

Use of this checklist by authors and reviewers will improve quality and transparency in reporting randomized clinical trials of pain treatments.
Keyword: Reporting checklist
Abstract
Introduction:
Randomized clinical trials (RCTs) are considered the gold standard when assessing the efficacy of interventions because randomization of treatment assignment minimizes bias in treatment effect estimates. However, if RCTs are not performed with methodological rigor, many opportunities for bias in treatment effect estimates remain. Clear and transparent reporting of RCTs is essential to allow the reader to consider the opportunities for bias when critically evaluating the results. To promote such transparent reporting, the Consolidated Standards of Reporting Trials (CONSORT) group has published a series of recommendations starting in 1996. However, a decade after the publication of the first CONSORT guidelines, systematic reviews of clinical trials in the pain field identified a number of common deficiencies in reporting (eg, failure to identify primary outcome measures and analyses, indicate clearly the numbers of participants who completed the trial and were included in the analyses, or report harms adequately).
Objectives:
To provide a reporting checklist specific to pain clinical trials that can be used in conjunction with the CONSORT guidelines to optimize RCT reporting.
Methods:
Qualitative review of a diverse set of published recommendations and systematic reviews that addressed the reporting of clinical trials, including those related to all therapeutic indications (eg, CONSORT) and those specific to pain clinical trials.
Results:
A checklist designed to supplement the content covered in the CONSORT checklist with added details relating to challenges specific to pain trials or found to be poorly reported in recent pain trials was developed.
Conclusion:
Authors and reviewers of analgesic RCTs should consult the CONSORT guidelines and this checklist to ensure that the issues most pertinent to pain RCTs are reported with transparency.
1. Introduction
Randomized clinical trials (RCTs) are considered the gold standard when assessing the efficacy of interventions because randomization of treatment assignment minimizes bias in treatment effect estimates. However, depending on the methodological rigor, many opportunities for bias in RCTs remain.24,34 The opportunities for such bias should be considered when evaluating and interpreting results of RCTs. This critical evaluation depends on the transparent reporting of clinical trial methods and results in the peer-reviewed literature. To promote such transparent reporting, the Consolidated Standards of Reporting Trials (CONSORT) group has published a series of recommendations starting in 1996. These recommendations cover a wide range of factors including randomization and blinding methods, statistical details, and participant flow, as well as guidance on which details should be covered in various sections of the article.29 Table 1 outlines the categories covered in CONSORT.
Table 1.
CONSORT headings.
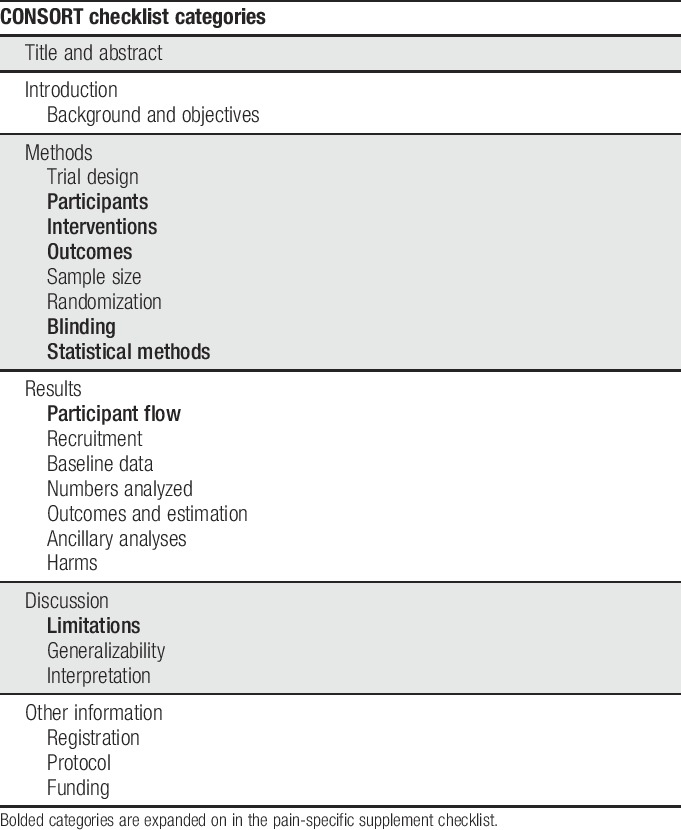
A decade after the publication of the first CONSORT guidelines, systematic reviews of clinical trials in the pain field identified a number of common deficiencies in reporting of clinical trials, including failure to identify primary outcome measures and analyses, indicate clearly the numbers of participants who completed the trial and were included in the analyses, or report harms adequately.8,15,16,18,19,21,38–40 In this article, we describe a checklist (Table 2) designed to supplement the content covered in the CONSORT checklist with added details relating to challenges specific to pain trials or found to be poorly reported in recent pain trials. We have not included areas for which reporting has been found to be poor in pain trials when further expansion of the CONSORT checklist seems unlikely to improve reporting (eg, harms reporting23). Although some discussion of various trial design issues as they relate to reporting is inevitable, the purpose of this checklist and accompanying article is not to inform pain trial design. For recommendations regarding study design and outcome measures for various types of pain trials, please see the other articles in this series. We believe that the use of this checklist by authors and reviewers in conjunction with the CONSORT statement29 will improve the reporting and enhance the interpretability of RCTs of pain treatments.
Table 2.
Pain-specific CONSORT supplement checklist.
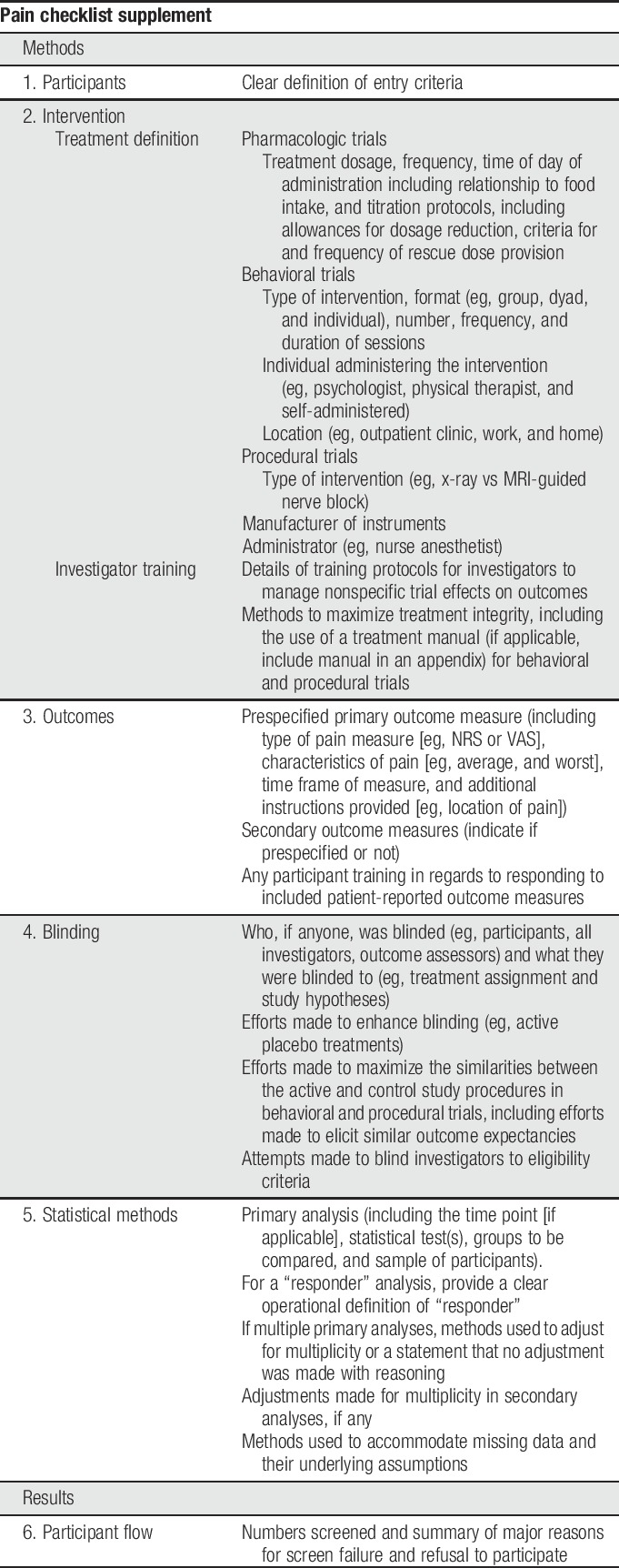
2. Methods
In preparation for developing this checklist, the authors reviewed a diverse set of published recommendations and systematic reviews that addressed the reporting of clinical trials, including those related to all therapeutic indications1,12,13,22,30,33,34,43,44 and those specific to pain clinical trials.8,10,11,14,19,21,37–41 The checklist was modified multiple times based on the input from all authors as well as the editors of this series. Examples were developed based on hypothetical protocols; although they may include elements of existing studies, they are not based largely on any particular example from the literature.
3. Checklist items
In this section, we provide an explanation for our reasoning as to why each item is particularly relevant for RCTs of pain treatments. Examples are provided for different types of interventions (ie, pharmacologic, behavioral, or interventional) or study designs where warranted. These examples are not meant to be inclusive of all possible design features, but rather an example of the types of details that are necessary when reporting.
3.1. Participants
Clear definitions of eligibility criteria are imperative for understanding the study design and for evaluating the generalizability of the study results. A clear definition of eligible participants is important in all trials; however, in pain trials, it is particularly important when patients with comorbid pain conditions or mental health conditions are excluded. These patients will certainly be assessed clinically for the pain conditions being studied and may not respond as well to treatment or be at higher risk for adverse events in response to treatment with certain pharmacologic classes often used for pain. In addition, results from baseline screening periods are often used as a part of eligibility criteria in pain trials (eg, requiring response to open-label treatment with the experimental drug in Enriched Enrollment Randomized Withdrawal (EERW) trials or excluding participants who fail to respond to 2 treatments with known efficacy in the condition of interest)26,28,32 and clear description of such eligibility criteria is imperative.
Example—Eligible patients were aged at least 18 years, had confirmed diabetes (ie, HbA1C ≥ 7) and a diagnosis of diabetic peripheral neuropathy, confirmed by a neurologist study investigator. They reported having pain associated with diabetes in their lower limbs for at least 3 months before the enrollment visit, which occurred 1 week before randomization. Patients were ineligible if they had a documented history of major depressive disorder or suicidal thoughts in their medical record or were unwilling to abstain from starting any new pain medication or altering dosages of any current pain medication for the duration of the trial. Patients were not excluded for comorbid pain conditions unless they caused pain in the lower limbs that may have been difficult to discern from diabetic peripheral neuropathy pain in self-report ratings. At the screening visit, participants were given a week-long daily diary that asked them to rate their average pain intensity and their worst pain intensity on a 0 to 10 numeric rating scale (NRS) (0 = no pain, 10 = worst pain imaginable). The following criteria were required for trial eligibility after the baseline week: (1) a minimum of 4 of 7 diary entries were completed, (2) the mean average pain score for the week was ≥4 of 10, and (3) average pain ratings were less than or equal to worst pain ratings for each day of ratings that were provided.
Example (same patient eligibility as above example, with the exception of this alternative screening requirement substituted for the one above)—All enrolled participants were treated with treatment A for 4 weeks in a run-in period. Treatment A was started at 15 mg BID with food for week 1 and increased to 30 mg BID for week 2 and 45 mg BID for weeks 3 and 4. If participants experienced adverse events while taking 45 mg BID that would otherwise cause them to discontinue treatment, they were allowed to revert to 30 mg BID. Participants who (1) could tolerate a minimum of 30 mg BID for the final 3 weeks of the run-in period, (2) experienced at least a 30% decrease in pain from baseline to the end of the 4-week run-in period, and (3) had a mean pain score of ≤4 out of 10 during the last week of the run-in period were randomized to continue on their maximum tolerated dosage or to placebo treatment.
3.2. Interventions
3.2.1. Treatment definition
Careful reporting of details is necessary when describing behavioral interventions, interventions involving invasive procedures, and pharmacologic interventions involving complicated titration protocols, all of which are commonly evaluated in pain trials. Full understanding of the intervention(s) is required not only for future research that will replicate and extend the RCT findings but also for translation into clinical practice. Treatment descriptions should include what the intervention consists of, at what schedule and for how long they will receive it, and what modifications to the treatment are allowed within the protocol (if applicable).
3.2.1.1. Pharmacologic example
Participants were randomly assigned to receive either treatment or matching placebo for 12 weeks. Participants were given 1 capsule of treatment A (30 mg/d) or matching placebo for the first week and 2 capsules (30 mg/d) taken once in the morning for the remaining 11 weeks of the trial. Participants were instructed to take all treatments with food. Participants experiencing adverse events that would otherwise lead to discontinuation were allowed to revert back to 1 capsule per day with approval from the investigator. If participants could not tolerate 1 capsule per day, they were withdrawn from the study. Acetaminophen (up to 3 g/d) was used as rescue therapy if the participants felt it was necessary. Use of all other rescue medications was not permitted. Whether to discontinue the assigned treatment due to adverse events was at the discretion of the participant. Investigators could discontinue treatment for a participant if they felt it was medically necessary in response to an adverse event.
3.2.1.2. Behavioral example
Participants were randomized to receive the strength training intervention alone or as part of a dyad including a relative or friend. The participants (including partners in the dyad treatment group) attended weekly 45-minute training sessions with a physical therapist at the study site for 12 weeks. In these sessions, they performed strength training exercises for the lower extremities including walking, air squats, stair climbing, and lunges (an Appendix can be provided where a complete description of exercises could be presented). In addition, participants were instructed to perform the same exercises at home 2 times per week, either alone or with their partner depending on their group assignment. Participants were required to have the ability to perform a minimum amount of each of these exercises for entry in the study (see manual and eligibility criteria); however, if on a certain day during the study the participant did not feel that they could perform some of the exercises safely, they were allowed to skip the exercise and this was documented.
3.2.1.3. Procedural example
Participants were randomized to use a temporary spinal cord stimulator for 12 weeks or to the wait-list control group. The spinal cord stimulator (authors should provide manufacturer and model number) was implanted by a neurologist. Participants were positioned prone on the procedure table. The interlaminar space was identified in the midline under fluoroscopy. Landmarks for a paramedian approach were identified under fluoroscopy to address pain in the affected lower limb for each participant. Using a 14-gauge Tuohy needle, the bevel was advanced from the medial aspect of the pedicle after local anesthesia was administered using standardized needles and local anesthetic medication with modifications based on body habitus. The Tuohy needle was advanced in the midline until there was a clear loss of resistance to saline. The leads were placed along the span of the thoracic segments corresponding to the affected segments and lateralization of target symptoms. The stimulator positioning was tested after initiation of the stimulus by discussion with the participant regarding the stimulus coverage. If necessary, the leads were repositioned to optimize coverage. The programming process used a standardized algorithm outlined in the device programming guide and training materials provided by the manufacturer (the authors should provide the detailed algorithm in a supplemental appendix). Once lead positioning was confirmed, the Tuohy needle and style were removed. The lead was secured to the participants' lumbar regions using sterile dressing with Steri-Strips and Tegaderm for the duration of the 12-week trial. A research coordinator reviewed the available preset stimulation program options with the participants in the recovery room in different positions (ie, reclined, sitting, and standing). At this time, the participants identified the programs that provided the best coverage and pain relief. The participants were instructed to use the programs that worked best for them and adjust the stimulators to an intensity that they could easily feel but were not uncomfortable. Participants were instructed to turn on the system for at least 1 hour in the morning, 1 hour in the middle of the day, and 1 hour in the evening; however, they were also free to use it as frequently as they would like during the day or night.
3.2.2. Investigator training
3.2.2.1. Participant interaction to minimize nonspecific trial effects on outcomes
Detecting differences in treatment effects using subjective patient-reported symptoms such as pain can be complicated by multiple factors. Pain ratings are susceptible to expectations and nonspecific effects such as attention received during clinical trial visits. These factors, among others, likely contribute to the large placebo responses that often occur in modern chronic pain trials.42 To demonstrate a difference in the effects between treatments being studied, it is helpful to minimize the nonspecific responses in all treatment groups. Training trial investigators to minimize participant expectations for the experimental treatment by explaining that the efficacy of the treatment is unknown may decrease the placebo response in both treatment groups. In addition, explaining to participants that being as accurate as possible in their pain ratings is important.11
Example—Investigators and research staff were trained in strategies to minimize the placebo effect when interacting with patients (eg, managing expectations about treatment efficacy and minimizing excess social interaction). A video training module was used to teach research staff how to deliver instructions to patients in a standardized fashion (an Appendix can be provided where authors provide the training video).
3.2.2.2. Treatment integrity
To ensure that the treatment was administered in a manner consistent with the treatment manual, investigators should be adequately trained to deliver the intervention and treatment integrity4 should be assessed and reported. This is particularly important for behavioral interventions which are commonly used for pain.
Example—Ten physical therapists were trained by 4 highly experienced clinical psychologists to deliver the mindfulness meditation treatment portion of the intervention at a 2-day workshop facilitated by the principal investigator (author(s) can provide an Appendix where they give a more detailed description of the course). In brief, the course included didactic presentations to describe the theory underlying the intervention followed by a role play demonstration for treatment delivery and then practice sessions in which pairs of physical therapists practiced delivering the treatment to one another with observation and feedback from the workshop facilitators. In addition, each physical therapist delivered the intervention to study participants at least 2 times in the presence of an experienced clinical psychologist who monitored the delivery for fidelity to the treatment manual and provided feedback. Further rounds of observation were made if deemed necessary by the clinical psychologist. In addition, all interventions were audio recorded and ongoing supervision was provided by a clinical psychologist based on these audio recordings. Finally, a random selection of 20% of the audio tapes were reviewed by 2 clinical psychologists not involved in the study and coded to assess both treatment integrity and therapist competence. Half of the recordings were coded by both psychologists; these were compared to assess reliability of coding (kappa coefficient = 0.82, indicating a high degree of reliability). Coding items included those for inclusion of essential content (ie, treatment integrity, eg, teaching and encouraging incorporation of mindfulness practices in everyday situations for the mindfulness condition) and therapist competence (ie, delivering treatment components in a skillful and responsive way, eg, using appropriate language and examples, with a patient with low health literacy).
3.3. Outcomes
Outcome measures in pain trials are often self-report measures of pain intensity or related domains (eg, physical function, mood, and sleep).10 Many factors exist that can affect the way in which participants interpret the 0 to 10 pain intensity NRS. For example, the common anchor of “worst pain imaginable” for the 10 rating is likely interpreted variably by different participants, and to our knowledge few instances occur in the published literature where researchers provide participants with any direction as to how to interpret this anchor.37 Furthermore, participants are often asked to rate their “average pain” over the past day. Yet, they are not provided any instructions regarding how to derive their rating (eg, should participants with highly fluctuating pain consider periods without any pain in their “average” pain score? Should they include their pain during sleep in their estimate?).9 Finally, patients often consider their pain interference with function and affective components of pain when completing their NRS ratings of pain intensity. Enhanced instructions to focus on pain intensity independent of mood and pain interference with activities could minimize the inclusion of these related constructs in NRS pain intensity ratings.9,37 Currently, little research is available regarding the optimal instructions for participants pertaining to these details of the NRS and certainly no consensus exists; however, clear reporting of instructions provided to participants in RCTs will provide data on which to base future research. Of note, clear reporting of pain intensity measures has been deficient in recent clinical trials of pain treatments.39
Example—The prespecified primary outcome measure was a 0 to 10 NRS (0 = no pain, 10 = worst pain imaginable) for average pain over the last 24 hours. Research staff administered training to the participants on completing the pain diary. In brief, participants were asked to (1) complete pain ratings on their own before bedtime; (2) focus on the pain in their legs and feet throughout the entire day considering the intensities felt during different activities when determining their average pain; and (3) avoid considering pain from other sources such as a headache when rating their pain (note: author(s) can refer to an Appendix here where they provide the complete training manual). Prespecified secondary outcome measures included the pain interference question from the Brief Pain Inventory—Short form [BPI-SF]6 and the Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC).2 The BPI interference question asks patients to circle the number that best describes how, during the past 24 hours, pain has interfered with the following symptoms on a 0 to 10 NRS (0 = does not interfere, 10 = completely interferes): general activity, mood, walking ability, normal work, relations with other people, sleep, and enjoyment of life. The WOMAC is a self-report scale that has items that fall within 3 domains: pain, stiffness, and physical function. It asks patients to rate their difficulty with each item on a 0 to 4 scale (0 = none, 1 = slight, 2 = moderate, 3 = very, and 4 = extremely).
3.4. Blinding
Double blinding can be challenging in many pain trials because pharmacologic pain treatments often have recognizable side effects and full blinding of investigators or patients can be impossible with certain behavioral or procedural treatments. Clear reporting of efforts made to maximize blinding and to control for effects not related to the active treatment in behavioral trials (eg, attention received during study visits) allows the reader to evaluate the methods used in the trial.
Example—This trial compared a physical therapy intervention group with an educational information comparison group. (note: the active intervention would be described here, see Section 3.2.1 for examples). The educational information comparison group received informational packets outlining similar exercises to the ones performed with the physical therapist in the active treatment group. Participants in this group met with a study therapist to discuss their progress for the same amount of time at the same frequency with which the participants in the active group met with the physical therapist. The study participants were blind to the research hypotheses. They were told that it was unknown whether receipt of an educational packet providing exercise instructions or visits to a physical therapist to perform those exercises was more effective. After the study, the participants were informed of the real study hypothesis and consent to use their data was obtained. Participants were asked to rate their expectations for the outcome of their treatment condition after treatment assignment, but before their first treatment session to examine whether expectations between the 2 groups were similar. In addition, the research coordinator administering the outcome measures was blinded with respect to the treatment assignment and the participants were asked not to discuss their study activities with her.
3.5. Statistical methods
Prespecification of the primary analysis including identification of the measure(s), time point (if applicable), description of the statistical model and statistical test, groups to be compared, methods for handling multiplicity, methods for accommodating missing data, and sample to be used (eg, all randomized participants vs only those who completed the trial according to the protocol) is necessary to enhance trial credibility and minimize the probability of a type I error.18,22,41 Multiple outcomes are often important in pain conditions. For example, investigators may prioritize improvement of pain intensity and physical function equally in an osteoarthritis trial or improvements in pain intensity and fatigue in a trial of fibromyalgia. Furthermore, it is often of interest to evaluate the effects of a treatment on acute and chronic pain or compare more than 2 treatments, both of which also may lead to multiple analyses of equal importance. If more than 1 analysis is declared primary, prespecified methods should be used to adjust for multiplicity, especially in later phase trials that are designed to evaluate efficacy.18 These methods should be clearly reported. For example, when authors state that there are co-primary analyses, it should be reported whether the protocol specified that the trial would be concluded a success if both analyses yielded a result with P < 0.05 or if either analysis yielded a result with P < 0.025. A recent systematic review found deficiencies in the identification of primary analyses and methods to adjust for multiplicity in pain trials.18
In most RCTs, some participants discontinue before the end of the study leading to missing data. Others might remain enrolled in the study, but might not provide some data at some assessment points for other reasons (eg, missed visit). As a result, missing data are a common problem in many chronic pain trials.16,25 A large amount of missing data can lead to bias in treatment effect estimates. Using a statistician-recommended strategy to accommodate missing data, rather than excluding participants whose data are missing, can minimize bias. Such strategies include using multiple, prespecified methods to accommodate missing data that make different assumptions about the patterns of missingness.27,31 Methods to accommodate missing data were reported in fewer than half of pain trials reviewed in a recent systematic review.16
Example—The co-primary outcome measures were the average pain NRS and the Fibromyalgia Impact Questionnaire score5 measured at 12 weeks after randomization. For each outcome measure, cognitive behavioral therapy was compared with education control using an analysis of covariance (ANCOVA) model that included a treatment group as the factor of interest and the corresponding baseline symptom score as a covariate. The primary analyses included all available data from all randomized participants. Missing data were accommodated using the technique of multiple imputations. The imputation procedure for each outcome variable used a treatment group and outcomes at all time points, along with the Markov chain Monte Carlo simulation, to produce 20 complete data sets. These data sets were analyzed separately using ANCOVA and the results were combined across data sets using Rubin's rules.26 A Bonferroni correction was used to adjust for multiplicity; a P-value less than 0.025 for either analysis was considered significant to preserve the overall significance level at 0.05. A secondary analysis compared the percentage of “responders” between groups using a χ2 test. A “responder” was defined as a participant whose NRS pain scores at 12 weeks (1) decreased by at least 30% from baseline and (2) was below 4 of 10. Participants who prematurely discontinued were defined as nonresponders. Prespecified analyses of secondary outcome variables used similar ANCOVA models to those of the primary analyses. No adjustment for multiplicity was made in the secondary analyses, as they were considered exploratory and hypothesis generating.
3.6. Participant flow
As clearly outlined in the CONSORT guidelines,30 it is imperative for the evaluation of any trial data that the number of participants who were randomized, completed, and whose data were included in the analyses as well as reasons for dropout are outlined for each group. To enhance understanding of generalizability of the trial, we also recommend reporting the numbers of participants who were screened before randomization and the reasons for exclusion.
Example—In total, 650 patients were screened for study enrollment; 200 did not meet initial eligibility criteria (authors can refer to the CONSORT diagram for reasons). Another 30 participants were eliminated after the baseline week for the following reasons: participant's mean pain score was <4 (n = 18), participant did not complete at least 4 diary entries (n = 7), and participant did not return for the randomization visit (n = 5). See CONSORT guidelines for an example of the remainder of the participant flow reporting (items 13 a and b).30
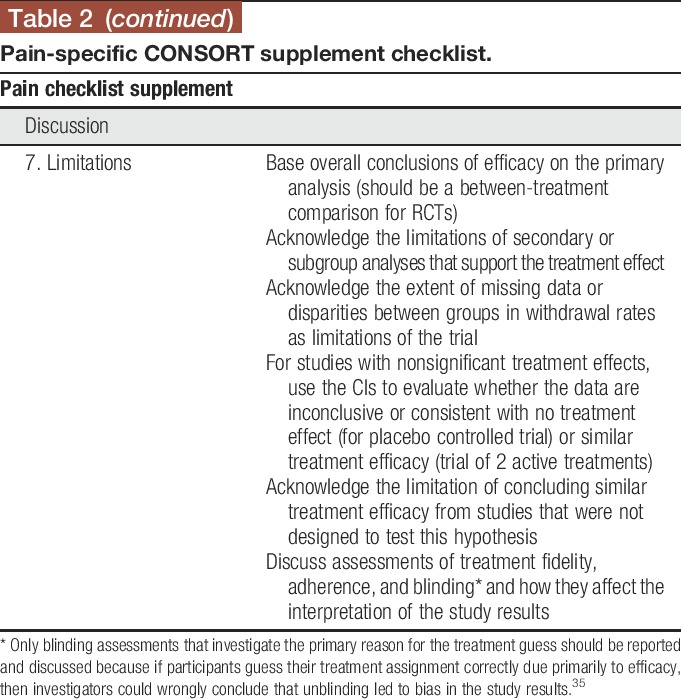
3.7. Limitations
It is important to interpret the results of RCTs appropriately based on the statistical analyses performed and overall context of the trial. Overall conclusions of efficacy should be based on the primary between-treatment comparisons. Discussions pertaining to potential efficacy based on changes from baseline in each treatment group in the absence of statistically significant between-treatment differences are discouraged, but if included must be accompanied by an acknowledgment that such analyses do not reflect the level of evidence provided by an RCT and that such effects are possibly due to placebo and other nonspecific effects as well as regression to the mean.4,16 It is important to outline the limitations of secondary and subgroup analyses that support a treatment effect in the absence of support by the primary analysis. The degree to which interpretation of these analyses is limited depends on whether there were a limited number of prespecified secondary and subgroup analyses as compared to many post hoc analyses and whether attempts were made to control the probability of a type I error in the secondary analyses.22 It is important to note that post hoc analyses are valuable for hypothesis generation and, when of interest, should be included in RCT reports with the appropriate caveats. For studies yielding nonsignificant results, the confidence interval (CI) for the treatment effect should be considered when determining whether the data are consistent with the absence of a clinically meaningful treatment effect or are inconclusive.7,20,37 Although CIs can be used to evaluate the possibility that the data support comparable efficacy of 2 interventions, formal prospective noninferiority or equivalence studies are necessary to confirm the result.44 Poor treatment integrity or adherence to the protocol or the absence (or compromise) of blinding can lead to biased treatment effect estimates. A clear discussion of potential effects of low treatment integrity, adherence, or blinding in conjunction with appropriate interpretation of the statistical analyses will provide a balanced interpretation for readers.
Example—The estimated treatment effect from the primary efficacy analysis that compared pain severity between the treatment A and placebo groups was not significant. However, the CI for the treatment effect included a difference of 3 points on the pain NRS in favor of treatment A, suggesting that the results of this study cannot rule out a potentially clinically meaningful effect for treatment A. In addition, nominally significant differences (ie, P < 0.05) between groups were obtained in secondary analyses comparing the treatment groups with respect to measures of pain interference with function and sleep using items from the BPI interference question. Although these secondary analyses cannot be considered confirmatory, these results in combination with the inconclusive result from the primary analysis suggest that further research may be warranted to determine whether treatment A is effective for chronic low back pain.
Example—This study failed to demonstrate a significant difference between treatments A and B with respect to mean pain NRS score in patients with chronic low back pain. The 95% CI for the group difference excluded differences larger than 1.0 NRS points in favor of either treatment, suggesting that there is no clinically meaningful difference between the treatments. It should be acknowledged, however, that currently no consensus exists for the minimal clinically meaningful between-group treatment difference in pain scores. In addition, a study with a prespecified hypothesis designed to evaluate the equivalence of the 2 treatments is required to confirm this conclusion. The endpoint pain score was significantly lower than baseline pain score in both groups, suggesting the possibility of some benefit for each treatment. However, the absence of a placebo group makes it impossible to determine whether the apparent effects of either treatment are due only to placebo effects (eg, effects from expectation or increased attention received during a clinical trial), natural history, or regression to the mean.
Example 3—The overall (average) fidelity to the treatment protocol among the study clinicians was 94% (range 85%–100%), suggesting that the social workers delivered the intervention as it was intended. Thus, deviations from the protocol did not seem to contribute to the lack of treatment efficacy observed for the pain coping skills treatment in this study.
4. Conclusions
To maximize readers' ability to critically evaluate the results and conclusions drawn based on RCTs, it is imperative that authors clearly report the methods and results of those RCTs and carefully interpret those results within the limits of the designs and analyses of the trials. Authors and reviewers of analgesic RCTs should consult the CONSORT guidelines and this checklist to ensure that the issues most pertinent to pain trials are reported with transparency. Although these recommendations are focused on reporting of RCTs, reviewers and readers can also use the information presented here to evaluate the quality of the design and the validity of the results when reading articles reporting the findings from RCTs.
Disclosures
Dr. Gross has received grants from NIH for educational support, an editorial stipend from the American Academy of Neurology, and royalties from Blackwell and Wiley. All other authors have no relevant conflicts of interest to declare.
The views expressed in this article are those of the authors and no official endorsement by the Food and Drug Administration (FDA) or the pharmaceutical and device companies that provided unrestricted grants to support the activities of the Analgesic, Anesthetic, and Addiction Clinical Trial Translations, Innovations, Opportunities, and Networks (ACTTION) public-private partnership should be inferred. Financial support for this project was provided by the ACTTION public-private partnership which has received research contracts, grants, or other revenue from the FDA, multiple pharmaceutical and device companies, philanthropy, and other sources.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
References
- [1].Altman D, Bland M. Absence of evidence is not evidence of absence. BMJ 1995;311:485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bellamy N, Buchanan WW, Goldsmith CH, Campbell J, Stitt LW. Validation study of WOMAC: a health status instrument for measuring clinically important patient relevant outcomes to antirheumatic drug therapy in patients with osteoarthritis of the hip or knee. J Rheumatol 1988;15:1833–40. [PubMed] [Google Scholar]
- [3].Borrelli B. The assessment, monitoring, and enhancement of treatment fidelity in public health clinical trials. J Public Health Dent 2011;71:S52–63. [PubMed] [Google Scholar]
- [4].Boutron I, Dutton S, Ravaud P, Altman DG. Reporting and interpretation of randomized controlled trials with statistically nonsignificant results for primary outcomes. JAMA 2010;303:2058–64. [DOI] [PubMed] [Google Scholar]
- [5].Buckhardt CS, Clark SR, Bennet RM. The Fibromyalgia Impact Questionnaire: development and validation. J Rheumatol 1991;18:728–33. [PubMed] [Google Scholar]
- [6].Cleeland C. Brief pain inventory, 1991. Available at: http://www.mdanderson.org/education-and-research/departments-programs-and-labs/departments-and-divisions/symptom-research/symptom-assessment-tools/brief-pain-inventory.html. Accessed February 15, 2017. [Google Scholar]
- [7].Detsky AS, Sackett DL. When was a “negative” clinical trial big enough? How many patients you needed depends on what you found. Arch Intern Med 1985;145:709–12. [PubMed] [Google Scholar]
- [8].Dworkin JD, Mckeown A, Farrar JT, Gilron I, Hunsinger M, Kerns RD, McDermott MP, Rappaport BA, Turk DC, Dworkin RH, Gewandter JS. Deficiencies in reporting of statistical methodology in recent randomized trials of nonpharmacologic pain treatments: ACTTION systematic review. J Clin Epidemiol 2016;72:56–65. [DOI] [PubMed] [Google Scholar]
- [9].Dworkin RH, Burke LB, Gewandter JS, Smith SM. Reliability is necessary but far from sufficient. How might the validity of pain ratings be improved? CJP 2015;31:599–602. [DOI] [PubMed] [Google Scholar]
- [10].Dworkin RH, Turk DC, Farrar JT, Haythornthwaite JA, Jensen MP, Katz NP, Kerns RD, Stucki G, Allen RR, Bellamy N, Carr DB, Chandler J, Cowan P, Dionne R, Galer BS, Hertz S, Jadad AR, Kramer LD, Manning DC, Martin S, McCormick CG, McDermott MP, McGrath P, Quessy S, Rappaport BA, Robbins W, Robinson JP, Rothman M, Royal MA, Simon L, Stauffer JW, Stein W, Tollett J, Wernicke J, Witter J; Immpact. Core outcome measures for chronic pain clinical trials: IMMPACT recommendations. PAIN 2005;113:9–19. [DOI] [PubMed] [Google Scholar]
- [11].Dworkin RH, Turk DC, Peirce-Sandner S, Burke LB, Farrar JT, Gilron I, Jensen MP, Katz NP, Raja SN, Rappaport BA, Rowbotham MC, Backonja MM, Baron R, Bellamy N, Bhagwagar Z, Costello A, Cowan P, Fang WC, Hertz S, Jay GW, Junor R, Kerns RD, Kerwin R, Kopecky EA, Lissin D, Malamut R, Markman JD, McDermott MP, Munera C, Porter L, Rauschkolb C, Rice AS, Sampaio C, Skljarevski V, Sommerville K, Stacey BR, Steigerwald I, Tobias J, Trentacosti AM, Wasan AD, Wells GA, Williams J, Witter J, Ziegler D. Considerations for improving assay sensitivity in chronic pain clinical trials: IMMPACT recommendations. PAIN 2012;153:1148–58. [DOI] [PubMed] [Google Scholar]
- [12].European Medicines Agency. Guideline on choice of non-inferiority margin. Stat Med 2006;25:1628–38. [DOI] [PubMed] [Google Scholar]
- [13].Gallo P, Chuang-Stein C, Dragalin V, Gaydos B, Krams M, Pinheiro J. Adaptive designs in clinical drug development–an Executive Summary of the PhRMA Working Group. J Biopharm Stat 2006;16:275–83; discussion 285–91, 293–8, 311–2. [DOI] [PubMed] [Google Scholar]
- [14].Gewandter JS, Dworkin RH, Turk DC, McDermott MP, Baron R, Gastonguay MR, Gilron I, Katz NP, Mehta C, Raja SN, Senn S, Taylor C, Cowan P, Desjardins P, Dimitrova R, Dionne R, Farrar JT, Hewitt DJ, Iyengar S, Jay GW, Kalso E, Kerns RD, Leff R, Leong M, Petersen KL, Ravina BM, Rauschkolb C, Rice AS, Rowbotham MC, Sampaio C, Sindrup SH, Stauffer JW, Steigerwald I, Stewart J, Tobias J, Treede RD, Wallace M, White RE. Research designs for proof-of-concept chronic pain clinical trials: IMMPACT recommendations. PAIN 2014;155:1683–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gewandter JS, McDermott MP, Mckeown A, Smith SM, Pawlowski JR, Poli JJ, Rothstein D, Williams MR, Bujanover S, Farrar JT, Gilron I, Katz NP, Rowbotham MC, Turk DC, Dworkin RH. Reporting of intention-to-treat analyses in recent analgesic clinical trials: ACTTION systematic review and recommendations. PAIN 2014;155:2714–9. [DOI] [PubMed] [Google Scholar]
- [16].Gewandter JS, McDermott MP, Mckeown A, Smith SM, Williams MR, Hunsinger M, Farrar J, Turk DC, Dworkin RH. Reporting of missing data and methods used to accommodate them in recent analgesic clinical trials: ACTTION systematic review and recommendations. PAIN 2014;155:1871–7. [DOI] [PubMed] [Google Scholar]
- [17].Gewandter JS, Mckeown A, McDermott MP, Dworkin JD, Smith SM, Gross RA, Hunsinger M, Lin AH, Rappaport BA, Rice AS, Rowbotham MC, Williams MR, Turk DC, Dworkin RH. Data interpretation in analgesic clinical trials with statistically nonsignificant primary analyses: an ACTTION systematic review. J Pain 2015;16:3–10. [DOI] [PubMed] [Google Scholar]
- [18].Gewandter JS, Smith SM, Mckeown A, Burke LB, Hertz SH, Hunsinger M, Katz NP, Lin AH, McDermott MP, Rappaport BA, Williams MR, Turk DC, Dworkin RH. Reporting of primary analyses and multiplicity adjustment in recent analgesic clinical trials: ACTTION systematic review and recommendations. PAIN 2014;155:461–6. [DOI] [PubMed] [Google Scholar]
- [19].Gewandter JS, Smith SM, Mckeown A, Edwards K, Narula A, Pawlowski JR, Rothstein D, Desjardins PJ, Dworkin SF, Gross RA, Ohrbach R, Rappaport BA, Sessle BJ, Turk DC, Dworkin RH. Reporting of adverse events and statistical details of efficacy estimates in randomized clinical trials of pain in temporomandibular disorders: analgesic, Anesthetic, and Addiction Clinical Trial Translations, Innovations, Opportunities, and Networks systematic review. J Am Dent Assoc 2015;146:246–54.e6. [DOI] [PubMed] [Google Scholar]
- [20].Goodman SN, Berlin JA. The use of predicted confidence intervals when planning experiments and the misuse of power when interpreting results. Ann Intern Med 1994;121:200–6. [DOI] [PubMed] [Google Scholar]
- [21].Hunsinger M, Smith SM, Rothstein D, Mckeown A, Parkhurst M, Hertz S, Katz NP, Lin AH, McDermott MP, Rappaport BA, Turk DC, Dworkin RH. Adverse event reporting in nonpharmacologic, noninterventional pain clinical trials: ACTTION systematic review. PAIN 2014;155:2253–62. [DOI] [PubMed] [Google Scholar]
- [22].Ioannidis JP. Why most published research findings are false. PLoS Med 2005;2:e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ioannidis JP, Evans SJ, Gøtzsche PC, O'neill RT, Altman DG, Schulz K, Moher D. Better reporting of harms in randomized trials: an extension of the CONSORT statement. Ann Intern Med 2004;141:781–8. [DOI] [PubMed] [Google Scholar]
- [24].Juni P, Altman DG, Egger M. Systematic reviews in health care: assessing the quality of controlled clinical trials. BMJ 2001;323:42–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Katz N. Methodological issues in clinical trials of opioids for chronic pain. Neurology 2005;65:S32–49. [DOI] [PubMed] [Google Scholar]
- [26].Katz N. Enriched enrollment randomized withdrawal trial designs of analgesics: focus on methodology. Clin J Pain 2009;25:797–807. [DOI] [PubMed] [Google Scholar]
- [27].Little RJA, Rubin DB. Statistical analyses with missing data. New York: John Wiley & Sons, Inc, 1987. [Google Scholar]
- [28].Mcquay HJ, Derry S, Moore RA, Poulain P, Legout V. Enriched Enrolment with Randomised Withdrawal (EERW): time for a new look at clinical trial design in chronic pain. PAIN 2008;135:217–20. [DOI] [PubMed] [Google Scholar]
- [29].Moher D, Hopewell S, Schulz KF, Montori V, Gøtzsche PC, Devereaux PJ, Elbourne D, Egger M, Altman DG. CONSORT 2010 explanation and elaboration: updated guidelines for reporting parallel group randomised trials. BMJ 2010;340:c869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Moher D, Schulz KF, Altman DG. The CONSORT statement: revised recommendations for improving the quality of reports of parallel-group randomized trials. Ann Intern Med 2001;134:657–62. [DOI] [PubMed] [Google Scholar]
- [31].Molenbergs G, Kenward MG. Missing data in clincal studies. Chichester, United Kingdom: John Wiley & Sons Ltd., 2007. [Google Scholar]
- [32].Moore RA, Wiffen PJ, Eccleston C, Derry S, Baron R, Bell RF, Furlan AD, Gilron I, Haroutounian S, Katz NP, Lipman AG, Morley S, Peloso PM, Quessy SN, Seers K, Strassels SA, Straube S. Systematic review of enriched enrolment, randomised withdrawal trial designs in chronic pain: a new framework for design and reporting. PAIN 2015;156:1382–95. [DOI] [PubMed] [Google Scholar]
- [33].Piaggio G, Elbourne DR, Pocock SJ, Evans SJ, Altman DG. Reporting of noninferiority and equivalence randomized trials: extension of the CONSORT 2010 statement. JAMA 2012;308:2594–604. [DOI] [PubMed] [Google Scholar]
- [34].Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias. Dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273:408–12. [DOI] [PubMed] [Google Scholar]
- [35].Senn SJ. Turning a blind eye: authors have blinkered view of blinding. BMJ 2004;328:1135–6; author reply 1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Simon R. Confidence intervals for reporting results of clinical trials. Ann Intern Med 1986;105:429–35. [DOI] [PubMed] [Google Scholar]
- [37].Smith SM, Amtmann D, Askew RL, Gewandter JS, Hunsinger M, Jensen MP, McDermott MP, Patel KV, Williams M, Bacci ED, Burke LB, Chambers CT, Cooper SA, Cowan P, Desjardins P, Etropolski M, Farrar JT, Gilron I, Huang IZ, Katz M, Kerns RD, Kopecky EA, Rappaport BA, Resnick M, Strand V, Vanhove GF, Veasley C, Versavel M, Wasan AD, Turk DC, Dworkin RH. Pain intensity rating training: results from an exploratory study of the ACTTION PROTECCT system. PAIN 2016;157:1056–64. [DOI] [PubMed] [Google Scholar]
- [38].Smith SM, Chang RD, Pereira A, Shah N, Gilron I, Katz NP, Lin AH, McDermott MP, Rappaport BA, Rowbotham MC, Sampaio C, Turk DC, Dworkin RH. Adherence to CONSORT harms-reporting recommendations in publications of recent analgesic clinical trials: an ACTTION systematic review. PAIN 2012;153:2415–21. [DOI] [PubMed] [Google Scholar]
- [39].Smith SM, Hunsinger M, Mckeown A, Parkhurst M, Allen R, Kopko S, Lu Y, Wilson HD, Burke LB, Desjardins P, McDermott MP, Rappaport BA, Turk DC, Dworkin RH. Quality of pain intensity assessment reporting: ACTTION systematic review and recommendations. J Pain 2015;16:299–305. [DOI] [PubMed] [Google Scholar]
- [40].Smith SM, Wang AT, Katz NP, McDermott MP, Burke LB, Coplan P, Gilron I, Hertz SH, Lin AH, Rappaport BA, Rowbotham MC, Sampaio C, Sweeney M, Turk DC, Dworkin RH. Adverse event assessment, analysis, and reporting in recent published analgesic clinical trials: ACTTION systematic review and recommendations. PAIN 2013;154:997–1008. [DOI] [PubMed] [Google Scholar]
- [41].Turk DC, Dworkin RH, Mcdermott MP, Bellamy N, Burke LB, Chandler JM, Cleeland CS, Cowan P, Dimitrova R, Farrar JT, Hertz S, Heyse JF, Iyengar S, Jadad AR, Jay GW, Jermano JA, Katz NP, Manning DC, Martin S, Max MB, Mcgrath P, Mcquay HJ, Quessy S, Rappaport BA, Revicki DA, Rothman M, Stauffer JW, Svensson O, White RE, Witter J. Analyzing multiple endpoints in clinical trials of pain treatments: IMMPACT recommendations. Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials. PAIN 2008;139:485–93. [DOI] [PubMed] [Google Scholar]
- [42].Tuttle AH, Tohyama S, Ramsay T, Kimmelman J, Schweinhardt P, Bennett GJ, Mogil JS. Increasing placebo responses over time in U.S. clinical trials of neuropathic pain. PAIN 2015;156:2616–26. [DOI] [PubMed] [Google Scholar]
- [43].U.S. Health and Human Services. Adaptive design clinical trials for drugs and biologicals: draft guidance, 2010. Available at: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm201790.pdf. Accessed November 22, 2016. [Google Scholar]
- [44].U.S. Health and Human Services. Guidance for industry: non-inferiority trials, 2010. Available at: http://www.fda.gov/downloads/Drugs/.../Guidances/UCM202140.pdf. Accessed November 22, 2016. [Google Scholar]