

Abstract
Glucose is a crucial substrate essential for cell survival and function. Changes in glucose levels impact neuronal activity and glucose deprivation increases feeding. Several brain regions have been shown to respond to glucoprivation, including the nucleus of the solitary tract (NTS) in the brain stem. The NTS is the primary site in the brain that receives visceral afferent information from the gastrointestinal tract. The catecholaminergic (CA) subpopulation within the NTS modulates many homeostatic functions including cardiovascular reflexes, respiration, food intake, arousal, and stress. However, it is not known if they respond to changes in glucose. Here we determined whether NTS-CA neurons respond to changes in glucose concentration and the mechanism involved. We found that decreasing glucose concentrations from 5 mM to 2 mM to 1 mM, significantly decreased action potential firing in a cell-attached preparation, whereas increasing it back to 5 mM increased the firing rate. This effect was dependent on glutamate release from afferent terminals and required presynaptic 5-HT3Rs. Decreasing the glucose concentration also decreased both basal and 5-HT3R agonist-induced increase in the frequency of spontaneous glutamate inputs onto NTS-CA neurons. Low glucose also blunted 5-HT-induced inward currents in nodose ganglia neurons, which are the cell bodies of vagal afferents. The effect of low glucose in both nodose ganglia cells and in NTS slices was mimicked by the glucokinase inhibitor glucosamine. This study suggests that NTS-CA neurons are glucosensing through a presynaptic mechanism that is dependent on vagal glutamate release, 5-HT3R activity, and glucokinase.
Keywords: glucose, catecholamine, serotonin, vagus, NTS
glucose is essential for cell survival and plays a critical role in metabolic processes. Multiple brain regions contain glucose-sensing neurons, including the hypothalamus and hindbrain (9, 30–33, 66). The nucleus of the solitary tract (NTS) is one brain region identified as responsive to glucoprivation, as when the glucose metabolism inhibitor 5TG is injected into the NTS, it elicits a feeding response (4, 13, 32). The NTS impacts multiple homeostatic functions and is the primary site by which visceral afferent information concerning cardiovascular, respiratory, and gastrointestinal systems enters the brain (22, 25, 40, 53, 55, 58). This provides a mechanism by which changes in glucose could impact these homeostatic responses; however, the neuronal phenotype that responds to changes in glucose, and the cellular mechanisms involved, are not well understood.
The A2/C2 group of catecholamine (CA) neurons is located in the NTS and is directly activated by incoming visceral afferents of the solitary tract (ST) (1). They are also activated by signals that inhibit food intake and inhibited by compounds that stimulate food intake, suggesting they lie downstream of a reflex satiety pathway (1, 10–12, 21, 26, 38, 44, 68, 69). Furthermore, optogenetic and chemogenetic activation of these neurons inhibits food intake, with prolonged activation reducing body weight (50). NTS-CA neurons form extensive projections, including to the hypothalamus, amygdala, nucleus accumbens, ventral tegmental area, parabrachial nucleus (PBN), and other brain stem nuclei (3, 42, 43, 50, 54, 61, 63, 67), making them ideally suited to send visceral afferent information in a coordinated manner to multiple brain regions. NTS-CA neurons express norepinephrine, epinephrine, and glutamate and the release of these neurotransmitters at these target nuclei can affect many behaviors, including stress, arousal, anxiety, reward, food intake, and cardiovascular function (8, 22, 25, 28, 40, 44, 50, 58–60).
Glucose plays a critical role in metabolic processes, and hindbrain catecholaminergic neurons are involved in hypoglycemia-induced food intake and other homeostatic functions modulated by glucose concentrations (18, 47, 52). Strong evidence exists suggesting that A1/C1 catecholamine neurons of the ventral lateral medulla (VLM) are glucosensing, but the role of the A2/C2 neurons is less clear and it is not known whether they respond to changes in glucose concentration (32, 48).
The goal of these studies was to determine whether NTS-CA neuronal activity is altered by changes in glucose concentration. We report here that the majority of NTS-CA neurons are glucose excitatory and that glucose has presynaptic effects on visceral afferent terminals to alter 5-HT-induced glutamate release. Furthermore, inhibition of glucokinase mimics the effects of glucose on 5-HT signaling, suggesting that glucokinase is involved in the glucosensing mechanism.
MATERIALS AND METHODS
NTS slices.
All animal procedures were conducted with the approval of the Animal Care and Use Committees at Washington State University (Pullman, WA) and in accordance with the United States Public Health Service Policy on Humane Care and Use of Laboratory Animals (PHS Policy) and the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH). Eight to sixteen-week-old mice (both males and females) were used for these studies. No difference was noted between male and female mice for the parameters studied and so the data were pooled. The hindbrain was removed and placed for 1 min in cold (0–4°C) artificial cerebral spinal fluid (aCSF). The cutting plane was aligned vertically, sectioning caudal to rostral when mounted in a vibrating microtome (Lieva VT-1000S). Coronal slices (250 μm thick) from the TH-EGFP mice (Matsushita et al. 2002) were cut with a sapphire knife (Delaware Diamond Knives) and preserved the raphe nucleus and caudal NTS. This allowed us to conserve serotonergic inputs into the NTS. Slices were submerged in a perfusion chamber and all recording were performed at 31–35°C.
Whole cell recordings were made using an external aCSF bath solution containing (in mM): 125 NaCl, 3 KCl, 1.2 KH2PO4, 1.2 MgSO4, NaHCO3, 10, 5, 2, or 1 dextrose, and 2 CaCl2, bubbled with 95% O2-5% CO2; 30–34°C; pH = 7.3, adjusted to 300–310 mosmol using sucrose. Internal recording solution contained (in mM) 10 NaCl, 125 KCl, 11 EGTA, 1 CaCl2, 2 MgCl2, and 10 HEPES, pH = 7.3, 295–300 mosmol. External bath solution was used in patch electrodes for cell-attached recordings. Neurons were recorded from NTS within 200 μm rostral or caudal from obex and medial to the ST. Patch electrodes, 3–5 MΩ, were guided to neurons using both fluorescence (FITC) and differential interference contrast (DIC) optics (Olympus BX51). Voltage clamp and current clamp recordings were made with an Axopatch 700B and pClamp10 software (Axon Instruments). Only neurons not exceeding holding currents of 100 pA at holding potential (VH) = −60 mV for the 10 min control period (input resistance > 120 MΩ) were studied further. Current clamp recordings were made at resting membrane potentials, and current injections were not used to hold the membrane at set potentials. Only neurons with an access resistance less than 20 MΩ were used for analysis. All membrane potentials reported were corrected for junction potential (~14 mV). All drugs were obtained from Tocris Cookson or Sigma Aldrich.
Cell cultures.
Nodose ganglion neurons were dissected from male and female mice (6–16 wk) and cultured similar to previously reported protocols (24). Cells were maintained in Neurobasal A media (Invitrogen). Nodose cells were recorded on days 1–2 at room temperature. Locally administered drugs were applied to nodose cells for 2 s with a pressure of 2–3 psi using a picospritzer III (Parker). Input resistance ranged from 0.9 to 18.5 MΩ. The response rate of neurons to 5-HT was highly variable, ranging from 10 to 70% of the neurons tested, depending on the culture.
Statistics.
All data are presented as means with errors bars as SE. Differences in drug effects were tested by repeated measured ANOVA, using Tukey’s post hoc analysis unless otherwise noted. Differences were considered statistically significant for P values <0.05 unless otherwise stated (Sigmaplot 11.2).
Mice.
TH-EGFP mice were on a C57Bl/6J background. Transgenic mice were housed on a 12-h light/12-h dark cycle at room temperature in the Department of Integrative Physiology and Neuroscience animal vivarium. Mouse chow and water were provided ad libitum. Genotyping and breeding of mice were as described previously (1). All animal procedures were conducted with the approval of the Institutional Animal Care and Use Co mMittee at WSU in accordance with the US Public Health Service Policy on Humane Care and Use of Laboratory Animals (PHS Policy) and the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH).
RESULTS
Low glucose decreases action potential firing of TH-EGFP neurons in NTS.
TH-EGFP neurons were easily identified and visualized for recordings (Fig. 1A). We have previously demonstrated a greater than 88% colocalization of EGFP with TH in the medial and caudal NTS of these mice (1, 10) To determine whether changes in glucose concentration alter action potential (AP) firing rates in NTS TH-EGFP neurons, we used cell attached recordings to preserve the internal conditions of the neurons and measured AP firing rate in different glucose concentrations. Decreasing the aCSF glucose concentration from 5 mM (1.87 ± 0.74 Hz) to 2 mM (1.23 ± 0.45 Hz) to 1 mM (0.43 ± 0.27 Hz) gradually decreased the action potential firing rate in TH-EGFP neurons (n = 7). This effect was reversible, as action potential firing rate increased again when glucose concentrations were returned to 5 mM glucose (2.85 ± 0.31 Hz; Fig. 1, B and C).
Fig. 1.

Low glucose decreases action potential (AP) firing of nucleus of the solitary tract (NTS) TH-EGFP neurons in the NTS. A: visualization of NTS coronal brain slice taken from a TH-EGFP mouse using differential interference contrast (DIC) (top left) and fluorescence (bottom left), scale bar, 1 mM. CC: central canal; ST: solitary tract. Visualization of patched neuron using DIC (top right) and patched neuron in fluorescence (bottom right). Scale bar, 10 μm. B: average AP firing rate from a cell-attached recording of a TH-EGFP neuron in lowered glucose concentrations binned into 60-s periods. C: average fold change of cell-attached AP firing rate in lowered glucose concentrations from 5 mM (1.0 ± 0.7 intrinsic variance), 2 mM (0.8 ± 0.2), 1 mM (0.3 ± 0.2), and then increased to 2 mM (0.3 ± 0.3) and 5 mM (1.2 ± 0.3; n = 7). D: average fold change of cell-attached AP firing rate in lowered glucose concentrations with a synaptic cocktail starting in 5 mM (1.0 ± 0.2 intrinsic variance), to 5 mM+Cocktail (1.1 ± 0.1) to 2 mM+Cocktail (1.1 ± 0.1) to 1 mM+Cocktail (0.9 ± 0.1), and then raised to 2 mM+Cocktail (1.3 ± 0.2) to 5 mM+Cocktail (0.8 ± 0.1). Error bars indicate SE; *P < 0.05 denotes a significant change in AP firing rate compared with 5 mM glucose, one-way ANOVA.
Effect of glucose on NTS TH-EGFP neurons is presynaptic.
To determine whether the effects of glucose were direct or indirect (e.g., pre- or postsynaptic), we tested the effect of glucose after blocking GABAA, ionotropic glutamate [α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)/kainate and N-methyl-d-aspartate (NMDA) and glycine receptors using GABAzine (0.5 μM), 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione (NBQX, 50 μM), (2R)-amino-5-phosphonopentanoate (AP-5, 50 μM), and strychnine (1 μM), respectively (Cocktail). Using a cell-attached preparation, we again recorded the AP firing rate of NTS TH-EGFP neurons, at a starting glucose concentration of 5 mM (6.45 ± 1.05 Hz), to 5 mM+Cocktail (5.92 ± 1.94 Hz) to 2 mM+Cocktail (6.32 ± 2.17 Hz), to 1 mM+Cocktail (4.87 ± 1.5 Hz), and then raised to 2 mM+Cocktail (6.81 ± 2.35 Hz) to 5 mM+Cocktail (3.71 ± 1.34 Hz; Fig. 1D). In contrast to the control experiment described above, in the presence of GABAA, AMPA/kainate, NMDA, and glycine receptor antagonists, lowering glucose concentration no longer decreased AP firing rate, suggesting glucose actions are presynaptic and dependent on the release of GABA, glutamate, and/or glycine. Interestingly, the combination of blocking both excitatory (glutamate) and inhibitory (GABA and glycine) inputs did not significantly alter basal AP firing rate (n = 5; one-way ANOVA).
Effect of glucose on NTS TH-EGFP neurons is mediated by changes in presynaptic glutamate release.
We have previously shown that the firing rate of NTS TH-EGFP neurons is dependent on glutamate inputs (10). As glucose has been shown to alter both the excitability of vagal afferents and nodose ganglion cells (17) and the release of glutamate from presynaptic terminals (64, 65), we tested whether the effects of altering glucose concentration were dependent on presynaptic glutamate release. Using a cell-attached preparation, we again recorded the AP firing rate of NTS TH-EGFP neurons at a starting glucose concentration of 5 mM (1.97 ± 1.06 Hz) to 2 mM (1.4 ± 1.09 Hz) to 1 mM (1.03 ± 0.86 Hz), verifying that the cells were glucose excitatory. We then bath applied the non-NMDA ionotropic glutamate receptor antagonist NBQX (50 μM) before raising the glucose concentration back from 1 mM (0.99 ± 0.81 Hz) to 2 mM (0.06 ± 0.04 Hz) and finally 5 mM (0.03 ± 0.01 Hz; n = 6). In contrast to the control experiments (Fig. 1, B and C), AP firing rate did not return after raising glucose concentrations in the presence of NBQX, suggesting that the effects of glucose depends on glutamate inputs. The effects of NBQX were not rapidly washed out in 5 mM glucose (0.02 ± 0.01 Hz; Fig. 2, A and B), likely due to the high affinity of NBQX for AMPA receptor.
Fig. 2.
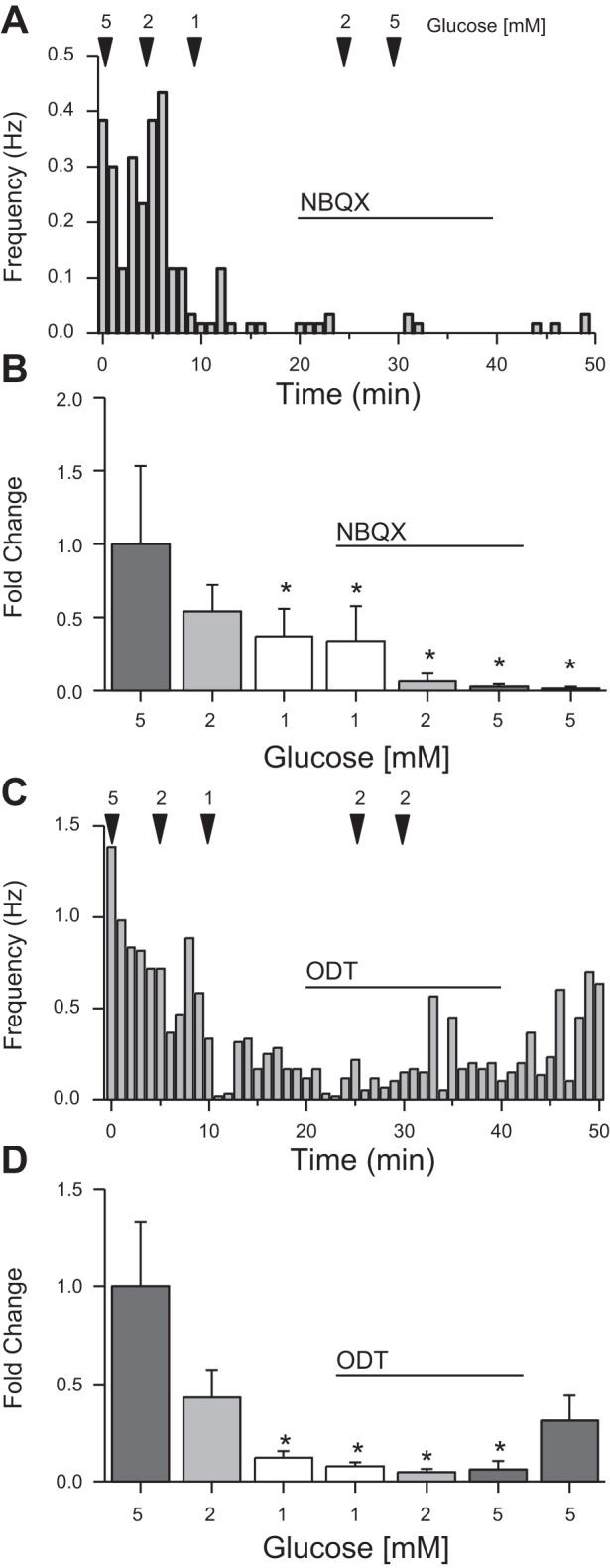
Effect of glucose on NTS TH-EGFP neurons is mediated by presynaptic glutamate release and the 5-HT3R. A: average AP firing rate from a representative cell-attached recording of a TH-EGFP neuron in lowered glucose concentrations after application of the non-N-methyl-d-aspartate (NMDA) ionotropic glutamate receptor antagonist 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione (NBQX, 50 μM), binned into 60-s periods. B: average fold change of cell-attached AP firing rate in lowered glucose concentrations from 5 mM (1.0 ± 0.5 intrinsic variance), 2 mM (0.5 ± 0.2) 1 mM (0.4 ± 0.2), and after application of NBQX (50 μM) in 1 mM (0.3 ± 0.2), 2 mM (0.1 ± 0.05), 5 mM (0.03 ± 0.02) followed by a 5 mM wash (0.02 ± 0.01; n = 6). C: AP firing rate from a representative cell-attached recording of a TH-EGFP neuron in lowered glucose concentrations with ondansetron (ODT; 0.5 μM) treatment, binned into 60-s periods. D: average fold change of cell-attached AP firing rate in lowered glucose concentrations from 5 mM (1.0 ± 0.3 intrinsic variance), 2 mM (0.4 ± 0.1), 1 mM (0.13 ± 0.04), and after application of ODT (0.5 μM) in 1 mM (0.08 ± 0.02), 2 mM (0.05 ± 0.02), and 5 mM (0.1 ± 0.05) followed by a 5 mM wash (0.3 ± 0.1; n = 6/8). Error bars indicate SE; *P < 0.05 denotes a significant change compared with baseline (aCSF/control), one-way ANOVA.
Effect of glucose on NTS TH-EGFP neurons requires 5-HT3Rs.
The 5-HT3R is a potent modulator of presynaptic glutamate release in the NTS, and the size of 5-HT3R-mediated 5-HT currents are dependent on glucose concentration (2, 12, 65). To test whether glucose effects are dependent on 5-HT3R function we lowered the glucose concentration from 5 mM (1.17 ± 0.28 Hz) to 2 mM (0.76 ± 0.27 Hz) to 1 mM (0.18Hz ± 0.06 Hz; n = 6/8) and then bath applied the 5-HT3R antagonist ondansetron (ODT, 0.5 μM) and raised the glucose concentration from 1 mM (0.44 ± 0.32 Hz), 2 mM (0.02 ± 0.02 Hz), to 5 mM (0.05 ± 0.04 Hz) in the presence of ODT. ODT blocked the return of AP firing after increasing glucose concentrations suggesting that glucose actions on NTS TH-EGFP neurons require 5-HT3Rs. After ODT was washed out for 15 min in 5 mM glucose concentration, the firing rate of the neurons began to increase again (0.25 ± 0.11 Hz; ~21% of control. Fig. 2, C and D).
5-HT3R-stimulated increase in sEPSC frequency in NTS TH-EGFP-positive neurons is blunted in low glucose concentrations.
As we had determined that the effects of glucose were indirect we next used whole cell patch-clamp techniques in the coronal brain slice to determine whether glucose affects basal 5-HT3R modulation of spontaneous excitatory postsynaptic current (sEPSC) frequency in NTS TH-EGFP neurons as this has been demonstrated to be a potential mechanism for changes in glucose to alter glutamate inputs onto unidentified NTS neurons (65).
We have previously shown that 5-HT increases sEPSC frequency in horizontal slices, but that there was no effect of the 5-HT antagonist alone in that orientation, suggesting that we had severed the serotonergic inputs (12). In contrast, 5-HT has been shown to provide a basal tone in coronal slices to increase sEPSCs frequency (65), suggesting that the serotonergic inputs are intact in the coronal slice. To confirm that we also see an endogenous basal 5-HT tone in our coronal slices we applied ODT (0.5μM) in 10 mM glucose; sEPSC frequency decreased from 4.29 ± 1.01 Hz during the control period to 3.27 ± 0.63 Hz in the presence of ODT. After the wash, sEPSC frequency returned to 4.30 ± 0.08 Hz (Fig. 3, A and B). In the presence of ODT, sEPSC amplitude did not change (−33.2 ± 6.23 pA) compared with control (−34.91 ± 6.15 pA; P < 0.05, one-way ANOVA; Fig. 3C).
Fig. 3.
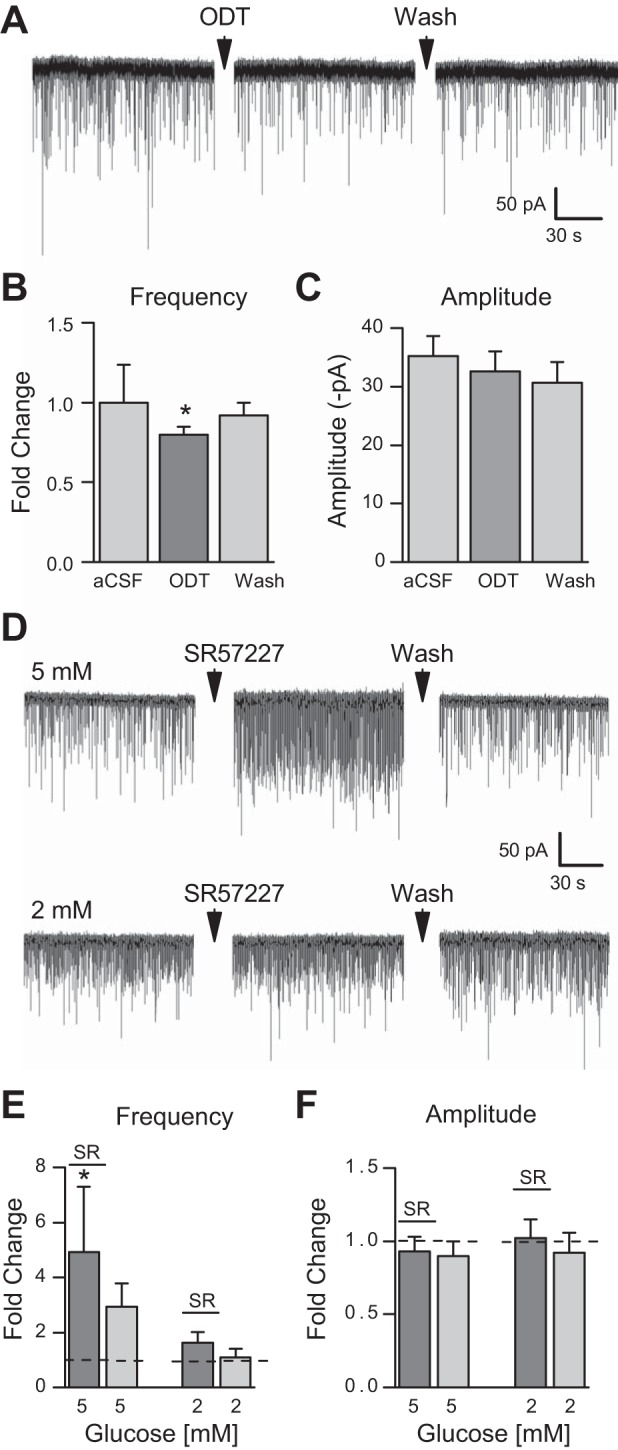
5-HT3R activity on NTS TH-EGF-positive neurons is blunted in low glucose concentrations. A: representative (single) trace from a TH-EGFP-positive neuron in a coronal brain slice following bath application of the 5-HT3R antagonist ODT (0.5 μM) in 10 mM glucose. Average fold change of sEPSCs frequency (control: 1 ± 0.2 intrinsic variance; ODT 0.8 ± 0.1; Wash: 0.9 ± 0.1) (B) and change in amplitude (C) following bath application of ODT (0.5 μM) in 10 mM glucose (n = 8) are shown. D: representative (single) trace from a TH-EGFP-positive neuron in a coronal brain slice following bath application of the 5-HT3R antagonist SR57227 (30 μM) in 5 mM and 2 mM glucose. Fold change of sEPSC frequency in 5 mM (5 mM+SR: 4.9 ± 2.4, Wash: 2.9 ± 0.8) and 2 mM glucose concentrations (2 mM+SR: 1.6 ± 0.4; 2 mM Wash: 1.1 ± 0.3) (E) and change in amplitude (F) following bath application of SR57227 (30 μM) in 5 mM (n = 8) compared with 2 mM glucose (n = 6) after SR57227 application in 5 mM compared with 2 mM glucose concentration. Error bars indicate SE. *P < 0.05 denotes a significant change compared with baseline (aCSF/control), one-way ANOVA.
To determine whether the effect of 5-HT on the sEPSC frequency of NTS TH-EGFP neurons is dependent on glucose concentration, we used whole cell patch clamp techniques to record from NTS TH-EGFP neurons. In 5 mM glucose, bath application of SR57227 (30 μM), a 5-HT3R agonist, increased sEPSC frequency from 7.14 ± 1.75 Hz to 18.42 ± 4.6 Hz (Fig. 3, D and E). After a 10-min wash, sEPSC frequency returned to control (15.58 ± 5.63 Hz). In 2 mM glucose, bath application of SR57227 did not change sEPSC frequency (5 mM: 5.5 ± 1.62 Hz, 2 mM: 6.76 ± 1.98 Hz, 2 mM+SR: 13.5 ± 6.29 Hz, 2 mM: 10.15 ± 4.19, 5 mM: 8.02 ± 4.03 Hz; Fig. 3E). sEPSC amplitude did not change after application of SR57227 in 5 mM (Control: −31.8 ± 3.8 pA; SR: −30.4 ± 4.4 pA; Wash: −27.7 ± 3.2 pA) or 2 mM glucose concentrations (Control: −35.48 ± 5.38 pA; SR: −33.77 ± 3.22 pA; Wash: −34.5 ± 5.25 pA; P < 0.05, one-way ANOVA; Fig. 3F).
Low glucose blunts 5-HT inward current in dissociated nodose ganglion neurons.
To determine the mechanism by which glucose acts we performed whole cell patch-clamp recordings in dissociated nodose ganglion neurons, which are the cell bodies of the presynaptic afferents providing inputs onto the TH-EGFP neurons in our NTS slices. We started by testing the effect of 5-HT on the holding current of nodose ganglion neurons. We locally applied 5-HT from a glass pipette with a 2-s puff from a picospritzer. To determine the effective 5-HT concentration, we tested 5-HT at two concentrations, 100 μM and 300 μM. The 100 μM concentration of 5-HT induced a large inward current (−312.54 ± 132.73 pA; n = 5) with it being further increased at 300 μM (−1063.77 ± 275.02 pA; n = 11; Fig. 4B). For 100 μM and 300 μM 5-HT concentrations, decay time was 10.92 ± 2.69 s and 43.04 ± 9.19 s, respectively (data not shown). To control for cell size and capacitance differences, we also calculated the current density induced by both 100 μM 5-HT as −5.63 ± 2.88 pA/pF [membrane capacitance (Cm): 62.52 ± 3.9 pF] and 300 μM 5-HT as −23.68 ± 7.0 pA/pF (Cm: 52.44 ± 3.38 pF). We used 100 μM 5-HT for the remainder of our nodose recordings as this produced an optimal response.
Fig. 4.

Low glucose blunts 5-HT inward current in dissociated nodose ganglion cells. A: representative (single) traces from cultured nodose ganglion neurons following local application of 5-HT (300 μM) in 10 mM (top) and 2 mM (bottom) glucose concentrations, respectively. B: effect of 5-HT at 100 μM (n = 5) and 300 μM (n = 11) concentrations in 10 mM glucose. Average current density (C) and decay time (D) of recorded nodose ganglion cells after direct 5-HT (100 μM) treatment in 10 mM (n = 9), 5 mM (n = 6), and 2 mM (n = 8) glucose concentrations. E: current density after local application of capsaicin (CAP, 100 μM) in 10 mM (n = 6) and 2 mM (n = 8) glucose concentrations. Error bars indicate SE; *P < 0.05 denotes a significant change compared with 10 mM glucose, one-way ANOVA.
We next tested 100 μM 5-HT in 10 mM, 5 mM, and 2 mM glucose concentrations. In 10 mM glucose, 5-HT induced a large inward current (−585.88 ± 131.17 pA, Fig. 4A), which was significantly larger than at 2 mM glucose (−15.25 ± 4.42 pA; Fig. 4A) where 5-HT-induced inward currents were minimal. The effect was intermediate at 5 mM (trace not shown, −159.5 ± 55.26 pA). Current density was −8.91 ± 2.03 pA/pF (Cm: 66.49 ± 3.0 pF; n = 9) at 10 mM, −4.04 ± 1.22 pA/pF (Cm: 38.72 ± 2.93 pF; n = 6) at 5 mM, and −0.3 ± 0.07 pA/pF (Cm: 48.6 ± 6.03 pF; n = 8) at 2 mM glucose concentrations (Fig. 4C). Decay times at 10, 5, and 2 mM were 33.2 ± 10.95 s, 10.3 ± 1.82 s, and 6.36 ± 1.91 s, respectively (Fig. 4D).
To determine whether glucose had nonselective effects on all ligand gated ion channels in nodose neurons, we also tested the response of the TrpV1 ligand capsaicin (CAP). In contrast to 5-HT, altering glucose concentration did not significantly change CAP-induced inward currents (100 μM) at 10 mM glucose (−1605.8 ± 450.37 pA; CD: −30.31 ± 8.29 pA/pF; Cm: 51.64 ± 6.93 pF; n = 6) and 2 mM glucose (−1187.26 ± 352.65 pA; CD: −20.54 ± 4.61 pA/pF; Cm: 51.55 ± 8.24 pF; n = 8; Fig. 4E).
Glucokinase inhibition blunts 5-HT-induced inward current in dissociated nodose ganglia.
Glucokinase has been shown to be required for the effects of glucose on nodose neuronal excitability (17). To determine whether it is also required for glucose effects on 5-HT3R activity, we tested the glucokinase inhibitors; if glucokinase is required, inhibiting it should mimic the effect of lowering glucose concentrations. We tested the effects of the inhibitors in 10 mM glucose concentration, as the 5-HT response is largest at this concentration.
Pretreating neurons with glucosamine (GSM, 100 μM) significantly decreased the amplitude of 5-HT-induced inward currents (−333.48 ± 186.42 pA; n = 6) compared with control (−1237.98 ± 565.79 pA; n = 6) as fold change (one-way ANOVA, Tukey post hoc, P < 0.01; Fig. 5, A and B). Fold change of current density was significantly less in GSM (−8.56 ± 4.46 pA/pF) compared with control (−35.32 ± 18.79 pA/pF; Fig. 5, A and B). These data suggest that glucose actions on 5-HT signaling are dependent on glucokinase activity. As a control we again tested CAP, a TRPV1 agonist, in 10 mM glucose before (−20.45 ± 9.52 pA/pF; −1091.27 ± 572.0 pA, n = 6) and after pretreatment of GSM (−19.8 ± 8.76 pA/pF; −1042.62 ± 527.26 pA n = 6; Fig. 5B) and saw no significant change. The decay time was also not significantly different (CAP in control: 36.25 ± 14.48 s, n = 6; CAP in GSM: 31.53 ± 12.93 s, n = 6 cells from 3 cultures; Fig. 5D).
Fig. 5.
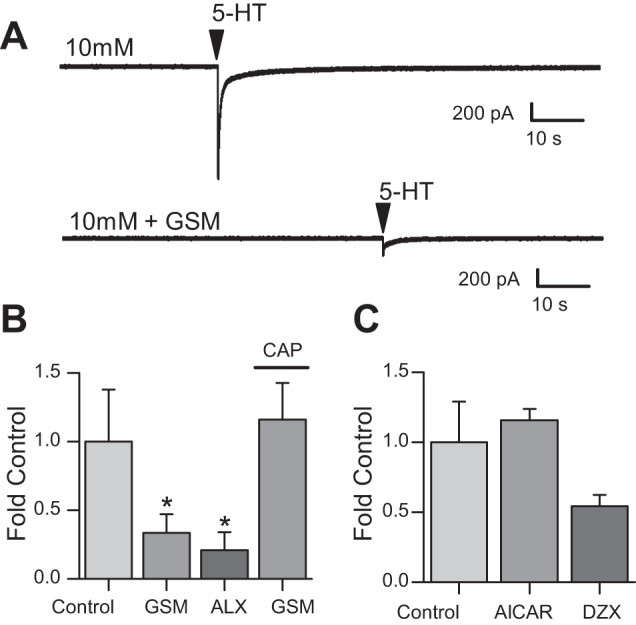
Glucokinase inhibition and ATP-sensitive potassium (KATP) channel activation blunts 5-HT inward current in dissociated nodose ganglia. A: representative (single) traces from cultured nodose ganglion neurons during 10 mM glucose concentration control (top) and the glucosamine (GSM, 100 μM; bottom) following local application of 5-HT (100 μM). B: average fold change in current after 5-HT (100 μM) application in control (1.0 ± 0.41 intrinsic variance) the presence of GSM (100 μM; 0.3 ± 0.1; n = 6), alloxan (ALX; 400 μM; 0.2 ± 0.1; n = 5), and GSM (100 μM) with local CAP (100 μM; 1.2 ± 0.2; n = 6) treatment. C: average fold change in current after 5-HT application in control (1 ± 0.35 intrinsic variance), the presence of 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR; 1 mM; 1.2 ± 0.1; n = 5), and diazoxide (DZX; 0.5 ± 0.1; n = 5). Error bars indicate SE; *P < 0.05 denotes a significant change compared with 5-HT in 10 mM glucose, one-way ANOVA.
To confirm this finding we also tested another glucokinase inhibitor, alloxan. Pretreating neurons with alloxan (400 μM) for 10 min significantly decreased the amplitude of 5-HT-induced inward currents (−57.06 ± 24.05 pA; n = 5) compared with control (−749.14 ± 211.24 pA; n = 5) as fold change (one-way ANOVA, Tukey post hoc, P < 0.01). 5-HT current density was significantly less with pretreatment of alloxan (−1.07 ± 0.52 pA/pF) compared with control (−13.34 ± 3.63 pA/pF; Fig. 5B). Decay time was also significantly different between control (37.39 ± 15.5 s) and GSM (15.65 ± 6.4 s) or alloxan (17.37 ± 8.44 s) pretreatment preceding local application of 5-HT (Fig. 5D).
KATP channels, but not AMPK, influence 5-HT currents in dissociated nodose ganglion neurons.
We next determined whether the effects of glucose involved AMP kinase (AMPK) as AMPK activity is positively correlated with the AMP-to-ATP ratio. As AMPK is activated by low glucose, if AMPK were required, activating it should decrease 5-HT-induced currents. Bath application of the AMPK activator 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR, 1 mM) onto dissociated nodose ganglion neurons did not alter 5-HT-induced inward currents (−331.22 ± 111.17 pA) when compared with control (−280.44 ± 86.75 pA; n = 5). There was also no change in 5-HT current density before (−6.73 ± 1.37 pA/pF; Cm: 55.63 ± 3.61 pF; n = 5) or after AICAR application (−7.86 ± 1.74 pA/pF; Fig. 5C) or decay time (control: 45.76 ± 10.73 s and AICAR: 42.44 ± 9.7 s; n = 5 cells from 3 cultures; P < 0.05, one-way ANOVA). This suggests that the effect of glucose concentration on 5-HT-induced inward currents is independent of AMPK.
Next, we determined whether ATP-sensitive potassium channel (KATP) channels were involved in the glucose-sensing mechanism. KATP channels are open when glucose concentrations, and thus intracellular ATP levels, are low. The KATP channel activator diazoxide (300 μM), which would be expected to mimic low glucose, significantly decreased the amplitude of the inward current induced by 5-HT by ~40% (Control: −1217.1 ± 680.48 pA; diazoxide: −810.92 ± 560.93 pA; 0.54 ± 0.08-fold control, n = 5 from 3 cultures; P < 0.05, one-way ANOVA; Fig. 5C).
Glucokinase inhibition blunts 5-HT effects on sEPSCs in NTS TH-EGFP-positive neurons.
The glucokinase inhibitor GSM blunted the effect of 5-HT-induced currents in nodose ganglion neurons. We next tested whether this effect was also observed in the brain slice (Fig. 5, A and B). In 5 mM glucose, 5-HT (30 μM) increased control sEPSC frequency from 4.6 ± 1.37 Hz to 15.27 ± 4.59 Hz (4.2 ± 1.5-fold control; n = 15). GSM treatment (300 μM; 0.7 ± 0.2-fold; 3.09 ± 1.17 Hz; n = 7) decreased control sEPSCs frequency (4.97 ± 1.92 Hz; n = 7). In the presence of GSM, 5-HT did not increase sEPSCs frequency (3.7 ± 1.4-fold; 7.38 ± 2.68 Hz; n = 7) when normalized to control (P < 0.05, one-way ANOVA; data not shown). sEPSC amplitude was not altered after 5-HT application with (−34.91 ± 7.74 pA vs. −27.31 ± 5.1 pA, respectively; n = 7) or without GSM pretreatment (−31.23 ± 3.8 pA vs. −36.86 ± 4.41 pA, respectively, data not shown).
While there was a trend for GSM to inhibit 5-HT effects on sEPSC frequency in 5 mM glucose, which is apparent on the scatter plot, it did not reach statistical significance (Fig. 6A). To increase our ability to detect an inhibition we recorded 5-HT responses in 10 mM glucose as they are larger. In 10 mM glucose 5-HT (30 μM) increased control sEPSC frequency from 4.18 ± 1.21 Hz to 16.64 ± 5.43 Hz (n = 8). GSM treatment (300 μM; 2.43 ± 0.52 Hz) had no effect on control sEPSCs frequency (2.97 ± 1.92 Hz; n = 8), but significantly attenuated the 5-HT-induced increase sEPSCs frequency (7.38 ± 2.68 Hz; n = 8) when normalized to control (Fig. 6, D and E). sEPSC amplitude was not altered after 5-HT application with (−24.26 ± 2.79 pA vs. −31.51 ± 7.38 pA, respectively) or without GSM pretreatment (−23.93 ± 3.53 pA vs. −25.31 ± 3.96 pA, respectively; Fig. 6F). 5-HT application compared with 5-HT application after pretreating was significantly decreased in 10 mM glucose (P < 0.05, one-way ANOVA). A comparison of GSM effects in 5 mM versus 10 mM glucose concentrations is shown in Fig. 6, A and B.
Fig. 6.
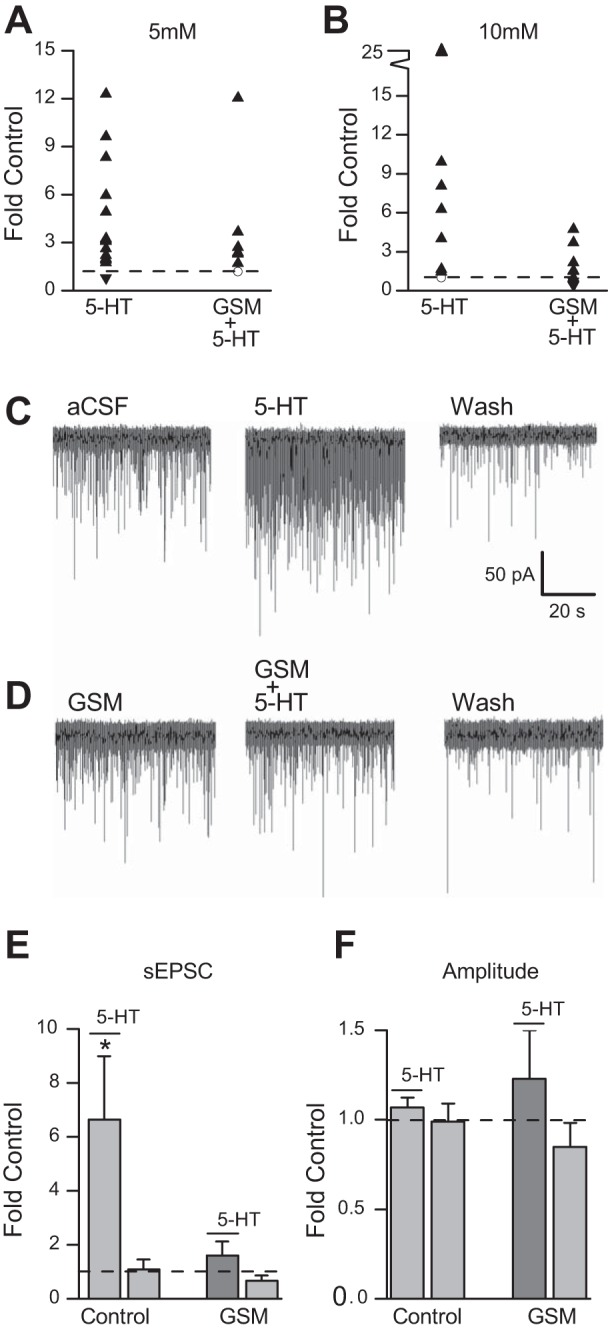
GSM blunts 5-HT effects on spontaneous excitatory postsynaptic current (sEPSC) frequency in NTS TH-EGFP neurons in slices. A: scatterplot showing sEPSCs frequency in individual neurons after 5-HT application with (n = 7) and without (n = 15) pretreatment of GSM (300 μM) in 5 mM glucose. B: scatterplot showing individual cell distribution in response to 5-HT (30 μM) with (n = 8) and without (n = 8) pretreatment of GSM (100 μM) in 10 mM glucose (▲, significant increase; ▼, significant decrease; ☐, not significant). C: representative (single) trace from a TH-EGFP-positive neuron from a coronal brain slice in 10 mM glucose with a bath application of 5-HT (30 μM); D: EGFP-positive neuron from a coronal brain slice in the presence of GSM (300 μM) with a bath application of 5-HT. E: effect of 5-HT (30 μM) on sEPSC frequency in 10 mM glucose concentration control (5-HT: 6.7 ± 2.4 Hz; Wash: 1.1 ± 0.9 Hz), and GSM (GSM+5-HT: 3.4 ± 0.4; Wash: 0.6 ± 0.2) and F, amplitude in control (5-HT: 1.1 ± 0.5; Wash: 1.0 ± 0.1) and in the presence of GSM (GSM+5-HT: 1.1 ± 0.2; Wash: 0.8 ± 0.1) expressed as fold change. Error bars indicate SE; *P < 0.05 denotes a significant change, one-way ANOVA.
DISCUSSION
Catecholamine neurons in the NTS are implicated in a broad number of homeostatic functions including the control of food intake, reward, stress, and cardiovascular reflexes (22, 23, 25, 38, 44, 45, 50, 56, 58, 59, 69). Changes in glucose concentration can have a large impact on neuronal activity (27, 64) and also affect homeostatic functions (15). Hindbrain catecholamine neurons are critical for glucoprivic responses and A1/C1 catecholamine neurons respond to changes in glucose (46). However, it was not known whether changes in glucose concentration alter the activity of NTS-CA neurons. Here we report four key new findings. First, the majority of NTS-CA neurons fire more action potentials in high glucose (5 mM) concentrations than in low glucose (1 mM) concentrations. Second, the effect of glucose on action potential firing rate is dependent on glutamate inputs. Third, the effect of glucose on glutamate release requires presynaptic 5-HT3R. Fourth, inhibition of glucokinase mimics the effect of low glucose to inhibit both 5-HT3R-stimulated currents in nodose neurons and glutamate release in the NTS.
Majority of NTS TH-EGFP neurons are excited in higher glucose concentrations.
Here we show for the first time that glucose concentration has a direct relationship with action potential firing in NTS-CA neurons. The finding that 86% of NTS-CA neurons are excited by high glucose shows a remarkably homogenous response. In comparison, a survey of all NTS neurons (of unidentified phenotype) showed that 35% were excited and 21% inhibited by higher glucose concentrations, with the rest being nonresponsive (36). The response of GABAergic NTS neurons to changes in glucose concentration were also very heterogeneous, with 40% being glucose excited (GE), 33% glucose inhibited (GI), and 27% nonresponsive (7). Our results therefore identify NTS-CA neurons as a unique population of neurons predominately excited in high glucose concentrations. The fact that changes in glucose increase the firing rate of almost all of these neurons provides a mechanism for the coordinated modulation of the majority of downstream targets of NTS-CA neurons, which include multiple brain regions (3, 42, 43, 54, 61, 63, 67). It is possible that the 14% of CA neurons that decrease their firing in higher glucose, or are nonresponsive, represent a specific subpopulation that underlie a discrete function and project to different brain regions than glucose-excitatory CA neurons.
Our finding that NTS-CA neurons decrease their firing rate in lower glucose concentrations is also consistent with the fact that glucoprivation does not increase fos-ir in the majority of A2/C2 neurons, in contrast to the large activation of A1/C1 neurons (48). If A2/C2 neurons are glucose excitatory (albeit indirectly), their activity would be predicted to decrease in a glucoprivic state, and therefore c-fos is unlikely to be activated in these neurons.
Glucose excites NTS-CA neurons by increasing glutamate inputs onto NTS TH-EGFP-positive neurons through the 5-HT3R.
Our results show that the effect of glucose concentration on NTS-CA neuronal firing is dependent on glutamate inputs as it is blocked by the ionotropic AMPA/kainate glutamate receptor antagonist NBQX. This suggests that the effects of glucose are indirect on glutamate terminals in the NTS, with higher concentrations of glucose leading to a higher probability of glutamate release. Consistent with this model we found that spontaneous glutamate inputs onto NTS-CA neurons are also dependent on glucose concentration, with glucose inputs being increased in higher glucose. This finding is consistent with previous reports that glucose concentration impacts the frequency of glutamate inputs onto unidentified rat NTS neurons (64) and that the firing rate of NTS-CA neurons is dependent on spontaneous glutamate inputs (10, 12).
The effect of glucose on both NTS-CA neuronal firing rate and spontaneous glutamate inputs is also dependent on 5-HT3Rs as it was blocked by the 5-HT3R antagonist ODT. The size of the effect of the 5-HT3R agonist SR57227 on sEPSC frequency in NTS-CA neurons was also positively correlated with aCSF glucose concentrations. This suggests that increasing glucose concentrations increases serotonin 5-HT3R signaling in vagal afferent terminals to increase glutamate release onto NTS-CA neurons. Consistent with this we show that the size of the 5-HT3R-mediated current in mouse vagal afferent nodose neurons is dependent on glucose concentration, as has been reported previously for the rat (2). Browning and her colleagues showed that this is likely due to changes in the surface expression of 5-HT3Rs, as surface expression is dependent on glucose concentration (2, 65). Here we extend those findings to the mouse and identify NTS-CA neurons as a critical downstream population of neurons that are excited by glucose in a 5-HT3R-dependent manner.
NTS neurons receive a basal serotonergic tone onto 5-HT3Rs that contributes to the normal frequency of release of glutamate as ODT application decreases sEPSC frequency in the general population of NTS neurons (65). This effect is positively correlated with aCSF glucose concentrations. Our findings suggest that this basal serotonin tone also exists on afferent inputs specifically onto NTS-CA neurons. One implication of this result is that it suggests that serotonergic terminals release 5-HT onto vagal afferent terminals to control glutamate activation of NTS-CA neurons independently of vagal afferent firing. Furthermore, as this tone is lost in lower glucose concentrations it suggests a mechanism by which changes in glucose can modulate the firing rate of NTS-CA neurons, and presumably the catecholaminergic tone at downstream target nuclei, again independently of changes in vagal afferent firing. Interestingly, we do not see this basal serotonin tone in a horizontal slice preparation, which preserves the connections in the rostral caudal orientation, but doesn’t retain the dorsal ventral connections (12). This suggests that the basal tone of serotonin might originate from the raphe obscurus, which lies ventral to the NTS, is present in the coronal, but not the horizontal slice, and is an endogenous source of 5-HT producing neurons located ventral to the NTS (41). Interestingly, stimulation of the raphe obscurus produces significant 5-HT release into the dorsal vagal complex (DVC; i.e., the NTS and dorsal motor nucleus of the vagus), in fed, but not fasted rats (37, 65). In addition, high-fat diet blunts the ability of glucose to increase 5-HT3R responses (62). Therefore, different energy states could potentially influence excitation of this pathway by altering both the size of serotonin release and activation of 5-HT3R at the level of the NTS.
Glucokinase activity contributes to presynaptic 5-HT signaling.
Here we report that the reduction in 5-HT3R function in vagal afferents seen in low glucose is mimicked by two different glucokinase inhibitors. Glucokinase is the enzyme responsible for converting glucose to glucose-6-phosphate and acts as a glucose sensor, shifting cellular function and metabolic processes based on fluctuating glucose concentrations (29). Glucokinase has been shown to respond to the glucose concentrations we use here in other brain regions (4, 13, 16, 17). It is present in nodose ganglia neurons and knockdown of its expression by short hairpin RNAs abolishes glucose excitation of nodose neurons (17). It is also required for glucose sensing by GABAergic neurons in the NTS (7) and its expression decreases following chronic hyperglysemia induced by streptozotocin-induced diabetes (20). Taken together these results suggest that glucokinase provides one mechanism by which vagal afferents “sense” changes in glucose concentration to alter serotonin receptor function. Critically, inhibition of glucokinase did not alter the current mediated by the vallinoid receptor VR1; suggesting the effect is selective to the 5-HT3R and it is not a global effect of the drugs on excitability.
The glucokinase inhibitor GSM also blunted 5-HT3R-mediated glutamate release in the coronal brain slice, suggesting that glucokinase affects 5-HT3R signaling in vagal afferent terminals to modulate glutamate release onto NTS-CA neurons. This effect was most robust at high concentrations of glucose (10 mM), likely because 5-HT3R expression is already decreased in lower glucose concentrations, making it hard to measure a further reduction by glucokinase inhibition.
KATP channel influences 5-HT currents in vagal afferents.
KATP channels, which are inhibited by high levels of intracellular ATP, are also found in the NTS (34, 70) and in nodose ganglia neurons (17). High concentrations of glucose increase intracellular ATP (6) and this has been shown to inhibit KATP channels to increase neuronal firing rate in some neurons (14, 17). We found that the KATP channel activator diazoxide, which mimics low glucose concentrations and activates KATP channels, significantly decreased the 5-HT-induced inward currents even in 10 mM glucose concentrations. This suggests that KATP channels also play a role in modulating 5-HT currents in vagal afferents. KATP channels have previously been shown to be important for glucose-induced firing of gastric afferents (16, 17) and for mediating the GE effect in an unidentified subpopulation of caudal NTS neurons (14) as well as the GE response of medial NTS GABA neurons (7). Our data suggests that at least some of these GE neurons could be NTS-CA neurons that respond to changes in glucose concentration.
Our predicted model is that in high glucose, a combination of activation of glucokinase and inhibition of KATP channels depolarizes vagal afferent terminals and increases glutamate release onto NTS-CA neurons. In contrast, we did not find evidence that AMPK, whose activity is also regulated by changes in the AMP-to-ATP ratio in neurons, plays a role in the expression of the 5-HT3R on nodose ganglion neurons. This is contrary to other neuronal populations, including in the hypothalamus, hippocampus, and hindbrain, where AMPK has been shown to be critical (5, 39, 51). However, this finding is consistent with the finding that glucoprivation does not activate AMPK in A2/C2 neurons, in contrast to the A1/C1 neurons (33).
Physiological implications.
NTS-CA neurons have been widely implicated in the control of homeostatic functions, including the control of food intake. The finding that alterations in glucose concentrations impacts NTS-CA neuronal activity identifies one potential mechanism by which changes in glucose levels can modulate feeding behavior (4, 46, 49, 52). Brain glucose levels don’t fluctuate as much as blood glucose; with brain glucose concentrations reported to range from ~0.2 mM in a state of hypoglycemia to ~4 mM in a state of hyperglycemia, with normoglycemic levels at ~2.5 mM in rats (51, 57). In contrast, blood glucose concentrations in rats can range from ~3 mM to ~16 mM (51, 57). For our brain slice studies we see changes in glutamate inputs and NTS-CA neuronal activity with changes in glucose concentrations from 5 mM to 2 mM, within these physiological ranges. In addition, increasing the glucose concentration to 10 mM further increases the size of the 5-HT3R response (as was also shown by Refs. 2 and 64); suggesting that if the NTS sees higher glucose concentrations the neurons will further increase their firing rate. Our data suggests that hyperglycemia will increase the effect of serotonin to increase NTS-CA neuronal firing rate through increased glutamate inputs, presumably leading to increased transmitter release at their downstream targets. In contrast, during low-to-normal glucose conditions this drive will be blunted and the activity (firing rate) of these neurons is likely to be lower. The exact concentration of glucose that NTS-CA neurons are exposed to in vivo is not known. The NTS lies directly below the area postrema, a circumventricular organ that lies outside of the blood brain barrier (BBB) and portions of the medial/commissural NTS have been reported to contain some fenestrated capillaries (19, 35). If NTS-CA neurons are exposed to glucose concentrations closer to blood glucose levels, then there is likely a serotonergic drive that increases glutamate inputs onto these neurons, even during normal glucose concentrations. However, parts of the NTS are not exposed to the BBB and likely see glucose concentrations closer to those in other brain regions. The nodose ganglia, which contain the cell bodies of vagal afferents, lie outside the BBB and are therefore exposed to blood glucose concentrations It is possible that prolonged high blood glucose could influence the longer-term expression of 5-HT3Rs on vagal afferents.
Perspectives and Significance
In summary, we show that the firing rate of NTS-CA neurons is impacted by changes in glucose concentration, with their firing rate being higher in higher glucose concentrations. We show that this is an indirect (presynaptic) mechanism through potentiation of 5-HT3R activation of vagal afferent terminals to increase glutamate release. Additionally, we show that glucose effects on 5-HT3R function are dependent on glucokinase activity and KATP channels and are independent of AMP kinase. Blood and brain glucose concentrations are altered in hypo- and hyperglycemia. Given the crucial role of NTS-CA neurons in the control of food intake, stress responses, and motivational behaviors, these studies identify a potential mechanism by which high glucose concentrations could influence these behaviors (Fig. 7). Thus they contribute to our understanding of how glucose can impact neuronal circuits involved in the control of food intake and the mechanisms by which physiological glucose levels can influence neuronal activity.
Fig. 7.
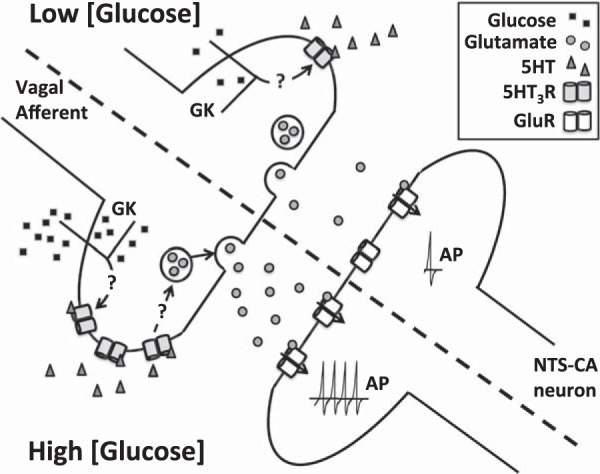
A model of how different glucose concentrations impact NTS-CA neuronal activity. Our findings suggest that changes in glucose concentration alter presynaptic vagal afferent glutamate release onto NTS-CA neurons through modulation of the 5-HT3R. In high glucose concentrations (bottom), 5-HT3R activity increases glutamate release onto NTS-CA neurons, which increases action potential firing in these neurons. Low glucose concentrations (top) decrease 5-HT3R activity and blunt the effects of 5-HT on NTS-CA neuron activity. Glucokinase (GK) appears to be important for the effects of changes in glucose.
GRANTS
This work was supported by National Institute of Diabetes and Digestion and Kidney Disease (NIDDKD) Grant DK083452 (to S. Appleyard). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIDDKD or the National Institutes for Health. This work was also supported by a training grant from the Poncin Trust (Washington) to B. Roberts.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
B.L.R., M.Z., and S.M.A conceived and designed the experiments; B.L.R., M.Z., H.Z., and C.D. performed experiments; B.L.R. and M.Z. analyzed data; B.L.R. and S.M.A. interpreted results of experiments; B.L.R. prepared figures; B.L.R. and S.M.A. drafted manuscript; B.L.R. and S.M.A. edited and revised manuscript; B.L.R., M.Z., H.Z., C.D., and S.M.A. approved final version of manuscript.
ACKNOWLEDGMENTS
Current address of B. L. Roberts is the Division of Diabetes, Obesity, and Metabolism, Oregon National Primate Research Center, Beaverton, OR 97006. Current address of H. Zhao is School of Life Sciences, University of Science & Technology of China, Hefei City, Anhui 230027, P.R. China.
REFERENCES
- 1.Appleyard SM, Marks D, Kobayashi K, Okano H, Low MJ, Andresen MC. Visceral afferents directly activate catecholamine neurons in the solitary tract nucleus. J Neurosci 27: 13292–13302, 2007. doi: 10.1523/JNEUROSCI.3502-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Babic T, Troy AE, Fortna SR, Browning KN. . Glucose-dependent trafficking of 5–HT3 receptors in rat gastrointestinal vagal afferent neurons. Neurogastroenterol Motil 24: e476–e488, 2012. doi: 10.1111/j.1365-2982.2012.01987.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Balcita-Pedicino JJ, Rinaman L. Noradrenergic axon terminals contact gastric preautonomic neurons in the paraventricular nucleus of the hypothalamus in rats. J Comp Neurol 501: 608–618, 2007. doi: 10.1002/cne.21267. [DOI] [PubMed] [Google Scholar]
- 4.Balfour RH, Hansen AM, Trapp S. Neuronal responses to transient hypoglycaemia in the dorsal vagal complex of the rat brainstem. J Physiol 570: 469–484, 2006. doi: 10.1113/jphysiol.2005.098822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beall C, Hamilton DL, Gallagher J, Logie L, Wright K, Soutar MP, Dadak S, Ashford FB, Haythorne E, Du Q, Jovanović A, McCrimmon RJ, Ashford ML. Mouse hypothalamic GT1-7 cells demonstrate AMPK-dependent intrinsic glucose-sensing behaviour. Diabetologia 55: 2432–2444, 2012. doi: 10.1007/s00125-012-2617-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Berg JM, Tymoczko JL, Stryer L. Biochemistry. London: Palgrave MacMillan, 2011. [Google Scholar]
- 7.Boychuk CR, Gyarmati P, Xu H, Smith BN. Glucose sensing by GABAergic neurons in the mouse nucleus tractus solitarii. J Neurophysiol 114: 999–1007, 2015. doi: 10.1152/jn.00310.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cole RL, Sawchenko PE. Neurotransmitter regulation of cellular activation and neuropeptide gene expression in the paraventricular nucleus of the hypothalamus. J Neurosci 22: 959–969, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cotero VE, Zhang BB, Routh VH. The response of glucose-excited neurones in the ventromedial hypothalamus to decreased glucose is enhanced in a murine model of type 2 diabetes mellitus. J Neuroendocrinol 22: 65–74, 2010. doi: 10.1111/j.1365-2826.2009.01938.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cui RJ, Li X, Appleyard SM. Ghrelin inhibits visceral afferent activation of catecholamine neurons in the solitary tract nucleus. J Neurosci 31: 3484–3492, 2011. doi: 10.1523/JNEUROSCI.3187-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cui RJ, Roberts BL, Zhao H, Andresen MC, Appleyard SM. Opioids inhibit visceral afferent activation of catecholamine neurons in the solitary tract nucleus. Neuroscience 222: 181–190, 2012. doi: 10.1016/j.neuroscience.2012.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cui RJ, Roberts BL, Zhao H, Zhu M, Appleyard SM. Serotonin activates catecholamine neurons in the solitary tract nucleus by increasing spontaneous glutamate inputs. J Neurosci 32: 16530–16538, 2012. doi: 10.1523/JNEUROSCI.1372-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dallaporta M, Himmi T, Perrin J, Orsini JC. Solitary tract nucleus sensitivity to moderate changes in glucose level. Neuroreport 10: 2657–2660, 1999. doi: 10.1097/00001756-199908200-00040. [DOI] [PubMed] [Google Scholar]
- 14.Dallaporta M, Perrin J, Orsini JC. Involvement of adenosine triphosphate-sensitive K+ channels in glucose-sensing in the rat solitary tract nucleus. Neurosci Lett 278: 77–80, 2000. doi: 10.1016/S0304-3940(99)00898-8. [DOI] [PubMed] [Google Scholar]
- 15.Elmquist JK, Coppari R, Balthasar N, Ichinose M, Lowell BB. Identifying hypothalamic pathways controlling food intake, body weight, and glucose homeostasis. J Comp Neurol 493: 63–71, 2005. doi: 10.1002/cne.20786. [DOI] [PubMed] [Google Scholar]
- 16.Grabauskas G, Song I, Zhou S, Owyang C. Electrophysiological identification of glucose-sensing neurons in rat nodose ganglia. J Physiol 588: 617–632, 2010. doi: 10.1113/jphysiol.2009.182147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grabauskas G, Zhou S-YY, Lu Y, Song I, Owyang C. Essential elements for glucosensing by gastric vagal afferents: immunocytochemistry and electrophysiology studies in the rat. Endocrinology 154: 296–307, 2013. doi: 10.1210/en.2012-1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grill HJ, Hayes MR. Hindbrain neurons as an essential hub in the neuroanatomically distributed control of energy balance. Cell Metab 16: 296–309, 2012. doi: 10.1016/j.cmet.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gross PM, Wall KM, Pang JJ, Shaver SW, Wainman DS. Microvascular specializations promoting rapid interstitial solute dispersion in nucleus tractus solitarius. Am J Physiol Regul Integr Comp Physiol 259: R1131–R1138, 1990. [DOI] [PubMed] [Google Scholar]
- 20.Halmos KC, Gyarmati P, Xu H, Maimaiti S, Jancsó G, Benedek G, Smith BN. Molecular and functional changes in glucokinase expression in the brainstem dorsal vagal complex in a murine model of type 1 diabetes. Neuroscience 306: 115–122, 2015. doi: 10.1016/j.neuroscience.2015.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ho JM, Anekonda VT, Thompson BW, Zhu M, Curry RW, Hwang BH, Morton GJ, Schwartz MW, Baskin DG, Appleyard SM, Blevins JE. Hindbrain oxytocin receptors contribute to the effects of circulating oxytocin on food intake in male rats. Endocrinology 155: 2845–2857, 2014. doi: 10.1210/en.2014-1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Itoh H, Buñag RD. Age-related reduction of reflex bradycardia in conscious rats by catecholaminergic nucleus tractus solitarius lesions. Mech Ageing Dev 67: 47–63, 1993. doi: 10.1016/0047-6374(93)90111-4. [DOI] [PubMed] [Google Scholar]
- 23.Kenny PJ. Common cellular and molecular mechanisms in obesity and drug addiction. Nat Rev Neurosci 12: 638–651, 2011. doi: 10.1038/nrn3105. [DOI] [PubMed] [Google Scholar]
- 24.Kinch DC, Peters JH, Simasko SM. Comparative pharmacology of cholecystokinin induced activation of cultured vagal afferent neurons from rats and mice. PLoS One 7: e34755, 2012. doi: 10.1371/journal.pone.0034755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kubo T, Goshima Y, Hata H, Misu Y. Evidence that endogenous catecholamines are involved in alpha 2-adrenoceptor-mediated modulation of the aortic baroreceptor reflex in the nucleus tractus solitarii of the rat. Brain Res 526: 313–317, 1990. doi: 10.1016/0006-8993(90)91238-C. [DOI] [PubMed] [Google Scholar]
- 26.Lam DD, Zhou L, Vegge A, Xiu PY, Christensen BT, Osundiji MA, Yueh CY, Evans ML, Heisler LK. Distribution and neurochemical characterization of neurons within the nucleus of the solitary tract responsive to serotonin agonist-induced hypophagia. Behav Brain Res 196: 139–143, 2009. doi: 10.1016/j.bbr.2008.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee DK, Jeong JH, Chun SK, Chua S Jr, Jo YH. Interplay between glucose and leptin signalling determines the strength of GABAergic synapses at POMC neurons. Nat Commun 6: 6618, 2015. doi: 10.1038/ncomms7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leibowitz SF. Central neurotransmitters and control of specific appetite for the macronutrients [in French]. Ann Endocrinol (Paris) 49: 133–140, 1988. [PubMed] [Google Scholar]
- 29.Lenzen S. A fresh view of glycolysis and glucokinase regulation: history and current status. J Biol Chem 289: 12189–12194, 2014. doi: 10.1074/jbc.R114.557314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Levin BE, Dunn-Meynell AA, Routh VH. CNS sensing and regulation of peripheral glucose levels. Int Rev Neurobiol 51: 219–258, 2002. doi: 10.1016/S0074-7742(02)51007-2. [DOI] [PubMed] [Google Scholar]
- 31.Levin BE, Routh VH, Kang L, Sanders NM, Dunn-Meynell AA. Neuronal glucosensing: what do we know after 50 years? Diabetes 53: 2521–2528, 2004. doi: 10.2337/diabetes.53.10.2521. [DOI] [PubMed] [Google Scholar]
- 32.Li A-JJ, Wang Q, Dinh TT, Powers BR, Ritter S. Stimulation of feeding by three different glucose-sensing mechanisms requires hindbrain catecholamine neurons. Am J Physiol Regul Integr Comp Physiol 306: R257–R264, 2014. doi: 10.1152/ajpregu.00451.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li AJ, Wang Q, Ritter S. Participation of hindbrain AMP-activated protein kinase in glucoprivic feeding. Diabetes 60: 436–442, 2011. doi: 10.2337/db10-0352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malik R, Ferguson AV. Hydrogen sulfide depolarizes neurons in the nucleus of the solitary tract of the rat. Brain Res 1633: 1–9, 2016. doi: 10.1016/j.brainres.2015.12.029. [DOI] [PubMed] [Google Scholar]
- 35.Merchenthaler I. Neurons with access to the general circulation in the central nervous system of the rat: a retrograde tracing study with fluoro-gold. Neuroscience 44: 655–662, 1991. doi: 10.1016/0306-4522(91)90085-3. [DOI] [PubMed] [Google Scholar]
- 36.Mimee A, Ferguson AV. Glycemic state regulates melanocortin, but not nesfatin-1, responsiveness of glucose-sensing neurons in the nucleus of the solitary tract. Am J Physiol Regul Integr Comp Physiol 308: R690–R699, 2015. doi: 10.1152/ajpregu.00477.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mohammed JR, Saska TA, Chi J, Stephens RL Jr. Stimulation of the nucleus raphe obscurus produces marked serotonin release into the dorsal medulla of fed but not fasted rats–glutamatergic dependence. Brain Res 695: 100–103, 1995. doi: 10.1016/0006-8993(95)00505-K. [DOI] [PubMed] [Google Scholar]
- 38.Mönnikes H, Lauer G, Arnold R. Peripheral administration of cholecystokinin activates c-fos expression in the locus coeruleus/subcoeruleus nucleus, dorsal vagal complex and paraventricular nucleus via capsaicin-sensitive vagal afferents and CCK-A receptors in the rat. Brain Res 770: 277–288, 1997. doi: 10.1016/S0006-8993(97)00865-2. [DOI] [PubMed] [Google Scholar]
- 39.Murphy BA, Fakira KA, Song Z, Beuve A, Routh VH. AMP-activated protein kinase and nitric oxide regulate the glucose sensitivity of ventromedial hypothalamic glucose-inhibited neurons. Am J Physiol Cell Physiol 297: C750–C758, 2009. doi: 10.1152/ajpcell.00127.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olson VG, Heusner CL, Bland RJ, During MJ, Weinshenker D, Palmiter RD. Role of noradrenergic signaling by the nucleus tractus solitarius in mediating opiate reward. Science 311: 1017–1020, 2006. doi: 10.1126/science.1119311. [DOI] [PubMed] [Google Scholar]
- 41.Pickel VM, Joh TH, Chan J, Beaudet A. Serotoninergic terminals: ultrastructure and synaptic interaction with catecholamine-containing neurons in the medial nuclei of the solitary tracts. J Comp Neurol 225: 291–301, 1984. doi: 10.1002/cne.902250212. [DOI] [PubMed] [Google Scholar]
- 42.Reyes BA, Van Bockstaele EJ. Divergent projections of catecholaminergic neurons in the nucleus of the solitary tract to limbic forebrain and medullary autonomic brain regions. Brain Res 1117: 69–79, 2006. doi: 10.1016/j.brainres.2006.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Riche D, De Pommery J, Menetrey D. Neuropeptides and catecholamines in efferent projections of the nuclei of the solitary tract in the rat. J Comp Neurol 293: 399–424, 1990. doi: 10.1002/cne.902930306. [DOI] [PubMed] [Google Scholar]
- 44.Rinaman L. Hindbrain noradrenergic A2 neurons: diverse roles in autonomic, endocrine, cognitive, and behavioral functions. Am J Physiol Regul Integr Comp Physiol 300: R222–R235, 2011. doi: 10.1152/ajpregu.00556.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rinaman L, Baker EA, Hoffman GE, Stricker EM, Verbalis JG. Medullary c-Fos activation in rats after ingestion of a satiating meal. Am J Physiol Regul Integr Comp Physiol 275: R262–R268, 1998. [DOI] [PubMed] [Google Scholar]
- 46.Ritter S, Dinh TT, Li AJ. Hindbrain catecholamine neurons control multiple glucoregulatory responses. Physiol Behav 89: 490–500, 2006. doi: 10.1016/j.physbeh.2006.05.036. [DOI] [PubMed] [Google Scholar]
- 47.Ritter S, Li AJ, Wang Q, Dinh TT. Minireview: The value of looking backward: the essential role of the hindbrain in counterregulatory responses to glucose deficit. Endocrinology 152: 4019–4032, 2011. doi: 10.1210/en.2010-1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ritter S, Llewellyn-Smith I, Dinh TT. Subgroups of hindbrain catecholamine neurons are selectively activated by 2-deoxy-D-glucose induced metabolic challenge. Brain Res 805: 41–54, 1998. doi: 10.1016/S0006-8993(98)00655-6. [DOI] [PubMed] [Google Scholar]
- 49.Ritter S, Taylor JS. Vagal sensory neurons are required for lipoprivic but not glucoprivic feeding in rats. Am J Physiol Regul Integr Comp Physiol 258: R1395–R1401, 1990. [DOI] [PubMed] [Google Scholar]
- 50.Roman CW, Derkach VA, Palmiter RD. Genetically and functionally defined NTS to PBN brain circuits mediating anorexia. Nat Commun 7: 11905, 2016. doi: 10.1038/ncomms11905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Routh VH. Glucose-sensing neurons: are they physiologically relevant? Physiol Behav 76: 403–413, 2002. doi: 10.1016/S0031-9384(02)00761-8. [DOI] [PubMed] [Google Scholar]
- 52.Sanders NM, Ritter S. Repeated 2-deoxy-D-glucose-induced glucoprivation attenuates Fos expression and glucoregulatory responses during subsequent glucoprivation. Diabetes 49: 1865–1874, 2000. doi: 10.2337/diabetes.49.11.1865. [DOI] [PubMed] [Google Scholar]
- 53.Saper CB, Chou TC, Elmquist JK. The need to feed: homeostatic and hedonic control of eating. Neuron 36: 199–211, 2002. doi: 10.1016/S0896-6273(02)00969-8. [DOI] [PubMed] [Google Scholar]
- 54.Sawchenko PE, Swanson LW. Central noradrenergic pathways for the integration of hypothalamic neuroendocrine and autonomic responses. Science 214: 685–687, 1981. doi: 10.1126/science.7292008. [DOI] [PubMed] [Google Scholar]
- 55.Schild JH, Clark JW, Hay M, Mendelowitz D, Andresen MC, Kunze DL. A- and C-type rat nodose sensory neurons: model interpretations of dynamic discharge characteristics. J Neurophysiol 71: 2338–2358, 1994. [DOI] [PubMed] [Google Scholar]
- 56.Schiltz JC, Sawchenko PE. Specificity and generality of the involvement of catecholaminergic afferents in hypothalamic responses to immune insults. J Comp Neurol 502: 455–467, 2007. doi: 10.1002/cne.21329. [DOI] [PubMed] [Google Scholar]
- 57.Silver IA, Erecińska M. Extracellular glucose concentration in mammalian brain: continuous monitoring of changes during increased neuronal activity and upon limitation in oxygen supply in normo-, hypo-, and hyperglycemic animals. J Neurosci 14: 5068–5076, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Simon OR, Basuray BN, West WL, Copeland R. Interaction between the baroreflex and anterior hypothalamic stimulation. Demonstration of a noradrenergic involvement. Neuropharmacology 24: 665–675, 1985. doi: 10.1016/0028-3908(85)90110-8. [DOI] [PubMed] [Google Scholar]
- 59.Smith RJ, Aston-Jones G. Noradrenergic transmission in the extended amygdala: role in increased drug-seeking and relapse during protracted drug abstinence. Brain Struct Funct 213: 43–61, 2008. doi: 10.1007/s00429-008-0191-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stornetta RL, Sevigny CP, Guyenet PG. Vesicular glutamate transporter DNPI/VGLUT2 mRNA is present in C1 and several other groups of brainstem catecholaminergic neurons. J Comp Neurol 444: 191–206, 2002. doi: 10.1002/cne.10141. [DOI] [PubMed] [Google Scholar]
- 61.Travagli RA, Hermann GE, Browning KN, Rogers RC. Brainstem circuits regulating gastric function. Annu Rev Physiol 68: 279–305, 2006. doi: 10.1146/annurev.physiol.68.040504.094635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Troy AE, Simmonds SS, Stocker SD, Browning KN. High fat diet attenuates glucose-dependent facilitation of 5-HT3-mediated responses in rat gastric vagal afferents. J Physiol 594: 99–114, 2016. doi: 10.1113/JP271558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ueta Y, Kannan H, Higuchi T, Negoro H, Yamaguchi K, Yamashita H. Activation of gastric afferents increases noradrenaline release in the paraventricular nucleus and plasma oxytocin level. J Auton Nerv Syst 78: 69–76, 2000. doi: 10.1016/S0165-1838(99)00049-1. [DOI] [PubMed] [Google Scholar]
- 64.Wan S, Browning KN. d-Glucose modulates synaptic transmission from the central terminals of vagal afferent fibers. Am J Physiol Gastrointest Liver Physiol 294: G757–G763, 2008. doi: 10.1152/ajpgi.00576.2007. [DOI] [PubMed] [Google Scholar]
- 65.Wan S, Browning KN. Glucose increases synaptic transmission from vagal afferent central nerve terminals via modulation of 5-HT3 receptors. Am J Physiol Gastrointest Liver Physiol 295: G1050–G1057, 2008. doi: 10.1152/ajpgi.90288.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang R, Liu X, Hentges ST, Dunn-Meynell AA, Levin BE, Wang W, Routh VH. The regulation of glucose-excited neurons in the hypothalamic arcuate nucleus by glucose and feeding-relevant peptides. Diabetes 53: 1959–1965, 2004. doi: 10.2337/diabetes.53.8.1959. [DOI] [PubMed] [Google Scholar]
- 67.Wang ZJ, Rao ZR, Shi JW. Tyrosine hydroxylase-, neurotensin-, or cholecystokinin-containing neurons in the nucleus tractus solitarii send projection fibers to the nucleus accumbens in the rat. Brain Res 578: 347–350, 1992. doi: 10.1016/0006-8993(92)90269-F. [DOI] [PubMed] [Google Scholar]
- 68.Williams DL, Schwartz MW, Bastian LS, Blevins JE, Baskin DG. . Immunocytochemistry and laser capture microdissection for real-time quantitative PCR identify hindbrain neurons activated by interaction between leptin and cholecystokinin. J Histochem Cytochem 56: 285–293, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Willing AE, Berthoud HR. Gastric distension-induced c-fos expression in catecholaminergic neurons of rat dorsal vagal complex. Am J Physiol Regul Integr Comp Physiol 272: R59–R67, 1997. [DOI] [PubMed] [Google Scholar]
- 70.Zhang W, Carreño FR, Cunningham JT, Mifflin SW. Chronic sustained and intermittent hypoxia reduce function of ATP-sensitive potassium channels in nucleus of the solitary tract. Am J Physiol Regul Integr Comp Physiol 295: R1555–R1562, 2008. doi: 10.1152/ajpregu.90390.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]