

Abstract
Recent technological advances in gene sequencing have led to a rapid increase in gene discovery in epilepsy. However, the ability to assess pathogenicity of variants, provide functional analysis, and develop targeted therapies has not kept pace with rapid advances in sequencing technology. Thus, although clinical genetic testing may lead to a specific molecular diagnosis for some patients, test results often lead to more questions than answers. As the field begins to focus on therapeutic applications of genetic diagnoses using precision medicine, developing processes that offer more than equivocal test results is essential. The success of precision medicine in epilepsy relies on establishing a correct genetic diagnosis, analyzing functional consequences of genetic variants, screening potential therapeutics in the preclinical laboratory setting, and initiating targeted therapy trials for patients. We describe the structure of a comprehensive, pediatric Epilepsy Genetics Program that can serve as a model for translational medicine in epilepsy.
Keywords: Epilepsy genetics, precision medicine, translational medicine
Introduction
A 10-month-old girl is admitted to the hospital for the second time with clusters of seizures. According to her parents, she is otherwise healthy and has met all of her developmental milestones. The inpatient team sends DNA for testing with an epilepsy gene panel, and 3 weeks later results reveal a variant of uncertain significance in the PCDH19 gene.
The parents, already coping with the fear and uncertainty surrounding their infant daughter’s seizures, are understandably mired in a fresh round of unanswered questions. Does our daughter have PCDH19-related epilepsy? Will she have developmental delays? Will she learn to talk? Will she have autism? Are our other children at risk for having seizures, and should we have them tested? Should we avoid vaccinations for our children? If we have more children, how likely is it they would have PCDH19-related epilepsy? How can our child have a genetic condition even though we have no family history of anything genetic? Is there a specific treatment for PCDH19-related epilepsy? We read about a clinical trial online—can our child participate?
These are a few examples of the types of questions that families ask when faced with a new or uncertain genetic finding. Our Epilepsy Genetics Program’s mission is to help families understand the complexities of genetic test results and what they mean for their children, to offer tailored treatment when available, to make referrals to ongoing research studies, and to connect them with appropriate support organizations.
In recent years, recognition of the genetic contributions to epilepsy has moved beyond the research realm and into clinical practice.1–8 The advent of next-generation sequencing has led to the identification of many genetic causes of epilepsy.9,10 However, investigations into the mechanisms by which these genes confer susceptibility to seizures or epilepsy, variant classification, and the translation of these findings to inform clinical care, lag behind the rate of gene discovery. With today’s increased knowledge of the genetic underpinnings of epilepsy, efforts are now shifting from gene discovery to application. Functional analysis and identification of more targeted therapies are moving to the forefront of research in epilepsy genetics7. Precision medicine efforts in epilepsy fall under the auspices of a national movement toward precision medicine, defined by the National Institutes of Health as “an emerging approach for disease treatment and prevention that takes into account individual variability in genes, environment, and lifestyle for each person.”
As we focus efforts more toward precision medicine in the field of epilepsy with the ultimate goal of administering targeted therapies based on an individual’s underlying genetic etiology, including gene-specific clinical trials, it is imperative that patients receive the correct molecular diagnosis and that appropriate patients are referred to available research studies and clinical trials. To that end, we have created a translational Epilepsy Genetics Program with those aims. Here we present our overall approach to epilepsy genetics as one example of integrating the rapidly evolving genetic and functional data that are emerging in the field into clinical practice. Our program features a multidisciplinary Epilepsy Genetics Clinic staffed by epileptologists with a special interest in genetics and expertise in genetic epilepsy syndromes who partner with licensed genetic counselors uniquely trained in epilepsy genetics. Together, we aim to systematically evaluate, and provide accurate diagnoses for, children with a presumed genetic etiology for their epilepsy. Our clinical team works to collect thorough medical and family histories, interpret genetic test results, and carefully disseminate those results while clearly outlining the current limitations of genetic testing and diagnoses.
When a molecular diagnosis is achieved, we provide recommendations for changes in medical management, identify family members who may be at risk, provide information regarding recurrence risk and options for prenatal testing for future pregnancies, and direct families to available gene-specific support resources. Our clinical team works closely with our own research laboratory, as well as those of collaborators, to evaluate functional consequences and assess genetic variants in animal models, with the long-term goal of screening drugs for potential precision medicine trials.
Additionally, patients identified in our clinic may be directed to ongoing clinical trials as they become available over time. Here, we outline the structure of our Epilepsy Genetics Program and describe how each component is integral to providing the most innovative care possible in the era of precision medicine in epilepsy.
History of the Epilepsy Genetics Program
The Epilepsy Genetics Program was developed in 2010 as Boston Children’s Hospital began participating as an enrollment site for the Epilepsy Phenome/Genome Project.11 Our clinic was formally launched in 2011 by an epileptologist (AP) and a licensed genetic counselor (BRS), each with clinical and research-based expertise in neurogenetics. Since then, we have trained 2 additional physicians (already trained in child neurology and clinical electrophysiology) in epilepsy genetics, and they have joined our faculty (HEO, CA). Two additional licensed genetic counselors (including LS) joined the program as well.
Clinical research was a component from inception and now includes everyone on the clinical team as well as a research assistant (MK), who enrolls individual families, collects phenotyping information on research participants, and serves as a liaison with collaborative studies, such as Epi4K12 and Epi25, a multicenter collaboration between the epilepsy genetics community and the Broad Institute of Harvard and the Massachusetts Institute of Technology.
The basic research component of the program formally complemented the clinical research focus when the independent Poduri laboratory was launched in 2013, which includes research fellows (GT, JFPU) and research assistants. We were inspired in part by the epilepsy genetics clinical and research group of Ingrid Scheffer and Samuel Berkovic in Australia, where the doctors are the researchers and promote synergy between the clinical and research realms, and also by our numerous mentors in translational science.
Epilepsy Genetics Clinic
Our multidisciplinary Epilepsy Genetics Clinic provides consultations for patients with a variety of indications and at various stages throughout the diagnostic odyssey. Our patient population consists primarily of children with early onset refractory epilepsy who often have other neurological comorbidities such as developmental delay, intellectual disability, and/or autism spectrum disorder. The children who come to our clinic are often severely affected, and their parents are looking for an explanation and better treatment options to help their child.
We use a multidisciplinary approach within the Epilepsy Genetics Clinic wherein each family meets with both an epileptologist and a genetic counselor. The epileptologist takes a thorough seizure and neurodevelopmental history and performs a neurological exam. The genetic counselor obtains a complete, syndrome-specific family history, explains genetics concepts and inheritance patterns, describes genetic testing options and potential results of genetic testing, reviews the benefits and limitations of genetic testing, discusses the possibility of uncovering incidental or secondary findings as recommended by the American College of Medical Genetics and Genomics13,14 and obtains informed consent for genetic testing. Additionally, the genetic counselor ensures that the families have a reasonable expectation of the ways in which a genetic test result may or may not inform clinical care, and addresses the psychosocial implications of genetic testing.15 The epileptologist and genetic counselor work together to design and implement a plan for genetic testing, interpret genetic test results, and make recommendations for follow up.
The families we see generally fit into one of 3 main categories: 1) families seeking a genetic diagnosis for their children, 2) families whose children have a genetic diagnosis that may not be definitive, and 3) families of patients who have an established genetic diagnosis, for whom we could offer insight into treatment or enroll in a precision medicine trial.
Families Seeking a Diagnosis
A 2-year-old previously healthy and developmentally normal boy suddenly experiences episodes of generalized tonic-clonic seizures with dramatic post-ictal loss of speech and ataxia lasting several days to weeks. While his recovery to baseline was reassuring, the acute convulsions and post-ictal periods were understandably distressing. An initial genetic evaluation for episodic ataxia and seizures was unrevealing, and treatment was initiated for epilepsy (levetiracetam as a broad-spectrum agent given the generalized-appearing onset and generalized slowing on EEG) as well as a possible channelopathy (acetazolamide). The episodes continued, each time with complete recovery. He subsequently went on to have episodes that were diagnosed on video as paroxysmal kinesigenic dyskinesia. Carbamazepine was added to his regimen at the same time that an expanded genetic panel that had become available for clinical testing in the interim revealed a pathogenic variant in the PRRT2 gene, as well as a variant of uncertain significance in the same gene. While carbamazepine was clinically indicated based on symptomatology, the genetic diagnosis provided diagnostic certainty regarding the other more dramatic symptoms and afforded the parents confidence in withdrawing the other medications. Had PRRT2 testing been available at the time of the first convulsive episode, treatment would have been simplified to carbamazepine monotherapy and anxiety regarding progressive, metabolic disorders would have been allayed much earlier.
Many families come to the Epilepsy Genetics Clinic seeking a genetic diagnosis. Some children are referred without completing any genetic testing. For these patients, we perform a complete consult as described above, including recommendations for specific genetic tests based on the medical and family history of the patient. Such recommendations may include a chromosomal microarray, single gene analysis if a specific etiology is suspected (eg, SCN1A variant suspected for classic Dravet syndrome), or a gene panel that includes particular genes of interest. If such testing yields no diagnosis, we consider whole exome sequencing. Our clinical strategy for these patients is outlined in Figure 1.
Figure 1.
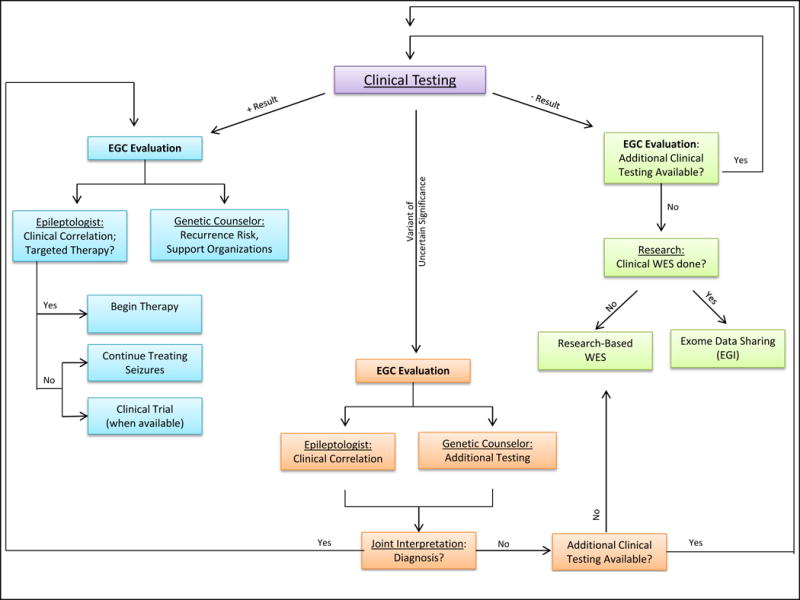
Schematic of Epilepsy Genetics Clinic consult of a patient who has had previous genetic testing. Includes potential pathways based on clinical findings, treatment options, and research enrollment.
Abbreviations: EGC, Epilepsy Genetics Clinic; EGI, Epilepsy Genetics Initiative; WES, whole exome sequencing.
Some patients are referred having already had some level of genetic testing, which often includes a chromosomal microarray or gene panel. These patients are often referred to us to consult upon unclear findings or genetic variants of uncertain significance. Prior to meeting with these patients, our epileptologists and genetic counselors take a systematic approach to variant interpretation that includes interrogating databases such as ClinVar, the Exome Aggregation Consortium (ExAC) database, and DECIPHER,16–18 reviewing the available published literature and gene-specific databases as available, considering whether testing other family members would provide additional evidence in the interpretation of the clinical significance of a particular genetic finding, and facilitating any additional testing. When faced with variants in genes that are relatively newly reported in association with epilepsy, we may also consult clinical and research colleagues at other centers who have familiarity or expertise with these genes. In some circumstances, we also consult colleagues in clinical genetic testing laboratories to ask if they have previously reported clinical variants in a given gene associated with epilepsy. There are efforts underway to consolidate some of these types of resources (e.g., Rare Epilepsy Network, International League Against Epilepsy Genetics Commission, and ClinVar). Our approach at present is individualized for each patient, accessing multiple resources as well as collective experience to arrive at a synthesis regarding the relevance of a given gene and the likelihood of pathogenicity of a given variant. We use as a general guideline the ACMG guidelines regarding gene-level and variant-level significance.13
Once all results are available, the Epilepsy Genetics Clinic team meets to review medical records and genetic test results at a weekly case conference to generate a plan for the full consultation. In some cases, we conclude that previous genetic testing has not provided a diagnosis and may suggest additional testing for the patient prior to a full consultation with the epileptologist.
Some patients, despite having exhausted all appropriate clinical testing, are left without a diagnosis. There are many patients and families who are well-known to the Epilepsy Genetics Clinic for whom we have not been able to identify a molecular etiology. Due to the translational nature of the Epilepsy Genetics Program, we are able to offer enrollment in one or more research studies. These may include research whole exome sequencing with subsequent confirmation in a Clinical Laboratory Improvement Amendments (CLIA)-certified laboratory for patients who are unable to obtain whole exome sequencing on a clinical basis, or enrollment in data-sharing collaborations focused on variant interpretation and identification of new candidate genes. Research opportunities such as these are clearly helpful from a medical perspective as families may eventually obtain a diagnosis, but may also be important in helping families to be proactive in the search for an answer for their child’s condition.
Patients Whose Diagnoses are in Question
For 2 years Patient A had a presumed diagnosis of PCDH19-related epilepsy, until the diagnostic lab that had performed the testing reclassified the variant based on results from other patients. Whole exome sequencing identified a new variant of uncertain significance in a different gene, and her parents struggled to make sense of this new information.
Sometimes families come to our clinic believing their child has a particular genetic diagnosis because of the result of a clinical test, when in fact the particular finding does not adequately establish a diagnosis for the patient. Examples include genetic variants that are more likely to be benign than pathogenic, inherited variants in which the family history is not significant for features associated with the genetic condition, variants of uncertain significance in which the patient’s phenotype is not consistent with what has been reported in the literature, variants in genes that later become associated with new phenotypes as their associated clinical spectra are broadened, and heterozygous variants in genes associated with autosomal recessive conditions.
For patients who fall into this category, our team evaluates the genetic finding in the context of the patient’s medical and family history and communicates any discrepancies in a manner that is informative while keeping in mind the potential psychological impact on a patient or family. In other cases, variants are reclassified from likely pathogenic to likely benign or benign based on new evidence, through which a genetic diagnosis may be retracted. This can be especially challenging when a family has established a sense of identity around a particular diagnosis. In such scenarios, we work with families to help them understand the new interpretation as well as help them navigate the changing roles in their communities.
Patients Who Have an Established Diagnosis
Patient M was a 5-year-old male who had been diagnosed with Dravet syndrome and had a documented pathogenic variant in the SCN1A gene. An international patient, he had been followed in his country of origin but his parents were not satisfied with the information they had been given regarding prognosis and treatment options. They sought a second opinion.
In the Epilepsy Genetics Clinic, we also follow patients who have an established genetic diagnosis. This diagnosis may have been achieved through the Epilepsy Genetics Clinic or may have been obtained previously from an outside provider. Such patients are typically followed by a primary neurologist and come to the Epilepsy Genetics Clinic on a yearly basis to discuss new knowledge about the condition or new guidelines for medical management, as well as ongoing research studies and clinical trials for which they may be eligible.
Ideally, when a causative genetic variant is identified, there would be a formal clinical trial in which the patient could enroll. However, given the inundation of genetic variants associated with epilepsy, to date formal clinical trials are available for only a handful of genes. Of note, we must also be aware that different variants in the same gene may have different functional consequences (such as a gain or loss of protein function) and thus require alternative therapies. In such cases, determining the functional consequence of a particular variant is imperative prior to starting a trial of the drug.
One could imagine that the future of clinical trials in epilepsy may take one step beyond “gene-specific” and thus become “variant-specific.” Patients in this category may have the opportunity to have their genetic variants functionally analyzed in a laboratory, and based on either mechanism or through drug screening, a potential therapy could be identified. From there, patients may be started on a medication and their progress may be documented in an “N of 1” trial. While this mechanism by no means replaces a proper clinical trial, such cases might provide the preliminary indications that would encourage a group of investigators to engage in the lengthy but necessary process of setting up a clinical trial.
As researchers, we think of these present and future scenarios and the scientific potential therein. For families, however, the most important aspect of having a genetic diagnosis is what it means for their child or their family today. For some genetic forms of epilepsy, a diagnosis can directly alter treatment. Examples of such targeted therapies are listed in Table 1, including some that have become conventional and others that are rational in basis but not as well established.6,19–29 Additionally, a diagnosis may allow us to prognosticate based on what has been reported in the literature, provide an understanding of any associated medical issues and eliminate the need for additional medical evaluations, provide genetic counseling regarding recurrence risk and testing options for future pregnancies, and provide resources for family support organizations.
Table 1.
Treatment Considerations for Genetic Forms of Epilepsy.
| Gene | Syndrome(s) | Treatment considerations |
|---|---|---|
| ALDH7A1 | Pyridoxine-dependent epilepsy | pyridoxine |
| GRIN2A | GRIN2A-related epilepsy | memantine or dextromethorphan for gain-of-function variants |
| KCNQ2 | Benign familial neonatal/infantile seizures; KCNQ2-related epileptic encephalopathy | ezogabine for loss-of-function variants |
| KCNT1 | Epilepsy of infancy with migrating focal seizures | Quinidine for gain-of-function variants |
| PNPO | Pyridoxal 5′-phosphate-dependent epilepsy | pyridoxal 5′phosphate |
| PRRT2 | Infantile convulsions; paroxysmal kinesigenic dyskinesia | oxcarbazepine; carbamazepine |
| SCN1A | Dravet syndrome; SCN1A-related epilepsy | avoid sodium channel blockers |
| SCN2A | SCN2A-related epilepsy | phenytoin; high-dose carbamazepine |
| SCN8A | SCN8A-related epilepsy | phenytoin; high-dose carbamazepine |
| SLC2A1 | Glucose transporter deficiency | ketogenic diet |
| TSC1; TSC2 | Tuberous sclerosis complex (spasms) | vigabatrin |
Clinical Research
The Epilepsy Genetics Program is involved in several clinical research endeavors, including independent projects and collaborative efforts. Such projects include research-based whole exome sequencing and whole exome sequencing data sharing, participation in tissue and data repositories, formal clinical trials, and “N of 1” precision medicine trials. Epileptologists, genetic counselors, and research coordinators work together to identify eligible participants and enroll them into one or more research studies.
More specifically, we enroll patients into our own Genetics of Epilepsy and Related Disorders research study, a protocol under which we have several ongoing research projects. We send out samples for whole exome sequencing on a research basis or independently collect and analyze raw data for patients who have had clinical whole exome sequencing, with the aim of identifying new candidate genes for epilepsy. Our sequencing efforts are ongoing, and we are engaged in collaborations with the Epi25 community and the Broad Institute community. For patients in the Epilepsy Genetics Clinic for whom insurance has denied clinical genetic testing, we are able to offer research-based whole exome sequencing. Our protocol allows us to return research results to the families once the findings have been confirmed in a CLIA-certified laboratory.
We also enroll patients with documented genetic findings for phenotyping and natural history studies, and patients for whom we hope to pursue functional analysis, either through our own research laboratory or by sending samples or data to collaborating laboratories. For some cases, we have had the opportunity to report both genetic findings as well as expanded genotype-phenotype spectra with our collaborators in the epilepsy genetics community.26,30–32
For patients who have had clinical whole exome sequencing, we participate as a site for the Epilepsy Genetics Initiative ([http://www.cureepilepsy.org/egi] sponsored by Citizens United for Research in Epilepsy and the National Institute of Neurological Disorders and Stroke). Through this initiative, our patients’ data will be reanalyzed on a regular basis, allowing updated analysis that will incorporate novel gene discovery and recognition that a given gene may be important for epilepsy.
We are also launching a pilot epilepsy genetics initiative in conjunction with the Boston Children’s Hospital Biobank for Health Discovery as part of the multicenter Genomics Research and Innovation Network. We created a PCDH19-related epilepsy patient registry, in collaboration with the University of California, San Francisco. The goal of this registry is to collect genetic and clinical information for individuals who harbor a variant in the PCDH19 gene to inform and encourage research into PCDH19-related epilepsy. We recently enrolled patients who are followed in the Epilepsy Genetics Center in a multicenter Phase I clinical trial that aimed to determine the safety and efficacy of an adjunctive therapy for patients with a specific genetic form of epilepsy.
Laboratory Research
A significant component of our Epilepsy Genetics Program is to model genetic variants to identify gene dysfunction, and then perform precision drug screening with the hope of translating findings back to the clinic. With the hundreds of established genes associated with epilepsy, it is not possible for one program to have the bandwidth to provide functional analysis or drug screening capabilities for all of them. While we continue working on a small subset of these genes within our Program, we work in collaboration with other investigators who are performing functional studies on other genes using various models.
In our Epilepsy Genetics Program, we are using the zebrafish as a model system. Zebrafish are an established in vivo model for neuroscience and medical research.33 They possess a high genetic homology (80%–85%) to humans,34 a central nervous system with a similar brain archetype,35,36 and conserved neurochemistry.37 Zebrafish were first established as a model for epilepsy in 2005.38 Since this initial work, numerous studies have shown zebrafish are amenable to the same convulsants and antiepileptic medications as humans,39,40 and the model has shown to be a promising system for drug discovery for genetic epilepsy.41
We are using a multifaceted approach to conduct functional analyses on a variety of epilepsy-associated genes. We are employing CRISPR and CRISPR-associated proteins to rapidly and efficiently develop knockout zebrafish models. Second, we are performing high-throughput phenotyping to detect spontaneous seizures and seizure susceptibility. Third, in collaboration with the Neuromodulation Program at Boston Children’s Hospital, we are using noninvasive recording of extracellular field potentials (multi-unit activity) to validate models by demonstrating electrophysiological abnormalities.42 Fourth, we are crossing transgenic lines expressing fluorescent markers with our knockout lines to identify potential mechanisms behind altered excitability. Finally, we intend to perform high-throughput drug screening to test the wide range of FDA-approved drugs as well as newly developed drugs with our long-term goal of translating these discoveries into clinical trials. In addition to our zebrafish model, we are collaborating on projects to create induced pluripotent stem cells from fibroblasts and lymphocytes collected from patients with specific genetic forms of epilepsy, to create an in vitro model for additional functional analysis.
Conclusion
The success of precision medicine in epilepsy relies on accurate genetic diagnoses, open collaboration in research efforts, and efficient translation of research findings directly back into the clinic. We have highlighted the components of our own translational Epilepsy Genetics Program that we have developed to drive these aims forward. The multidisciplinary approach of our Epilepsy Genetics Clinic, which includes the unique expertise of epileptologists and of genetic counselors, allows us to address essentially all clinically relevant aspects of genetic testing in epilepsy. In one clinic, families are able to receive an interpretation and thorough explanation of genetic test results and what it means for their child as well as what the results could mean for future children and other family members. Additionally, they have immediate access to research studies and information pertaining to gene and/or syndrome support groups. Concurrently, our Program’s research efforts are enabling us to analyze the functional consequence of specific variants identified in patients and directly translate these findings to inform clinical care.
Although we highlight the integration of clinical evaluation, clinical research, and laboratory research, and hope the Epilepsy Genetics Program serves as a model for the development of similar programs, we recognize this may not be possible for many providers and/or researchers. Professionals involved in epilepsy genetics should not be daunted, as each of the components we have described may exist independently. For example, a comprehensive epilepsy genetics clinic can stand alone and interested patients and families may be referred to outside research studies. Conversely, researchers may alert the clinical epilepsy community of ongoing projects for which they may be actively recruiting participants. An active connection between the clinical, clinical research, and scientific communities has been fostered by numerous professional organizations with a growing emphasis on translational approaches to epilepsy genetics. Additionally, many connections have been initiated by parent-run advocacy groups. The importance of collaborations and shared efforts cannot be overstated. Continuing efforts with collaborators with whom we have worked for years, as well as establishing new collaborations with clinicians and researchers, will be essential as we progress in the era of precision medicine in epilepsy.
Acknowledgments
We thank the patients and families with whom we have the privilege to work. We thank our clinical colleagues at Boston Children’s Hospital and other institutions and our research collaborators in the epilepsy genetics community. We thank Scott Pomeroy, MD, PhD, at Boston Children’s Hospital and Daniel Lowenstein, MD, at the University of California San Francisco for their encouragement as we started our Epilepsy Genetics Program, and for sending colleagues to us. We are grateful to Phillip Pearl, MD, at Boston Children’s Hospital for ongoing support from the Division of Epilepsy. We thank Brandy Fureman, PhD, formerly at the National Institute of Neurological Disorders and Stroke and now at the Epilepsy Foundation, for encouraging us to tell our story in the form of a manuscript. AP has been supported by the National Institute of Neurological Disorders and Stroke (K23NS069784 and EPGP) and is a co-investigator of Epi4K and EpiPM. She receives funding for translational research from the Boston Children’s Hospital Translational Research Program and the Charles H. Hood Foundation. AP and BRS participate in the Epilepsy Genetics Initiative and receive funding from the National Institute of Neurological Disorders and Stroke and CURE for this collaborative project. Our research has additionally been supported by the Dravet Syndrome Foundation, The Cute Syndrome Foundation, The PCDH19 Alliance, Aaron’s Ohtahara, and gifts from patient families, to all of whom we extend our gratitude. We also wish to thank Melanie Fridl Ross, MSJ, ELS, for editing an initial draft of this manuscript.
Footnotes
Declaration of Conflicting Interests
AP serves on the Scientific Advisory Board of the Dravet Syndrome Foundation and the Professional Advisory Board of the Patient Centered Outcomes Research Institute (PCORI)-funded Rare Epilepsy Network.
References
- 1.Mefford HC, Yendle SC, Hsu C, et al. Rare copy number variants are an important cause of epileptic encephalopathies. Annals of neurology. 2011;70:974–85. doi: 10.1002/ana.22645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Veeramah KR, Johnstone L, Karafet TM, et al. Exome sequencing reveals new causal mutations in children with epileptic encephalopathies. Epilepsia. 2013;54:1270–81. doi: 10.1111/epi.12201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allen AS, Berkovic SF, Cossette P, et al. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–21. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Novo Mutations in Synaptic Transmission Genes Including DNM1 Cause Epileptic Encephalopathies. Am J Hum Genet. 2014;95:360–70. doi: 10.1016/j.ajhg.2014.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Olson H, Shen Y, Avallone J, et al. Copy number variation plays an important role in clinical epilepsy. Annals of neurology. 2014;75:943–58. doi: 10.1002/ana.24178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Poduri A, Sheidley BR, Shostak S, Ottman R. Genetic testing in the epilepsies-developments and dilemmas. Nat Rev Neurol. 2014;10:293–9. doi: 10.1038/nrneurol.2014.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.A roadmap for precision medicine in the epilepsies. The Lancet Neurology. 2015;14:1219–28. doi: 10.1016/S1474-4422(15)00199-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Helbig KL, Farwell Hagman KD, Shinde DN, et al. Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genet Med. 2016 doi: 10.1038/gim.2015.186. [DOI] [PubMed] [Google Scholar]
- 9.Poduri A, Lowenstein D. Epilepsy genetics–past, present, and future. Current opinion in genetics & development. 2011;21:325–32. doi: 10.1016/j.gde.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Helbig I, Lowenstein DH. Genetics of the epilepsies: where are we and where are we going? Curr Opin Neurol. 2013;26:179–85. doi: 10.1097/WCO.0b013e32835ee6ff. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abou-Khalil B, Alldredge B, Bautista J, et al. The epilepsy phenome/genome project. Clin Trials. 2013;10:568–86. doi: 10.1177/1740774513484392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Epi4K: Gene discovery in 4,000 genomes. Epilepsia. 2012 doi: 10.1111/j.1528-1167.2012.03511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Green RC, Berg JS, Grody WW, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–74. doi: 10.1038/gim.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krabbenborg L, Vissers LE, Schieving J, et al. Understanding the Psychosocial Effects of WES Test Results on Parents of Children with Rare Diseases. J Genet Couns. 2016 doi: 10.1007/s10897-016-9958-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Landrum MJ, Lee JM, Benson M, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–8. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Firth HV, Richards SM, Bevan AP, et al. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet. 2009;84:524–33. doi: 10.1016/j.ajhg.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mills PB, Struys E, Jakobs C, et al. Mutations in antiquitin in individuals with pyridoxine-dependent seizures. Nat Med. 2006;12:307–9. doi: 10.1038/nm1366. [DOI] [PubMed] [Google Scholar]
- 20.Pierson TM, Yuan H, Marsh ED, et al. mutation and early-onset epileptic encephalopathy: personalized therapy with memantine. Ann Clin Transl Neurol. 2014;1:190–8. doi: 10.1002/acn3.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gunthorpe MJ, Large CH, Sankar R. The mechanism of action of retigabine (ezogabine), a first-in-class K+ channel opener for the treatment of epilepsy. Epilepsia. 2012;53:412–24. doi: 10.1111/j.1528-1167.2011.03365.x. [DOI] [PubMed] [Google Scholar]
- 22.Bearden Dea. Targeted Treatment of Migrating Partial Seizures of Infancy with Quinidine. Annals of neurology. 2014 doi: 10.1002/ana.24229. [DOI] [PubMed] [Google Scholar]
- 23.Guerin A, Aziz AS, Mutch C, Lewis J, Go CY, Mercimek-Mahmutoglu S. Pyridox(am)ine-5-Phosphate Oxidase Deficiency Treatable Cause of Neonatal Epileptic Encephalopathy With Burst Suppression: Case Report and Review of the Literature. Journal of child neurology. 2015;30:1218–25. doi: 10.1177/0883073814550829. [DOI] [PubMed] [Google Scholar]
- 24.Torisu H, Watanabe K, Shimojima K, et al. Girl with a PRRT2 mutation and infantile focal epilepsy with bilateral spikes. Brain & development. 2014;36:342–5. doi: 10.1016/j.braindev.2013.05.009. [DOI] [PubMed] [Google Scholar]
- 25.Lamperti C, Invernizzi F, Solazzi R, et al. Clinical and genetic features of paroxysmal kinesigenic dyskinesia in Italian patients. Eur J Paediatr Neurol. 2016;20:152–7. doi: 10.1016/j.ejpn.2015.08.006. [DOI] [PubMed] [Google Scholar]
- 26.Howell KB, McMahon JM, Carvill GL, et al. SCN2A encephalopathy: A major cause of epilepsy of infancy with migrating focal seizures. Neurology. 2015;85:958–66. doi: 10.1212/WNL.0000000000001926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boerma RS, Braun KP, van den Broek MP, et al. Remarkable Phenytoin Sensitivity in 4 Children with SCN8A-related Epilepsy: A Molecular Neuropharmacological Approach. Neurotherapeutics. 2016;13:192–7. doi: 10.1007/s13311-015-0372-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang D, Pascual JM, De Vivo D. Glucose Transporter Type 1 Deficiency Syndrome. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews(R) Seattle (WA): 1993. [Google Scholar]
- 29.Overwater IE, Bindels-de Heus K, Rietman AB, et al. Epilepsy in children with tuberous sclerosis complex: Chance of remission and response to antiepileptic drugs. Epilepsia. 2015;56:1239–45. doi: 10.1111/epi.13050. [DOI] [PubMed] [Google Scholar]
- 30.Moller RS, Heron SE, Larsen LH, et al. Mutations in KCNT1 cause a spectrum of focal epilepsies. Epilepsia. 2015;56:e114–20. doi: 10.1111/epi.13071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pescosolido MF, Stein DM, Schmidt M, et al. Genetic and phenotypic diversity of NHE6 mutations in Christianson syndrome. Annals of neurology. 2014;76:581–93. doi: 10.1002/ana.24225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weckhuysen S, Ivanovic V, Hendrickx R, et al. Extending the KCNQ2 encephalopathy spectrum: clinical and neuroimaging findings in 17 patients. Neurology. 2013;81:1697–703. doi: 10.1212/01.wnl.0000435296.72400.a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zon LI. Zebrafish: a new model for human disease. Genome Res. 1999;9:99–100. [PubMed] [Google Scholar]
- 34.Howe K, Clark MD, Torroja CF, et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature. 2013;496:498–503. doi: 10.1038/nature12111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ullmann JF, Calamante F, Collin SP, Reutens DC, Kurniawan ND. Enhanced characterization of the zebrafish brain as revealed by super-resolution track-density imaging. Brain Struct Funct. 2015;220:457–68. doi: 10.1007/s00429-013-0667-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ullmann JF, Cowin G, Kurniawan ND, Collin SP. A three-dimensional digital atlas of the zebrafish brain. NeuroImage. 2010;51:76–82. doi: 10.1016/j.neuroimage.2010.01.086. [DOI] [PubMed] [Google Scholar]
- 37.Stewart AM, Ullmann JF, Norton WH, et al. Molecular psychiatry of zebrafish. Mol Psychiatry. 2015;20:2–17. doi: 10.1038/mp.2014.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baraban SC, Taylor MR, Castro PA, Baier H. Pentylenetetrazole induced changes in zebrafish behavior, neural activity and c-fos expression. Neuroscience. 2005;131:759–68. doi: 10.1016/j.neuroscience.2004.11.031. [DOI] [PubMed] [Google Scholar]
- 39.Berghmans S, Hunt J, Roach A, Goldsmith P. Zebrafish offer the potential for a primary screen to identify a wide variety of potential anticonvulsants. Epilepsy research. 2007;75:18–28. doi: 10.1016/j.eplepsyres.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 40.Afrikanova T, Serruys AS, Buenafe OE, et al. Validation of the zebrafish pentylenetetrazol seizure model: locomotor versus electrographic responses to antiepileptic drugs. PLoS One. 2013;8:e54166. doi: 10.1371/journal.pone.0054166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Baraban SC, Dinday MT, Hortopan GA. Drug screening in Scn1a zebrafish mutant identifies clemizole as a potential Dravet syndrome treatment. Nat Commun. 2013;4:2410. doi: 10.1038/ncomms3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meyer M, Dhamne S, LaCoursiere CM, Tambunan DE, Poduri A, Rotenberg A. Microarray noninvasive neuronal seizure recordings from intact larval zebrafish. PLoS One. 2016 doi: 10.1371/journal.pone.0156498. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]