabilized lung tissue sections are incubated with anti-Smad antibodies directly conjugated with complementary DNA fragments. Only when the antibodies are held in close proximity (<40 nm) by the complexed Smad antigens, their bound DNA fragments can be annealed. When that occurs, the DNA fragments can be assembled into a circular template using a ligase. This template is then amplified using an isothermal enzyme-mediated rolling circle DNA replication in the tissue section, as described previously (55), and the product can then be detected with a histochemical probe. In the absence of complexed Smad, antibodies bound with respective Smads are not proximate, and the complexed DNA cannot be circularized, amplified, or detected. As shown in the Fig. 3B, this assay detected mixed TGF-β and BMP R-Smad complexes in the developing lung tissue. The complexes were observed in bronchial smooth muscle and epithelial cells, SMC in the wall of pulmonary arteries (as identified by their perivascular orientation between birefringent internal and external elastic laminae), and in interstitial cells within the peripheral lung. Moreover, a majority of the complexes resided in a cytosolic or perinuclear distribution. In studies that control for nonspecific endogenous DNA amplification and histochemical probe binding (Fig. 3C), the tissues are exposed to antibodies with only one DNA fragment. No complexes were detected in these control assays.
Fig. 3.

Mixed TGFβ and BMP R-Smad complexes are detected in the newborn mouse lung. Mixed Smad complexes were identified in inflation-fixed lung sections using a proximity ligation assay (PLA), as described in methods, before hematoxylin counterstain. A: tissues are exposed to polyclonal rabbit (Rb) anti-Smad1/5/8 and Smad2/3 antibodies (ATB) with conjugated complementary cDNA (gray and black lines). The DNA only hybridizes when the mixed Smad complexes bring the antibodies into close proximity, as illustrated on the left, but not when the antibodies are separate because they are bound to uncomplexed Smads, shown on the right. Following rolling circle amplification (RCA), hybridization with a peroxidase-conjugated probe, and reaction with a colorimetric substrate, the mixed Smad complexes are revealed as red dots (positive signal). In the absence of complexes, the antibodies are not in close proximity, so the RCA cannot occur, and no signal is generated. B: mixed Smad complexes are detected in epithelial cells (*) and associated smooth muscle cells (SMC) of the bronchi (B), mouse pup pulmonary artery smooth muscle cells (PASMC) (arrow) residing between the inner and outer elastic laminae of pulmonary arteries (PA) and in lung parenchymal cells (arrowheads) distal to terminal bronchi (TB) and associated with respiratory bronchi (RB), and forming alveoli (Alv) near the pleura. Lower panels are at a higher magnification images. EC, endothelial cells. C: in contrast, no PLA signal was identified in lung sections reacted only with the anti-Smad2/3 antibody probe processed as described above. Typical lung images shown from n = 3 or 4 per group; the index bars are 10 µm long.
TGF-β stimulates nuclear pSmad1/5 localization and ID gene expression in mPASMC.
Past work in cell lines suggested that TGF-β-stimulated Smad1/5 and Smad2 complexes do not transduce nuclear BMP signaling (12). However, studies in other cell types have shown that TGF-β can stimulate nuclear BMP R-Smad localization and activity (20, 34). Reasoning that downstream BMP R-Smad signaling following TGF-β stimulation might be cell type-dependent, we next tested whether TGF-β increases nuclear pSmad1/5 localization and the expression of genes that are associated with BMP R-Smad signaling in the mPASMC. As shown in Fig. 4, TGF-β1 increased pSmad1/5 nuclear compartmentation in mPASMC. Although a small amount of pSmad1/5 immunoreactivity was detected in untreated mPASMC, the level of pSmad1/5 in the nucleus was increased significantly by TGF-β1 treatment. As expected, BMP4 treatment also increased pSmad1/5 nuclear localization in these cells.
Fig. 4.
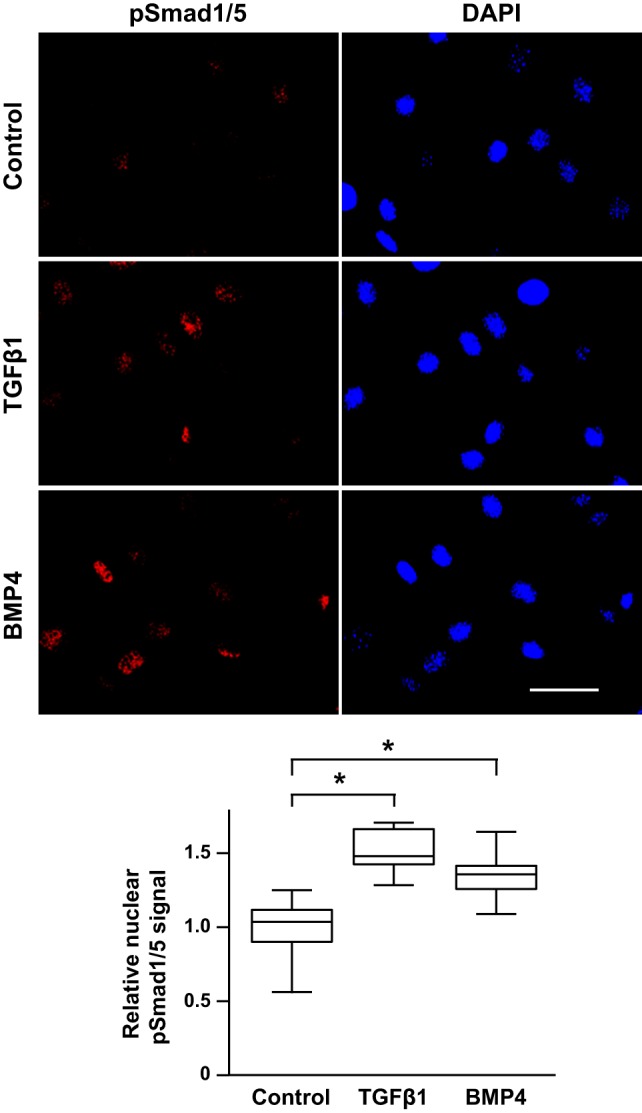
TGFβ increases pSmad1/5 nuclear compartmentation in mPASMC. Serum-starved cells were treated with 2.5 ng/ml TGFβ1, 50 ng/ml BMP4, or DMSO control for 1 h. Then the cells were fixed and permeabilized, and pSmad1/5 was detected using antibodies and wide-field epifluorescence microscopy. 4,6-diamidino-2-phenylindole (DAPI) was used to identify the nuclear compartment. Uncalibrated fluorescence signals corresponding to pSmad1/5 immunoreactivity in nuclear regions of interest were obtained and then referenced to the average nuclear pSmad1/5 fluorescence signal for the control cells. Index bar is 50 µm long. n = ~100 cells in each group; *P < 0.05.
To determine whether the TGF-β-stimulated nuclear pSmad1/5 localization was associated with increased expression of prototypical BMP-regulated genes, we tested the effect of TGF-β treatment on expression of inhibitor of DNA binding (ID; also known as inhibitor of differentiation) isoform mRNA and protein in mPASMC. The ID genes are helix-loop-helix proteins that are regulated by BMP in an activated Smad1/5-dependent manner (27, 35, 38). As shown in Fig. 5A, TGF-β1 treatment increased ID gene expression in the mPASMC. TGF-β exposure increased ID1 and ID3 mRNA levels in the cells as early as 1 h. Moreover, the TGF-β-stimulated increase of ID1 mRNA expression was found to be associated with an elevation of ID1 protein levels by 2 h (Fig. 5B). As expected, BMP4 treatment also increased the level of the ID isoform mRNA and protein expression.
Fig. 5.

TGFβ stimulates BMP R-Smad downstream signaling in mPASMC. A: TGFβ1 treatment caused increased mRNA expression of BMP-regulated genes ID1 and ID3, but not ID2. Serum-starved cells were treated with 2.5 ng/ml TGFβ1, 50 ng/ml BMP4, or DMSO control. One hour later, the cells were lysed, and RNA was collected. The mRNA expression level of the indicated genes was detected using end-point RT-PCR, as described in methods. Data shown are representative of at least three independent experiments. *P < 0.05; n = 4. B: TGFβ treatment caused increased ID1 protein expression. Serum-starved cells were either not treated or treated with 2.5 ng/ml TGFβ1 or 50 ng/ml BMP4 for the indicated times. Subsequently, the cells were lysed, and ID1 and GAPDH protein levels were determined using immunoblotting. A representative immunoblot from three independent experiments with similar results is shown.
TGF-β stimulates Smad1/5 phosphorylation via TGFβR1 but not BMPR1 in mPASMC.
To better define the mechanisms through which TGF-β regulates Smad1/5 phosphorylation in mPASMC, we used small-molecule inhibitors of the type 1 receptors for TGF-β and BMP. To inhibit TGFβR1, we used 1 µM SB505124, a dose previously shown to specifically inhibit TGFβR1, with no effect on BMPR1 activity (66). We also used 10 µM dorsomorphin, determined before to be a specific inhibitor of BMPR1 (66). As shown in Fig. 6, SB505124 completely inhibited TGF-β1-induced Smad1/5 phosphorylation in the mPASMC as well as Smad2 phosphorylation. Dorsomorphin treatment also partially inhibited TGF-β1-mediated Smad1/5 phosphorylation, while extinguishing the phosphorylation of those Smads typically stimulated by BMP4 treatment. Although this might suggest that TGF-β is acting through the BMPR1 to phosphorylate Smad1/5, others report that 10 µM dorsomorphin inhibits several kinases in addition to the BMP-activated ALKs (66). Therefore, we also employed LDN212854. Recent studies indicate that this compound is a more selective inhibitor of ALK2 over other BMPR1s (39). As shown in Fig. 7, while LDN212854 inhibited BMP4-mediated Smad1/5 phosphorylation, it did not decrease that stimulated by TGF-β treatment. Because dorsomorphin inhibits ALK1/2/3/6 and LDN212854 inhibits ALK1 to a lower degree (39), these results raise the possibility that ALK1 might play a role in TGF-β-regulated BMP signaling in mPASMC. This notion is supported by previous reports in which TGF-β was observed to directly, although weakly, interact with ALK1 (5, 44), particularly when coexpressed in cells with TGFβR2 (7). Moreover, when overexpressed in cells, ALK1 stimulates BMP R-Smad phosphorylation (10) and mediates Smad1/5 signaling in the presence of a constitutively active TGFβR1 mutant (44).
Fig. 6.
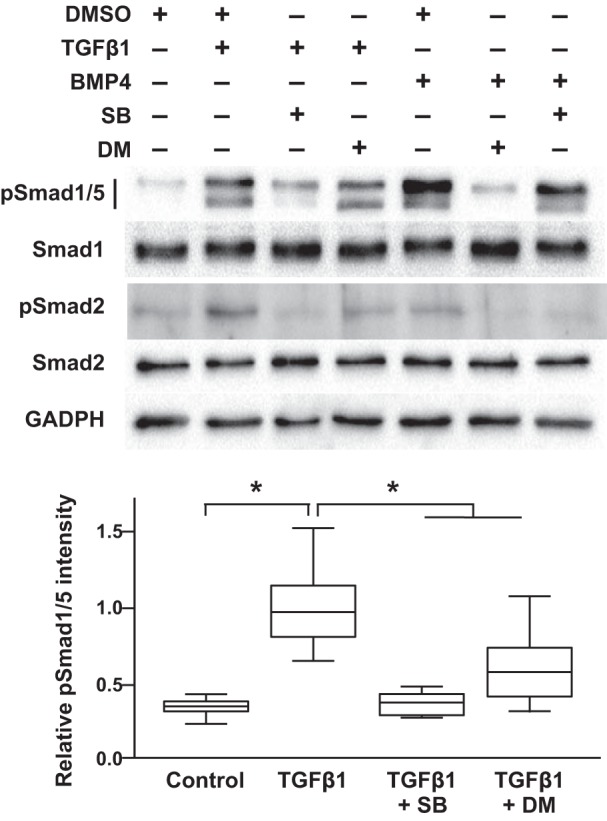
TGFβR1 mediates TGFβ-induced Smad1/5 phosphorylation in mPASMC. SB505124 (SB), an ALK4/5/7 inhibitor, prevented TGFβ-induced Smad1/5 phosphorylation, while dorsomorphin (DM), an ALK1/2/3/6 inhibitor, partly decreased TGFβ-induced Smad1/5 phosphorylation. Serum-starved cells were treated with 1 μM SB, 10 μM DM, or DMSO vehicle for 1 h before additional treatment with 2.5 ng/ml TGFβ1 or 20 ng/ml BMP4 for 1 h. Cell lysates were collected, and the protein expression level of the indicated Smads and GAPDH were detected using immunoblotting. Immunoblot images shown are representative of at least three independent experiments. For the densitometry analysis, n = 4 in each group; *P < 0.05.
Fig. 7.
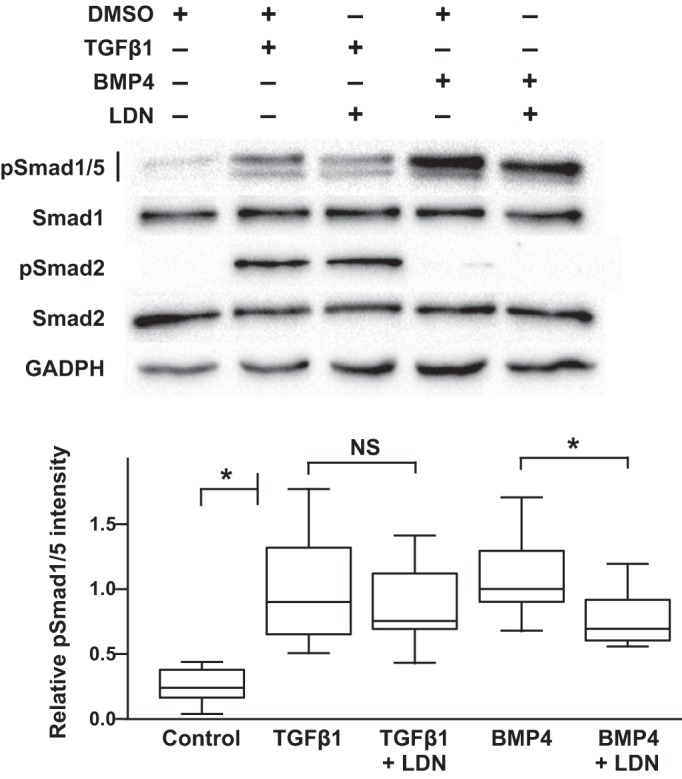
ALK2 does not inhibit TGFβ-induced BMP R-Smad phosphorylation in mPASMC. Although LDN212854 (LDN), an ALK2 inhibitor, prevented BMP-mediated Smad1/5 phosphorylation, it did not prevent the phosphorylation of these Smads by TGFβ. Serum-starved cells were treated with 125 nM LDN for 1 h before additional treatment with 2.5 ng/ml TGFβ1, 20 ng/ml BMP4, or DMSO vehicle for 1 h. Cell lysates were collected, and then the expression level of indicated Smads and GAPDH were detected using immunoblotting. Immunoblot images shown are representative of at least three independent experiments. For the densitometry analysis, n = 4 in each group; *P < 0.05.
ALK1 is expressed in the newborn lung tissue and PASMC and fibroblasts.
The role of ALK1 in mediating TGF-β-stimulated BMP R-Smad signaling in SMC is unknown. Some studies suggested that ALK1 is expressed solely within the endothelial cells but not other cells in the blood vessel wall (37, 51, 52). However, in other studies, ALK1 expression has been detected in cells residing in the wall of developing pulmonary arteries (45, 48) and in cultured aortic SMC (58, 77). As shown in Fig. 8A, we detected ALK1 mRNA in mPASMC and mFibroblasts, as well as in P10 mouse pup and adult lung samples. The specificity of the oligonucleotide probes is supported by the detection of a single amplicon of the expected size. Moreover, ALK1 immunoreactivity was detected in the cells and tissues (Fig. 8B). Equal amounts of protein were resolved using PAGE for this analysis. The Ponceau S protein blot staining confirmed effective transfer of the protein onto the membrane. The variation in the distribution of stained proteins between tissues and cells is expected. Similar to observations by others (45), the expression level of ALK1 is decreased in adult liver in comparison with that of the adult lung. In addition, the ALK1 immunoreactivity mapped to PASMC in the pup lung (Fig. 8C). Immunohistochemical studies of P10 mouse pup lung tissue sections revealed ALK1 expression in PASMC within the wall of large pulmonary arteries, as well as in interstitial cells in the peripheral lung.
Fig. 8.
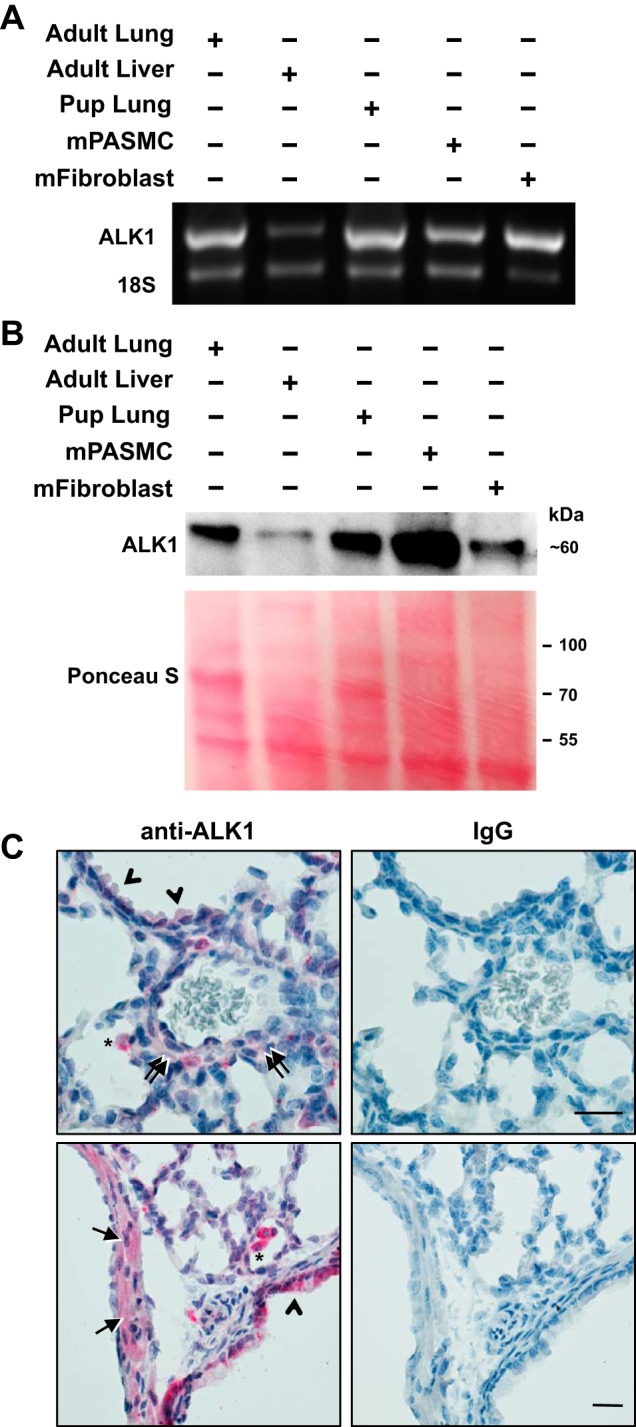
ALK1 is expressed in mPASMC, mFibroblasts, and the mouse pup lung. A: ALK1 mRNA expression in the indicated adult and P10 mouse tissues and cells detected using RT-PCR. B: ALK1 protein expression in the indicated tissues and cell lysates detected using immunoblotting. Equal amounts of proteins were resolved using PAGE; effective protein transfer to the immunoblot membrane was demonstrated using Ponceau S staining. ALK1 protein expression data shown are representative of at least three independent experiments. C: ALK1 immunoreactivity (red) was detected in smooth muscle cells in pulmonary arteries (arrows), epithelial cells (arrowheads), interstitial cells (double arrows), and macrophages (*) in P10 mouse pup lungs, counterstained with hematoxylin. Representative images from three pups; scale bar is 25 µm long.
ALK1 mediates TGF-β-stimulated BMP R-Smad signaling in mPASMC.
To test whether ALK1 mediates TGF-β-stimulated nuclear pSmad1/5 localization, we attempted to knock down ALK1 protein expression to inhibit ALK1 activity in mPASMC. However, using esiRNA to reduce ALK1 expression in mPASMC was unsuccessful because ALK1 protein levels proved very stable, despite effective abrogation of ALK1 mRNA levels. Therefore, we tested whether an anti-mALK1 antibody, which was generated using the extracellular domain of murine ALK1 as an immunogen (AA 23-119), would bind ALK1 and inhibit TGF-β-mediated Smad1/5 signaling. Others showed that an anti-human ALK1 antibody (e.g., PF-03446962) blocks BMP9-mediated signaling in cultured endothelial cells (64). However, that antibody was also found not to cross-react with mouse ALK1. To characterize the anti-mALK1, we assessed whether it binds to ALK1 on the surface of the mPASMC. As shown in Fig. 9A, the anti-mALK1 binds to live, nonpermeabilized mPASMC. Moreover, preadsorption of anti-mALK1 with mALK1-Fc inhibited the interaction of anti-mALK1 antibody with the cells, demonstrating the specificity of the antibody for ALK1 residing on the surface of the cells.
Fig. 9.

ALK1 regulates TGFβ-stimulated pSmad1/5 nuclear localization in mPASMC. A: ALK1 immunoreactivity is detected on the surface of mPASMC unless the antibody is preadsorbed with solubilized extracellular ALK1 domain (mALK1-Fc). Live cells were reacted with an anti-mALK1 antibody, an IgG2b monoclonal rat antibody that was generated against an extracellular fragment of mouse ALK1, without and with previous exposure to mALK1-Fc, or rat IgG2b, washed, fixed, and then reacted with a fluorescently labeled secondary antibody and DAPI. Subsequently, epifluorescence microscopy was performed. Typical images of two or three independent studies are shown. B: pretreatment with an anti-mALK1 antibody inhibits TGFβ-mediated BMP R-Smad phosphorylation. Cells were treated with 0 or 15 µg/ml anti-mALK1 and then incubated with 0 or 2.5 ng/ml TGFβ1 for 1 h. Subsequently, the cells were fixed and pSmad1/5 immunoreactivity, and DAPI reactivity was assessed. In additional immunoreactivity control studies, the cells were reacted with an isotype control instead of the anti-pSmad1/5 antibody. The nuclear immunoreactivity intensity signal in a region of interest identified by the DAPI reactivity was quantified and normalized with the mean level detected in the control cells. n = 45 per group; *P < 0.05. Scale bars = 50 µm.
We then tested whether this mALK1-binding antibody could block TGF-β-mediated pSmad1/5 nuclear localization. As shown in representative images in Fig. 9B, and in agreement with our previous results (Fig. 4), TGF-β1 increased nuclear pSmad1/5 immunoreactivity. However, exposing cells to anti-mALK1 before TGF-β1 treatment inhibited this increase in nuclear pSmad1/5 immunoreactivity. These observations were substantiated by quantitative analysis of nuclear pSmad1/5 signal intensity during immuno-epifluorescence microscopy. In additional control studies, preadsorption of the anti-mALK1 antibody with mALK1-Fc prevented its reduction of TGF-β-mediated Smad1/5 activation, confirming the specificity of the anti-ALK1 antibody for ALK1 (nuclear pSmad1/5 fluorescence intensity, arbitrary units: anti-mALK1 + TGF-β1: 2384 ± 466, anti-mALK1 and ALK1-Fc + TGF-β1: 2611 ± 523; n = 50, P < 0.05). Moreover, in the absence of TGF-β1 exposure, treatment of the cells with the anti-mALK1 antibody alone did not change nuclear pSmad1/5 immunoreactivity (nuclear pSmad1/5 fluorescence intensity, arbitrary units: control: 2148 ± 337, anti-mALK1: 2158 ± 369; n = 50, P > 0.05). Together, these data support the notion that ALK1 plays a role in regulating TGF-β-stimulated BMP R-Smad signaling in mPASMC.
DISCUSSION
TGF-β is a pleiotropic cytokine that plays an important role in regulating the differentiation and growth of cells that is required for the formation and maturation of several organs. TGF-β has an important function in regulating pulmonary morphogenesis. During early embryonic lung development, TGF-β helps regulate the branching of the conducting airway structures. During late lung development, studies suggest that excessive TGF-β can inhibit pulmonary microvascular assembly and alveolar development, contributing to newborn lung disease. However, surprisingly little is known about the intracellular pathways through which TGF-β regulates cells in the developing lung. Here, we characterized TGF-β-stimulated Smad signaling pathways in interstitial cells of the late-developing mouse pup lung.
TGF-β is well known to stimulate Smad2/3 phosphorylation and nuclear localization and, thereby, the expression of a select group of genes, such as plasminogen activator inhibitor-1 and connective tissue growth factor. But, recent data also suggest that TGF-β activates Smad1/5, which is typically associated with BMP signaling (8, 12, 20, 34, 69). For example, in a human breast cancer cell line, Liu et al. (34) showed that TGF-β stimulates Smad1 phosphorylation in as little as 10 min. However, this mechanism has also been found to be highly variable in various cell lines (8, 22), and it has not been reported in cells derived from the newborn lung. While detailing mechanisms through which TGF-β regulates gene expression in the developing lung, we unexpectedly observed that TGF-β stimulates Smad1/5 phosphorylation in PASMC and lung fibroblasts derived from mouse pups. This effect of TGF-β was robust; it was detected through a wide range of TGF-β levels in the mouse pup lung cells, including those levels detected in tracheal aspirates from babies with and without lung disease (28). Our results are similar to those observed in vascular smooth muscle cells obtained from developed organs. For example, TGF-β-stimulated Smad1/5 phosphorylation has been detected in aortic SMC (58), and PASMC derived from the lungs of adult human beings (13, 50, 73). In those studies, TGF-β-induced Smad1/5 phosphorylation was increased by 0.5–1 h of treatment. The increase in pSmad1/5 by TGF-β has also been shown to be transient. In cell lines, TGF-β-induced Smad1/5 phosphorylation lasted 1–2 h (12, 22, 69). In bovine arterial endothelial cells, TGF-β-stimulated Smad1/5 phosphorylation lasted 90 min, whereas Smad2 phosphorylation lasted longer but also diminished at ~2 h (20). Despite the relatively short duration of TGF-β-stimulated Smad1/5 phosphorylation, there appeared to be a persistent change in expression of BMP R-Smad-regulated genes. For example, TGF-β-stimulated ID1 protein expression was found to be increased in the endothelial cells for up to 30 h (20). Moreover, in adult PASMC, although TGF-β increased ID isoform mRNA levels for only about 1 h, the ID1 and ID2 protein levels remained elevated at 24 h posttreatment (60).
This TGF-β-stimulated BMP R-Smad phosphorylation detected in the newborn PASMC and lung fibroblasts likely has in vivo relevance in the developing lung. This is because we detected mixed Smad1/5/8·Smad2/3 complexes indicative of this cross-talk signaling mechanism in mouse pup lungs. Previous work demonstrated the specificity of the antibodies used here (15). In those studies, the mixed-Smad complex detection using PLA was decreased when the relevant Smads were knocked down in cells or the antibody probes were preadsorbed with the antigenic peptide. Moreover, the efficiency of this mixed-Smad detection technique was demonstrated by their ability to detect increased mixed-Smad complexes in tissues of transgenic mice with overexpression of active TGF-β1. In our work, the mixed-Smad complexes were detected primarily in the cytosol or perinuclear region of smooth muscle cells residing within the pulmonary arteries, cells within the peripheral lung parenchyma, and in epithelial cells and SMC in the conducting airways. These results suggest that TGF-β-mediated BMP R-Smad signaling might have a role in regulating several different cell types during late lung development.
Past work in cultured epithelial cells showed that the TGF-β-stimulated mixed R-Smad complexes do not include Smad4, which is required for nuclear translocation of the activated Smad complex, and do not stimulate BMP-regulated gene expression (12). However, studies in another cell type (e.g., Hs578T, a TGF-β-sensitive human breast cancer cell line), using heterologous Smad1 and Smad4 expression showed that TGF-β stimulated the formation of Smad1·and Smad4 complexes and the nuclear translocation of Smad1 (34). To test the functional significance of the TGF-β-stimulated Smad1/5 phosphorylation in mouse pup PASMC, we investigated whether TGF-β regulated downstream BMP R-Smad signaling. TGF-β treatment increased nuclear localization of phosphorylated Smad1/5 and unregulated ID isoform mRNA levels, which are indicative of nuclear BMP R-Smad signaling. These results suggest that TGF-β may control expression of PASMC genes previously thought to be regulated only through BMP signaling.
Although our kinase inhibitor data suggested that ALK1 might play a role in TGF-β signaling in mPASMC, several investigators have previously indicated that ALK1 is not expressed in vascular smooth muscle cells (37, 51, 52). However, their work employed methods that might not be sensitive enough to map ALK1 expression to cells residing in the vascular wall or lung interstitium. In other studies, ALK1 transcripts were detected within vascular SMC using in situ hybridization (45, 48). There might also be differential expression of ALK1 in smooth muscle cells based on the vascular origin; although ALK1 has been detected in SMC in the aorta (58, 77), it was not detected in SMC in colonic arteries and veins (20). We detected ALK1 protein expression in mouse pup PASMC and other lung cells in vivo, and ALK1 mRNA and protein were detected in PASMC derived from these animals. Pulmonary expression of ALK1 may also be species-specific. ALK1 mRNA has been detected in the lung homogenates of newborn mice (2) and rats (24) and mapped to PASMC of babies (2). But, ALK1 expression has been found not to be localized to SMC of newborn lamb pulmonary arteries (37).
We also determined that ALK1 mediates TGF-β-stimulated BMP R-Smad activation in mouse pup PASMC. For these studies, we used an antibody that was generated against the extracellular domain of mouse ALK1 (anti-mALK1) to test the role of ALK1 in regulating TGF-β-Smad1/5 signaling. We demonstrated that the antibody binds to the extracellular domains of ALK1 and inhibited TGF-β-mediated Smad1/5 phosphorylation and nuclear localization in mPASMC. Our data suggesting that ALK1 mediates TGF-β-stimulated Smad1/5 phosphorylation in mPASMC is in contrast with previous observations in adult aortic SMC (58). In that study, human aortic SMC transduced with a dominant-negative ALK1 mutant retained Smad1/5 phosphorylation when treated with TGF-β. Several studies suggest that ALK1 has a role in regulating angiogenesis and vasculogenesis. ALK1 is detected in highly vascularized tissue (e.g., lung and placenta), particularly, in endothelial cells (48, 51, 52, 70). ALK1 KO mice die at midgestation with defects in vascularization, including dysregulated arteriovenous development and fusion of major arteries and veins (44, 62). Moreover, studies suggest that the proangiogenic effects of TGF-β and BMPs are mediated by endothelial cell ALK1 (19). In microvascular endothelial cells from a variety of sources, studies suggest that ALK1 inhibits cell proliferation and migration, which might facilitate its role in angiogenesis (30). Our detection of ALK1 in PASMC suggests that it might also have a role in regulating vascular development through its effects in these cells.
Although our work identified an ALK1-dependent TGF-β-stimulated BMP R-Smad signaling pathway in mouse PASMC, we cannot yet determine how this mechanism might regulate lung vascular development. Previous studies using knockout mice suggest that ALK1 plays a critical role in regulating angiogenesis during gestation (44, 62). In particular, ALK1 appears to regulate the cellular assembly of the arterial wall. This is because histological examination of the ALK1-deficient mouse embryos generated during those studies revealed that SM22α- and αSMA-expressing cells, which are indicative of SMC and fibroblasts, do not invest within the vascular wall, subjacent to the endothelial cells. However, it is unclear whether these vascular defects were reflective of disrupted TGF-β-mediated or BMP-stimulated ALK1 signaling, or both. This is because ALK1 is a target of both TGF-β and BMP signaling in SMC. Currently, tools are not available to selectively inhibit TGF-β, but not BMP, signaling through ALK1 in vivo. Therefore, it is not yet possible to define the role of TGF-β-stimulated BMP R-Smad signaling in vascular development.
In conclusion, our studies indicate that it is important to consider both TGF-β- and BMP R-Smad signaling systems in determining how TGF-β regulates PASMC gene expression and function in the developing lung.
GRANTS
This work was supported by a grant from the National Institutes of Health (HL-125715, to J. D. Roberts, Jr.) and the Massachusetts General Hospital Department of Anesthesia, Critical Care, and Pain Medicine.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
H.Z., L.D., Y.Z., K.C.F., and J.D.R. conceived and designed research; H.Z., L.D., Y.Z., K.C.F., and J.D.R. performed experiments; H.Z., L.D., Y.Z., K.C.F., and J.D.R. analyzed data; H.Z., L.D., Y.Z., K.C.F., and J.D.R. interpreted results of experiments; H.Z., L.D., Y.Z., K.C.F., and J.D.R. prepared figures; H.Z. and J.D.R. drafted manuscript; H.Z., L.D., Y.Z., K.C.F., and J.D.R. edited and revised manuscript; H.Z., L.D., Y.Z., K.C.F., and J.D.R. approved final version of manuscript.
ACKNOWLEDGMENTS
We acknowledge the helpful suggestions of Drs. Rajeev Malhotra, Division of Cardiology Massachusetts General Hospital, and Lalage Wakefield, National Cancer Institute. This work was submitted in partial fulfillment of requirements for a Ph.D. degree at Guangzhou Medical University (H. Zhang).
REFERENCES
- 1.Alejandre-Alcázar MA, Kwapiszewska G, Reiss I, Amarie OV, Marsh LM, Sevilla-Pérez J, Wygrecka M, Eul B, Köbrich S, Hesse M, Schermuly RT, Seeger W, Eickelberg O, Morty RE. Hyperoxia modulates TGF-β/BMP signaling in a mouse model of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 292: L537–L549, 2007. doi: 10.1152/ajplung.00050.2006. [DOI] [PubMed] [Google Scholar]
- 2.Alejandre-Alcázar MA, Michiels-Corsten M, Vicencio AG, Reiss I, Ryu J, de Krijger RR, Haddad GG, Tibboel D, Seeger W, Eickelberg O, Morty RE. TGF-β signaling is dynamically regulated during the alveolarization of rodent and human lungs. Dev Dyn 237: 259–269, 2008. doi: 10.1002/dvdy.21403. [DOI] [PubMed] [Google Scholar]
- 3.Amy RW, Bowes D, Burri PH, Haines J, Thurlbeck WM. Postnatal growth of the mouse lung. J Anat 124: 131–151, 1977. [PMC free article] [PubMed] [Google Scholar]
- 4.Aschner Y, Downey GP. Transforming growth factor-β: master regulator of the respiratory system in health and disease. Am J Respir Cell Mol Biol 54: 647–655, 2016. doi: 10.1165/rcmb.2015-0391TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Attisano L, Cárcamo J, Ventura F, Weis FM, Massagué J, Wrana JL. Identification of human activin and TGF beta type I receptors that form heteromeric kinase complexes with type II receptors. Cell 75: 671–680, 1993. doi: 10.1016/0092-8674(93)90488-C. [DOI] [PubMed] [Google Scholar]
- 6.Bachiller PR, Nakanishi H, Roberts JD Jr. Transforming growth factor-β modulates the expression of nitric oxide signaling enzymes in the injured developing lung and in vascular smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 298: L324–L334, 2010. doi: 10.1152/ajplung.00181.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bassing CH, Yingling JM, Howe DJ, Wang T, He WW, Gustafson ML, Shah P, Donahoe PK, Wang XF. A transforming growth factor β type I receptor that signals to activate gene expression. Science 263: 87–89, 1994. doi: 10.1126/science.8272871. [DOI] [PubMed] [Google Scholar]
- 8.Bharathy S, Xie W, Yingling JM, Reiss M. Cancer-associated transforming growth factor β type II receptor gene mutant causes activation of bone morphogenic protein-Smads and invasive phenotype. Cancer Res 68: 1656–1666, 2008. doi: 10.1158/0008-5472.CAN-07-5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boström H, Willetts K, Pekny M, Levéen P, Lindahl P, Hedstrand H, Pekna M, Hellström M, Gebre-Medhin S, Schalling M, Nilsson M, Kurland S, Törnell J, Heath JK, Betsholtz C. PDGF-A signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell 85: 863–873, 1996. doi: 10.1016/S0092-8674(00)81270-2. [DOI] [PubMed] [Google Scholar]
- 10.Chen YG, Massagué J. Smad1 recognition and activation by the ALK1 group of transforming growth factor-β family receptors. J Biol Chem 274: 3672–3677, 1999. doi: 10.1074/jbc.274.6.3672. [DOI] [PubMed] [Google Scholar]
- 11.Czarkowska-Paczek B, Bartlomiejczyk I, Przybylski J. The serum levels of growth factors: PDGF, TGF-beta and VEGF are increased after strenuous physical exercise. J Physiol Pharmacol 57: 189–197, 2006. [PubMed] [Google Scholar]
- 12.Daly AC, Randall RA, Hill CS. Transforming growth factor beta-induced Smad1/5 phosphorylation in epithelial cells is mediated by novel receptor complexes and is essential for anchorage-independent growth. Mol Cell Biol 28: 6889–6902, 2008. doi: 10.1128/MCB.01192-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davies RJ, Holmes AM, Deighton J, Long L, Yang X, Barker L, Walker C, Budd DC, Upton PD, Morrell NW. BMP type II receptor deficiency confers resistance to growth inhibition by TGF-β in pulmonary artery smooth muscle cells: role of proinflammatory cytokines. Am J Physiol Lung Cell Mol Physiol 302: L604–L615, 2012. doi: 10.1152/ajplung.00309.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 425: 577–584, 2003. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 15.Flanders KC, Heger CD, Conway C, Tang B, Sato M, Dengler SL, Goldsmith PK, Hewitt SM, Wakefield LM. Brightfield proximity ligation assay reveals both canonical and mixed transforming growth factor-β/bone morphogenetic protein Smad signaling complexes in tissue sections. J Histochem Cytochem 62: 846–863, 2014. doi: 10.1369/0022155414550163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Forster B, Van De Ville D, Berent J, Sage D, Unser M. Complex wavelets for extended depth-of-field: a new method for the fusion of multichannel microscopy images. Microsc Res Tech 65: 33–42, 2004. doi: 10.1002/jemt.20092. [DOI] [PubMed] [Google Scholar]
- 17.Fredriksson S, Gullberg M, Jarvius J, Olsson C, Pietras K, Gústafsdóttir SM, Ostman A, Landegren U. Protein detection using proximity-dependent DNA ligation assays. Nat Biotechnol 20: 473–477, 2002. doi: 10.1038/nbt0502-473. [DOI] [PubMed] [Google Scholar]
- 18.Gauldie J, Galt T, Bonniaud P, Robbins C, Kelly M, Warburton D. Transfer of the active form of transforming growth factor-β1 gene to newborn rat lung induces changes consistent with bronchopulmonary dysplasia. Am J Pathol 163: 2575–2584, 2003. doi: 10.1016/S0002-9440(10)63612-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C, Karlsson S, ten Dijke P. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFβ/ALK5 signaling. Mol Cell 12: 817–828, 2003. doi: 10.1016/S1097-2765(03)00386-1. [DOI] [PubMed] [Google Scholar]
- 20.Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P. Balancing the activation state of the endothelium via two distinct TGF-β type I receptors. EMBO J 21: 1743–1753, 2002. doi: 10.1093/emboj/21.7.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hinck AP. Structural studies of the TGF-βs and their receptors—insights into evolution of the TGF-β superfamily. FEBS Lett 586: 1860–1870, 2012. doi: 10.1016/j.febslet.2012.05.028. [DOI] [PubMed] [Google Scholar]
- 22.Holtzhausen A, Golzio C, How T, Lee YH, Schiemann WP, Katsanis N, Blobe GC. Novel bone morphogenetic protein signaling through Smad2 and Smad3 to regulate cancer progression and development. FASEB J 28: 1248–1267, 2014. doi: 10.1096/fj.13-239178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ihaka R, Gentleman R. R: A language for data analysis and graphics. J Comput Graph Stat 5: 299–314, 1996. [Google Scholar]
- 24.Jin M, Lee J, Lee KY, Jin Z, Pak JH, Kim HS. Alteration of TGF-β-ALK-Smad signaling in hyperoxia-induced bronchopulmonary dysplasia model of newborn rats. Exp Lung Res 42: 354–364, 2016. doi: 10.1080/01902148.2016.1226448. [DOI] [PubMed] [Google Scholar]
- 25.Johnson BA, Lowenstein CJ, Schwarz MA, Nakayama DK, Pitt BR, Davies P. Culture of pulmonary microvascular smooth muscle cells from intraacinar arteries of the rat: characterization and inducible production of nitric oxide. Am J Respir Cell Mol Biol 10: 604–612, 1994. doi: 10.1165/ajrcmb.10.6.7516171. [DOI] [PubMed] [Google Scholar]
- 26.Khan SA, Joyce J, Tsuda T. Quantification of active and total transforming growth factor-β levels in serum and solid organ tissues by bioassay. BMC Res Notes 5: 636, 2012. doi: 10.1186/1756-0500-5-636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Korchynskyi O, ten Dijke P. Identification and functional characterization of distinct critically important bone morphogenetic protein-specific response elements in the Id1 promoter. J Biol Chem 277: 4883–4891, 2002. doi: 10.1074/jbc.M111023200. [DOI] [PubMed] [Google Scholar]
- 28.Kotecha S, Wangoo A, Silverman M, Shaw RJ. Increase in the concentration of transforming growth factor beta-1 in bronchoalveolar lavage fluid before development of chronic lung disease of prematurity. J Pediatr 128: 464–469, 1996. doi: 10.1016/S0022-3476(96)70355-4. [DOI] [PubMed] [Google Scholar]
- 29.Kumarasamy A, Schmitt I, Nave AH, Reiss I, van der Horst I, Dony E, Roberts JD Jr, de Krijger RR, Tibboel D, Seeger W, Schermuly RT, Eickelberg O, Morty RE. Lysyl oxidase activity is dysregulated during impaired alveolarization of mouse and human lungs. Am J Respir Crit Care Med 180: 1239–1252, 2009. doi: 10.1164/rccm.200902-0215OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lamouille S, Mallet C, Feige JJ, Bailly S. Activin receptor-like kinase 1 is implicated in the maturation phase of angiogenesis. Blood 100: 4495–4501, 2002. doi: 10.1182/blood.V100.13.4495. [DOI] [PubMed] [Google Scholar]
- 31.Lee KJ, Czech L, Waypa GB, Farrow KN. Isolation of pulmonary artery smooth muscle cells from neonatal mice. J Vis Exp 80: e50889, 2013 10.3791/50889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li A, Ma S, Smith SM, Lee MK, Fischer A, Borok Z, Bellusci S, Li C, Minoo P. Mesodermal ALK5 controls lung myofibroblast versus lipofibroblast cell fate. BMC Biol 14: 19, 2016. doi: 10.1186/s12915-016-0242-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu J, Tseu I, Wang J, Tanswell K, Post M. Transforming growth factor β2, but not β1 and β3, is critical for early rat lung branching. Dev Dyn 217: 343–360, 2000. doi:. [DOI] [PubMed] [Google Scholar]
- 34.Liu X, Yue J, Frey RS, Zhu Q, Mulder KM. Transforming growth factor β signaling through Smad1 in human breast cancer cells. Cancer Res 58: 4752–4757, 1998. [PubMed] [Google Scholar]
- 35.López-Rovira T, Chalaux E, Massagué J, Rosa JL, Ventura F. Direct binding of Smad1 and Smad4 to two distinct motifs mediates bone morphogenetic protein-specific transcriptional activation of Id1 gene. J Biol Chem 277: 3176–3185, 2002. doi: 10.1074/jbc.M106826200. [DOI] [PubMed] [Google Scholar]
- 36.Massagué J. TGF-β signal transduction. Annu Rev Biochem 67: 753–791, 1998. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 37.Mata-Greenwood E, Meyrick B, Steinhorn RH, Fineman JR, Black SM. Alterations in TGF-β1 expression in lambs with increased pulmonary blood flow and pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 285: L209–L221, 2003. doi: 10.1152/ajplung.00171.2002. [DOI] [PubMed] [Google Scholar]
- 38.Miyazono K, Miyazawa K. Id: a target of BMP signaling. Sci STKE 2002: pe40, 2002. doi: 10.1126/stke.2002.151.pe40. [DOI] [PubMed] [Google Scholar]
- 39.Mohedas AH, Xing X, Armstrong KA, Bullock AN, Cuny GD, Yu PB. Development of an ALK2-biased BMP type I receptor kinase inhibitor. ACS Chem Biol 8: 1291–1302, 2013. doi: 10.1021/cb300655w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mokres LM, Parai K, Hilgendorff A, Ertsey R, Alvira CM, Rabinovitch M, Bland RD. Prolonged mechanical ventilation with air induces apoptosis and causes failure of alveolar septation and angiogenesis in lungs of newborn mice. Am J Physiol Lung Cell Mol Physiol 298: L23–L35, 2010. doi: 10.1152/ajplung.00251.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morty RE, Königshoff M, Eickelberg O. Transforming growth factor-β signaling across ages: from distorted lung development to chronic obstructive pulmonary disease. Proc Am Thorac Soc 6: 607–613, 2009. doi: 10.1513/pats.200908-087RM. [DOI] [PubMed] [Google Scholar]
- 42.Nakanishi H, Sugiura T, Streisand JB, Lonning SM, Roberts JD Jr. TGF-β-neutralizing antibodies improve pulmonary alveologenesis and vasculogenesis in the injured newborn lung. Am J Physiol Lung Cell Mol Physiol 293: L151–L161, 2007. doi: 10.1152/ajplung.00389.2006. [DOI] [PubMed] [Google Scholar]
- 43.Nicola T, Hagood JS, James ML, Macewen MW, Williams TA, Hewitt MM, Schwiebert L, Bulger A, Oparil S, Chen YF, Ambalavanan N. Loss of Thy-1 inhibits alveolar development in the newborn mouse lung. Am J Physiol Lung Cell Mol Physiol 296: L738–L750, 2009. doi: 10.1152/ajplung.90603.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oh SP, Seki T, Goss KA, Imamura T, Yi Y, Donahoe PK, Li L, Miyazono K, ten Dijke P, Kim S, Li E. Activin receptor-like kinase 1 modulates transforming growth factor-β1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci USA 97: 2626–2631, 2000. doi: 10.1073/pnas.97.6.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Panchenko MP, Williams MC, Brody JS, Yu Q. Type I receptor serine-threonine kinase preferentially expressed in pulmonary blood vessels. Am J Physiol Lung Cell Mol Physiol 270: L547–L558, 1996. [DOI] [PubMed] [Google Scholar]
- 46.Pelton RW, Johnson MD, Perkett EA, Gold LI, Moses HL. Expression of transforming growth factor-β1, -β2, and -β3 mRNA and protein in the murine lung. Am J Respir Cell Mol Biol 5: 522–530, 1991. doi: 10.1165/ajrcmb/5.6.522. [DOI] [PubMed] [Google Scholar]
- 47.Pelton RW, Saxena B, Jones M, Moses HL, Gold LI. Immunohistochemical localization of TGF beta 1, TGF beta 2, and TGF beta 3 in the mouse embryo: expression patterns suggest multiple roles during embryonic development. J Cell Biol 115: 1091–1105, 1991. doi: 10.1083/jcb.115.4.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roelen BA, van Rooijen MA, Mummery CL. Expression of ALK-1, a type 1 serine/threonine kinase receptor, coincides with sites of vasculogenesis and angiogenesis in early mouse development. Dev Dyn 209: 418–430, 1997. doi:. [DOI] [PubMed] [Google Scholar]
- 49.Rothman A, Kulik TJ, Taubman MB, Berk BC, Smith CW, Nadal-Ginard B. Development and characterization of a cloned rat pulmonary arterial smooth muscle cell line that maintains differentiated properties through multiple subcultures. Circulation 86: 1977–1986, 1992. doi: 10.1161/01.CIR.86.6.1977. [DOI] [PubMed] [Google Scholar]
- 50.Schwartze JT, Becker S, Sakkas E, Wujak LA, Niess G, Usemann J, Reichenberger F, Herold S, Vadasz I, Mayer K, Seeger W, Morty RE. Glucocorticoids recruit TGFbr3 and Smad1 to shift transforming growth factor-beta signaling from the TGFbr1/Smad2/3 axis to the Acvrl1/Smad1 axis in lung fibroblasts. J Biol Chem 289: 3262–3275, 2014. doi: 10.1074/jbc.M113.541052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seki T, Hong KH, Oh SP. Nonoverlapping expression patterns of ALK1 and ALK5 reveal distinct roles of each receptor in vascular development. Lab Invest 86: 116–129, 2006. doi: 10.1038/labinvest.3700376. [DOI] [PubMed] [Google Scholar]
- 52.Seki T, Yun J, Oh SP. Arterial endothelium-specific activin receptor-like kinase 1 expression suggests its role in arterialization and vascular remodeling. Circ Res 93: 682–689, 2003. doi: 10.1161/01.RES.0000095246.40391.3B. [DOI] [PubMed] [Google Scholar]
- 53.Serra R, Pelton RW, Moses HL. TGFβ1 inhibits branching morphogenesis and N-myc expression in lung bud organ cultures. Development 120: 2153–2161, 1994. [DOI] [PubMed] [Google Scholar]
- 54.Silva DM, Nardiello C, Pozarska A, Morty RE. Recent advances in the mechanisms of lung alveolarization and the pathogenesis of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 309: L1239–L1272, 2015. doi: 10.1152/ajplung.00268.2015. [DOI] [PubMed] [Google Scholar]
- 55.Söderberg O, Gullberg M, Jarvius M, Ridderstråle K, Leuchowius KJ, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson LG, Landegren U. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 3: 995–1000, 2006. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- 56.Sureshbabu A, Syed MA, Boddupalli CS, Dhodapkar MV, Homer RJ, Minoo P, Bhandari V. Conditional overexpression of TGFβ1 promotes pulmonary inflammation, apoptosis and mortality via TGFβR2 in the developing mouse lung. Respir Res 16: 4, 2015. doi: 10.1186/s12931-014-0162-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Takeda M, Otsuka F, Nakamura K, Inagaki K, Suzuki J, Miura D, Fujio H, Matsubara H, Date H, Ohe T, Makino H. Characterization of the bone morphogenetic protein (BMP) system in human pulmonary arterial smooth muscle cells isolated from a sporadic case of primary pulmonary hypertension: roles of BMP type IB receptor (activin receptor-like kinase-6) in the mitotic action. Endocrinology 145: 4344–4354, 2004. doi: 10.1210/en.2004-0234. [DOI] [PubMed] [Google Scholar]
- 58.Tang Y, Yang X, Friesel RE, Vary CP, Liaw L. Mechanisms of TGF-β-induced differentiation in human vascular smooth muscle cells. J Vasc Res 48: 485–494, 2011. doi: 10.1159/000327776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Todaro GJ, Green H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J Cell Biol 17: 299–313, 1963. doi: 10.1083/jcb.17.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Upton PD, Davies RJ, Tajsic T, Morrell NW. Transforming growth factor-β1 represses bone morphogenetic protein-mediated Smad signaling in pulmonary artery smooth muscle cells via Smad3. Am J Respir Cell Mol Biol 49: 1135–1145, 2013. doi: 10.1165/rcmb.2012-0470OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Upton PD, Davies RJ, Trembath RC, Morrell NW. Bone morphogenetic protein (BMP) and activin type II receptors balance BMP9 signals mediated by activin receptor-like kinase-1 in human pulmonary artery endothelial cells. J Biol Chem 284: 15794–15804, 2009. doi: 10.1074/jbc.M109.002881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Urness LD, Sorensen LK, Li DY. Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nat Genet 26: 328–331, 2000. doi: 10.1038/81634. [DOI] [PubMed] [Google Scholar]
- 63.van Eys GJ, Niessen PM, Rensen SS. Smoothelin in vascular smooth muscle cells. Trends Cardiovasc Med 17: 26–30, 2007. doi: 10.1016/j.tcm.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 64.van Meeteren LA, Thorikay M, Bergqvist S, Pardali E, Gallo Stampino C, Hu-Lowe D, Goumans MJ, ten Dijke P. Anti-human activin receptor-like kinase 1 (ALK1) antibody attenuates bone morphogenetic protein 9 (BMP9)-induced ALK1 signaling and interferes with endothelial cell sprouting. J Biol Chem 287: 18551–18561, 2012. doi: 10.1074/jbc.M111.338103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vicencio AG, Lee CG, Cho SJ, Eickelberg O, Chuu Y, Haddad GG, Elias JA. Conditional overexpression of bioactive transforming growth factor-β1 in neonatal mouse lung: a new model for bronchopulmonary dysplasia? Am J Respir Cell Mol Biol 31: 650–656, 2004. doi: 10.1165/rcmb.2004-0092OC. [DOI] [PubMed] [Google Scholar]
- 66.Vogt J, Traynor R, Sapkota GP. The specificities of small molecule inhibitors of the TGFβ and BMP pathways. Cell Signal 23: 1831–1842, 2011. doi: 10.1016/j.cellsig.2011.06.019. [DOI] [PubMed] [Google Scholar]
- 67.Warburton D, Schwarz M, Tefft D, Flores-Delgado G, Anderson KD, Cardoso WV. The molecular basis of lung morphogenesis. Mech Dev 92: 55–81, 2000. doi: 10.1016/S0925-4773(99)00325-1. [DOI] [PubMed] [Google Scholar]
- 68.Witsch TJ, Turowski P, Sakkas E, Niess G, Becker S, Herold S, Mayer K, Vadász I, Roberts JD Jr, Seeger W, Morty RE. Deregulation of the lysyl hydroxylase matrix cross-linking system in experimental and clinical bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 306: L246–L259, 2014. doi: 10.1152/ajplung.00109.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wrighton KH, Lin X, Yu PB, Feng XH. Transforming growth factor β can stimulate Smad1 phosphorylation independently of bone morphogenic protein receptors. J Biol Chem 284: 9755–9763, 2009. doi: 10.1074/jbc.M809223200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu X, Robinson CE, Fong HW, Crabtree JS, Rodriguez BR, Roe BA, Gimble JM. Cloning and characterization of the murine activin receptor like kinase-1 (ALK-1) homolog. Biochem Biophys Res Commun 216: 78–83, 1995. doi: 10.1006/bbrc.1995.2594. [DOI] [PubMed] [Google Scholar]
- 71.Yang X, Long L, Southwood M, Rudarakanchana N, Upton PD, Jeffery TK, Atkinson C, Chen H, Trembath RC, Morrell NW. Dysfunctional Smad signaling contributes to abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circ Res 96: 1053–1063, 2005. doi: 10.1161/01.RES.0000166926.54293.68. [DOI] [PubMed] [Google Scholar]
- 72.Yu PB, Beppu H, Kawai N, Li E, Bloch KD. Bone morphogenetic protein (BMP) type II receptor deletion reveals BMP ligand-specific gain of signaling in pulmonary artery smooth muscle cells. J Biol Chem 280: 24443–24450, 2005. doi: 10.1074/jbc.M502825200. [DOI] [PubMed] [Google Scholar]
- 73.Yung LM, Nikolic I, Paskin-Flerlage SD, Pearsall RS, Kumar R, Yu PB. A selective transforming growth factor-β ligand trap attenuates pulmonary hypertension. Am J Respir Crit Care Med 194: 1140–1151, 2016. doi: 10.1164/rccm.201510-1955OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhao J, Sime PJ, Bringas P Jr, Gauldie J, Warburton D. Epithelium-specific adenoviral transfer of a dominant-negative mutant TGF-β type II receptor stimulates embryonic lung branching morphogenesis in culture and potentiates EGF and PDGF-AA. Mech Dev 72: 89–100, 1998. doi: 10.1016/S0925-4773(98)00019-7. [DOI] [PubMed] [Google Scholar]
- 75.Zhao J, Sime PJ, Bringas P Jr, Tefft JD, Buckley S, Bu D, Gauldie J, Warburton D. Spatial-specific TGF-β1 adenoviral expression determines morphogenetic phenotypes in embryonic mouse lung. Eur J Cell Biol 78: 715–725, 1999. doi: 10.1016/S0171-9335(99)80040-5. [DOI] [PubMed] [Google Scholar]
- 76.Zhou L, Dey CR, Wert SE, Whitsett JA. Arrested lung morphogenesis in transgenic mice bearing an SP-C-TGF-β1 chimeric gene. Dev Biol 175: 227–238, 1996. doi: 10.1006/dbio.1996.0110. [DOI] [PubMed] [Google Scholar]
- 77.Zhu D, Mackenzie NC, Shanahan CM, Shroff RC, Farquharson C, MacRae VE. BMP-9 regulates the osteoblastic differentiation and calcification of vascular smooth muscle cells through an ALK1-mediated pathway. J Cell Mol Med 19: 165–174, 2015. doi: 10.1111/jcmm.12373. [DOI] [PMC free article] [PubMed] [Google Scholar]