

ABSTRACT
While the RNA-dependent RNA polymerase L protein of rabies virus (RABV), a member of the genus Lyssavirus of the family Rhabdoviridae, has potential to be a therapeutic target for rabies, the molecular functions of this protein have remained largely unknown. In this study, to obtain a novel experimental tool for molecular function analysis of the RABV L protein, we established by using a reverse genetics approach an L gene-deficient RABV (Nishi-ΔL/Nluc), which infects, propagates, and correspondingly produces NanoLuc luciferase in cultured neuroblastoma cells transfected to express the L protein. trans-Complementation with wild-type L protein, but not that with a functionally defective L protein mutant, efficiently supported luciferase production by Nishi-ΔL/Nluc, confirming its potential for function analysis of the L protein. Based on the findings obtained from comprehensive genetic analyses of L genes from various RABV and other lyssavirus species, we examined the functional importance of a highly conserved L protein region at positions 1914 to 1933 by a trans-complementation assay with Nishi-ΔL/Nluc and a series of L protein mutants. The results revealed that the amino acid sequence at positions 1929 to 1933 (NPYNE) is functionally important, and this was supported by other findings that this sequence is critical for binding of the L protein with its essential cofactor, P protein, and thus also for L protein's RNA polymerase activity. Our findings provide useful information for the development of an anti-RABV drug targeting the L-P protein interaction.
IMPORTANCE To the best of our knowledge, this is the first report on the establishment of an L gene-deficient, reporter gene-expressing virus in all species of the order Mononegavirales, also highlighting its applicability to a trans-complementation assay, which is useful for molecular function analyses of their L proteins. Moreover, this study revealed for the first time that the NPYNE sequence at positions 1929 to 1933 in the RABV L protein is important for L protein's interaction with the P protein, consistent with and extending the results of a previous study showing that the P protein-binding domain in the L protein is located in its C-terminal region, at positions 1562 to 2127. This study indicates that the NPYNE sequence is a promising target for the development of an inhibitor of viral RNA synthesis, which has high potential as a therapeutic drug for rabies.
KEYWORDS: rabies virus, L gene-deficient virus, RNA polymerase, phosphoprotein
INTRODUCTION
Rabies is a viral zoonosis that is characterized by severe neurological signs and a high case mortality rate, almost 100%. Although several cases of recovery from rabies in humans after intensive medical care have been reported (1, 2), no definite therapy has yet been established, and there are approximately 59,000 human deaths per year worldwide, mainly in developing countries in Asia and Africa (3). The causative agent, rabies virus (RABV), is classified in the genus Lyssavirus of the family Rhabdoviridae within the order Mononegavirales. The genome of RABV is a nonsegmented negative-sense (NNS) RNA encoding five viral proteins: nucleoprotein (N protein), phosphoprotein (P protein), matrix (M) protein, glycoprotein (G protein), and large (L) protein. To establish an effective therapy for rabies, it is important to elucidate the molecular functions of these RABV proteins, all of which play indispensable roles in the viral propagation cycle, and then to evaluate the potential of each of them as a therapeutic target.
The RABV L protein (2,127 amino acids, ca. 244 kDa, in the strain Nishigahara [GenBank accession no. AB044824.1]) is a promising candidate for a therapeutic molecular target, since this protein functions as an RNA-dependent RNA polymerase (RdRp) and is therefore essential for the production of all viral proteins, including itself: the L protein participates in transcription of viral mRNAs by effecting its multiple enzymatic functions (transcriptase, capping, and polyadenylation enzymes) (4). The L protein also conducts replication of viral genomic RNA via its replicase activity. Importantly, there is accumulating evidence indicating or suggesting that these functions of the RABV L protein require interaction with other viral proteins. Specifically, to act as a viral RdRp, the L protein needs to bind with its essential cofactor, P protein, existing in a viral ribonucleoprotein (RNP) complex which is formed by encapsidation of viral genomic RNA by N proteins (4). The results of a recent study using a cell-free in vitro RNA synthesis assay suggest that interaction of the L protein with the N protein in an RNP complex is important for initiation of genomic RNA synthesis (5). Furthermore, it was previously reported that the balance of transcription and replication in RABV-infected cells is regulated by the M protein (6), suggesting direct or indirect interaction between L and M proteins. Interaction of the RABV L protein with host cellular molecules also plays an important role in the propagation cycle: Bauer et al. (7) recently reported that the RABV L protein has a dynein light chain 1 binding motif that mediates the interaction of the L protein with cytoskeletal microtubules and also that the interaction is involved in the efficient primary transcription by RABV.
Considering the multifunctionality of the L protein as well as its requirement of molecular interactions with other viral and cellular molecules, as described above, molecular function analysis of the L protein would provide useful information for establishing an effective therapy for rabies. However, there have been only a few studies that were carried with the aim of elucidating RABV L protein functions (8–11). Hence, further accumulation of knowledge about its molecular functions will be required to establish a therapeutic method for rabies.
The propagation modes of all viruses within the order Mononegavirales (NNS RNA viruses) in infected cells are believed to resemble each other, leading to the general idea that their L proteins have similar molecular functions. Consistent with this idea, a previous study showed by comparing amino acid sequences of L proteins from various species of NNS RNA viruses that there is a total of six highly conserved regions (CRs I to VI) (12). The following function analyses have revealed important roles for some of the CRs: CR III contains an important motif for RdRp activity (13), and CRs V and VI are both responsible for mRNA cap enzyme activities (14–17). Those studies have provided significant findings on molecular functions conserved among the NNS RNA virus L proteins. Meanwhile, considering that the order Mononegavirales consists of various virus species that have different host ranges and tissue tropisms, it is highly possible that the respective species have evolved unique structures and functions in their L proteins. However, the molecular functions of the L protein specific for the respective viruses, including RABV, have not been extensively examined so far.
Many previous studies on L protein functions of NNS RNA viruses have exploited the minigenome assay system, which is based on the transcription/replication of an artificial minigenome RNA driven by coexpression of the recombinant L protein with N and P proteins (18–23). This system enables indirect evaluation of the transcription/replication efficiency by measuring the expression level of a reporter gene (e.g., firefly luciferase gene) encoded in the minigenome RNA, providing a useful tool for examining molecular functions of the L protein. However, this system fully depends on a plasmid-based expression system to supply all of the N, P, and L proteins as well as, in many cases, minigenome RNA. This raises the problem that the data obtained from the minigenome system might not always reproduce the actual viral RNA synthesis in infected cells. Notably, data obtained in previous studies indicated that supplementation of N, P, and L proteins by helper RABV infection hardly induced reporter gene expression in cells transfected to express minigenome RNA (24), whereas supplementation of those proteins by plasmid transfection induced a high level of a reporter gene expression from the minigenome RNA (25), implying that the mechanism of the replication complex formation differs in RABV-infected cells and transfected cells in the minigenome system. This indicates the necessity to develop a new experimental system to evaluate the functions of the RABV L protein under a condition better reflecting actual infection.
In the present study, to develop a new experimental tool for studying molecular functions of the RABV L protein, we established by using a reverse genetics system of RABV strain Nishigahara an L gene-deficient virus expressing NanoLuc luciferase (Nishi-ΔL/Nluc), which propagates in cultured neuroblastoma cells by complementation with the L protein in trans. After confirming the applicability of Nishi-ΔL/Nluc to a trans-complementation assay, we sought to identify a novel functional domain in the RABV L protein by using this assay system. Following a comprehensive genetic analysis of L genes from RABV and other lyssavirus species, we identified a region highly conserved among these species at positions 1914 to 1933 in the RABV L protein and then analyzed the functional importance of this region by using Nishi-ΔL/Nluc and a series of L protein mutants. The results demonstrated that the amino acid sequence at positions 1929 to 1933 (NPYNE) on the RABV L protein is important for its RdRp function. We also revealed that the NPYNE sequence is important for L protein's interaction with its essential cofactor, P protein, and thus also for its RdRp activity.
RESULTS
Propagation of Nishi-ΔL/Nluc in neuroblastoma cells transfected to express the L protein.
Using a reverse genetics system of RABV strain Nishigahara (26), we successfully established L gene-deficient Nishi-ΔL/Nluc, which possesses, instead of the L gene, a NanoLuc luciferase gene in the genome (Fig. 1A). First, we checked whether Nishi-ΔL/Nluc propagates and spreads in mouse neuroblastoma NA cells transiently expressing the recombinant L protein. We found that Nishi-ΔL/Nluc formed a large number of viral N protein-positive foci in NA cells transfected with pCNiL plasmid expressing wild-type L protein (nontagged wtL) (Fig. 2A) but not in empty vector-transfected cells (Fig. 1BI and BII). Notably, in the N protein-positive cells, many cytoplasmic inclusion bodies, which are typically found in RABV-infected cells, were observed (Fig. 1BIII).
FIG 1.
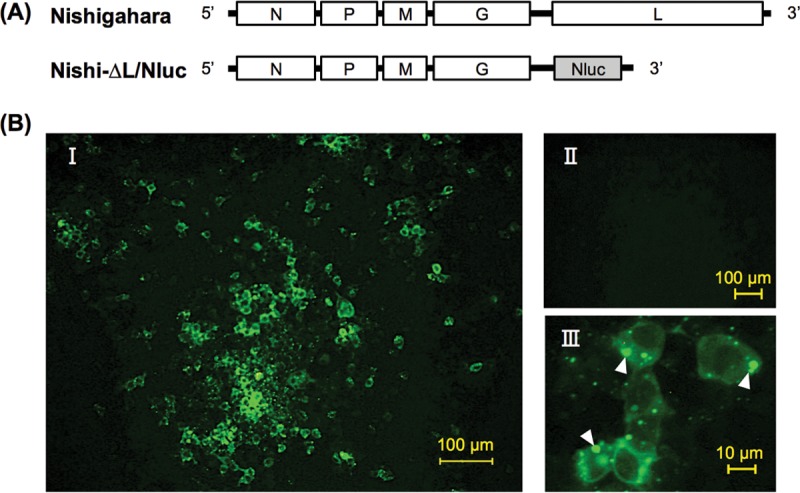
Propagation of the L gene-deficient Nishi-ΔL/Nluc in NA cells transfected to express the L protein. (A) Schematic diagram of the genome organization of Nishi-ΔL/Nluc. The white and gray bars represent viral genes and the NanoLuc luciferase gene, respectively. (B) At 2 days after inoculation with Nishi-ΔL/Nluc at an MOI of 0.01, the cells, which had been transfected with pCNiL plasmid or empty vector 2 days before the inoculation, were fixed and immunostained with an anti-RABV N protein monoclonal antibody. (BI and BIII) Images of the Nishi-ΔL/Nluc-infected cells, in which the recombinant L protein was complemented in trans by transfection, at low (BI) and high (BIII) magnifications. White arrowheads show cytoplasmic inclusion bodies. (BII) Low-magnification image of the Nishi-ΔL/Nluc-infected cells without complementation with L protein in trans (transfected with an empty vector).
FIG 2.
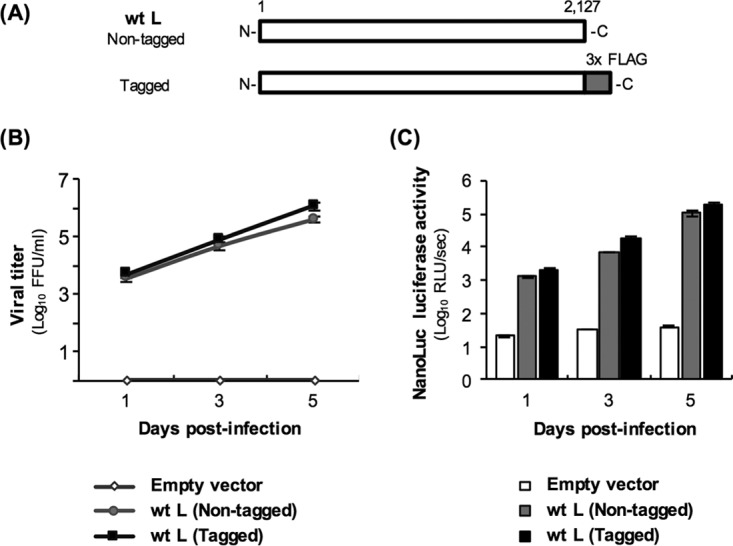
Growth curve of and NanoLuc luciferase production by Nishi-ΔL/Nluc in NA cells expressing the L protein. (A) Schematic diagrams of primary structures of nontagged and tagged wild-type L proteins (wtLs). The white and gray bars represent Nishigahara L protein and 3× FLAG tag, respectively. (B and C) At 2 days after transfection of NA cells with a plasmid expressing either wtL (nontagged) or wtL (tagged) or with an empty vector, Nishi-ΔL/Nluc was inoculated at an MOI of 0.01 into the cells. (B) Culture supernatants of the Nishi-ΔL/Nluc-infected cells were harvested at 1, 3, and 5 dpi. The viruses in supernatants were titrated in TetOff-NiL-NA cells by focus assays. (C) Lysates of the same infected cells were prepared at 1, 3, and 5 dpi before measuring the luciferase activities. The activities were calculated as relative light units (RLU) per second. All assays were carried out in triplicate, and the values in both graphs are means ± standard errors of the means.
Next, we examined the growth efficiency of Nishi-ΔL/Nluc in NA cells transiently expressing wtL. After inoculation of Nishi-ΔL/Nluc into NA cells transfected with pCNiL at a multiplicity of infection (MOI) of 0.01, the infectious titers in the culture supernatant constantly increased and reached 3.9 × 105 focus-forming units (FFU)/ml at 5 days postinfection (dpi) (Fig. 2B). In contrast, transfection with an empty plasmid did not support propagation of Nishi-ΔL/Nluc in NA cells. These results indicate that Nishi-ΔL/Nluc grows exponentially in NA cells expressing the L protein.
To check whether the expression level of NanoLuc luciferase corresponds to the growth efficiency of Nishi-ΔL/Nluc, we measured the luciferase activity in L protein-expressing, Nishi-ΔL/Nluc-infected NA cells, which had been used for the growth experiments described above. We found that the activity of luciferase produced by Nishi-ΔL/Nluc exponentially increased over time in the L protein-expressing cells but not in empty vector-transfected cells (Fig. 2C). These results indicate that the luciferase activity reflects the growth efficiency of Nishi-ΔL/Nluc in L protein-expressing NA cells.
Taken together, these findings demonstrate that Nishi-ΔL/Nluc has the abilities to propagate exponentially and to produce NanoLuc luciferase, of which the activity reflects its growth efficiency, in NA cells by complementation with the L protein in trans.
Potential of Nishi-ΔL/Nluc for molecular function analysis of the L protein.
The findings described above indicated the strong possibility that Nishi-ΔL/Nluc is applicable for the trans-complementation assay, which will be useful for studying molecular functions of the L protein. More specifically, this assay system would enable easy evaluation of how mutagenesis of the recombinant L protein influences its RdRp function by checking the activity of NanoLuc luciferase produced by Nishi-ΔL/Nluc. However, since the mutagenesis could affect not only the functionality but also the stability of the recombinant L protein, the expression level of the protein needs to be monitored in this system. We therefore constructed pCNiL-3×FLAG plasmid expressing wtL fused with a 3× FLAG tag (tagged wtL) (Fig. 2A) so that expression of the recombinant protein could be detected by using an anti-FLAG antibody. We found that addition of the tag did not affect the L protein's RdRp function (Fig. 2B and C). Also, expression of the tagged wtL was detected by Western blotting with an anti-FLAG antibody (Fig. 3, lane 2).
FIG 3.
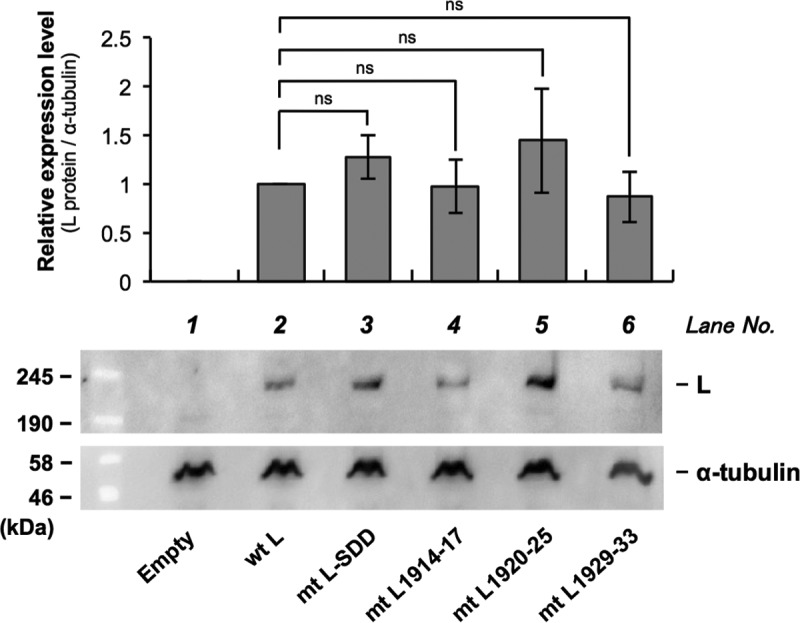
Analysis of the expression levels of a series of recombinant L proteins in Nishi-ΔL/Nluc-infected NA cells by Western blotting. The recombinant L proteins fused with a 3× FLAG tag were detected by using an anti-FLAG antibody. Alpha-tubulin in each sample was also detected as a loading control. The Western blott shows a representative result. The graph shows the relative expression levels of the respective L proteins standardized by the levels of alpha-tubulin, which were determined by quantification of the band intensities. The values are ratios, considering the standardized expression level of wtL as 1.0. All assays were carried out in triplicate, and the data are means ± standard errors of the means of the relative expression levels of the L proteins. ns, not significant (P ≥ 0.05).
Next, we tested the applicability of Nishi-ΔL/Nluc to the trans-complementation assay by using a functionally defective L protein mutant. It was previously reported that the GDN motif located in CR III is important for RdRp activity of L proteins of NNS RNA viruses (13, 27, 28). We therefore modified pCNiL-3×FLAG to express the functionally defective L protein mutant (mtL-SDD) by replacing the GDN motif with the SDD sequence at amino acid positions 728 to 730 (Fig. 4A), as previously reported (13). The conventional minigenome assay revealed that the mutations indeed disrupted the RdRp function of the recombinant L protein (Fig. 4B). We then examined the functionality of mtL-SDD by using Nishi-ΔL/Nluc: we measured luciferase activity produced by Nishi-ΔL/Nluc at 9 h after inoculation at an MOI of 0.01, when release of infectious Nishi-ΔL/Nluc progeny was not detected in wtL-expressing NA cells (data not shown). Thus, at this time point, we were able to evaluate the efficiency of viral RNA synthesis in the primary infected cells. Consistent with the results of the minigenome assay, complementation with mtL-SDD in trans induced luciferase activity significantly less efficiently than that of wtL did (P < 0.01) (Fig. 4C), while there was no statistically significant difference between the expression levels of the two proteins (Fig. 3, lanes 2 and 3). Similar results were obtained from the same assay at 48 h postinoculation (hpi) (Fig. 4D), when release of Nishi-ΔL/Nluc progeny and the following spread of infection were observed in wtL-expressing NA cells (Fig. 1B and 2B): the difference between the luciferase activities in the wtL- and mtL-SDD-expressing cells became more prominent than that at 9 hpi, probably due to distinct efficiencies of multiple-step growth of Nishi-ΔL/Nluc in the respective cells. These findings confirmed the functional defect of mtL-SDD, indicating that Nishi-ΔL/Nluc is applicable to the trans-complementation assay for exploring a novel functional domain in the RABV L protein.
FIG 4.
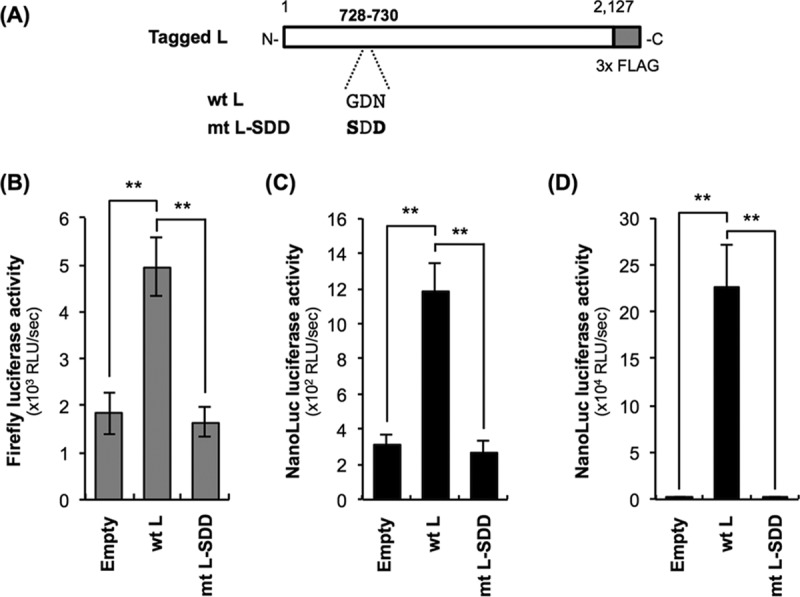
Examination of the applicability of Nishi-ΔL/Nluc to molecular function analysis of the RABV L protein by using a functionally defective L protein mutant, mtL-SDD. (A) Schematic diagram of primary structures of wtL and mtL-SDD. The white and gray bars represent Nishigahara L protein and 3× FLAG tag, respectively. Amino acid sequences at positions 728 to 730 of the respective L proteins are also indicated. The amino acid residues that are substituted from those at the same positions in wtL are shown in bold. (B) Conventional minigenome assay to confirm the functional defect of mtL-SDD. NA cells were transfected with an empty vector, pCNiL-3× FLAG, or pCNiL-SDD together with plasmids expressing the firefly luciferase-encoding minigenome RNA and viral N and P proteins. At 48 hpt, luciferase activities in cell lysates were determined. (C and D) trans-Complementation assay by using Nishi-ΔL/Nluc to assess the functionality of mtL-SDD. Nishi-ΔL/Nluc-infected NA cells, which had been transfected with a plasmid expressing either wtL or mtL-SDD or an with empty vector 2 days before the inoculation, were lysed at 9 hpi (C) or 48 hpi (D) for NanoLuc luciferase assays. All assays were carried out in triplicate and repeated three times independently. The values in graphs are means ± standard errors of the means from three independent experiments. **, significant difference at a P value of <0.01.
Genetic analyses of RABV and lyssavirus L genes.
We next searched for the RABV L protein region(s) that has high potential to be a novel functional domain, before evaluating its functional importance by the trans-complementation assay with Nishi-ΔL/Nluc. In this study, we aimed to identify a functionally important domain that is exposed to the surface of the L protein molecule, thus potentially being an interface with other viral or cellular molecules. We therefore performed a search for an L protein region(s) that satisfies the following two conditions: (i) the rate of amino acid substitutions (i.e., nonsynonymous mutations at the nucleotide level) is suppressed as a result of strong negative selection, so-called functional constraint, and (ii) amino acid residues composing the region are highly hydrophilic. To roughly identify such an L protein region(s), we carried out sliding-window analyses of the dN/dS ratio (proportion of the nonsynonymous mutation rate to the synonymous mutation rate) and of the hydropathy score by using the open reading frame sequences of L genes from a total of 15 representative RABV strains (Table 1). The values in the graphs in Fig. 5A represent averages of the dN/dS ratio and the hydropathy score in a window composed of 20 codon sites, when the window slides downward in a codon-by-codon manner. After searching for L protein regions with low values for the dN/dS ratio and hydropathy score, we found that the region around amino acid positions 1870 to 1930, which is clearly distinct from CRs I to VI conserved among NSS RNA viruses, fulfilled the above-mentioned two conditions. According to the hydropathy score, the region is roughly separated into two regions located around positions 1870 to 1890 and 1900 to 1930 (Fig. 5B). In this study, we decided to focus on the latter region, where the values of dN/dS ratio and hydropathy score were also observed to be low in similar sliding-window analyses for L genes from a total of 12 lyssavirus species (Table 2; Fig. 5C).
TABLE 1.
Rabies virus strains used for the genetic analyses in this study
| GenBank accession no. | Strain | Host | Country | Collection year |
|---|---|---|---|---|
| EU643590 | HN10 | Homo sapiens (human) | China | 2006 |
| KC595280 | Rus(Lipetsk)8052f_2011 | Vulpes vulpes (red fox) | Russia | 2011 |
| EU311738 | RRV ON-99-2 | Procyon lotor (raccoon) | Canada | 1999 |
| KM594036 | IP 542/10 | Nyctinomops laticaudatus (broad-eared bat) | Brazil | 2010 |
| KM594024 | IP 1170/12 | Callithrix jacchus (common marmoset) | Brazil | 2012 |
| KT336437 | 34312 | Canis lupus familiaris (dog) | South Africa | 2012 |
| AB519642 | BR-DR1 | Desmodus rotundus (common vampire bat) | Brazil | 2000 |
| KF155000 | RV2516 | Bos taurus (cattle) | Iraq | 2010 |
| JQ685952 | A02-2971 | Parastrellus hesperus (canyon bat) | USA | 2002 |
| JQ685947 | TN310 | Lasiurus cinereus (hoary bat) | USA | 2004 |
| JQ685942 | AZBAT-65094 | Eptesicus fuscus (big brown bat) | USA | 1981 |
| JQ685929 | MEXSK3644 | Spilogale putorius (spotted skunk) | Mexico | 2009 |
| JQ685916 | FL1010 | Lasiurus intermedius (northern yellow bat) | USA | 2002 |
| AB128149 | Ni-CE | Laboratory strain | Japan | |
| AB645847 | 1088 | Marmota monax (woodchuck) | USA | Unknown |
FIG 5.

Sliding-window analyses of dN/dS ratio and Kyte-Doolittle hydropathy score along the open reading frame sequences of L genes from a total of 15 representative RABV strains (A and B) or 12 lyssavirus species (C). Solid red and blue lines show the average values for dN/dS ratio and hydropathy score, respectively, in a window composed of 20 codon sites. The red and blue longitudinal bars on the solid lines show the standard deviations of dN/dS ratio and hydropathy score, respectively. Dashed red and blue lines show the average value for dN/dS ratio (0.07 in RABVs and 0.15 in lyssaviruses) and cutoff value for hydrophobic (hydropathy score > 0) and hydrophilic (hydropathy score < 0) regions, respectively. Yellow bars represent CRs I to VI. Blue bars indicate the regions with low values for dN/dS ratio and hydropathy score revealed in this study. aa, amino acids.
TABLE 2.
Lyssavirus species used for genetic analyses in this study
| GenBank accession no. | Species name (abbreviation) | Genotype | Country | Collection year |
|---|---|---|---|---|
| AB128149 | Rabies virus, Ni-CE strain (RABV) | 1 | Japan | Not applicable (laboratory strain) |
| EU293108 | Lagos bat virus (LBV) | 2 | Senegal | 1985 |
| NC_006429 | Mokola virus (MOKV) | 3 | Unknown | Unknown |
| EU293119 | Duvenhage virus (DUVV) | 4 | South Africa | 1971 |
| NC_009527 | European bat lyssavirus 1 (EBLV1) | 5 | Germany | 1968 |
| NC_009528 | European bat lyssavirus 2 (EBLV2) | 6 | United Kingdom | 2002 |
| NC_003243 | Australian bat lyssavirus (ABLV) | 7 | Australia | 1996 |
| EF614259 | Aravan virus (ARAV) | Not classified | Kyrgyzstan | 1991 |
| EF614261 | Khujand virus (KHUV) | Not classified | Tajikistan | 2001 |
| EF614260 | Irkut virus (IRKV) | Not classified | Russia | 2002 |
| EF614258 | West Caucasian bat virus (WCBV) | Not classified | Russia | 2002 |
| GU170201 | Shimoni bat virus (SHIBV) | Not classified | Kenya | 2009 |
To further specify the L protein region of our interest, we aligned and compared amino acid sequences of the L protein regions at amino acid positions 1890 to 1940, which originated from the 12 lyssavirus species described above (Fig. 6). We found that the region at positions 1914 to 1933 is highly conserved among these species and highly hydrophilic, indicating the possibility that this region has an important role in L protein function on the surface of the protein molecule. We therefore decided to consider this region as a target of molecular function analysis of the RABV L protein by the trans-complementation assay.
FIG 6.

Alignment of amino acid sequences of the L protein region at amino acid positions 1890 to 1940 in 12 lyssavirus species. Dots indicate amino acid residues that are identical to those at the same positions of the RABV L protein. Blue boxes show hydrophilic amino acid residues (Kyte-Doolittle hydropathy score: −3.2 to −4.5). Red asterisks indicate hydrophilic amino acid residues that are completely conserved among these lyssaviruses within the region at positions 1914 to 1933. RABV, rabies virus; DUVV, Duvenhage lyssavirus; EBLV1, European bat lyssavirus 1; IRKV, Irkut lyssavirus; EBLV2, European bat lyssavirus 2; KHUV, Khujand lyssavirus; ARAV, Aravan virus; ABLV, Australian bat lyssavirus; MOKV, Mokola lyssavirus; LBV, Lagos bat lyssavirus; SHIBV, Shimoni bat lyssavirus; WCBV, West Caucasian bat virus.
Importance of the L protein region at positions 1914 to 1933 in its RdRp function.
To examine the importance of the L protein region at positions 1914 to 1933 in its RdRp function, we constructed a total of three plasmids (pCNiL-mt1914-17, pCNiL-mt1920-25, and pCNiL-mt1929-33) to express a series of L protein mutants (mtL1914-17, mtL1920-25, and mtL1929-33, respectively) which have hydrophobic Ala residues, instead of hydrophilic residues that are completely conserved among lyssaviruses, in this region (Fig. 6 and 7A). Examination of the functionality of the respective mutants by the trans-complementation assay revealed that activities at 9 hpi of luciferase produced in infected NA cells expressing mtL1914-17 and mtL1920-25 were comparable to the activity in wtL-expressing cells (Fig. 7B). In contrast, the activity in NA cells expressing mtL1929-33 was significantly lower than that in NA cells expressing wtL (P < 0.05). Similar results were obtained from the equivalent assay at 48 hpi (Fig. 7C). Western blot analysis revealed that expression levels of all of these L protein mutants were statistically indistinguishable from the level of wtL (Fig. 3, lanes 2 and 4 to 6). Furthermore, the functional defect of mtL1929-33 was also confirmed by the conventional minigenome assay (data not shown). These data strongly suggest that mtL1929-33, but not mtL1914-17 or mtL1920-25, has a defect in RdRp function.
FIG 7.
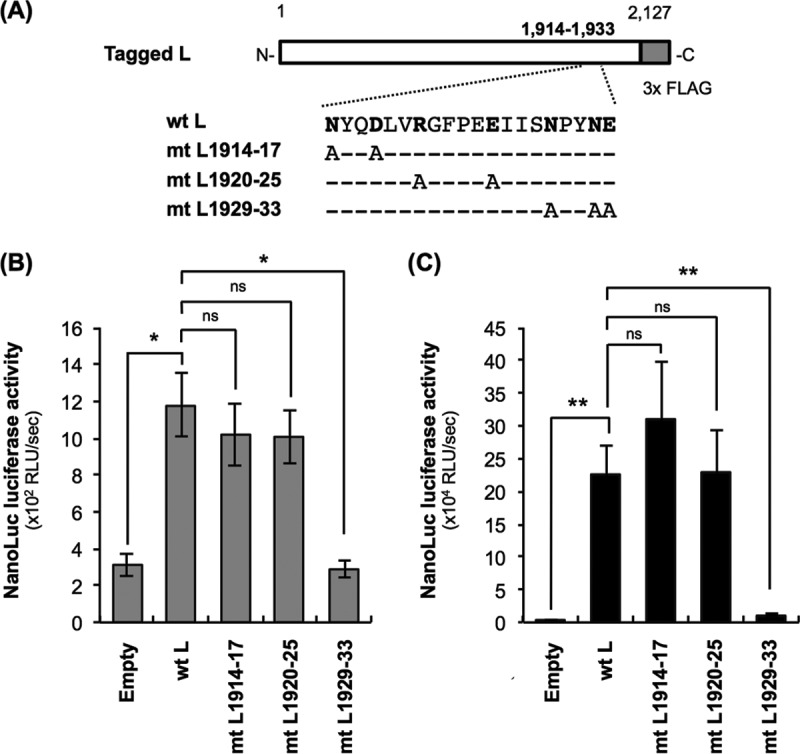
Examination of the functionality of L protein mutants by the trans-complementation assay. (A) Schematic diagram of primary structures of L protein mutants used in this study. The white and gray bars represent Nishigahara L protein and 3× FLAG tag, respectively. Dashes represent the residues that are identical to those at the same positions of wtL. Hydrophilic amino acids in the region at positions 1914 to 1933, which are perfectly conserved among L proteins from a total of 11 lyssavirus species analyzed in this study (see Fig. 6), are shown in bold. (B and C) NA cells were transfected to express wtL or a series of L protein mutants (mtL1914-17, mtL1920-25, and mtL1929-33) before inoculation with Nishi-ΔL/Nluc at an MOI of 0.01. The infected cells were lysed at 9 hpi (B) or 48 hpi (C) for NanoLuc luciferase assays. All assays were carried out in triplicate and repeated three times independently. The values are means ± standard errors of the means from the three independent experiments. *, significant difference at a P value of <0.05; **, significant difference at a P value of <0.01. ns, not significant (P ≥ 0.05).
Next, we examined whether an increase of mtL1929-33 expression level compensates the functional defect: we transfected NA cells with 0.5 μg and 1.0 μg of pCNiL-3×FLAG or pCNiL-mt1929-33 before inoculating them with Nishi-ΔL/Nluc at an MOI of 0.01 and then checking the luciferase activity at 9 hpi. We confirmed by Western blotting that the increase in amounts of the transfecting plasmids indeed enhanced expression levels of wtL and mtL1929-33 (Fig. 8A). However, there was no difference between the luciferase activities in NA cells transfected with 0.5 μg and 1.0 μg of pCNiL-mt1929-33, indicating that the increased mtL1929-33 expression did not compensate its functional defect (Fig. 8B).
FIG 8.

Effects of increased expression of L proteins on NanoLuc luciferase activities produced by Nishi-ΔL/Nluc in a trans-complementation assay. NA cells grown in a 24-well tissue culture plate were transfected with 0.5 μg and 1.0 μg of pCNiL-3×FLAG or pCNiL-mt1929-33 before inoculation with Nishi-ΔL/Nluc at an MOI of 0.01. The infected cells were lysed at 9 hpi, and luciferase activities were determined. (A) Increases in expression levels of L proteins were confirmed by Western blotting with an anti-FLAG antibody. Alpha-tubulin in each sample was also detected as a loading control. (B) Luciferase activities in the cell lysates at 9 hpi were determined by a NanoLuc luciferase assay and were calculated as RLU per second. All assays were carried out in triplicate, and the values are means ± standard errors of the means. **, significant difference at a P value of <0.01. ns, not significant (P ≥ 0.05).
Transcription and replication efficiencies of Nishi-ΔL/Nluc in mtL1929-33-expressing NA cells.
To determine whether the mutations in mtL1929-33 decrease its ability to synthesize viral RNA, we quantified the N gene mRNA (N mRNA) and the genomic RNA in mtL1929-33-expressing, Nishi-ΔL/Nluc-infected NA cells by real-time reverse transcription-PCR (RT-PCR) (Fig. 9). At 9 hpi, the expression level of the mRNA in mtL1929-33-expressing cells was significantly lower than the level in wtL-expressing cells (P < 0.01) (Fig. 9A). On the other hand, the amount of genomic RNA in mtL1929-33-expressing cells was statistically indistinguishable from that in wtL-expressing cells (Fig. 9B), probably due to the presence of nonreplicating genomic RNA derived from the inoculated virus and also due to very low replication efficiency at this early time point even in wtL-expressing cells. At 48 hpi, decreased mRNA expression was also observed in mtL1929-33-expressing cells (Fig. 9C). Notably, at this time point, the expression level of the genomic RNA in mtL1929-33-expressing cells was significantly lower than the level in wtL-expressing cells (P < 0.05) (Fig. 9D). These results indicate that the mutations in mtL1929-33 negatively impact viral RNA synthesis in Nishi-ΔL/Nluc-infected NA cells.
FIG 9.

Examination of transcription and replication efficiencies of Nishi-ΔL/Nluc in wtL- and mtL1929-33-expressing NA cells by real-time RT-PCR. NA cells were transfected to express wtL or mtL1929-33 before inoculation with Nishi-ΔL/Nluc at an MOI of 0.01. At 9 hpi (A and B) or 48 hpi (C, D, and E), the infected cells were collected and used for RNA extraction. The expression levels of N mRNA (A and C) and genomic RNA (B and D) in each cell lysate were measured by real-time RT-PCR. The levels are the number of copies of the respective RNAs per copy of mouse 18S rRNA. (E) The ratios of expression level of N mRNA to the level of genomic RNA were determined on the basis of their expression levels at 48 hpi. All assays were carried out in triplicate, and the values are means ± standard errors of the means. *, significant difference at a P value of <0.05; **, significant difference at a P value of < 0.01. ns, not significant (P ≥ 0.05).
Next, we tested the possibility that the mutations in mtL1929-33 preferentially disrupt either transcription or replication in Nishi-ΔL/Nluc-infected NA cells. More specifically, we compared the mRNA/genome ratios (the relative mRNA levels normalized by the genomic RNA levels) in the wtL-expressing and mtL1929-33-expressing NA cells at 48 hpi (Fig. 9E). The results showed that the mRNA/genome ratio in mtL1929-33-expressing NA cells was comparable to the ratio in wtL-expressing cells. Although we cannot exclude the possibility that transcription of low efficiency caused by the mutations indirectly lowers replication efficiency and vice versa, these data suggest that the mutations in mtL1929-33 do not preferentially inhibit either transcription or replication in Nishi-ΔL/Nluc-infected NA cells; in other words, the mutations inhibit the general synthesis of viral RNA by the L protein.
Importance of the newly identified NPYNE sequence for binding of the L protein with the P protein.
Chenik et al. (29) reported that the P protein-binding domain is located in the C-terminal region of the L protein, at positions 1562 to 2127. Based on the fact that this C-terminal region contains the NPYNE sequence at positions 1929 to 1933, we hypothesized that this sequence may be critical for interaction between the L and P proteins. To test this hypothesis, we analyzed the L-P protein interaction in 293T cells transfected to express wtL or mtL1929-33 together with the recombinant P protein by coimmunoprecipitation (co-IP). After co-IP of wtL with an anti-FLAG antibody, a substantial amount of the P protein was detected in the precipitate, whereas after co-IP of mtL1929-33, only a very small amount of the P protein was precipitated (Fig. 10). In the control experiments with an irrelevant mouse IgG, the P protein in the precipitate was below the detectable level (data not shown). These results were confirmed by an independently repeated experiment (data not shown). These data demonstrate that mtL1929-33 has an impaired ability to bind the P protein, indicating the importance of the NPYNE sequence for L protein's interaction with the P protein.
FIG 10.
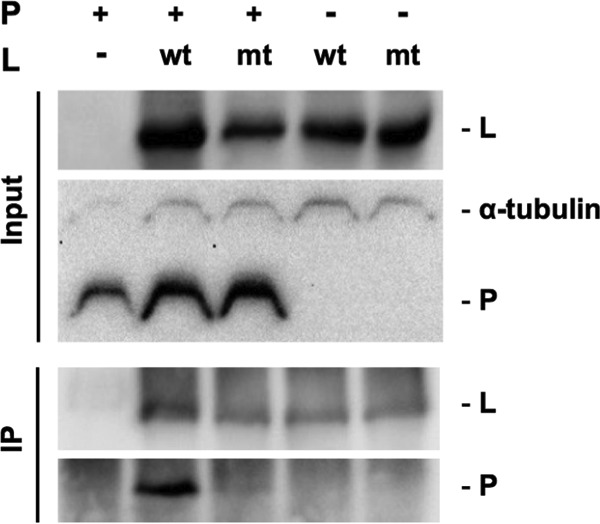
Comparison of the P protein-binding abilities of wtL and mtL1929-33 by co-IP analysis. 293T cells were transfected to express FLAG-tagged wtL or mtL1929-33 together with recombinant P protein. At 48 hpt, the cells were lysed and subjected to co-IP with an anti-FLAG antibody. The L protein and P protein in each sample were detected by Western blotting with an anti-FLAG antibody and anti-RABV P protein rabbit serum, respectively. Alpha-tubulin in each input sample was also detected as a loading control. wt, wtL; mt, mtL1929-33.
Taken together, the findings obtained in this study indicated that the NPYNE sequence at positions 1929 to 1933 in the RABV L protein, which is highly conserved among RABV and other lyssavirus species, has an important function in binding with its essential cofactor, P protein.
DISCUSSION
To the best of our knowledge, this is the first report on the establishment of an L gene-deficient virus in all NNS RNA virus species, including RABV. In addition, our findings have emphasized the utility of the virus for molecular function analysis of the viral RdRp L protein.
We demonstrated that an L gene-deficient RABV, Nishi-ΔL/Nluc, grows exponentially in NA cells transfected to express the L protein (Fig. 2B), similar to the genetically intact Nishigahara strain in nontransfected, normal NA cells (26, 30). In addition, after infection with Nishi-ΔL/Nluc, a large number of inclusion bodies, which are probably identical to Negri body-like structures known as replication complexes of RABV (31), was formed in the cytoplasm of NA cells expressing the L protein (Fig. 1BIII). These findings strongly suggest that trans-complementation with the L protein is sufficient for Nishi-ΔL/Nluc to complete the propagation cycle that models well the cycle in Nishigahara-infected cells. Our data also indicated that Nishi-ΔL/Nluc produces NanoLuc luciferase, the activity of which corresponds to its growth efficiency, in the presence of the recombinant L protein (Fig. 2C), indicating the possibility that Nishi-ΔL/Nluc is applicable to a trans-complementation assay system that enables easy evaluation of the functionality of the L protein based on luciferase activity. This possibility was strongly supported by the finding that expression of a functionally defective L protein mutant, mtL-SDD, failed to induce luciferase production by Nishi-ΔL/Nluc (Fig. 4C and D). Based on these findings, we conclude that Nishi-ΔL/Nluc provides a useful experimental tool for molecular function analysis of the L protein.
In this study, we examined the functionality of the respective L protein mutants (mtL-SDD, mtL1914-17, mtL1920-25, and mtL1929-33) by using two distinct assays, namely, the conventional minigenome assay and the novel trans-complementation assay with Nishi-ΔL/Nluc. All of the results obtained from these two assays were consistent with each other (Fig. 4 and data not shown), except for the results for mtL1914-17: data obtained from the minigenome assays indicated that this L protein mutant has a functional defect (data not shown), whereas data from the trans-complementation assay indicated that this mutant has integrity in its function (Fig. 7B and C). Although the reason for this contradiction is not clear, we believe that the trans-complementation assay reflects more closely L protein function in actual RABV-infected cells because, in contrast to the minigenome assay system, this assay only minimally requires an artificial plasmid expression system to support propagation of Nishi-ΔL/Nluc by complementation with the L protein. Further studies will be needed to elucidate the mechanism underlying the above-described contradiction in the results of the two assays.
For sliding-window analyses of the dN/dS ratio and hydropathy score (Fig. 5), we chose L gene sequences from a total of 15 representative RABV strains, which belong to different phylogenetic lineages (data not shown). Importantly, the results of these analyses were very similar to those of identical analyses based on L gene sequences from a total of 197 RABV strains (data not shown). Therefore, we believe that the data obtained from the sliding-window analyses in this study illustrate the general features of L genes/proteins from all RABV strains in nature. The results indicate that the L protein regions around positions 1870 to 1890 and 1900 to 1930 are highly conserved among RABV strains and are highly hydrophilic (Fig. 5B). In this study, we focused on the region around positions 1900 to 1930 as a target of molecular function analysis of the L protein since this region is highly conserved among lyssavirus species, including RABV (Fig. 5C). The other region around positions 1870 to 1890, which is conserved specifically among RABV strains (Fig. 5B), would be another interesting target for function analysis in the future. The findings would be able to highlight the functional differences between RABV and other lyssavirus L proteins.
The trans-complementation assay with Nishi-ΔL/Nluc demonstrated that the NPYNE sequence at positions 1929 to 1933, which is perfectly conserved among lyssavirus species, including RABV (Fig. 6), is important for viral RNA synthesis (Fig. 7 and 9). Further examinations revealed that the NPYNE sequence is critically involved in binding of the L protein with its essential cofactor, P protein (Fig. 10). This is consistent with the results of a previous study showing that the L protein interacts with the P protein via its C-terminal region at positions 1562 to 2127 (29). Since the interaction between L and P proteins is indispensable for L protein's RdRp activity, it appears reasonable that mtL1929-33, lacking the NPYNE sequence, has a defect in both the abilities to synthesize viral mRNA and genomic RNA (Fig. 9). Considering the fact that the sequence is located in a highly hydrophilic region (Fig. 6), there is a strong possibility that the NPYNE sequence forms a part of the interface with the P protein. Notably, the L protein-binding domain of the RABV P protein has already been identified in its N-terminal region at positions 1 to 19 (29). Thus, it would be interesting to examine whether and how this L protein-binding domain in the P protein interacts with the NPYNE sequence in the L protein. Interestingly, Lys and Arg residues at positions 3 and 12 in the P protein, respectively, are perfectly conserved among the 12 lyssavirus species examined in this study (data not shown), leading to the speculation that one of these positively charged residues might form an ionic bond with the negatively charged Glu residue at position 1933, the last residue of the NPYNE sequence, in the L protein. Molecular structural studies will be required to fully elucidate the mode of interaction between the L and P proteins.
While the mutations in mtL1929-33 significantly impaired the interaction between L and P proteins, these mutations did not completely destroy the P protein-binding ability of the L protein: a very small amount of P protein was observed in the precipitate after co-IP of mtL1929-33 (Fig. 10). In addition, expression of mtL1929-33 supported the viral RNA synthesis by Nishi-ΔL/Nluc with very low but certain efficiency (Fig. 9C and D). All of these findings suggest that the mutations in mtL1929-33 cause a decrease rather than a loss of RdRp function. However, this decrease of the function was not compensated by increasing the expression level of mtL1929-33 (Fig. 8), suggesting a complicated mechanism by which the L-P protein interaction impaired by those mutations negatively impacts L protein's RdRp function. One possibility for the mechanism is that the mutations might significantly reduce the speed of RNA polymerization by the L protein through abnormal interaction between the L and P proteins.
In conclusion, this study has demonstrated that Nishi-ΔL/Nluc provides a useful experimental tool for molecular functional studies of the RdRp L protein of RABV. Importantly, the utility of this experimental tool has been confirmed by an actual example: the trans-complementation assay with Nishi-ΔL/Nluc revealed for the first time that the NPYNE sequence at positions 1929 to 1933 in the RABV L protein is functionally important. Furthermore, the results of further examinations indicated that this NPYNE sequence is critically involved in the L protein's interaction with its essential cofactor, P protein, thus also in the L protein's RdRp activity. We believe that these findings are of great value for the development of a therapeutic drug for rabies targeting the L-P protein interaction to inhibit viral RNA synthesis.
MATERIALS AND METHODS
Cells.
Cells of a baby hamster kidney (BHK) cell clone, BHK/T7-9 (32), which constitutively express T7 RNA polymerase, were maintained in Eagle's minimal essential medium (EMEM) supplemented with 10% tryptose phosphate broth and 5% fetal calf serum (FCS). Mouse neuroblastoma NA cells were grown in EMEM containing 10% FCS. Human embryonic kidney (HEK) 293T cells were maintained in Dulbecco's modified EMEM supplemented with 10% FCS. An NA cell clone constitutively expressing the L protein of RABV strain Nishigahara (TetOff-NiL-NA cells) was established by using the Retro-X Tet-off advanced inducible expression system (Clontech, Mountain View, CA). In this study, TetOff-NiL-NA cells were cultured in the absence of tetracycline or its derivative so that expression of the L protein was always induced.
Construction of plasmids.
To construct the pCNiL plasmid expressing the Nishigahara L protein (designated nontagged wtL), we amplified a cDNA fragment including the open reading frame of the Nishigahara L gene by PCR from a full-length genome plasmid of RABV strain Nishigahara (26) and then cloned it into pCAGGS/MCS (kindly provided by Yoshihiro Kawaoka). We also generated pCNiL-3×FLAG expressing the Nishigahara L protein C-terminally fused with a 3× FLAG tag (designated tagged wtL) (Fig. 2A). pCNiL-3×FLAG was modified to obtain pCNiL-SDD expressing a functionally defective mutant mtL-SDD harboring Gly-to-Ser and Asn-to-Asp mutations at amino acid positions 728 and 730, respectively (Fig. 4A). In addition, we constructed a total of three plasmids (pCNiL-mt1914-17, pCNiL-mt1920-25, and pCNiL-mt1929-33) expressing L protein mutants (mtL1914-17, mtL1920-25, and mtL1929-33, respectively) having hydrophobic Ala residues, instead of hydrophilic residues that are completely conserved among lyssaviruses, in the region from positions 1914 to 1933 (Fig. 7A).
The minigenome plasmid pC-NiDI-Luc was also constructed to express a negative-sense luciferase-encoding minigenome RNA. Specifically, a cDNA fragment which consisted of a hammerhead ribozyme (33), the 5′-terminal noncoding region of the Nishigahara genome, a firefly luciferase gene, the 3′-terminal noncoding region of the Nishigahara genome, and a hepatitis delta virus antigenomic ribozyme was cloned downstream of a CAG promoter in pCAGGS/MCS.
The genome plasmid pNishi-ΔL/Nluc was established by modification of the full-length genome plasmid (pNi-Luc) of the recombinant Nishigahara strain expressing firefly luciferase (34): the cDNA region containing a firefly luciferase gene and the L gene was replaced with a cDNA fragment containing the NanoLuc luciferase gene, which had been amplified by PCR from the pNL1.1.CMV vector (Promega, Madison, WI), resulting in complete deletion of the L gene coding region.
Details of the construction of all plasmids described above are available on request.
Establishment of L gene-deficient Nishi-ΔL/Nluc.
To obtain L gene-deficient Nishi-ΔL/Nluc, we transfected BHK/T7-9 cells grown in a 24-well tissue culture plate with pNishi-ΔL/Nluc (2.0 μg/well), together with pT7IRES-RN (0.4 μg/well), -RP (0.1 μg/well), and -RL (0.2 μg/well) expressing viral N, P, and L proteins, respectively, as previously reported (32). At 3 days posttransfection (dpt), the cells were harvested together with the culture medium by scraping and were then overlaid onto L protein-expressing TetOff-NiL-NA cells, which were grown in a 6-well tissue culture plate. After 6 days, the culture supernatant was collected and stored as a viral stock at −80°C. The infectious titer of the viral stock was determined by a focus assay with TetOff-NiL-NA cells as described below.
Immunostaining of NA cells infected with Nishi-ΔL/Nluc.
NA cells grown in a 24-well tissue culture plate were transfected by using TransIT-2020 reagent (Mirus, Madison, WI) with 0.5 μg/well of pCAGGS/MCS or pCNiL. At 2 dpt, the cells were inoculated with Nishi-ΔL/Nluc at an MOI of 0.01. At 2 dpi, the infected cells were fixed with 2% paraformaldehyde for 1 h and 100% methanol for 1 min and then immunostained with anti-N protein mouse monoclonal antibody 13-27 (35) and fluorescein isothiocyanate (FITC)-labeled anti-mouse IgG (Cappel, West Chester, PA), followed by observation with a Biozero fluorescence microscope (BZ-8000 series; Keyence).
Growth of Nishi-ΔL/Nluc in NA cells transfected to express L protein.
NA cells grown in a 24-well tissue culture plate were transfected by using TransIT-2020 reagent with 0.5 μg of pCAGGS/MCS, pCNiL, or pCNiL-3×FLAG. At 2 dpt, the cells were inoculated with Nishi-ΔL/Nluc at an MOI of 0.01. The culture supernatants were collected at 1, 3, and 5 dpi, and viral titers (calculated as FFU per milliliter) of the respective supernatants were determined by focus assays as previously reported (34), with the following modification: instead of NA cells, TetOff-NiL NA cells were infected before fixation at 4 dpi. After collection of the supernatant samples, the cells infected with Nishi-ΔL/Nluc were subjected to a NanoLuc luciferase assay. Specifically, the cells were lysed with 100 μl of passive lysis buffer (Promega) before luciferase activities in the lysates were determined by using the NanoGlo luciferase reporter assay system (Promega).
Minigenome assay.
NA cells grown in a 24-well tissue culture plate were transfected by using Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) with 0.4 μg of pC-NiDI-Luc and 0.4 μg of pCAGGS/MCS, pCNiL-3×FLAG, or pCNiL-SDD, together with 0.4 μg of pCAGGS-NiN and 0.06 μg of pCAGGS-NiP, which were previously constructed to express the N protein and P protein of Nishigahara, respectively (36). After 48 h, the cells were lysed to determine firefly luciferase activity by using the luciferase assay system (Promega).
trans-Complementation assay with Nishi-ΔL/Nluc.
NA cells grown in a 24-well tissue culture plate were transfected by using TransIT-2020 reagent with 0.5 μg or 1.0 μg of pCNiL-3×FLAG, pCNiL-SDD, pCNiL-mt1914-17, pCNiL-mt1920-25, and pCNiL-mt1929-33. At 2 dpt, the cells were inoculated with Nishi-ΔL/Nluc at an MOI of 0.01 and then subjected to NanoLuc luciferase assay at 9 and 48 hpi as described above.
Western blotting.
Cell lysate samples prepared for the function analysis described above were separated by sodium dodecyl sulfate–5% polyacrylamide gel electrophoresis (5% SDS-PAGE) before transfer to a polyvinylidene difluoride membrane (Millipore, Billerica, MA). After blocking with phosphate-buffered saline (PBS) containing 0.1% Tween 20 and 5% nonfat dry milk, the bands of FLAG-tagged L protein and alpha-tubulin on the membrane were visualized with anti-FLAG M2 monoclonal antibody (Sigma-Aldrich, St. Louis, MO) and anti-alpha-tubulin antibody (Sigma-Aldrich), respectively. Chemiluminescent signal detection and densitometry analysis were carried out by using ChemiDoc XRS+ and Image Lab software (Bio-Rad, Hercules, CA).
Genetic analysis of RABV and lyssavirus L genes.
Complete nucleotide sequences of L genes from a total of 15 RABV strains and 11 other lyssavirus species were retrieved from the GenBank database. Detailed information on the L sequences is shown in Tables 1 and 2. Multiple alignments of nucleotide and amino acid sequences for RABV and lyssavirus L genes were undertaken using the computer program MAFFT (37). One amino acid insertion observed at position 23 in the Australian bat lyssavirus L protein was removed from the alignment.
Sliding-window analyses of the dN/dS ratio in the L gene were conducted between strain Ni-CE (GenBank accession number AB128149) and each of the other RABV or lyssavirus species using the program ADAPTSITE (38); the window was 20 codons wide and moved in steps of one codon. Hydropathy scores along the amino acid sequences of the L protein were calculated as the Kyte and Doolittle index with a window size of 20 amino acids and a step size of 1 amino acid residue using GENETYX (version 10.1.1; Genetyx, Tokyo, Japan).
Real-time RT-PCR.
NA cells grown in a 24-well tissue culture plate were transfected with 0.5 μg of pCAGGS/MCS, pCNiL-3×FLAG, or pCNiL-mt1929-33. After 2 days, the cells were infected with Nishi-ΔL/Nluc at an MOI of 0.01. At 9 hpi, total cellular RNA was extracted by using an RNeasy Plus minikit (Qiagen, Hilden, Germany). The extracted RNA was transcribed into cDNAs by using SuperScript IV reverse transcriptase (Invitrogen) and an RABV-specific primer (Table 3), oligo(dT)20 (Invitrogen), or a random hexamer (Invitrogen) to detect the RABV genomic RNA, N mRNA, or 18S rRNA, respectively. Real-time PCR was performed by using a StepOnePlus real-time PCR system (Applied Biosystems, Carlsbad, CA) and Power SYBR green PCR master mix (Applied Biosystems). PCR conditions were as follows: 95°C for 10 min and 40 cycles of 95°C for 15 s and 60°C for 1 min. The primer pairs used for real-time PCR and their targets are listed in Table 3.
TABLE 3.
Sequences of the primers used in this study
| Purpose | Primer (sense) | Sequence (5′–3′) | Annealing site positionsa |
|---|---|---|---|
| RT | RT for genome (+) | AGGCAGCTGAACTGACAAAGACTGA | 1179–1203 |
| PCR | |||
| Genomic RNA | Rabies genome F (+) | GTGTGCCGGAAATCTATTGATTGTGTA | 1438–1464 |
| Rabies genome R (−) | CTGATAGCACTCGGATTGACGAAGA | 1525–1549 | |
| N mRNA | Rabies N mRNA F (+) | ATGAAGACTGTTCAGGGCTGGTAT | 768–791 |
| Rabies N mRNA R (−) | CCCTGGCTCGAACATCCTTCTTA | 876–898 | |
| 18S rRNA | Mouse 18S rRNA F (+) | GACTCAACACGGGAAACCTCAC | |
| Mouse 18s rRNA R (−) | CAGACAAATCGCTCCACCAAC |
Site from the 3′-terminal region of the full-genome sequence of the RABV Ni strain (GenBank accession no. AB044824.1).
Coimmunoprecipitation.
HEK 293T cells grown in a 6-well tissue culture plate were transfected by using TransIT-X2 (Mirus) with 2.5 μg of pCAGGS-NiP and 7.5 μg of pCNiL-3×FLAG or pCNiL-mt1929-33. At 48 h posttransfection (hpt), the cells were washed with PBS and then lysed with lysis buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1% NP-40) containing cOmplete Mini protease inhibitor (Roche, Switzerland). The cell lysates were centrifuged at 15,000 × g for 10 min at 4°C to remove large debris. Then supernatants were collected and precleared by incubation with 20 μl of protein A/G plus agarose immunoprecipitation reagent (Santa Cruz Biotechnology, Dallas, TX) and 2 μg of IgG from mouse normal IgG (Santa Cruz) at 4°C for 30 min. Following centrifugation at 1,000 × g for 1 min at 4°C, supernatants containing FLAG-tagged L protein were incubated with 4 μg of anti-FLAG M2 antibody or mouse normal IgG for 2 h at 4°C and then further incubated with 20 μl of protein A/G plus agarose beads overnight with rotation at 4°C. After centrifugation at 1,000 × g for 1 min at 4°C, the agarose beads containing immunoprecipitated complex were washed five times with the above-described lysis buffer and boiled with SDS sample buffer for 30 min at 37°C. The precipitated proteins were analyzed by Western blotting using an anti-RABV P protein rabbit serum, which had been raised by immunization with purified recombinant P protein of RABV strain RC-HL. The anti-FLAG M2 antibody was also used to detect the tagged L protein in the immunoprecipitates.
Statistical analysis.
One-way analysis of variance (ANOVA) with Dunnett's multiple-comparison test and Tukey's multiple-comparison test was conducted to determine statistical significance. P values of <0.05 were considered statistically significant.
ACKNOWLEDGMENTS
We are grateful to Yoshihiro Kawaoka (University of Tokyo, Tokyo, Japan) for kindly providing the pCAGGS/MCS plasmid. We also thank Kentaro Yamada (Oita University, Oita, Japan) and Wataru Kamitani (Osaka University, Osaka, Japan) for valuable technical advice.
This study was partially supported by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (no. 25660225 and 15K08500) and a grant from the Ministry of Education, Culture, Sports, Science and Technology, Japan, for the Joint Research Program of the Research Center for Zoonosis Control, Hokkaido University.
REFERENCES
- 1.Willoughby RE, Tieves KS, Hoffman GM, Ghanayem NS, Amlie-Lefond CM, Schwabe MJ, Chusid MJ, Rupprecht CE. 2005. Survival after treatment of rabies with induction of coma. N Engl J Med 352:2508–2514. doi: 10.1056/NEJMoa050382. [DOI] [PubMed] [Google Scholar]
- 2.Jackson AC. 2013. Therapy of human rabies, p 575–589. In Jackson AC. (ed), Rabies, 3rd ed Academic Press, London, United Kingdom. [Google Scholar]
- 3.Hampson K, Coudeville L, Lembo T, Sambo M, Kieffer A, Attlan M, Barrat J, Blanton JD, Briggs DJ, Cleaveland S, Costa P, Freuling CM, Hiby E, Knopf L, Leanes F, Meslin FX, Metlin A, Miranda ME, Müller T, Nel LH, Recuenco S, Rupprecht CE, Schumacher C, Taylor L, Vigilato MAN, Zinsstag J, Dushoff J. 2015. Estimating the global burden of endemic canine rabies. PLoS Negl Trop Dis 9(4):e0003709. doi: 10.1371/journal.pntd.0003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.William HW, Conzelmann KK. 2013. Rabies virus, p 17–60. In Jackson AC. (ed), Rabies, 3rd ed Academic Press, London, United Kingdom. [Google Scholar]
- 5.Morin B, Liang B, Gardner E, Ross RA, Whelan SPJ. 2017. An in vitro RNA synthesis assay for rabies virus defines ribonucleoprotein interactions critical for polymerase activity. J Virol 91:e01508-16. doi: 10.1128/JVI.01508-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Finke S, Mueller-Waldeck R, Conzelmann KK. 2003. Rabies virus matrix protein regulates the balance of virus transcription and replication. J Gen Virol 84:1613–1621. doi: 10.1099/vir.0.19128-0. [DOI] [PubMed] [Google Scholar]
- 7.Bauer A, Nolden T, Nemitz S, Perlson E, Finke S. 2015. A dynein light chain 1 binding motif in rabies virus polymerase L protein plays a role in microtubule reorganization and viral primary transcription. J Virol 89:9591–9600. doi: 10.1128/JVI.01298-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morimoto K, Akamine T, Takamatsu F, Kawai A. 1998. Studies on rabies virus RNA polymerase: 1. cDNA cloning of the catalytic subunit (L protein) of avirulent HEP-Flury strain and its expression in animal cells. Microbiol Immunol 42:485–496. doi: 10.1111/j.1348-0421.1998.tb02314.x. [DOI] [PubMed] [Google Scholar]
- 9.Tao L, Ge J, Wang X, Zhai H, Hua T, Zhao B, Kong D, Yang C, Chen H, Bu Z. 2010. Molecular basis of neurovirulence of Flury rabies virus vaccine strains: importance of the polymerase and the glycoprotein R333Q mutation. J Virol 84:8926–8936. doi: 10.1128/JVI.00787-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ogino M, Ito N, Sugiyama M, Ogino T. 2016. The rabies virus L protein catalyzes mRNA capping with GDP polyribonucleotidyltransferase activity. Viruses 8(5):144. doi: 10.3390/v8050144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tian D, Luo Z, Zhou M, Li M, Yu L, Wang C, Yuan J, Li F, Tian B, Sui BK, Chen H, Fu ZF, Zhao L. 2016. Critical role of K1685 and K1829 in the large protein of rabies virus in viral pathogenicity and immune evasion. J Virol 90:232–244. doi: 10.1128/JVI.02050-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poch O, Blumberg BM, Bougueleret L, Tordo N. 1990. Sequence comparison of five polymerases (L proteins) of unsegmented negative-strand RNA viruses: theoretical assignment of functional domains. J Gen Virol 71:1153–1162. doi: 10.1099/0022-1317-71-5-1153. [DOI] [PubMed] [Google Scholar]
- 13.Schnell MJ, Conzelmann KK. 1995. Polymerase activity of in vitro mutated rabies virus L protein. Virology 214:522–530. doi: 10.1006/viro.1995.0063. [DOI] [PubMed] [Google Scholar]
- 14.Li J, Rahmeh A, Morelli M, Whelan SPJ. 2008. A conserved motif in region V of the large polymerase proteins of nonsegmented negative-sense RNA viruses that is essential for mRNA capping. J Virol 82:775–784. doi: 10.1128/JVI.02107-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neubauer J, Ogino M, Green TJ, Ogino T. 2016. Signature motifs of GDP polyribonucleotidyltransferase, a non-segmented negative strand RNA viral mRNA capping enzyme, domain in the L protein are required for covalent enzyme-pRNA intermediate formation. Nucleic Acids Res 44:330–341. doi: 10.1093/nar/gkv1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rahmeh A, Li J, Kranzusch PJ, Whelan SPJ. 2009. Ribose 2′-O methylation of the vesicular stomatitis virus mRNA cap precedes and facilitates subsequent guanine-N-7 methylation by the large polymerase protein. J Virol 83:11043–11050. doi: 10.1128/JVI.01426-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma Y, Wei Y, Zhang X, Zhang Y, Cai H, Zhu Y, Shilo K, Oglesbee M, Krakowka S, Whelan SPJ, Li J. 2014. mRNA cap methylation influences pathogenesis of vesicular stomatitis virus in vivo. J Virol 88:2913–2926. doi: 10.1128/JVI.03420-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruedas JB, Perrault J. 2014. Putative domain-domain interactions in the vesicular stomatitis virus L polymerase protein appendage region. J Virol 88:14458–14466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bankamp B, Kearney SP, Liu X, Bellini WJ, Rota PA. 2002. Activity of polymerase proteins of vaccine and wild-type measles virus strains in a minigenome replication assay. J Virol 76:7073–7081. doi: 10.1128/JVI.76.14.7073-7081.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feller JA, Smallwood S, Skiadopoulos MH, Murphy BR, Moyer SA. 2000. Comparison of identical temperature-sensitive mutations in the L polymerase proteins of Sendai and parainfluenza3 viruses. Virology 276:190–201. doi: 10.1006/viro.2000.0535. [DOI] [PubMed] [Google Scholar]
- 21.Trunschke M, Conrad D, Enterlein S, Olejnik J, Brauburger K, Mühlberger E. 2013. The L-VP35 and L-L interaction domains reside in the amino terminus of the Ebola virus L protein and are potential targets for antivirals. Virology 441:135–145. doi: 10.1016/j.virol.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu X, Cheng J, Xue J, Jin J, Song Y, Zhao J, Zhang G. 2017. Roles of the polymerase-associated protein genes in Newcastle disease virus virulence. Front Microbiol 8:161. doi: 10.3389/fmicb.2017.00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruedas JB, Perrault J. 2009. Insertion of enhanced green fluorescent protein in a hinge region of vesicular stomatitis virus L polymerase protein creates a temperature-sensitive virus that displays no virion-associated polymerase activity in vitro. J Virol 83:12241–12252. doi: 10.1128/JVI.01273-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Finke S, Conzelmann K-K. 1999. Virus promoters determine interference by defective RNAs: selective amplification of mini-RNA vectors and rescue from cDNA by a 3′ copy-back ambisense rabies virus. J Virol 73:3818–3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Conzelmann KK, Schnell M. 1994. Rescue of synthetic genomic RNA analogs of rabies virus by plasmid-encoded proteins. J Virol 68:713–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamada K, Ito N, Takayama-ito M, Sugiyama M, Minamoto N. 2006. Multigenic relation to the attenuation of rabies virus. Microbiol Immunol 50:25–32. doi: 10.1111/j.1348-0421.2006.tb03767.x. [DOI] [PubMed] [Google Scholar]
- 27.Matsumoto Y, Ohta K, Yumine N, Goto H, Nishio M. 2015. Identification of two essential aspartates for polymerase activity in parainfluenza virus L protein by a minireplicon system expressing secretory luciferase. Microbiol Immunol 59:676–683. doi: 10.1111/1348-0421.12329. [DOI] [PubMed] [Google Scholar]
- 28.Sleat DE, Banerjee AK. 1993. Transcriptional activity and mutational analysis of recombinant vesicular stomatitis virus RNA polymerase. J Virol 67:1334–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chenik M, Schnell M, Conzelmann KK, Blondel D. 1998. Mapping the interacting domains between the rabies virus polymerase and phosphoprotein. J Virol 72:1925–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ito N, Takayama M, Yamada K, Sugiyama M, Minamoto N. 2001. Rescue of rabies virus from cloned cDNA and identification of the pathogenicity-related gene: glycoprotein gene is associated with virulence for adult mice. J Virol 75:9121–9128. doi: 10.1128/JVI.75.19.9121-9128.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lahaye X, Vidy A, Pomier C, Obiang L, Harper F, Gaudin Y, Blondel D. 2009. Functional characterization of Negri bodies (NBs) in rabies virus-infected cells: evidence that NBs are sites of viral transcription and replication. J Virol 83:7948–7958. doi: 10.1128/JVI.00554-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ito N, Takayama-Ito M, Yamada K, Hosokawa J, Sugiyama M, Minamoto N. 2003. Improved recovery of rabies virus from cloned cDNA using a vaccinia virus-free reverse genetics system. Microbiol Immunol 47:613–617. doi: 10.1111/j.1348-0421.2003.tb03424.x. [DOI] [PubMed] [Google Scholar]
- 33.Ruffner DE, Stormo GD, Uhlenbeck OC. 1990. Sequence requirements of the hammerhead RNA self-cleavage reaction. Biochemistry 29:10695–10702. [DOI] [PubMed] [Google Scholar]
- 34.Yamaoka S, Ito N, Ohka S, Kaneda S, Nakamura H, Agari T, Masatani T, Nakagawa K, Okada K, Okadera K, Mitake H, Fujii T, Sugiyama M. 2013. Involvement of the rabies virus phosphoprotein gene in neuroinvasiveness. J Virol 87:12327–12338. doi: 10.1128/JVI.02132-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Minamoto N, Tanaka H, Hishida M, Goto H, Ito H, Naruse S, Yamamoto K, Sugiyama M, Kinjo T, Mannen K, Mifune K. 1994. Linear and conformation-dependent antigenic sites on the nucleoprotein of rabies virus. Microbiol Immunol 38:449–455. [DOI] [PubMed] [Google Scholar]
- 36.Masatani T, Ito N, Shimizu K, Ito Y, Nakagawa K, Sawaki Y, Koyama H, Sugiyama M. 2010. Rabies virus nucleoprotein functions to evade activation of the RIG-I-mediated antiviral response. J Virol 84:4002–4012. doi: 10.1128/JVI.02220-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Katoh K, Misawa K, Kuma K, Miyata T. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suzuki Y, Gojobori T, Nei M. 2001. ADAPTSITE: detecting natural selection at single amino acid sites. Bioinformatics 17:660–661. doi: 10.1093/bioinformatics/17.7.660. [DOI] [PubMed] [Google Scholar]