

ABSTRACT
The discovery that adeno-associated virus 2 (AAV2) encodes an eighth protein, called assembly-activating protein (AAP), transformed our understanding of wild-type AAV biology. Concurrently, it raised questions about the role of AAP during production of recombinant vectors based on natural or molecularly engineered AAV capsids. Here, we show that AAP is indeed essential for generation of functional recombinant AAV2 vectors in both mammalian and insect cell-based vector production systems. Surprisingly, we observed that AAV2 capsid proteins VP1 to -3 are unstable in the absence of AAP2, likely due to rapid proteasomal degradation. Inhibition of the proteasome led to an increase of intracellular VP1 to -3 but neither triggered assembly of functional capsids nor promoted nuclear localization of the capsid proteins. Together, this underscores the crucial and unique role of AAP in the AAV life cycle, where it rapidly chaperones capsid assembly, thus preventing degradation of free capsid proteins. An expanded analysis comprising nine alternative AAV serotypes (1, 3 to 9, and rh10) showed that vector production always depends on the presence of AAP, with the exceptions of AAV4 and AAV5, which exhibited AAP-independent, albeit low-level, particle assembly. Interestingly, AAPs from all 10 serotypes could cross-complement AAP-depleted helper plasmids during vector production, despite there being distinct intracellular AAP localization patterns. These were most pronounced for AAP4 and AAP5, congruent with their inability to rescue an AAV2/AAP2 knockout. We conclude that AAP is key for assembly of genuine capsids from at least 10 different AAV serotypes, which has implications for vectors derived from wild-type or synthetic AAV capsids.
IMPORTANCE Assembly of adeno-associated virus 2 (AAV2) is regulated by the assembly-activating protein (AAP), whose open reading frame overlaps with that of the viral capsid proteins. As the majority of evidence was obtained using virus-like particles composed solely of the major capsid protein VP3, AAP's role in and relevance for assembly of genuine AAV capsids have remained largely unclear. Thus, we established a trans-complementation assay permitting assessment of AAP functionality during production of recombinant vectors based on complete AAV capsids and derived from any serotype. We find that AAP is indeed a critical factor not only for AAV2, but also for generation of vectors derived from nine other AAV serotypes. Moreover, we identify a new role of AAP in maintaining capsid protein stability in mammalian and insect cells. Thereby, our study expands our current understanding of AAV/AAP biology, and it concomitantly provides insights into the importance of AAP for AAV vector production.
KEYWORDS: AAP, AAV, capsid assembly, adeno-associated virus, assembly-activating protein, parvovirus
INTRODUCTION
Fifty years after their discovery (1), adeno-associated viruses (AAVs), which are small, nonpathogenic members of the family Parvoviridae and the genus Dependovirus, have become stars among all viral vectors that are currently in development and use for human gene therapy (2). Their attractiveness is partly due to (i) the existence of multiple natural serotypes with broad tissue tropism and (ii) the ease with which the AAV capsid can be engineered. This allows repurposing of the capsid and enhancement of essential vector properties, such as cell specificity or evasion of neutralizing antibodies that preexist in humans or are induced by vector administration. The AAV capsid consists of 60 viral proteins (VPs) which form an 18- to 26-nm icosahedral particle with T=1 symmetry (3) that encapsidates a 4.7-kb-long, single-stranded viral DNA. The three proteins VP1, VP2, and VP3 are represented in the capsid in an approximate ratio of 1:1:10, which is determined by the RNA splicing frequency and by varying translation efficiencies due to alternative start codons within the AAV cap gene. To modify and improve the AAV capsid for gene therapy, a wide variety of techniques are available, from ancestral reconstruction and peptide display to directed molecular evolution (recently reviewed in references 2 and 4). Among the latter, a very powerful and versatile approach is DNA family shuffling, i.e., creation of chimeric AAV capsid sequences via cap gene fragmentation and reassembly based on partial homologies. In 2008, this technique was introduced into the AAV field by three independent groups (5–7). The first was Mark Kay's team, who reported AAVDJ, a shuffled hybrid of AAV serotypes 2, 8, and 9 that possesses high efficiency in the liver and other cell types (5). Since then, numerous other laboratories have harnessed this technology to enrich further novel AAV capsids with improved properties in, for instance, muscle, central nervous system (CNS), stem cells, or eye (see, e.g., references 8–16).
Despite this imposing track record, a 2010 study by the Kleinschmidt group (17) raised questions about a potential inherent drawback of molecular AAV capsid evolution technologies including DNA shuffling. In this seminal work, Sonntag et al. identified a previously overlooked protein of 23 to 26 kDa that is encoded in the second open reading frame (ORF) of the AAV2 cap gene and that overlaps with the VP2 and VP3 N termini. It uses a nonconventional CTG start codon that is embedded between the weak ACG start codon of VP2 and the strong VP3 ATG, suggesting a temporally coordinated expression pattern of AAP and VP proteins. As shown in the original work and solidified in two follow-up studies from the same group (18, 19), this protein plays a pivotal role in the assembly of AAV2 capsids; hence, it was dubbed assembly-activating protein (AAP). While the exact mechanisms have yet to be elucidated, it was proposed that AAP promotes translocation of AAV2 VP3 proteins to the nucleolus, where AAV2 capsid assembly occurs. In line with this, in 2015, Earley et al. identified multiple basic regions in the AAP2 C terminus that function as nuclear and/or nucleolar localization signals (20). Further data from Naumer et al. suggest that AAP induces a conformational alteration in VP3, indicating a function as scaffold that nucleates AAV capsid assembly (18). Deletions of either the hydrophobic AAP N terminus or of the C terminus of VP3 impact AAV2 capsid assembly, implying that these two domains mediate direct AAP-VP interaction. Still, it also remains possible that a key function of AAP is promotion of nucleolar VP transport to facilitate interaction with nucleolar proteins which in turn chaperone AAV capsid assembly. Arguing against this, at least as an exclusive mechanism, is that assembly of AAV serotypes other than AAV2 can occur outside the nucleolus and that AAP-independent targeting of AAV2 VP3 to the nucleolus via appropriate peptides did not foster AAV2 capsid assembly (17, 21).
As mentioned above, the discovery of AAP and of its critical role in the AAV life cycle could have major implications for AAV vector evolution via capsid modification, for two reasons. First, AAP is not unique to AAV2 but is well conserved among AAV serotypes 1 to 13, and homologous AAPs also exist in other dependoparvoviruses (but not autonomous parvoviruses) (19). Second, the AAP ORF fully overlaps that of the VP proteins in all AAV serotypes, implying that diversification of the VP ORF may inadvertently also yield nonnatural AAP proteins. If their function was impaired, this would hamper production of AAV capsid libraries and thereby limit AAV vector evolution. Curiously, although Sonntag and colleagues had implied this possibility in their original study (17), experimental confirmation is lacking to date. In fact, it remained unclear whether AAP is crucial for AAV vector production in mammalian cells at all, as nearly all data published thus far have been obtained with artificial AAV virus-like particles that were composed of only the major capsid protein VP3.
The role of AAP is similarly dubious in the case of a widely used alternative AAV production system, namely, insect cells and baculovirus expression vectors (BEV). In this system, which was first described by the Kotin group in 2002 (22), baculovirus promoters replace the native AAV promoters, resulting in modified regulation of rep and cap gene expression. The original protocol was further improved in 2009 by combining rep and cap genes in a single BEV, using two strong baculovirus late promoters (polyhedrin and P10, respectively) for their expression (23). Furthermore, the ATG codon of VP1 was mutated to ACG in order to produce VP1-VP2-VP3 proteins in insect cells in a stoichiometry close to 1:1:10. Owing to these and other optimizations (24, 25), insect cells can now be exploited to produce fully functional AAV particles with high yields, which makes the system interesting for large-scale vector manufacturing. Nonetheless, until now, the function of AAP during the assembly of recombinant AAV particles in insect cells has not been investigated. In fact, it is not even known whether AAP is expressed at all in this heterologous context and whether it is required for correct particle assembly.
Consequently, our aim in this study was to begin to dissect the still-enigmatic role of AAP for generation of recombinant AAV vectors in mammalian and insect cells, based on a panel of serotypes comprising AAV2 and nine others. To this end, we derived two sets of mammalian expression constructs from these 10 AAV serotypes in which we either knocked out AAP in a helper plasmid context or overexpressed each AAP from a strong promoter, respectively. In parallel, we also constructed BEV either lacking or overexpressing AAP2. Below, we describe our analysis of the ability of the different helper plasmids and viruses to produce AAV vectors in mammalian or insect cells in the presence or absence of AAP. The consistency of our data across two heterologous AAV vector manufacturing systems implies that our findings are broadly relevant for the understanding of AAP's role in the biology of wild-type and recombinant AAVs. Moreover, our findings on the interchangeability of AAPs between AAV serotypes have far-ranging consequences for work with synthetic AAV capsids derived by molecular evolution.
RESULTS
AAP is critical for AAV vector production and for VP protein stability.
Previous work has almost exclusively studied the role of AAP in AAV capsid assembly in the context of virus-like particles that were composed of merely the major capsid protein VP3 (17–21). In contrast, the central aim of the present work was to analyze AAP biology in mammalian and insect cells during production of recombinant AAV vectors based on complete viral capsids comprising all three capsid proteins, VP1 to VP3. To set up an appropriate assay system, we initially focused on the best-characterized AAV serotype 2 (AAV2) and created an AAV2 helper plasmid in which we knocked out AAP expression. To this end, we modified plasmid p5E18(2/2), comprising the AAV2 rep and cap genes but no inverted terminal repeats (ITRs) (26), by (i) changing the AAP start codon from CTG to CCG and (ii) introducing a stop codon 66 bp downstream of the modified start (Fig. 1A; see Data Sets S1 and S4 in the supplemental material for full sequences of wild-type and mutated AAV2 AAP, respectively). Importantly, these modifications did not change the AAV2 VP amino acid sequence in the resulting plasmid, AAV2mut.
FIG 1.

Generation of an AAP knockout AAV2 helper plasmid and recovery by AAP2 trans-complementation. (A) Modifications that were introduced into wild-type (wt) AAV2 cap to knock out AAP2. The AAP2 start codon CTG was changed to CCG (causing a switch from leucine [L] to proline [P]) and a stop codon inserted 66 bp downstream. Note that the protein sequences of VP1 to VP3 were not influenced by the mutations (not shown). (B) trans-complementation assay to measure rescue of the AAV2mut construct with different amounts of AAP2. (C) HEK293T cells were transfected in 6-well format to produce GFP-encoding AAV vectors (see Materials and Methods for details), using either a wild-type AAV2 helper (AAV2wt) or the AAP2 knockout mutant. The latter was supplemented with the indicated amounts of AAP2 expression plasmid per well. Equal amounts of crude cell lysates were subsequently used to transduce new HEK293T cells, and numbers of GFP-expressing cells were determined 48 h later via flow cytometry. Values were normalized to the AAV2wt control (set to 1.0). Shown are means from three independent biological replicates plus SD. Statistical analysis was by a one-way ANOVA with Bonferroni's multiple-comparison test. n.s., not significant; ***, P < 0.001. (D) Western blot analysis of cell lysates obtained 48 h after transfection as described for panel B. Next to the viral proteins VP1 to VP3, AAV Rep proteins as well as HA-tagged AAP2 were detected using the B1, 303.9, or anti-HA antibody, respectively. The spliced Rep variants Rep68 and Rep40 are typically much less abundant than Rep78 and Rep52. (E) Quantitative reverse transcription-PCR of all major AAV2 transcripts. RNA was isolated from HEK293T cells transfected as described in the text. Arrows indicate primer pairs. Shown are viral mRNA levels relative to those for wild-type AAV2 (black circles, set to 1). Data were calculated using the 2−ΔΔCT method and normalized to GAPDH as a housekeeper. Each symbol is from one experiment with three technical replicates. *, P < 0.05; **, P < 0.01.
Next, we investigated the ability of this AAP knockout construct to yield functional virions in mammalian cells by using it as a helper plasmid for production of recombinant AAV2 vector particles on a small scale. We therefore triple transfected human embryonic HEK293T cells with a green fluorescent protein (GFP)-encoding self-complementary AAV vector plasmid, an adenoviral helper construct, and either AAV2mut or, as positive control, the parental wild-type AAV2 helper plasmid (Fig. 1B). Two days later, cells were harvested into phosphate-buffered saline (PBS) and lysed by repeated freeze-thawing, and the resulting supernatants were used to transduce fresh HEK293T cells. Percentages of GFP-expressing cells were then used as a surrogate marker for vector production. Notably, the AAV2mut construct failed to yield any detectable functional AAV2 vector particles (Fig. 1C, compare first two data points). This suggested a critical role of AAP in AAV vector production, congruent with and extending previous data with VP3-only capsids (17–21).
To study whether the lack of AAV2 AAP (here called AAP2) could be complemented in trans, an AAP2 expression plasmid was generated. To this end, the AAP2 start codon was changed from CTG to ATG, and the cDNA was inserted behind a cytomegalovirus (CMV) promoter and N-terminally fused with a hemagglutinin (HA) tag. The functionality of the resulting plasmid was then assessed in small-scale vector productions as described above, except that the AAP2 expression plasmid was added as a fourth construct in increasing amounts (Fig. 1B). Notably, the smallest amount of AAP2 plasmid (0.063 μg) (light gray bar in Fig. 1C) already sufficed to rescue the production of infectious vector particles from the AAV2mut plasmid to roughly 50% of that for the wild-type AAV2 helper. Under our assay conditions, the efficiency of rescue by AAP2 trans-complementation was dose dependent, reaching a maximum at 0.5 μg AAP2 DNA, where the numbers of transduced and thus GFP-positive cells matched those for the wild-type AAV2 control. These findings were further validated by cesium chloride gradient-based segregation of functional or assembled AAV2 capsids from a large-scale preparation, which verified the inability of AAV2mut to produce AAV vector particles and showed potent rescue by AAP2 overexpression (data not shown).
Surprisingly, Western blot analysis of cell lysates from the samples for Fig. 1C revealed a pronounced reduction in the amounts of all three capsid proteins (VP1, VP2, and VP3) expressed from AAV2mut compared to the wild-type AAV2 control (Fig. 1D, top gels). This loss of expression could be restored to wild-type levels upon AAP2 complementation, again in a dose-dependent manner akin to the functional data in Fig. 1C. The AAP-dependent effect was specific for the VP proteins, since levels of Rep proteins remained constant under all conditions (Fig. 1D, middle gels). Expression of increasing amounts of HA-tagged AAP2 was confirmed using an anti-HA tag antibody (Fig. 1D, bottom gel).
To begin to dissect the mechanisms underlying the loss or increase in VP proteins, we analyzed levels of all major AAV mRNA transcripts. We therefore isolated total mRNA from HEK293T cells again transfected with all plasmids needed for AAV vector production, i.e., GFP vector, adenoviral helper, and wild-type or mutant AAV2 helper, with or without AAP2 supplied in trans. Quantitative reverse transcription-PCR (qRT-PCR) revealed only minor, mostly nonsignificant differences in the levels of rep or cap mRNAs after knockout or supplementation of AAP2, relative to those with the wild-type AAV2 control (Fig. 1E). This suggested that AAP2 knockout affects neither transcription nor steady-state levels of cap mRNAs and in turn implied that (de)stabilization occurs on the VP protein level.
AAP2 is also crucial for AAV2 vector production and VP protein stability in insect cells.
The finding that AAP2 not only controls AAV2 particle production but also affects levels of capsid proteins in transfected mammalian cells was unexpected. We therefore sought to determine whether these two phenotypes could be recapitulated in a heterologous AAV2 vector production system, namely, dual-baculovirus infection of insect cells. For this purpose, we introduced the exact same mutations that had resulted in the AAP2 knockout in our mammalian AAV2 helper plasmid into a recombinant baculovirus expressing AAV2 VP1 to VP3 and Rep78/52, yielding BEV-Rep2Cap2-AAP2mut (Fig. 2A). AAV2 vectors were then produced in spinner flasks after infection of Sf9 insect cells with two BEV: (i) BEV-AAV carrying an enhanced GFP (eGFP) or an alpha-1-antitrypsin (A1AT) transgene flanked by AAV2 ITR sequences and (ii) BEV-Rep2Cap2 expressing AAV2 Rep78/52 and the three VP proteins, with the AAP2 ORF either being intact (AAV2wt) or carrying the knockout mutations (AAV2mut). Interestingly, assembled AAV2 particles were undetectable by enzyme-linked immunosorbent assay (ELISA) in crude Sf9 cell lysates in the absence of AAP2 (Table 1). Additionally, a drastic, two-log drop in the number of vector genomes was observed at harvest when AAP2 was impaired (Fig. 2B, compare wt versus mut). We hypothesized that the low-level detection of AAV vector genomes with BEV-Rep2Cap2-AAP2mut (Fig. 2B, white bar) is partially due to contaminating baculoviral DNA in the crude cell lysates. Indeed, we measured an average of 2.65 × 107 BEV genome copies per ml in these samples (n = 3), using a qPCR targeting the baculoviral polymerase gene. Notably, identical to our observations in mammalian cells, we were unable to recover any functional AAV2 particles from insect cells without AAP after purification by cesium chloride density gradient ultracentrifugation (data not shown). Collectively, our results show that AAP2 knockout in the recombinant baculovirus Rep2Cap2 precludes capsid assembly and vector particle production in insect cells, validating and expanding our findings in mammalian cells.
FIG 2.

Role of AAP in AAV2 assembly in insect cells. (A) Generation of AAP2 knockout recombinant baculovirus. AAP2 was knocked out through mutation of the AAP2 start codon and insertion of a stop codon, impairing the AAP2 ORF but retaining the cap ORF, similarly to the case for AAP knockout AAV2 helper plasmid (Fig. 1A). BEV-Rep2Cap2-AAP2mut was generated as described in Materials and Methods using the Bac-to-Bac technology. (B) AAP2 trans-complementation and overexpression in the dual-baculovirus production system (n = 3). Sf9 cells were coinfected with BEV-AAV and BEV-RepCap at an MOI of 0.05 per baculovirus. Conditions “wt” and “mut” correspond to coinfection with BEV-eGFP-Puro and BEV-Rep2Cap2 or BEV-Rep2Cap2-AAPmut, respectively. Condition “trans” corresponds to infection with BEV-eGFP-Puro-p10AAP2 expressing AAP2 in trans and BEV-Rep2Cap2-AAP2mut. For the “over” condition, Sf9 cells were infected with BEV-Rep2Cap2 and BEV-eGFP-Puro-p10AAP2. Vector genomes in crude cell lysates were quantified at harvest by free-ITR qPCR (see Materials and Methods). Statistical analysis was by a one-way ANOVA with Bonferroni's multiple-comparison test. n.s., not significant; ***, P < 0.001. (C) Time course of AAP2 and VP expression during AAV2 vector production in insect cells. Sf9 cells were coinfected with BEV-eGFP and BEV-Rep2Cap2 (wt) or BEV-Rep2Cap2-AAPmut (m) at an MOI of 0.05 per baculovirus. Western blotting was performed from 10 μg of total proteins extracted from cell pellets of uninfected (c, control) or infected Sf9 cells recovered every 24 h postinfection. AAP2 or VP proteins were detected using the anti-AAP2 or polyclonal anti-VP antibodies, respectively. The asterisks indicate high-molecular-weight (*) or low-molecular-weight (**) signals from the anti-VP antibody; the latter may represent degraded VP proteins. (D) Time course of AAP and VP expression during AAV2 vector production with AAP2 overexpression. Sf9 cells were infected with BEV-Rep2Cap2 and BEV-eGFP-Puro (wt) or BEV-eGFP-Puro-p10AAP2 (o, overexpression) at an MOI of 0.05 per baculovirus. Western blotting was performed, and AAP2 and VP proteins were detected, as for panel C. SDS-polyacrylamide gel loading and BEV infection were verified with anti-GAPDH and anti-GP39 antibodies, respectively. (E) Detection of B1 antibody-reactive, high-molecular-weight bands in mammalian cells expressing AAV2 VP proteins in the absence of AAP2. Shown is a representative Western blot analysis of lysates from HEK293T cells transfected and treated as for Fig. 4B, left panel. The blot was overexposed to better illustrate the high-molecular-weight signal (asterisk) that is most pronounced in the AAV2mut sample in the absence of AAP2 (third lane).
TABLE 1.
Quantification of AAV2 vector genomes and assembled particles in crude lysates from BEV-infected insect cellsa
| AAV2 | Batch | Transgene | No./ml |
|
|---|---|---|---|---|
| Vector genomes | Particles | |||
| AAV2wt | 1 | eGFP | 2.84 × 1010 | 1.22 × 1011 |
| 2 | eGFP | 2.58 × 1010 | 7.83 × 1010 | |
| 3 | A1AT | 8.60 × 109 | 1.06 × 1011 | |
| Median (IQR) | 2.58 × 1010 (0.99)* | 1.06 × 1011 (0.16) | ||
| AAV2mut | 4 | eGFP | 1.84 × 108 | ND |
| 5 | eGFP | 1.90 × 108 | ND | |
| 6 | A1AT | 1.39 × 108 | ND | |
| Median (IQR) | 1.84 × 108 (0.26)* | NA | ||
Sf9 cells were coinfected with BEV-AAV carrying an eGFP (n = 2) or A1AT (n = 1) transgene and BEV-Rep2Cap2 expressing Rep78/52 and wild-type AAP2 (AAV2wt) or mutated AAP2 (AAV2mut) at an MOI of 0.05 per baculovirus. Crude cell lysates were prepared at 96 h postinfection after Triton cell lysis and low-speed centrifugation. Vector genomes and total particles were quantified at harvest by free-ITR qPCR and A20 ELISA, respectively. IQR, interquartile range; ND, not detected (AAV2 ELISA limit of detection, 4.53 × 108 particles); NA, not applicable. *, P < 0.05. Statistical analysis was by a one-tailed Mann-Whitney test.
From these results, we anticipated that AAP trans-complementation will be required in insect cells for production of AAV variants whose AAP gene is disrupted, e.g., as a consequence of AAV capsid gene shuffling. To verify this assumption, we cloned AAP2 into BEV-AAV downstream of the strong baculoviral P10 promoter and upstream of the AAV vector cassette. This design was chosen based on reports that the dual-BEV infection system is more efficient than a triple-BEV system for AAV vector production (23). As shown in Fig. 2B (compare “trans” to the first two bars), AAV2 vector yields from the AAP2-deficient BEV-Rep2Cap2 were indeed fully restored by BEV-AAV-p10AAP2 coinfection. This demonstrated the feasibility to efficiently trans-complement AAP in insect cells, akin to our findings in mammalian cells.
Based on our finding that AAP2 knockout led to a reduction of intracellular VP levels in mammalian cells (Fig. 1D), we also analyzed VP protein expression during AAV2 vector production in insect cells in the presence or absence of AAP2. Therefore, Sf9 cell pellets were collected after dual-BEV infection every day, and total protein was extracted and subjected to detection of AAP and VP proteins by Western blotting (Fig. 2C). Time course experiments showed that AAP2 was expressed from the AAV2 cap ORF as early as 24 h after BEV infection and reached a maximum at 48 h. AAP2 expression then declined at 72 h and became undetectable after 96 h, indicative of a temporally orchestrated expression and turnover of AAP2 during AAV2 vector production in insect cells. Proteins VP1 to VP3 were also expressed from the AAP2-deficient BEV but eventually declined, culminating in almost complete loss of VP-specific signals in the absence of AAP at harvest time (96 h postinfection). Taking the results together, we were able to confirm the control of AAV2 VP levels by AAP2 in two independent AAV production systems, mammalian cells (Fig. 1C and D) and insect cells (Fig. 2B and C).
To study whether, vice versa, AAV2 vector production can be boosted in insect cells by AAP2 overexpression or whether it would also be saturated by wild-type AAP2 as in mammalian cells (Fig. 1C and D), BEV-AAV-p10AAP2 was used for infection of Sf9 cells in combination with BEV-Rep2Cap2-AAP2wt. In this scenario, AAP2 is expressed from both baculoviruses, which should result in AAP2 overexpression, as confirmed by Western blotting (Fig. 2D). Bar “over” in Fig. 2B illustrates that excessive amounts of AAP2 did not improve AAV vector production at harvest, implying that AAP2 expression is rate-limiting in the baculovirus/AAV system but can also become saturated.
Interestingly, in insect cells infected with AAP2-depleted BEV-Rep2Cap2, we noted high-molecular-weight signals (marked with an asterisk in Fig. 2C) that were reminiscent of those observed in mammalian cells transduced with AAV vectors (27–29) and thus were possibly related to capsid protein ubiquitination. To test whether we could also find similar bands in mammalian cells, we repeated the experiment for Fig. 1D but now overexposed the Western blots. Indeed, as exemplified in Fig. 2E (also marked with an asterisk), we detected additional high-molecular-weight signals with the B1 antibody, which were most pronounced for the AAV2mut construct in the absence of extra AAP2. While additional studies are needed to identify the exact nature of these bands, we cautiously interpret their specific appearance in the absence of AAP as hints that posttranslational modification, possibly ubiquitination, may be involved in capsid protein destabilization during AAV vector production in insect and mammalian cells.
As a whole, our data demonstrate that in both mammalian and insect cells, (i) AAP2 is expressed from the AAV2 cap gene in similar manners, (ii) AAP2 is strictly required for AAV2 capsid assembly and vector particle production, (iii) AAP2 knockout phenotypes can be restored by AAP2 trans-complementation, and (iv) AAP2 overexpression does not boost AAV2 vector production.
Free AAV VP proteins undergo rapid turnover but become stabilized via AAP2-mediated AAV2 particle formation.
The data at this point showed not only that AAP2 is required for formation of functional AAV2 vector particles in two heterologous systems but that its presence also correlates with the detection of VP protein signals by Western blotting. Of note, the latter technique cannot distinguish free versus assembled capsid proteins, due to the use of denaturing conditions. We thus specifically quantified free VP proteins in cells 48 h after transfection by immunostaining with the B1 antibody. Its epitope is localized on the inside of assembled AAV2 virions (30), thus preventing detection of capsids in immunofluorescence stainings.
Interestingly, this analysis revealed comparable numbers of VP-positive cells and also similar mean intensities of antibody staining in all cases, i.e., wild-type or mutant AAV2 helper, with or without AAP2 supplied in trans (Fig. 3A and B). No AAV2 capsid-specific A20 signals were detected when AAV2mut was transfected in the absence of extra AAP2 (Fig. 3B), indicating the complete lack of assembled AAV2 particles in cells devoid of AAP2, consistent with our findings in insect cells (Table 1). Together, these data suggested that cells transfected with an AAV2 helper genome contain a low steady-state level of free VP proteins, irrespective of AAP2's presence or absence, and that the increased VP signals seen in Western blot analysis (Fig. 1D and 2C) in the presence of AAP were derived mostly from assembled capsids.
FIG 3.

Determination of the half-lives of AAV2 VP proteins and of AAP2. (A and B) Quantification of AAV2 VP proteins (A) or assembled particles (B) in microscopy pictures (not shown) of cells stained with the B1 or A20 antibody at 48 h posttransfection. Six pictures per condition were analyzed with the CellProfiler program to determine percentages of positive cells or mean integrated intensities of stained cells. **, P < 0.001; ****, P < 0.0001; n.s., not significant. (C and D) Determination of the VP protein half-life in mammalian cells. HEK293T cells were transfected with the AAV2mut helper plasmid with or without the AAP2 expression plasmid. Cycloheximide (CHX) treatment was started at 24 h after transfection, and samples were collected at 0, 0.5, 1, 2, 3, and 5 h after CHX treatment. Part of the sample was processed for Western blot analysis, while the remainder underwent freeze-thaw cycles for nondenaturing dot blot analysis. Dot blots were hybridized with the B1 antibody to detect unassembled, free VP proteins. The signal of each dot was quantified with Image J software and normalized to the untreated (no CHX) condition at each time point. A technical duplicate was performed and analyzed for each biological replicate (n = 3). Plotted on the left in panel D are the means with the standard errors of the mean (SEM). The dashed line indicates a 50% signal reduction. Shown on the right in panel D are representative Western blots stained for total VP proteins (free and assembled) with the B1 antibody. (E and F) Determination of the VP protein half-life in insect cells. Sf9 cells were infected with AAP2-depleted BEV-Rep2Cap2 and BEV-eGFP or BEV-eGFP-Puro-p10AAP2 (encoding AAP2) at an MOI of 0.05 per baculovirus. CHX chase started at 40 h after BEV infection, and samples were collected at 0, 0.5, 1, 2, 3, 5, and 8 h later. Native dot blotting and signal quantification were performed as described for HEK293T in panels C and D. The graph in panel F shows the means for three biological replicates with SD for each time point after normalization to the untreated condition (without CHX) and to t = 0 h. (G) Determination of the AAP2 half-life in insect cells. Sf9 cells were infected with BEV-Rep2Cap2 and BEV-eGFP at an MOI of 0.05 per baculovirus. CHX chase started at 40 h after BEV infection, and samples were collected at 0, 0.5, 1, 2, 3, 5, 8, 10, and 24 h later. The representative Western blots (from 10 μg of total proteins extracted from cell pellets at each time point) illustrate the rapid decline of AAP2 steady-state levels in the presence of CHX.
In turn, this implied that unassembled VP proteins may be unstable and undergo a rapid turnover. For experimental confirmation, we again transfected HEK293T cells as for Fig. 1B but now arrested protein translation by adding cycloheximide (CHX) 24 h after transfection (Fig. 3C). We then collected samples at various time points and used native dot blotting with the B1 antibody to detect free VP proteins or denaturing Western blotting for measurement of total VP proteins (free and assembled). As hypothesized, we found that free VP proteins are quickly turned over in mammalian cells, with an approximate half-life of 2 to 3 h (Fig. 3D, graph). These dot blot analyses also revealed that the intrinsic stability of free VP proteins is independent of AAP expression. This rapid loss of free VP proteins was further reflected in the Western blot analysis of cells treated with CHX but lacking AAP2 and thus unable to assemble capsids (Fig. 3D, upper left blot). In contrast, the decrease in free VP proteins was masked by the presence of assembled capsids when AAP was present in trans (Fig. 3D, two bottom blots). Likewise, total VP protein levels were stable in the absence of CHX, regardless of the presence or absence of AAP (Fig. 3D, two right blots).
Again, we made congruent observations in the insect cell system, where we also detected a rapid drop in free VP protein levels in cells infected with BEV-Rep2Cap2 encoding AAP2 or not and treated with CHX (Fig. 3E and F). In addition, the high stringency of our anti-AAP2 antibody in insect cell lysates allowed us also to study AAP2 stability in the presence of CHX. As exemplified in Fig. 3G, we found that AAP2 undergoes a rapid turnover also when protein synthesis is blocked. Collectively, our discoveries that both free AAV2 VP proteins and AAP2 are unstable in cells imply a very stringent timing of their expression and activity in the AAV life cycle and during vector production.
Artificial stabilization of free VP proteins is insufficient for particle production or VP trafficking.
The finding that free VP proteins are stabilized by AAP2-mediated capsid assembly raised the question whether, vice versa, artificially increasing free VP proteins beyond the low steady-state level would trigger assembly, even in the absence of AAP2. In other words, is a main function of AAP a direct stabilization of free VP proteins, and is the ensuing accumulation of assembled capsids only an indirect consequence of the elevated VP protein levels? To address these questions experimentally, we expanded on our hypothesis that free VP proteins might be degraded in the absence of AAP2 via the proteasome, as inferred from our data in Fig. 2C and E and from prior evidence for VP protein ubiquitination during AAV transduction (27–29). We thus repeated the small-scale vector productions but now added the proteasome inhibitor MG-132 at 5 h and/or 24 h after transfection to the cells (Fig. 4A). Indeed, Western blot analysis of cell lysates harvested at 48 h posttransfection showed a marked increase in VP levels expressed from the AAV2mut helper after MG-132 treatment compared to the same construct without MG-132 (Fig. 4B, third lane in each blot; also note that the left gel confirms the data in Fig. 1D). In fact, even without adding AAP2, VP protein amounts from AAV2mut became identical to those from wild-type AAV2 control after proteasome inhibition (5 and/or 24 h posttransfection). Taken together, these data and the extra B1 bands in insect cells, whose appearance coincided with progressive VP protein reduction (Fig. 2C), support the idea that free AAV2 capsid proteins are indeed ubiquitinated and degraded when AAP2 is absent.
FIG 4.

Evidence for proteasomal degradation of free VP proteins in the absence of AAP. (A) Workflow for AAV crude cell lysate production with MG-132 treatment at 5 and/or 24 h posttransfection. AAV, AAV2 helper plasmid expressing rep and cap; Ad, adenoviral helper plasmid; GFP, GFP-encoding AAV vector plasmid. (B) Western blot analysis of cell lysates obtained from the workflow in panel A. Cells were either left untreated (left blot) or incubated with MG-132 at 5 h (middle blot) and/or 24 h (right blot) after transfection. VP1 to VP3 proteins were detected with the B1 antibody. (C) Quantification of relative GFP expression in HEK293T cells at 48 h after transduction with AAV crude cell lysates (in principle, the same samples as for panel B). Positive cells were counted via flow cytometry and values normalized to the AAV2wt control without AAP2 (set to 1.0). Shown are means from three independent biological replicates plus SD. (D and E) Effect of proteasomal inhibition on AAP/VP/capsid localization. HEK293T cells were transfected with APP2mut (D) or cotransfected with AAP2mut and HA-tagged AAP2 (E) prior to staining with antibodies against free VP proteins (B1, green, upper panels) or assembled capsids (A20, green, lower panels) or DAPI (nuclei). The images illustrate the localization of AAP2 (red) and VP/capsid (B1 or A20, green) in the presence or absence of MG-132. Overlay images are shown on the right side of each panel. Arrowheads highlight examples of clear VP/capsid (B1/A20) and AAP2 colocalization in the nuclei of transfected cells. Insets show a higher magnification of selected areas. The images shown are maximum projections of deconvolved confocal optical sections spanning the entire volume of cells. All images are representative of three independent experiments. Scale bar, 5 μm; scale bar in insets, 2 μm.
Use of the lysates from the cells used for Fig. 4B for transduction of fresh cells showed that trans-complementation of AAV2mut with AAP2 had fully restored vector production (Fig. 4C), confirming our data in Fig. 1C. In contrast, none of the samples produced with AAV2mut in the absence of extra AAP2 contained detectable GFP-expressing vector particles, regardless of whether or not MG-132 was added during production (third bar in each of the graphs in Fig. 4C). We thus conclude that the mere increase in free VP proteins (even to wild-type levels [Fig. 4B]) achieved by inhibiting their proteasomal degradation was insufficient to trigger formation of infectious AAV2 vector particles.
We then asked whether inhibition of proteasomal degradation would affect VP protein localization to the nucleus and the nucleoli, where AAV2 capsid assembly occurs (30). To this end, we cotransfected HEK293T cells as before and then employed confocal microscopy to track AAP2 (via the HA tag), free VP proteins (B1 antibody), or assembled capsids (A20 antibody). As exemplified in Fig. 4D, B1 signals in cells transfected with the AAV2mut construct remained predominantly cytoplasmic, irrespective of the absence or presence of MG-132. As expected, A20 staining was negative due to the absence of capsid assembly without AAP2. When AAP2 was codelivered (Fig. 4E), the B1 signals showed a marked localization to nucleoli, which was again not influenced by MG-132 treatment. In the presence of AAP2, we also detected abundant A20 signals, indicating the expected assembly of intact capsids.
We thus conclude that although inhibition of the proteasome increases the levels of free VP proteins in cells, this does not suffice to promote AAP2-independent capsid assembly, nor does it affect VP protein localization in the cell. Instead, our data show that nuclear VP trafficking and VP1 to -3 particle formation strictly depend on the presence of AAP2.
AAP is also critical for production of vectors derived from nine alternative AAV serotypes and highly interchangeable.
Thus far, we had focused on wild-type AAV2 since AAP was originally discovered in this serotype (17), providing a firm basis to establish our assays. This now allowed us to pursue the major aim of this work, which was to unravel the role of AAP during production of AAV vectors derived from multiple natural serotypes and to study whether AAP is generally critical across AAV isolates. Curiously, data on non-AAV2 AAP proteins remain very sparse. In 2011, the Kleinschmidt group reported the capability of AAP1 or AAP5 to trans-complement AAV serotype 1, 2, or 5 (19). In the same study, AAP2 was also used to rescue capsid assembly from serotypes 1, 2, 5, 8, and 9. More recently, the Nakai group investigated the ability of AAP from AAV1 to AAV12 to support assembly of AAV serotypes 1 to 12 (21). Importantly, in nearly all of these prior experiments, virus-like particles were produced with VP3 alone, leaving the role of the different AAP variants in formation of genuine AAV capsids and functional vectors of multiple serotypes largely unclear.
To fill in this critical gap, we generated a set of nine additional AAVmut helper constructs based on serotypes 1, 3 to 9, and rh10, in which we knocked out AAP expression in a manner akin to our strategy for AAV2 (except for our AAV4 and AAV5 templates, which already contained AAP-debilitating mutations due to codon modification; see Materials and Methods for details and Data Set S4 in the supplemental material). In parallel, we cloned expression constructs for all nine corresponding AAP variants, also in a manner similar to that for the AAP2 plasmid (see Data Set S1 in the supplemental material). Correct and comparable expression of all AAP variants as well as successful AAP knockout (using serotypes 2, 8, and 9 as examples) were confirmed by Western blotting using an anti-HA antibody detecting an N-terminal HA tag that is present in all 10 AAP variants (Fig. 5A). Interestingly, we noted distinct sizes of the AAP variants despite their similar lengths (Data Set S1) as well as a double band for AAP8 and AAPrh10 (plus a faint lower band for AAP5), akin to the observation by Earley and colleagues for AAP10 (21).
FIG 5.

Broad interchangeability of AAP variants from multiple AAV serotypes. (A) Western blot analysis to confirm proper AAP expression from all constructs. HEK293T cells were transfected with the expression plasmids encoding AAP from AAV serotypes 1 to rh10 under control of a strong CMV promoter. AAP proteins were detected using an anti-HA antibody binding to an N-terminal HA tag present in all AAP expression constructs. The three lanes “mut” illustrate successful knockout of AAP following mutation of the start codon and introduction of a stop codon. To validate the effect of these changes, the mutated AAP sequences of AAV2, -8, and -9 (used as examples) were cloned into the identical expression plasmid as for all the wild-type AAPs. Note the complete absence of signals in these three lanes, verifying the full knockout. (B) trans-complementation assays in which AAP-deficient AAV1, AAV3 to -9, and AAVrh10 helpers were supplemented with their corresponding AAPs during vector production. The crude cell lysates produced were used for transduction of SF539 (AAV1, AAV3, AAV5 to -9, and AAVrh10) or MCF7 (AAV4) cells. Values were always normalized to the cognate wild-type (wt) controls (set to 1.0). (C) Cross-reactivities of 10 wild-type AAPs with the AAV2mut helper. The plasmid combinations shown were cotransfected to produce GFP-expressing vectors, which were then used to transduce HEK293T cells. Values were normalized to that for the wild-type AAV2 helper (set to 1.0). Shown are means from three independent biological replicates plus SD. Statistical analysis was by a one-way ANOVA with Bonferroni's multiple-comparison test (A/B). n.s., not significant; **, P < 0.01; ***, P < 0.001. (D) Sequence identity of AAPs of 10 different AAV serotypes. The heat map depicts the percentage of unique amino acids between a pair of two AAPs (see Materials and Methods for details). (E) trans-complementation of 10 AAP-depleted helper plasmids with 10 AAP variants. Shown are representative data from three independent crude lysate productions. In each row, the wild-type (wt) helper is set to 100%, and rescue potencies are color coded. Note that in some cases (‡), trans-complementation of the AAV3 mutant reached up to 150% of that of the wt helper.
Next, we tested whether like AAV2, serotypes 1, 3 to 9, and rh10 would also depend on their own AAP for AAV vector particle production. To do so, we again employed the trans-complementation assay, using the cognate AAP expression plasmid for rescue of each knockout mutant. Cell lysates were subsequently used to transduce cells that we had identified as susceptible for the various serotypes (see Materials and Methods), and the numbers of GFP-expressing cells were determined as a surrogate readout for production of functional vector particles.
Figure 5B exemplifies our finding that like AAV2, all of the other nine AAV serotypes also strictly depend on their cognate AAP for AAV vector production. Evidence is the highly significant drops in numbers of transduced cells and hence in vector production as soon as AAP was knocked out. The only exceptions were AAV serotypes 4 and 5, where the AAVmut constructs yielded transduction-competent vector particles even in the absence of AAP. However, values were about 10-fold lower than those obtained with the wild-type helpers, congruent with latest data from Earley et al. for AAV5 (21). Moreover, we observed that all nine serotypes could be efficiently trans-complemented with their corresponding AAPs, always restoring vector particle production to close-to-wild-type levels.
We next asked whether the ability of serotype-specific AAPs to rescue an AAP knockout mutant was indeed restricted to the cognate serotype or whether they could also cross-complement other AAP-depleted helper plasmids during vector production and, if so, to what degree. Therefore, we first cotransfected the AAV2mut helper with all 10 AAP variants (plus GFP vector and adenoviral helper) and then compared the activities of the resulting lysates to that of the wild-type AAV2 helper (Fig. 5C). Interestingly, eight of the 10 AAP variants (including AAP2) were capable of rescuing the AAP2 defect in the AAV2 helper by at least 45% under our assay conditions. The exceptions were again AAV serotypes 4 and 5, whose AAPs failed to cross-complement the AAP2 knockout helper plasmid, consistent with the poor homology of AAP4 and AAP5 with all other AAPs (Fig. 5D). Our finding, moreover, is in line with and extends data obtained by Naumer et al. (18) and Earley et al. (21), both of whom noted an inability of AAP5 to efficiently rescue assembly of AAV2 virus-like-particles composed of VP3.
We then expanded the analysis even further and tested all possible combinations of 10 AAP-depleted helpers and 10 AAP expression plasmids. From the data in Fig. 5E, several observations should be highlighted: (i) the vast majority of combinations of AAP-deficient helper and AAP expression plasmids resulted in functional vector particles, usually in a range comparable to that for the genuine helper-AAP pairs; (ii) AAP4 rescued only AAV4 and AAV5 and none of the other eight serotypes; (iii) similarly, AAP5 rescued AAV4 and AAV5, as well as AAV8 and AAV9, but was inert toward the remaining six serotypes; (iv) surprisingly, vice versa, the AAV4 and AAV5 AAP mutants were rescued by nearly all AAPs (possibly because they are less AAP dependent and tend to self-assemble), with the exception of the combination of AAV5 and AAP9; (v) rescue of AAVrh10 failed with four of the 10 AAPs, again including AAP9 (akin to the AAV5-AAP9 pair [see point iv]); and (vi) production of AAV3 vectors was enhanced by roughly 1.5-fold through coexpression of AAV3mut and AAP from AAV2, AAV3, AAV7, and AAVrh10 compared to the wild-type AAV3 helper. This is consistent with our data in Fig. 5B and may imply that AAV3 capsid assembly depends on the presence and the amount of AAP even more critically than that of the other serotypes.
Most AAV serotypes strictly require AAP for capsid assembly.
To corroborate these functional data with analyses of AAP-dependent assembly of physical particles derived from the different serotypes, we performed nondenaturing dot blotting (Fig. 6A). Using monoclonal antibodies that detect assembled capsids of AAV1 to -6, AAV8, or AAV9, we found that AAP trans-complementation of AAP knockout mutants efficiently recovered the formation of intact viral particles to wild-type levels, congruent with the transduction data in Fig. 5B. Also in line with these data, we noted that even in the absence of their cognate AAP, AAV4 and AAV5 gave pronounced signals with antibodies that bind to assembled AAV4 or AAV5 capsids, respectively (arrows in Fig. 6A). The same was observed for AAV1, albeit to a lesser extent, despite the absence of detectable functional particles (compare Fig. 5B). In the dot blot assay, we moreover aimed to detect free, nonassembled capsid proteins using the B1 antibody or a polyclonal anti-VP serum (for AAV4, as B1 does not bind AAV4 VP proteins). As expected, heat (95°C) denaturation of the samples produced with either the wild-type constructs or the mutants in the presence of extra AAP resulted in positive signals; vice versa, these were absent in the nondenatured samples. Together, these data imply that assembly of free VP proteins is very potent in the presence of AAP when it is expressed from the wild-type helper or supplied in trans. One exception may again be AAV4, for which we also detected traces of free VP proteins even in the presence of AAP4 (Fig. 6A and B) (although we cannot rule out the possibility that the polyclonal serum also weakly detects assembled capsids). A final striking observation was that heat denaturation of the AAV5 samples produced in the absence of AAP5 barely resulted in signals with the B1 antibody (which would have indicated the expected free AAV5 VP proteins) (arrowhead in Fig. 6A), not even under harsh denaturing conditions including SDS (Fig. 6B, arrowhead). Again, similar observations were made for AAV1 and AAV4 as well, but to a lesser extent (respective arrowheads in Fig. 6A). Possibly, in the absence of AAP5, AAV5 VP proteins can self-assemble into aggregates that expose the epitope of the ADK5 antibody (raised against assembled AAV5 capsids). However, unlike regular AAV5 capsids, these AAP5-independent structures seem to be highly resistant to various denaturing conditions (heat and SDS).
FIG 6.

Proof that AAP fosters assembly of capsids of multiple AAV serotypes. (A) Capsids of the eight AAV serotypes shown were produced by cotransfecting HEK293T cells with the corresponding wild-type or AAP-deficient (mutant) AAV helper, an AAV vector, and an adenoviral helper. Following freeze-thaw lysis at 3 days posttransfection, capsid-containing supernatants were spotted onto nitrocellulose membranes under nondenaturing conditions. In every other lane, the capsids were heat denatured via incubation at 95°C for 5 min. Intact capsids were detected with the ADK series of antibodies (ADK8 cross-reacts with AAV3) or A20 (for AAV2). For most of the shown AAV serotypes (AAV2, -3, -6, -8, and -9), lack of AAP prevented formation of assembled capsids. Arrows highlight exceptions, i.e., signals detected with the ADK antibodies for AAV1, -4, and -5 even in the absence of AAP. Note that low or no signals were detected with the VP serum or B1 antibody following heat denaturation of the same samples (arrowhead). The blots shown are representative of three independent experiments, which all gave similar results. (B) Further investigation of the three exceptions from panel A, i.e., AAV serotypes 1, 4, and 5. In lanes S, the samples were not only heated but additionally treated with SDS (see Materials and Methods for details). The arrowhead highlights the particular resistance of the AAP-independent AAV5 structures even to these harsh denaturing conditions. (C) Stabilization of free VP proteins by MG-132 treatment does not enhance capsid assembly. Capsid were produced, blotted, and detected as described for panel A. Instead of heat and/or SDS denaturation, samples in lane MG were derived from cells that were treated with the proteasome inhibitor MG-132 during vector production. At 5 and 24 h after transfection, the medium was changed to fresh medium containing MG-132, and 48 h later, supernatants from the lysed cells were analyzed. Note that AAV4 and AAV5, as well as, to a lesser extent, AAV1 (and AAV6), gave signals even in the absence of AAP, independently confirming the data in panels A and B. The blots shown are representative of two independent experiments, which gave similar results.
Furthermore, we expanded the proteasome inhibition experiments to the other serotypes as well. The dot blot results shown in Fig. 6C independently validate those in Fig. 6A and additionally illustrate that akin to the case for AAV2, mere stabilization of free VP proteins by inhibition of proteasomal degradation during vector production is generally not sufficient to promote AAV capsid assembly.
AAP proteins of different AAV serotypes show distinct intracellular localization patterns.
Next, we also extended the immunofluorescence analysis from Fig. 3A and B to the other serotypes. Congruent with the case for AAV2, we found that the other seven tested serotypes (AAV1, AAV3 to -6, and AAV8 and -9) also expressed a steady-state level of VP proteins even in the absence of AAP (Fig. 7, left columns). Moreover, consistent with the dot blot data in Fig. 6, we readily obtained positive signals with the ADK4 and ADK5 antibodies and, to a lesser extent, also with the ADK1 antibody (Fig. 7, right columns). In contrast, ADK/A20 signals were hardly detected for the other five serotypes in the absence of their AAP. We conclude from the sum of our data from the immunofluorescence, dot blot, and transduction analyses that not only AAV2 but at least 10 different natural AAV serotypes strictly require AAP for proper assembly of intact and infectious particles, albeit the extent of this dependency may vary, as implied by the AAV4 and AAV5 data.
FIG 7.

Further evidence for the varying dependence of AAV serotypes on AAP for assembly of capsid-like structures. Capsids of the eight AAV serotypes shown were produced by cotransfecting HEK293T cells with the AAP-deficient AAV helper constructs and an adenoviral helper plasmid. Shown are stainings with the B1 antibody or the polyclonal anti-VP serum (AAV4) to detect free VP proteins (left columns) or with antibodies detecting assembled capsids (A20 or ADK series; right columns). Note the strong signals with the ADK4 or ADK5 antibodies (detecting assembled AAV4 or AAV5 capsids, respectively), substantiating the evidence for AAP-independent assembly of capsid-like, ADK-reactive structures. Fewer but similarly prominent signals were obtained with ADK1 (detecting assembled AAV1 capsids), in line with the results from the dot blot analyses in Fig. 6.
The discovery that the serotype-specific AAPs differ in their activity (Fig. 5) encouraged us to study whether they also display unique cellular localization patterns, perhaps distinct from the nucleolar signals reported for AAP2 (17). So far, there is indirect evidence for serotype-specific AAP localization obtained by Sonntag et al. (19), who used monoclonal antibodies to localize assembled VP3 particles of serotypes 1, 2, 5, 8, and 9 in the presence of AAP2. The authors noted strong nucleolar capsid signals for AAV1 and AAV2 versus a mixture of nucleolar and nuclear signals for AAV8 and AAV9 (and none for AAV5, as trans-complementation with AAP2 failed, in line with our data). Later, Earley et al. (20) speculated that AAP5 might lack a nucleolar localization signal due to an overall low net positive charge and that AAV5 assembly might thus occur outside the nucleolus. Indeed, in a follow-up study, that group experimentally confirmed that AAP5 and also AAP9 exhibit decreased nucleolar association (21).
Here, we exploited our new AAP expression plasmids derived from 10 different AAV serotypes to independently investigate differences in intracellular AAP localization. This was facilitated by the fact that all 10 AAP cDNAs were N-terminally fused with an HA tag, permitting detection with an anti-HA antibody. Our transduction data had already verified that this tag does not impact AAP functionality, congruent with studies using different tags (AU1 [17] or FLAG [21]). Furthermore, when we compared AAP2 localization in HEK293T cells transfected with HA-tagged AAP2 or AAP2 C-terminally fused to GFP, using an anti-HA antibody or our polyclonal anti-AAP2 antibody for detection of the HA-AAP2 protein, we observed identical, predominantly nucleolar localization in all cases (data not shown). Of note, this pattern is consistent with data from the Kleinschmidt group (17). Altogether, this provided high confidence that N-terminal fusion of the HA tag affects neither functionality nor localization of AAP2 and thus encouraged us to harness this tag for studies on the localization of the nine other AAPs.
In a first experiment, we transfected HeLa cells with our 10 AAP expression plasmids and then detected AAP localization by immunostaining with the anti-HA antibody at 42 h posttransfection (Fig. 8A). AAP2 again displayed a very distinct nucleolar localization, consistent with observations for this serotype by the Kleinschmidt and Nakai groups (17, 19, 21). The other nine AAPs were also detected in the nucleus but showed various localization patterns, confirming and extending recent data reported by Earley et al. for a different collection of AAV serotypes (21). While AAP1, AAP3, and AAP7 resembled AAP2, a mixed nucleolar and nuclear distribution was observed for AAP4, AAP6, and AAPrh10. Conversely, AAP5, AAP8, and AAP9 localized mostly in the nucleoplasm, without apparent accumulation in the nucleoli.
FIG 8.

Intracellular localization of AAP proteins of 10 different AAV serotypes and correlation with VP expression and capsid formation. (A) Expression plasmids encoding AAPs of the AAV serotypes shown were transfected together with a corresponding AAP-deficient helper plasmid into HeLa cells. AAP was detected by staining with an anti-HA tag antibody at 42 h posttransfection. Nuclei were stained with Hoechst stain. Transfections and immunostainings were performed in LabTek format. Images were captured with a Leica TCS SP5 microscope. Scale bar, 20 μm. (B and C) HEK293T cells were cotransfected in LabTek slides with AAP-deficient helpers of the serotypes shown, together with expression plasmids encoding cognate HA-tagged AAP, a luciferase vector, and an adenoviral helper. Free VP proteins (B) or assembled capsids (C) were detected using B1/VP or the A20/ADK antibodies, respectively. AAP was stained with an anti-HA tag antibody, and nuclei were stained with Hoechst stain. Pictures (not shown) were taken with an automated Olympus microscope at 20 h or 42 h posttransfection. Shown are normalized cross correlation coefficients (NC) that were determined as a measure of colocalization of the red (HA) and green (VP [B] or capsids [C]) color channel. An NC value of −1 indicates complete disagreement, and +1 indicates complete overlap of the two stains.
In a second experiment, we then cotransfected HEK293T cells with the 10 different AAP expression constructs in the presence of the cognate AAV helper plasmids, as well as a luciferase-encoding vector and adenoviral helper plasmid, i.e., a typical setting for AAV vector production. AAP localization was detected by staining the cells 20 h or 42 h later with the anti-HA antibody, while the B1 antibody (binding nine of the 10 AAV serotypes used, except for AAV4 [31]) or anti-VP serum was used to stain free VP proteins. Moreover, as for Fig. 7, assembled capsids were detected where antibodies were available, i.e., ADK1 (AAV1), A20 (AAV2), ADK8 (AAV3 and AAV8), ADK4 to ADK6 (AAV4 to AAV6), and ADK9 (AAV9) (19). As summarized in Fig. 8B and C, we generally detected a low percentage of cells by wide-field microscopy that were concurrently positive for AAP and free or assembled capsid proteins, respectively. The results depended not only on the serotype but also on the time point analyzed. Particularly notable is AAV2, for which we noted the highest correlation of AAP and A20 signals (Fig. 8C) among all serotypes and at both time points, in line with our earlier data (compare the insets in Fig. 4E). Also interesting is that for all serotypes, we observed a trend toward reduced AAP and ADK/A20 correlation (Fig. 8C) at 42 h compared to the earlier time point, while the AAP-B1/VP correlation (Fig. 8B) remained constant or even increased at 42 h.
Collectively, these data show that (i) AAPs of different AAV serotypes exhibit unique intracellular localization patterns and (ii) AAP is infrequently codetected in cells with abundant expression of free VP proteins or that contain assembled AAV capsids; especially at later stages of vector production, the AAP-capsid correlation decreases.
DISCUSSION
Our aim in this work was to address a curious question that has lingered in the AAV field ever since it was raised in 2010 in a seminal study from the Kleinschmidt group (17): does AAP and the overlap of its ORF with that of the capsid proteins restrict the production of AAV vectors? If so, should one in turn be concerned that inadvertent disruption of AAP during molecular capsid evolution, e.g., via DNA family shuffling of AAV capsid genes, may perturb the functionality of the resulting synthetic vectors? While these questions appear to be simple and straightforward, finding answers is challenging, for several reasons. One is that the biology of AAP in the AAV life cycle is still largely enigmatic, as only a few studies have investigated its role as a scaffold, chaperone, and/or transporter for AAV particle formation (17–21). An additional complication is that all prior studies had used artificial AAV capsids that were composed merely of the major capsid protein VP3 expressed from a robust CMV promoter (17–21). This was based on data that VP3 subunits can, in principle, form virus-like particles that are morphologically indistinguishable from wild-type capsids (32, 33). Despite the simplicity and usefulness of the VP3 model system, drawbacks are that it properly reflects neither (i) endogenous VP protein levels in mammalian cells, due to the strong ectopic promoter used for VP3 expression, nor (ii) the interplay of AAP with the three individual capsid proteins or with genuine capsids composed of VP1 to VP3. Moreover, because the resulting, VP1-/VP2-depleted virions are noninfectious, the role of AAP in functionality of full particles containing AAV DNA (wild type or recombinant) could not be investigated. In fact, to our best knowledge, there are only two reported data sets from the last 6 years where AAP was studied in the context of a full-length AAV capsid gene, including Fig. 3 in reference 17. There, however, the underlying construct was a replication- and packaging-competent wild-type AAV2 plasmid, leaving the role of AAP in AAV vector production, the topic of our own study, unclear. The second data set is from Earley et al. (21), who recently showed that AAV serotype 5 can assemble physical and functional VP1 to -3 particles even in the absence of AAP5, albeit at much lower efficiency compared to when in its presence.
Consequently, a prerequisite in this work was to de novo assemble AAP-dependent helper constructs and to set up new model systems to study AAP activity. To this end, we not only modified AAV2 but also generated AAP-deficient helper plasmids based on nine other frequently used serotypes, AAV1, AAV3, AAV6 to -9, and AAVrh10. We then employed these to produce recombinant AAV vectors and subsequently applied a battery of assays to measure the degree of trans-complementation with homo- or heterologous AAPs during particle production. These assays comprised the quantification of numbers of transduced cells, which is a different readout for AAP activity than in previous studies, which primarily used ELISA-based titration of assembled capsids as the endpoint. That was imposed by the focus of these studies on virus-like VP3 particles that were noninfectious due to the lack of VP1 (and VP2), thus preventing the analysis of functional virions. Our analysis of functional particles was facilitated by the identification of three cell lines that are transducible by all 10 AAV serotypes that were used in this study, HEK293T (AAV2), MCF7 (AAV4), and SF539 (AAV1, AAV3, AAV5 to -9, and AAVrh10). We note that SF539, a human gliosarcoma cell line that is part of the NCI-60 panel, should also be useful beyond our work due to its high susceptibility to transduction with numerous AAV serotypes.
In addition, we comprehensively studied AAP-dependent expression of AAV genes and proteins by reverse transcription-PCR, Western blotting, dot blotting, and immunofluorescence, and we quantified capsid assembly by immunostaining with capsid-specific antibodies. Still, we acknowledge that there are additional qualitative and quantitative technologies that are useful to dissect AAP biology and that we will employ in future work, e.g., electron microscopy and capsid ELISAs for AAV1 to -3, AAV5, and AAV8 (19) and AAV9. In particular, high-resolution visualization technologies should help to unravel the nature of the ADK-reactive structures that we noted for AAV4 and AAV5 in the absence of AAP (Fig. 6 and 7).
In the meantime, our present combination of complementary assays has already yielded a series of important new observations that expand pioneering work by the Kleinschmidt and Nakai labs and thus enhance our understanding of AAP biology (Fig. 9). We particularly consider our comparative studies between mammalian and insect cells as highly informative, since, to the best of our knowledge, the role of AAP in the context of baculoviruses and insect cells has never been investigated. From the sum of our experiments in these two heterologous systems, we consider the following key findings as particularly relevant for AAV biology and AAV vector production.
FIG 9.
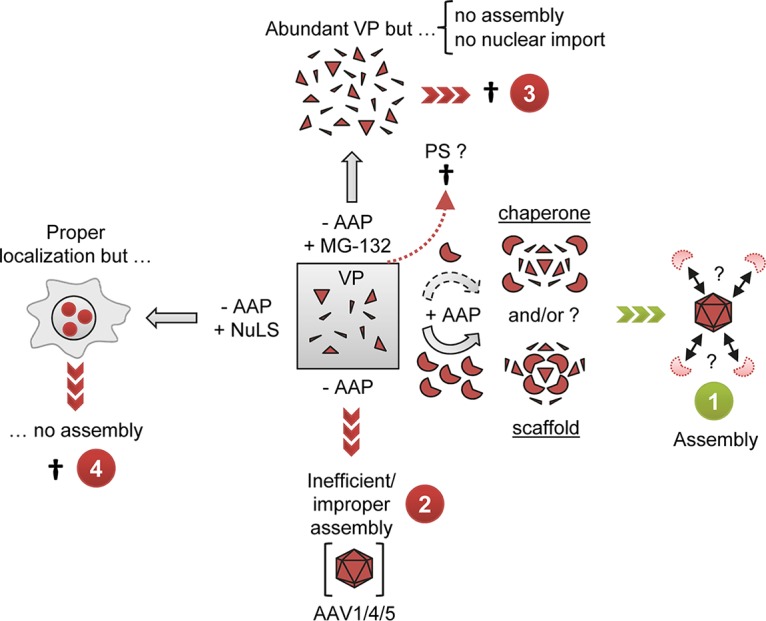
Model for the role of AAP and for its relationship with AAV capsid protein expression and assembly. Cells transfected with AAV helper plasmids contain a steady-state level of VP proteins (gray box), resulting from constant de novo expression and degradation likely involving the proteasome (PS). (1) When AAP is present, it acts in a dose-dependent manner as a chaperone and/or scaffold that mediates assembly of free VP proteins into capsids. Possibly, AAP is then degraded or displaced once capsid assembly has been completed. (2) Selected AAV serotypes, especially AAV4 and AAV5, are able to assemble capsid-like structures in the absence of AAP. However, this process is less efficient, and these structures also may differ from genuine capsids. (3) Inhibition of the proteasome, e.g., with MG-132, leads to an increase of free VP proteins but fosters neither capsid assembly nor nuclear/nucleolar trafficking. (4) Likewise, forced targeting of free VP proteins to the nucleolus via nucleolar localization signals (NuLS) also does not suffice to induce capsid assembly, as long as AAP is absent.
(i) Not only AAV2 but all 10 serotypes tested in this work critically depend on the presence of cognate AAPs for production of functional vector particles, experimentally validating prior notions that AAP is largely conserved across dependoparvoviruses (19). Moreover, our results independently verify recent data from Earley and colleagues (21), which they obtained mostly with VP3-only virions of multiple serotypes, and extend the overall conclusion to genuine AAV particles composed of VP1, VP2, and VP3.
(ii) The AAP ORF is also expressed in insect cells from cap-encoding constructs that are driven by the baculoviral P10 promoter.
(iii) For AAV2, the AAP dependence is equally pronounced in mammalian and insect cells. This supports the idea that AAP directly interacts with AAV capsid proteins, rather than translocating VP to specific cellular structures for interaction with other factors. Still, we note that we cannot formally exclude the existence of homologs of such factors in mammalian and insect cells.
(iv) AAP trans-complementation is dose dependent and saturable, implying that AAP is a rate-limiting factor but that overexpression will not boost AAV vector production. Evidence includes that in the dual-baculovirus/Sf9 production system, AAP expression driven by the strong P10 baculoviral promoter fully trans-complemented the mutant phenotype, but AAP overexpression did not improve vector genome yields. Likewise, excessive AAP expression did not boost production of individual serotypes that were already AAP competent in mammalian cells. An exception in our hands was AAV3, which seems to be more AAP dependent. Intriguingly, we observed an increase in AAV3 vector production only when the AAV3mut helper, and not the wild-type variant, was used and only when it was complemented with particular AAPs. While the reasons are unknown at this point, this observation may indicate a critical role of the AAP sequence and the levels and/or kinetics of AAP expression.
(v) AAP depletion results in a reduction of VP protein signals in Western blots (in both mammalian and insect cells) but not in immunofluorescence analyses (mammalian cells). Vice versa, AAP overexpression in trans fully restores VP steady-state levels from AAP-depleted helper plasmids. In line with this, Earley et al. have recently also documented an increase in VP levels when an AAP5-depleted helper construct was cotransfected with extra AAP5 (21). Moreover, we found that AAP knockout has no significant impact on cap mRNA levels. A unifying interpretation supported by our CHX chase experiments (Fig. 3C to F) is that free VP proteins are constantly produced but rapidly degraded in the cell unless AAP mediates their assembly into stable capsids. This can explain the dose-dependent increase in VP/B1 signals in Western blots from the AAV2mut plasmid in the presence of AAP2. We believe that this stems from VP stabilization through assembly; hence, the VP proteins detected by Western blotting in the presence of AAP2 were mostly derived from AAV2 particles that were denatured during sample preparation and thus regained reactivity with the B1 antibody. In contrast, they probably do not reflect higher total levels of free VP protein, because the latter would have been detected by B1 immunofluorescence analysis as well.
The conclusion that free VP proteins are rapidly turned over is further supported by our finding that proteasome inhibition during vector production with AAV2mut increased VP protein amounts to wild-type levels and triggered additional, high-molecular-weight bands. Together, these data suggest posttranslational modification, possibly ubiquitination, as a mechanism for degradation of free VP proteins, consistent with previous studies (27–29). These include seminal work from the Srivastava lab and others showing improved AAV transduction after elimination of surface tyrosines (34–36) that are phosphorylated and mediate capsid ubiquitination. Notably, the distinct high-molecular-weight bands detected here in MG-132-treated mammalian cells or in insect cells at 48 h after infection with the AAP-depleted BEV-Rep2Cap2 resemble the signals that Yan et al. observed for heat-denatured AAV particles (29). The former samples probably represented free VP proteins and were better substrates for ubiquitin conjugation than intact virions. To unravel whether the signals noted here truly reflect ubiquitinated VP proteins was beyond the scope of the present work and thus remains an interesting question for future investigations.
Finally, it is noteworthy that the MG-132-induced increase in VP protein levels did not result in intact vector particles for any of the serotypes studied here, implying that VP proteins do not spontaneously self-assemble and form infectious particles in cells once a certain steady-state level has been reached. Accordingly, we believe that the main function of AAP2 is not to directly stabilize VP proteins, because this alone would not suffice to foster particle assembly. Instead, a major function of AAP2 is probably to chaperone assembly of free VP proteins into capsids, which prevents ubiquitination and degradation via the proteasome and thereby indirectly enhances VP stability. Moreover, it is interesting to note that MG-132 treatment had no effect on intracellular VP localization, showing that VP protein stabilization per se is also not sufficient for nuclear or nucleolar AAP trafficking. Together with data from Sonntag et al., who forced nucleolar VP3 accumulation and also found no evidence for AAP-independent AAV2 capsid assembly (17), this implies that AAP exerts its main function by mechanisms different from mere stimulation of VP protein expression or intracellular targeting (see also point iii above and the next point). Still, it remains possible that VP protein levels are an important determinant of capsid assembly as well. This has also been implied by the Nakai lab, who interpreted their notion of AAP-independent AAV5 capsid assembly as a potential by-product of the superphysiological VP3 protein expression from their particular construct (21). Intriguingly, we have previously reported that a genuine AAV5 helper plasmid, expressing the three VP proteins from the physiological p40 promoter, also results in VP levels much higher than those for various other AAV serotypes (31, 37). Hence, efficient AAV capsid formation may ultimately rely on a combination of VP protein expression above a certain threshold followed by, or concurrent with, AAP-mediated assembly into intact and stable capsids.
(vi) While AAPs are required to assemble functional virions, they show distinct intracellular localization patterns that (i) vary between the different AAV serotypes and (ii) rarely overlap those of free VP proteins or assembled capsids. For AAP2, we confirmed nucleolar localization, in line with the presence of nuclear and nucleolar localization signals in AAP2 (20). Moreover, we noted that cells expressing AAP2 often also stained positive with the A20 antibody (indicative of AAV2 capsid assembly). Further of note, preliminary data (not shown) obtained with our polyclonal anti-AAP antibody imply that the amount of cells positive for nucleolar AAP2 decreases over time relative to that of cells containing assembled AAV2 capsids. These findings agree with the reduced AAP2 protein levels observed at late time points of AAV virus production in insect cells (Fig. 2C) and with the short AAP2 half-life (Fig. 3G), substantiating our conclusion that the viral mechanisms may be similar in both species. Together with data that steady-state AAP2 levels are influenced by coexpressed capsid proteins (19), a picture emerges where expression and localization of AAP and VP proteins are tightly coordinated and affect each other in a spatiotemporal yet transient manner (Fig. 9). As noted, the sum of our data and those from the Kleinschmidt and Nakai groups implies that these interactions differ between the serotypes and that the observations made with AAV2 may be unique rather than prototypic. Proof includes that several serotypes, such as AAV5, AAV8, or AAV9, do not accumulate in nucleoli but are instead found mostly in the nucleoplasm (Fig. 8) (21). This confirms a hypothesis by Earley et al. that nucleolar localization correlates with the electric charge of basic regions in AAP, i.e., above 12 for AAP2, which may trigger nucleolar localization, versus 8 for AAP5, which was the lowest charge of all analyzed AAPs and may cause nucleolar exclusion (20). Altogether, the distinct and often discordant localization patterns of AAP, VP, and capsids that have now been consistently observed in multiple independent studies imply that nucleolar factors are generally not essential for AAV capsid assembly. Instead, capsid assembly may rapidly occur or at least be triggered at the site of AAP localization, which itself is determined by the AAV serotype. This would be congruent with the concept of coevolution of the overlapping AAP/VP ORFs and functions that was proposed and experimentally studied by Kawano and colleagues (38). Finally notable in this context is that in insect cells, AAV2 capsid assembly seems to take place in vesicles located in the nuclear periphery, where all AAV components (Rep and VP proteins) accumulate (39), but not in nucleoli, despite a reorganization of nuclear compartments due to baculoviral replication (40).
As the exact mechanisms of action of AAP remain elusive and deserve further research, we believe it will be highly rewarding to draw analogies to other parvoviruses that are inherently AAP independent. We particularly note extensive, highly informative work from the Almendral group on the assembly of the evolutionarily related protoparvovirus minute virus of mice (MVM). As reported (41, 42), the VP1 and VP2 capsid proteins of MVM form cytoplasmic homotrimeric (3VP2) or heterotrimeric (2VP2/1VP1) assembly intermediates which then exploit a structured, β-stranded nuclear localization motif (NLM) for entry in the nucleus and completion of the assembly process. For the oligomerization of these translocation-competent trimers to occur, VP2 is most important, as it assists cytoplasmic folding and thus viral morphogenesis. Interestingly, the β-stranded configuration of the NLM is conserved across parvoviruses, as is the need for a contribution of additional host or viral chaperone-like activities supporting the final stages of capsid maturation (42). Also of note, nuclear translocation of MVM capsid subunits was found to highly depend on the cell cycle, especially the S phase, as well as the cell type (43). Finally, it is interesting that newly expressed MVM VP1 is ubiquitinated and degraded and that VP2 is capable of preventing these processes, implying a role as a chaperone that directly masks the ubiquitination domains in VP1 and/or assists capsid folding and thus indirectly blocks access for the E3 ubiquitin ligase (41). These data further support our present findings and conclusions that de novo-synthesized AAV VP proteins are also destabilized by posttranslational modification, possibly ubiquitination, and that this process is counteracted by the activity of AAP, perhaps in a manner akin to the function of VP2 for MVM. Thus, the AAV/AAP field should benefit from these lessons already learned from other parvoviruses and may be able to shed further light on AAP function by also focusing more on examining the structure and localization of AAV VP subunits in the presence or absence of AAP.
Importantly, our present work and a complementary study (unpublished data) have revealed several lines of evidence that despite the serotype-specific features of AAP, inadvertent AAP shuffling during molecular capsid gene evolution may be less detrimental than anticipated. This includes our finding that all AAPs could trans-complement AAP knockout mutants of a different serotype, with the exceptions of AAP4 and (partially) AAP5. Our data are in accordance with, and largely expand, those of Sonntag et al., who noted cross-complementation between AAV/AAP of serotypes 1, 2, 8, and 9 but not 5 and who postulated that AAP directly interacts with as-yet-unknown VP/capsid domains (19). As the capsid and AAP sequences of AAV4 and AAV5 are most divergent from all other serotypes, it is plausible that this interaction is perturbed as soon as one of the two partners is derived from these very heterologous serotypes. Our conclusions are likewise consistent with work from Earley and colleagues, who studied another set of AAV serotypes and also noted a high degree of AAP interchangeability (21). With respect to molecular AAV capsid evolution, this implies that even if there are sequences in a capsid library that comprise a defective AAP gene, other capsid/AAP variants within the same library may cross-complement and thus rescue this deficiency. Moreover, additional data that will be presented elsewhere show that most chimeric AAPs present in shuffled capsid libraries are actually functional to begin with and that production of such libraries does not depend on excessive amounts of AAP.
In conclusion, by being the first to address the role of AAP in infectious vector particles derived from 10 different AAV serotypes, with one of them produced in two heterologous systems, the present study advances our understanding of fundamental AAV/AAP biology and concomitantly delivers original insights into the relevance of AAP for the field of recombinant AAV and molecular capsid evolution.
MATERIALS AND METHODS
Cloning procedures.
To generate AAP expression vectors, AAP sequences (see Data Set S1 in the supplemental material) were amplified from wild-type AAV cap genes with primers 983 to 992 and 1292 to 1294 (see Data Sets S2 and S3 in the supplemental material for all primer sequences and combinations). These primers introduced a NotI restriction site in front of the AAP coding sequence and an EcoRI restriction site behind. This permitted cloning into plasmid pIRES-Ago2 (44), which contains a cytomegalovirus (CMV) promoter followed by a FLAG tag, an HA tag, and the same two restriction sites.
AAP2-GFP was generated by overlap extension PCR (OE-PCR) as described by Schürmann et al. (44) using primers 983 and 1161 to 1163. Thereby, the AAP2 stop codon and the GFP start codon were removed, and GFP was C-terminally fused in frame with AAP2. The AAP2-GFP expression vector was then cloned as described above for the wild-type AAP sequences.
AAV helper plasmids with an AAP knockout (AAVmut constructs; see Data Set S4 in the supplemental material) were generated through OE-PCR (see Data Sets S2 and S5 in the supplemental material for primer sequences and OE-PCR conditions). In total, eight AAV serotypes (AAV1, -2, -3[b; here simply called 3], -6 to -9, and -rh10) were mutated by (i) changing the AAP start codon to CCG and (ii) introducing a stop codon 66 bp downstream of the modified start. AAP knockout constructs for AAV serotypes 4 and 5 were derived from synthetic capsid genes AAV4O and AAV5O, respectively, which carry numerous debilitating mutations in AAP4/5 as a by-product of codon modification of the AAV4/5 cap open reading frames (ORFs). A more detailed characterization of these two cap variants will be provided elsewhere (unpublished data). Briefly, by codon adapting the VP ORF in these two genes, we had inadvertently introduced a variety of mutations in the other two reading frames. These included alterations of the AAP4 and AAP5 start codons (mutations of CTG to a nonfunctional CAG) as well as introduction of various stop codons (three in AAP4 and one in AAP5), serendipitously leading to AAP knockouts that were useful for the present work (see Data Set S4 in the supplemental material for the nature and location of all mutations in the final constructs). Primers 178 and 833 were employed as flanking primers during the second PCR and allowed for cloning of each cap gene as a HindIII/SpeI fragment into a helper plasmid carrying AAV2 rep. This plasmid is a derivative of p5E18 (26) carrying a SpeI site behind AAV2 cap, thus permitting excision and swapping of capsid genes via HindIII (cuts in AAV2 rep) and SpeI (cuts after cap).
To experimentally confirm the AAP knockout, three representative expression plasmids were cloned based on the mutated AAP2, AAP8, or AAP9 sequence. Therefore, they were PCR amplified from the mutated cap genes using the respective primers and combinations thereof shown in Data Sets S2 and S3 and then also cloned under control of a CMV promoter via NotI and EcoRI, identically to the wild-type AAP counterparts.
To decipher the role of AAP2 in insect cells, donor plasmid pSR-Rep2Cap2-AAPmut was constructed from plasmid pUC57-Rep2Cap2 (kindly provided by O. Merten, Genethon, France). The latter allows for expression of Rep78/52 and VP1/VP2/VP3 proteins in Sf9 cells from the AAV2 rep and cap ORFs, respectively, that were optimized as described by Smith et al. (23). AAP2 knockout was realized by mutating the AAP2 CTG start codon to CCG (T-to-C exchange at nucleotide position 528 of AAV2 cap) and by inserting a stop codon at nucleotide 594 of AAV2 cap via G-to-A mutation, akin to our strategy for the mammalian AAV2 helper plasmid. The two mutations were introduced by two successive PCR mutagenesis steps using primer pairs t528cfor/rev and g594afor/rev (Data Set S2). Following validation of the correct mutations in donor plasmid pSR-Rep2Cap2-AAPmut by Sanger sequencing, it was used to generate the recombinant baculovirus Rep2Cap2-AAP2mut (see below).
For AAP2 trans-complementation and overexpression studies in insect cells, a fragment containing the P10 baculoviral promoter followed by the AAP2 ORF was synthesized and cloned into plasmid pMB-eGFP-Puro between the gentamicin cassette and the AAV2 5′ inverted terminal repeat (ITR). Plasmid pMB-eGFP-Puro is derived from the pFastBac plasmid (Thermo Fisher Scientific) and contains a human CMV promoter, the enhanced green fluorescent protein (eGFP) reporter gene, followed by an encephalomyocarditis virus (EMCV) internal ribosome entry site (IRES), a puromycin resistance sequence, and the 3′ untranslated region of the human hemoglobin beta (HBB) gene. The recombinant AAV genome in pMB-eGFP-Puro is delimited by the wild-type flip and flop ITRs from AAV2. The resulting donor plasmid, pMB-eGFP-Puro-p10AAP2, was validated by Sanger sequencing and used to produce the corresponding baculovirus (see below).
Generation of recombinant baculoviruses.
For AAV vector production using the dual-baculovirus/Sf9 platform, the following BEV-AAVs were generated: (i) BEV-eGFP-Puro and BEV-eGFP-Puro-p10AAP2 from the pMB-eGFP-Puro and the pMB-eGFP-Puro-p10AAP2 donor plasmids, respectively; (ii) BEV-eGFP from the pFB-eGFP plasmid, which is identical to the pMB-eGFP-Puro plasmid but lacks the IRES and the puromycin cDNA and contains ITRs of AAV2 derived from plasmid pSub-201 (45); and (iii) BEV-A1AT, expressing alpha-1-antitrypsin (A1AT) from a CAG promoter (CMV enhancer fused to the chicken beta-actin promoter) and followed by a simian virus 40 (SV40) polyadenylation signal. BEV-Rep2Cap2 and BEV-Rep2Cap2-AAPmut were generated from the pUC57-Rep2Cap2 and the pSR-Rep2Cap2-AAPmut donor plasmids, respectively.
Tn7 site-specific transposition of the cassette of interest in the bacmid backbone was performed by transformation of 10 ng of each donor plasmid in Escherichia coli DH10Bac bacteria in accordance with the instructions in the Bac-to-Bac expression system manual (Thermo Fisher Scientific). The presence of the insert DNA in recombinant bacmids was validated by PCR using primers M13-pUC-F and M13-pUC-R (Data Set S2), which bind on either side of the insert, or BAC-G, which targets the gentamicin resistance sequence in the insertion cassette, as well as by Sanger sequencing. One microgram of each bacmid DNA was then transfected into 1 × 106 Spodoptera frugiperda Sf9 insect cells (Invitrogen) cultivated in 6-well plates using 9 μl Cellfectin II reagent (Thermo Fisher Scientific). The supernatants (P1 stocks) were recovered at 96 h posttransfection. P1p clones were isolated from the P1 stocks by plaque assay. One clone per recombinant baculovirus was selected based on the infectious titer in PFU and genetic stability of the insert after five passages. The BEV P2 stocks were then generated after amplification of the P1p stock in Sf9 cells seeded in spinner flasks and sequenced. The absence of AAP2 expression and correct VP expression profiles were confirmed by Western blotting after Sf9 infection with BEV-Rep2Cap2-AAP2mut and in comparison with BEV-Rep2Cap2 infection.
Cell culture.
All mammalian cell lines were cultured under standard sterile growth conditions and incubated at 37°C with 5% CO2. HEK293T, MCF7 and SF539 cells were cultured in Dulbecco's modified Eagle medium (DMEM) with GlutaMAX (Life Technologies) supplemented with 10% fetal bovine serum (FBS) (Sigma-Aldrich) and 100 U/ml penicillin-streptomycin (Life Technologies). Cells were split every 2 to 4 days when they had reached around 80% confluence. Sf9 insect cells were grown at 27°C in Sf-900 III SFM medium (Thermo Fisher Scientific) in spinner flasks.
AAV crude cell lysate production.
HEK293T cells were seeded in 6-well plates at a density of around 5 × 105 cells in 2 ml medium per well 1 day before transfection and grown to about 70% confluence, which we found to be optimal for transfection. For transfection of the cells in one well, a mixture of 22 μl polyethylenimine (PEI) (linear; molecular weight, ∼250,000; Polysciences), 27 μl H2O, and 49 μl 300 mM NaCl (Sigma-Aldrich) was prepared. Another mixture made in parallel contained a total of 2.6 μg DNA, 49 μl H2O, and 49 μl 300 mM NaCl. Subsequently, both mixes were combined, vortexed, incubated for 10 min at room temperature, and added dropwise to the cells. For recombinant AAV vector production, we transfected equal amounts (867 ng each per well) of an adenoviral helper construct (46), an AAV helper carrying AAV2 rep and the selected wild-type or mutated AAV capsid gene, and a plasmid carrying a CMV promoter-driven GFP gene cDNA flanked by two AAV ITRs (one of them mutated) that allow for packaging as self-complementary AAV vectors (47). When AAP was included during vector production, the DNA amount per construct was reduced to 690 ng, and either 530 ng of AAP plasmid or 530 ng of sheared salmon sperm stuffer DNA was added to the same total of 2.6 μg. Forty-eight hours after transfection, cells were harvested into the medium with a pipette and centrifuged for 10 min at 1,500 × g. The supernatant was discarded and the cell pellet resuspended in 1 ml 1× PBS. After a second identical centrifugation, the pellet was resuspended in 500 μl 1× PBS. Sixty microliters of this suspension was transferred to a new tube and diluted with 40 μl 1× PBS for later Western blot analysis. Cells in the remaining suspension (roughly 440 μl) were lysed by five freeze-thaw cycles (37°C/liquid nitrogen) and 1 min of sonication (Ultrasonic bath; Bandelin). Cell debris was removed by 10 min of centrifugation at 16,100 × g, and the AAV particle-containing supernatant was transferred to a new tube. The AAV crude cell lysates were stored at −20°C for later transduction experiments.
AAV2 vector production in insect cells, time course experiments, and particle titration.
Sf9 cells cultured at a density of 1 × 106 cells/ml were infected with a BEV expressing AAV2 rep and cap and a BEV-AAV at a multiplicity of infection (MOI) (here, infectious particle number per cell as determined by plaque assay) of 0.05 per baculovirus. Four days after infection, cells were lysed by agitating them in 0.5% Triton X-100 detergent (Sigma-Aldrich) for 2.5 h at 27°C, followed by 2 h of incubation at 37°C with 5 U/ml Benzonase (Merck) and 2 mM MgCl2. Cell debris was then removed by 5 min of centrifugation at 1,000 × g, and crude cell lysates were stored at −80°C for AAV2 particle quantification.
For time course experiments, 1-ml aliquots of the production bulk were harvested every day, and cells were pelleted by 5 min of centrifugation at 1,000 × g. Dry cell pellets were stored at −20°C for protein extraction.
Vector genomes were quantified in crude cell lysates by free-ITR quantitative PCR (qPCR) (48) and baculovirus genomes by a qPCR targeting the baculoviral DNA polymerase gene using primers Bac-F and Bac-R (Data Set S2) and probe FAM-CAA ACA CGC GCA TTA ACG AGA GCA CC-TAMRA. Assembled AAV2 capsid titers were determined in crude cell lysates using the A20 ELISA kit (Pratv; Progen Biotechnik) according to the manufacturer's manual.
Crude cell lysate transduction and flow cytometry analysis.
Cells were seeded in 96-well plates 24 h before AAV crude lysate transduction at the following densities (cells per well): 1.5 × 104 to 2.0 × 104 HEK293T cells, 0.5 × 104 MCF7 cells, and 0.4 × 104 SF539 cells. HEK293T and SF539 cells were used to test all vectors except AAV4, which was measured in MCF7 cells. Cells were transduced with 5 μl undiluted vector as well as with dilutions of 1:10 and 1:100. At 48 h postransduction, cells were processed for flow cytometry analysis of GFP expression as a surrogate marker of AAV vector transduction efficiency. Therefore, cell culture medium was discarded and cells were washed with 100 μl 1× PBS. After 5 min of incubation at 37°C with 30 μl trypsin (Life Technologies), cells were resuspended in 170 μl 1% bovine serum albumin (BSA) (Roth) in 1× PBS. Amounts of transduced, GFP-positive cells were measured with an FC500 MPL flow cytometer (Beckmann Coulter) in a 96-well plate format, and data were analyzed using the accompanying MXP software. For the final analysis, the crude lysate dilution in which the AAV wild-type control showed fewer than 95% positive cells was selected, in order to avoid artifacts from oversaturation. All values were then normalized to this control (set to 1) and compared with other measurements.
Proteasome inhibition experiments.
To inhibit proteasome activity during production of crude cell lysates, cell culture medium was removed at 5 and/or 24 h posttransfection, and fresh medium containing the proteasome inhibitor MG-132 (final concentration, 2 μM; Santa Cruz Biotechnology) was added. Following cell harvest, 100 μl of the cell suspension was collected separately for Western blot analysis. For dot blot studies, the remaining 400 μl was processed as described above to yield crude lysates.
CHX chase experiments.
HEK293T cells were seeded in a 6-well plate and transfected as stated above. After 24 h, the cells were washed with PBS, and medium either containing cycloheximide (CHX) (Applichem) at a concentration of 100 μg/ml or not containing CHX (untreated control samples) was added. At the indicated time points, samples were harvested by washing the wells with PBS and transferring them in 500 μl PBS to a 1.5-ml tube. The samples underwent freeze-thaw cycles as described above, and 50 μl was used for dot blot analysis (see below) using the B1 antibody to detect free, unassembled VP proteins. The images were developed by chemiluminescence using a ChemoCam (INTAS Science Imaging Instruments). For quantification, the intensity was measured using Fiji ImageJ (https://fiji.sc/), and the treated sample was compared to the untreated control. For each biological replicate, a technical duplicate was performed, and means with the standard errors of the mean (SEM) were calculated.
To perform a CHX chase in insect cells, Sf9 cells were infected with BEV-GFP and either BEV-Rep2Cap2 or BEV-Rep2Cap2-AAPmut at an MOI of 0.05 each. After 40 h (t = 0), CHX was added to the cells to a final concentration of 75 μg/ml. At the indicated time points after CHX addition, cells were harvested and prepared for native dot blot analysis (as described below for HEK293T cells) or for Western blotting.
Western blot analysis.
To detect viral proteins via Western blotting, 2× SDS sample buffer was added to the samples. After boiling for 5 min at 95°C, they were separated on either 8% (VP1, VP2, VP3, and actin) or 12% (AAP) resolving sodium dodecyl sulfate (SDS)-polyacrylamide gels together with a standard protein ladder (PageRuler Plus prestained protein ladder; Thermo Fisher Scientific). Subsequently, proteins were transferred to a nitrocellulose membrane (neoLab) through semidry blotting and blocked with either 5% milk (Roth) or 1% BSA in Tris-buffered saline with Tween 20 (TBS-T). Primary antibodies B1 (against free AAV capsid proteins VP1 to VP3) and 303.9 (against all four AAV Rep proteins) (both kind gifts from J. Kleinschmidt [49]), anti-HA (sc-7392; Santa Cruz Biotechnology), and anti-β-actin (sc-47778; Santa Cruz Biotechnology) were diluted 1:30, 1:10, 1:300, and 1:1,000, respectively, in blocking buffer. For detection, a horseradish peroxidase-conjugated secondary goat anti-mouse antibody (Jackson ImmunoResearch Laboratories) diluted 1:10,000 in blocking buffer was used. Western Lightning Plus-ECL reagent (PerkinElmer) was used to visualize bound antibodies. Blots were exposed to X-ray films (GE Healthcare) and developed in an X-Omat 2000 processor (Kodak). Membranes were cut around the 55-kDa ladder band before blocking in case VP proteins and actin were codetected on the same gel.
For analysis of proteins in insect cells, 10 μg total protein was extracted in radioimmunoprecipitation assay (RIPA) buffer and loaded onto Novex 10% Tris-glycine polyacrylamide gels (Thermo Fisher Scientific). After semidry transfer, the membrane was blocked with 1× PBS, 1% Tween 20, and 5% milk overnight at 4°C and then incubated with one of the following antibodies: anti-AAP2 antibody (produced during this study [see below]) at a 1:200 dilution, polyclonal rabbit anti-VP antibody at a 1:500 dilution (61084; Progen Biotechnik), mouse anti-Rep antibody 303.9 at a 1:20 dilution, polyclonal goat anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) antibody at a 1:10,000 dilution (IMG-3073; Novus Biologicals), or mouse anti-GP39 antibody at a 1:100 dilution (a kind gift from L. E. Volkman). For detection, the following horseradish peroxidase-conjugated secondary antibodies were used: donkey anti-rabbit antibody at a 1:20,000 dilution (711-035-152; Jackson ImmunoResearch Laboratories), goat anti-mouse antibody at a 1:2,000 dilution (P0447; Dako), or rabbit anti-goat antibody at a 1:2,000 dilution (P0449; Dako). For time course experiments, the membrane was stripped following AAP2 detection with 2% SDS, 62.5 mM Tris (pH 6.7), and 0.1 M 2-mercaptoethanol and subsequently hybridized with the anti-VP antibody, the anti-GAPDH antibody, and finally the anti-GP39 antibody.
Immunostaining and microscopy analysis.
HeLa or HEK293T cells were seeded 1 day prior to transfection in 8-chamber LabTek slides (Thermo Fisher Scientific) coated with polylysine (Sigma-Aldrich). One day later, cells were transfected using TurboFect transfection reagent (0.8 μl/well; Thermo Fisher Scientific). The total DNA amount in the final solution was 1 μg, which in the case of HEK293T cells comprised equal amounts of an adenoviral helper construct (46), an AAV helper carrying rep together with the mutated AAV capsid, the corresponding AAP plasmid or stuffer DNA, and plasmid pAAVpsi2, carrying Renilla and firefly luciferase transgenes (50) flanked by ITRs. At 20 or 42 h after transfection, cells were fixed with 4% paraformaldehyde (PFA) in PBS at room temperature for 30 min and then permeabilized for 10 min at room temperature using 0.1% Triton. After 60 min of incubation with IF blocking buffer (5% fetal calf serum [Sigma-Aldrich] in PBS), cells were incubated for 1 to 2 h at room temperature with 120 μl of the primary antibodies A20/ADK (detecting assembled AAV capsids; a kind gift from J. Kleinschmidt [49]) or B1 (detecting free capsid proteins of all AAV serotypes used here except AAV4). All anti-AAV antibodies were used as undiluted hybridoma supernatants. Anti-HA antibody (GTX29110; GeneTex) was diluted 1:500 in the hybridoma supernatants. Cells were then washed twice for 10 min each with IF blocking buffer and then incubated for 1 h at 37°C with secondary antibodies diluted 1:700 in blocking buffer. To quantify B1 and A20/ADK stainings, an anti-mouse AF488-labeled secondary antibody (Life Technologies) was used. The anti-HA antibody was detected with an anti-rabbit AF647-labeled secondary antibody (Life Technologies). To stain nuclei, Hoechst stain (Molecular Probes, Thermo Fisher Scientific) diluted 1:3,000 was added to all samples. Next, cells were washed with 1× PBS and stored at 4°C in the dark in 200 μl 1× PBS per well. HeLa cells were treated the same way, except that cells were analyzed at 42 h posttransfection and stained with the anti-HA antibody (and Hoechst stain). Microscopy pictures were taken with an Olympus IX-81 inverted fluorescence microscope (HEK293T cells) or a Leica TCS SP5 confocal microscope (HeLa cells). HeLa cells were preferred over HEK293T cells for detection of HA localization by confocal microscopy because of their more consistent cell morphology. Microscopy pictures were quantified and analyzed with the open-source software CellProfiler (http://cellprofiler.org/) (inverted microscope pictures) or with the Fiji program (confocal microscope pictures). Automated image segmentation was performed to determine B1 or A20/ADK mean intensities and to classify positive cells (51).
For analysis by confocal microscopy, HEK293 cells were seeded in glass-bottom 8-chamber LabTek slides. After 24 h, cells were transfected using TurboFect transfection reagent (0.8 μl/well; Thermo Fisher Scientific) and 1 μg total DNA consisting of equal amounts of the adenoviral helper construct, the AAV2mut helper, the AAP2 expression plasmid or stuffer DNA, and a luciferase-encoding AAV vector plasmid. Sixteen hours posttransfection, cells were treated with MG-132 (final concentration, 2 μM; Santa Cruz Biotechnology). After an additional 24 h, cells were fixed and stained with antibodies (either B1/A20 plus anti-mouse AF488-labeled secondary antibody or anti-HA antibody plus anti-rabbit AF568-labeled secondary antibody). Microscopy was performed on an inverted Leica SP8 confocal microscope using HyD detectors and a PL APO glycerol immersion objective (63× NA = 1.3). Optical sections were acquired in three channels sequentially: DAPI (4′,6′-diamidino-2-phenylindole) (405-nm laser; spectral detection band, 410 to 502 nm), Alexa 488 (488-nm laser; spectral detection band, 502 to 577 nm), and Alexa 568 (561-nm laser; spectral detection band, 577 to 749 nm). Optical sections spanning the entire volume of the cells were acquired with sampling fulfilling the Nyquist criteria (72 nm in the xy plane and 300 nm along the optical axis). At least three representative volumes were acquired per condition per replicate. Acquired images were deconvolved using the iterative constrained maximum-likelihood estimation algorithm and theoretical (but adaptive) PSF (“blind” mode) in Autoquant X3 (Media Cybernetics) software. Maximum projections along the z axes of all the acquired optical sections were generated using Fiji image analysis software.
Image processing for determination of normalized cross correlation coefficients.
Images were processed using the Konstanz Information Miner (KNIME) (www.knime.org) and the KNIME image processing plug-ins (KNIP). A workflow in which cellular objects were identified by background subtraction, automatic global thresholding, and connected component analysis was compiled. For each individual cellular object showing a green signal, the normalized cross correlation coefficient (NC) was determined as a measure of colocalization of the red and green color channel. A value of −1 indicates complete disagreement, and +1 indicates complete overlap of the two stains. These values were grouped together with additional information from the experiment and stored in a table. The distribution of these measures is shown as box plots.
RNA extraction and mRNA quantification.
For RNA extraction, HEK293T cells were seeded and transfected as described above in 6-well plate formats. At 48 h after transfection, cells were washed with 1× PBS, resuspended in 1 ml QIAzol lysis reagent (Qiagen), and transferred to 1.5-ml reaction tubes. Subsequently, RNA was purified according to the manufacturer's protocol (QIAzol handbook) with inclusion of a DNase I digestion step using the Turbo DNA-free kit (Thermo Fisher Scientific). RNA concentrations were determined using the NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). To measure mRNA copy numbers in the extracted RNA samples, 2.4 μg RNA was transcribed with the Tetro cDNA synthesis kit (Bioline) using the random hexamer primers according to the manufacturer's protocol. cDNA was then diluted 1:200, and 1.25 μl was used in 12.5-μl qPCR mixtures composed of 6.25 μl 2× SYBR Select master mix (Thermo Fisher Scientific), 0.75 μl 10 μM forward primer (1107, 1109, 1111, or 1113 [Data Set S2]), 0.75 μl 10 μM reverse primer (1108, 1110, 1112, or 1114), 1.25 μl cDNA template, and 3.75 μl H2O. Reactions were run in a StepOnePlus real-time PCR system (Thermo Fisher Scientific) using the following program: 95°C for 10 min, 40 cycles of 95°C for 15 s and 60°C for 1 min, and a final melting step from 60°C to 95°C in increments of 0.3°C. Samples were measured in duplicates and normalized to a GAPDH housekeeper (primer pair 1117/1118). For the final analysis, sample values were calculated according to the 2−ΔΔCT method (52).
Polyclonal anti-AAP2 antibody production.
The anti-AAP2 antibody was ordered from the company Perbio, Thermo Fisher Scientific, and produced by immunization of rabbits with peptide GKDSSTTTGDSDPRDSTS (AAP2 residues 121 to 138) (17). Affinity purification yielded the final polyclonal anti-AAP2 antibody (preserved in 1% BSA), which was used for Western blot analyses of insect cell samples and for immunostainings.
Dot blot analysis.
Crude cell lysates containing AAV particles were produced as stated above. A nitrocellulose membrane (Thermo Fisher Scientific) was cut to size, soaked in PBS for 10 min, and assembled into a Bio-Dot microfiltration apparatus (Bio-Rad) according to the manufacturer's protocol. Next, 15 μl of the indicated crude lysates was loaded. To promote capsid disassembly for detection of free, unassembled viral capsid proteins, 100 μl of each sample was heated to 95°C for 5 min, and 15 μl was loaded separately. In selected cases, 20 μl 6× SDS buffer was added prior to the heating step to create even harsher conditions for capsid disassembly, and 18 μl was loaded to compensate for the dilution with the buffer. Afterwards, vacuum was applied. Once all liquids had passed through the apparatus, the wells were washed twice with 100 μl PBS per well. The device was disassembled, and the membrane was blocked for 1 h at room temperature with 5% milk in TBS-T. Half of the membrane was then incubated with the B1 antibody (1:10 diluted) to detect free viral capsid proteins, while the other half was used to visualize assembled capsids. As noted above, the B1 antibody binds VP proteins from AAV serotypes 1 to 3 and 5 to 9 but not AAV4. Therefore, to also detect AAV4 VP proteins, a polyclonal anti-VP serum (Progen) was used at a 1:200 dilution. For detection of assembled capsids, the following serotype-specific antibodies were used (all were used at a 1:10 dilution): for AAV1, ADK1; for AAV2, A20; for AAV3/AAV8, ADK8; for AAV4, ADK4; for AAV5, ADK5; and for AAV9, ADK9 (19). The blots were developed in a manner akin to that for regular Western blots using secondary monoclonal peroxidase-coupled anti-mouse (against B1 and A20/ADK) or anti-rabbit (against the polyclonal anti-VP serum) antibodies for detection by chemiluminescence.
Statistics.
For analysis of the trans-complementation assays, samples were compared to the respective wild type. Typically shown are means from at least three independent experiments with standard deviations (SD). To assess statistical significance, a one-way analysis of variance (ANOVA) with Bonferroni's multiple-comparison test was performed. P values of less than 0.05 were considered statistically significant. To generate the heat map in Fig. 5D, we used a modified version of our in-house tool “Salanto” (reference 44 and unpublished data). Pairs of AAP variants were compared to each other at a time based on the function “Precise type assignment,” and the percentage of unique, nonidentical positions was depicted in a matrix.
Supplementary Material
ACKNOWLEDGMENTS
We thank various members of the Grimm and Ayuso labs for critically reading the manuscript. We are grateful to J. Kleinschmidt and L. E. Volkman for their kind gifts of antibodies and to O. Merten for providing an AAV2 BEV construct.
We declare no conflict of interest.
A.-K.H. and D.G. gratefully acknowledge support from Collaborative Research Center SFB1129 (project TP2, Deutsche Forschungsgemeinschaft [DFG]). D.G. and his lab are further thankful for support from Collaborative Research Center TRR179 (project TP18, DFG). S.G., A.-K.H., E.W., and D.G. are grateful for funding from the Cluster of Excellence CellNetworks at Heidelberg University (funded by the DFG [EXC81]). J.F. appreciates a PhD stipend from the Hartmut Hoffmann-Berling International Graduate School (HBIGS) at Heidelberg University. M.P.-B. and E.A. acknowledge support from Institut National de la Santé et la Recherche Médicale (INSERM), CHU Nantes, University of Nantes, and the Association Française contre les Myopathies (AFM). K.B., V.L. and D.G. gratefully acknowledge funding through the German Center for Infection Research (DZIF, TTU HIV). S.G., C.K., and D.G. further acknowledge support through a research collaboration with the company Baxalta Inc./Shire.
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.01198-17.
REFERENCES
- 1.Atchison RW, Casto BC, Hammon WM. 1965. Adenovirus-associated defective virus particles. Science 149:754–756. doi: 10.1126/science.149.3685.754. [DOI] [PubMed] [Google Scholar]
- 2.Grimm D, Zolotukhin S. 2015. E pluribus unum: 50 years of research, millions of viruses, and one goal—tailored acceleration of AAV evolution. Mol Ther 23:1819–1831. doi: 10.1038/mt.2015.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Agbandje-McKenna M, Kleinschmidt J. 2011. AAV capsid structure and cell interactions. Methods Mol Biol 807:47–92. doi: 10.1007/978-1-61779-370-7_3. [DOI] [PubMed] [Google Scholar]
- 4.Kotterman MA, Schaffer DV. 2014. Engineering adeno-associated viruses for clinical gene therapy. Nat Rev Genet 15:445–451. doi: 10.1038/nrg3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grimm D, Lee JS, Wang L, Desai T, Akache B, Storm TA, Kay MA. 2008. In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J Virol 82:5887–5911. doi: 10.1128/JVI.00254-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koerber JT, Jang JH, Schaffer DV. 2008. DNA shuffling of adeno-associated virus yields functionally diverse viral progeny. Mol Ther 16:1703–1709. doi: 10.1038/mt.2008.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li W, Asokan A, Wu Z, Van Dyke T, DiPrimio N, Johnson JS, Govindaswamy L, Agbandje-McKenna M, Leichtle S, Redmond DE Jr, McCown TJ, Petermann KB, Sharpless NE, Samulski RJ. 2008. Engineering and selection of shuffled AAV genomes: a new strategy for producing targeted biological nanoparticles. Mol Ther 16:1252–1260. doi: 10.1038/mt.2008.100. [DOI] [PubMed] [Google Scholar]
- 8.Gray SJ, Blake BL, Criswell HE, Nicolson SC, Samulski RJ, McCown TJ, Li W. 2010. Directed evolution of a novel adeno-associated virus (AAV) vector that crosses the seizure-compromised blood-brain barrier (BBB). Mol Ther 18:570–578. doi: 10.1038/mt.2009.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Powell SK, Khan N, Parker CL, Samulski RJ, Matsushima G, Gray SJ, McCown TJ. 2016. Characterization of a novel adeno-associated viral vector with preferential oligodendrocyte tropism. Gene Ther 23:807–814. doi: 10.1038/gt.2016.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang L, Jiang J, Drouin LM, Agbandje-McKenna M, Chen C, Qiao C, Pu D, Hu X, Wang DZ, Li J, Xiao X. 2009. A myocardium tropic adeno-associated virus (AAV) evolved by DNA shuffling and in vivo selection. Proc Natl Acad Sci U S A 106:3946–3951. doi: 10.1073/pnas.0813207106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Asuri P, Bartel MA, Vazin T, Jang JH, Wong TB, Schaffer DV. 2012. Directed evolution of adeno-associated virus for enhanced gene delivery and gene targeting in human pluripotent stem cells. Mol Ther 20:329–338. doi: 10.1038/mt.2011.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dalkara D, Byrne LC, Klimczak RR, Visel M, Yin L, Merigan WH, Flannery JG, Schaffer DV. 2013. In vitro and in vivo models for the study of human polyomavirus infection. Viruses 8:E292. doi: 10.3390/v8100292. [DOI] [Google Scholar]
- 13.Excoffon KJ, Koerber JT, Dickey DD, Murtha M, Keshavjee S, Kaspar BK, Zabner J, Schaffer DV. 2009. Directed evolution of adeno-associated virus to an infectious respiratory virus. Proc Natl Acad Sci U S A 106:3865–3870. doi: 10.1073/pnas.0813365106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jang JH, Koerber JT, Kim JS, Asuri P, Vazin T, Bartel M, Keung A, Kwon I, Park KI, Schaffer DV. 2011. An evolved adeno-associated viral variant enhances gene delivery and gene targeting in neural stem cells. Mol Ther 19:667–675. doi: 10.1038/mt.2010.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koerber JT, Klimczak R, Jang JH, Dalkara D, Flannery JG, Schaffer DV. 2009. Molecular evolution of adeno-associated virus for enhanced glial gene delivery. Mol Ther 17:2088–2095. doi: 10.1038/mt.2009.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ward P, Walsh CE. 2009. Chimeric AAV cap sequences alter gene transduction. Virology 386:237–248. doi: 10.1016/j.virol.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 17.Sonntag F, Schmidt K, Kleinschmidt JA. 2010. A viral assembly factor promotes AAV2 capsid formation in the nucleolus. Proc Natl Acad Sci U S A 107:10220–10225. doi: 10.1073/pnas.1001673107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Naumer M, Sonntag F, Schmidt K, Nieto K, Panke C, Davey NE, Popa-Wagner R, Kleinschmidt JA. 2012. Properties of the adeno-associated virus assembly-activating protein. J Virol 86:13038–13048. doi: 10.1128/JVI.01675-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sonntag F, Kother K, Schmidt K, Weghofer M, Raupp C, Nieto K, Kuck A, Gerlach B, Bottcher B, Muller OJ, Lux K, Horer M, Kleinschmidt JA. 2011. The assembly-activating protein promotes capsid assembly of different adeno-associated virus serotypes. J Virol 85:12686–12697. doi: 10.1128/JVI.05359-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Earley LF, Kawano Y, Adachi K, Sun XX, Dai MS, Nakai H. 2015. Identification and characterization of nuclear and nucleolar localization signals in the adeno-associated virus serotype 2 assembly-activating protein. J Virol 89:3038–3048. doi: 10.1128/JVI.03125-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Earley LF, Powers JM, Adachi K, Baumgart JT, Meyer NL, Xie Q, Chapman MS, Nakai H. 18 January 2017. Adeno-associated virus assembly-activating protein is not an essential requirement for capsid assembly of AAV serotypes 4, 5, and 11. J Virol. doi: 10.1128/JVI.01980-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Urabe M, Ding C, Kotin RM. 2002. Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum Gene Ther 13:1935–1943. doi: 10.1089/10430340260355347. [DOI] [PubMed] [Google Scholar]
- 23.Smith RH, Levy JR, Kotin RM. 2009. A simplified baculovirus-AAV expression vector system coupled with one-step affinity purification yields high-titer rAAV stocks from insect cells. Mol Ther 17:1888–1896. doi: 10.1038/mt.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aslanidi G, Lamb K, Zolotukhin S. 2009. An inducible system for highly efficient production of recombinant adeno-associated virus (rAAV) vectors in insect Sf9 cells. Proc Natl Acad Sci U S A 106:5059–5064. doi: 10.1073/pnas.0810614106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mietzsch M, Casteleyn V, Weger S, Zolotukhin S, Heilbronn R. 2015. OneBac 2.0: Sf9 cell lines for production of AAV5 vectors with enhanced infectivity and minimal encapsidation of foreign DNA. Hum Gene Ther 26:688–697. doi: 10.1089/hum.2015.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiao W, Chirmule N, Berta SC, McCullough B, Gao G, Wilson JM. 1999. Gene therapy vectors based on adeno-associated virus type 1. J Virol 73:3994–4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Douar AM, Poulard K, Stockholm D, Danos O. 2001. Intracellular trafficking of adeno-associated virus vectors: routing to the late endosomal compartment and proteasome degradation. J Virol 75:1824–1833. doi: 10.1128/JVI.75.4.1824-1833.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duan D, Yue Y, Yan Z, Yang J, Engelhardt JF. 2000. Endosomal processing limits gene transfer to polarized airway epithelia by adeno-associated virus. J Clin Invest 105:1573–1587. doi: 10.1172/JCI8317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yan Z, Zak R, Luxton GW, Ritchie TC, Bantel-Schaal U, Engelhardt JF. 2002. Ubiquitination of both adeno-associated virus type 2 and 5 capsid proteins affects the transduction efficiency of recombinant vectors. J Virol 76:2043–2053. doi: 10.1128/jvi.76.5.2043-2053.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wistuba A, Kern A, Weger S, Grimm D, Kleinschmidt JA. 1997. Subcellular compartmentalization of adeno-associated virus type 2 assembly. J Virol 71:1341–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grimm D, Pandey K, Nakai H, Storm TA, Kay MA. 2006. Liver transduction with recombinant adeno-associated virus is primarily restricted by capsid serotype not vector genotype. J Virol 80:426–439. doi: 10.1128/JVI.80.1.426-439.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rabinowitz JE, Xiao W, Samulski RJ. 1999. Insertional mutagenesis of AAV2 capsid and the production of recombinant virus. Virology 265:274–285. doi: 10.1006/viro.1999.0045. [DOI] [PubMed] [Google Scholar]
- 33.Warrington KH Jr, Gorbatyuk OS, Harrison JK, Opie SR, Zolotukhin S, Muzyczka N. 2004. Adeno-associated virus type 2 VP2 capsid protein is nonessential and can tolerate large peptide insertions at its N terminus. J Virol 78:6595–6609. doi: 10.1128/JVI.78.12.6595-6609.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petrs-Silva H, Dinculescu A, Li Q, Min SH, Chiodo V, Pang JJ, Zhong L, Zolotukhin S, Srivastava A, Lewin AS, Hauswirth WW. 2009. High-efficiency transduction of the mouse retina by tyrosine-mutant AAV serotype vectors. Mol Ther 17:463–471. doi: 10.1038/mt.2008.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qiao C, Zhang W, Yuan Z, Shin JH, Li J, Jayandharan GR, Zhong L, Srivastava A, Xiao X, Duan D. 2010. Adeno-associated virus serotype 6 capsid tyrosine-to-phenylalanine mutations improve gene transfer to skeletal muscle. Hum Gene Ther 21:1343–1348. doi: 10.1089/hum.2010.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhong L, Li B, Mah CS, Govindasamy L, Agbandje-McKenna M, Cooper M, Herzog RW, Zolotukhin I, Warrington KH Jr, Weigel-Van Aken KA, Hobbs JA, Zolotukhin S, Muzyczka N, Srivastava A. 2008. Next generation of adeno-associated virus 2 vectors: point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc Natl Acad Sci U S A 105:7827–7832. doi: 10.1073/pnas.0802866105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grimm D, Kay MA, Kleinschmidt JA. 2003. Helper virus-free, optically controllable, and two-plasmid-based production of adeno-associated virus vectors of serotypes 1 to 6. Mol Ther 7:839–850. doi: 10.1016/S1525-0016(03)00095-9. [DOI] [PubMed] [Google Scholar]
- 38.Kawano Y, Neeley S, Adachi K, Nakai H. 2013. An experimental and computational evolution-based method to study a mode of co-evolution of overlapping open reading frames in the AAV2 viral genome. PLoS One 8:e66211. doi: 10.1371/journal.pone.0066211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gallo-Ramirez LE, Ramirez OT, Palomares LA. 2011. Intracellular localization of adeno-associated viral proteins expressed in insect cells. Biotechnol Prog 27:483–493. doi: 10.1002/btpr.565. [DOI] [PubMed] [Google Scholar]
- 40.Rohrmann GF. 2013. Baculovirus molecular biology, 3rd ed National Center for Biotechnology Information, Bethesda, MD. [PubMed] [Google Scholar]
- 41.Lombardo E, Ramirez JC, Garcia J, Almendral JM. 2002. Complementary roles of multiple nuclear targeting signals in the capsid proteins of the parvovirus minute virus of mice during assembly and onset of infection. J Virol 76:7049–7059. doi: 10.1128/JVI.76.14.7049-7059.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Riolobos L, Reguera J, Mateu MG, Almendral JM. 2006. Nuclear transport of trimeric assembly intermediates exerts a morphogenetic control on the icosahedral parvovirus capsid. J Mol Biol 357:1026–1038. doi: 10.1016/j.jmb.2006.01.019. [DOI] [PubMed] [Google Scholar]
- 43.Gil-Ranedo J, Hernando E, Riolobos L, Dominguez C, Kann M, Almendral JM. 2015. The mammalian cell cycle regulates parvovirus nuclear capsid assembly. PLoS Pathog 11:e1004920. doi: 10.1371/journal.ppat.1004920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schurmann N, Trabuco LG, Bender C, Russell RB, Grimm D. 2013. Molecular dissection of human Argonaute proteins by DNA shuffling. Nat Struct Mol Biol 20:818–826. doi: 10.1038/nsmb.2607. [DOI] [PubMed] [Google Scholar]
- 45.Samulski RJ, Chang LS, Shenk T. 1987. A recombinant plasmid from which an infectious adeno-associated virus genome can be excised in vitro and its use to study viral replication. J Virol 61:3096–3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matsushita T, Elliger S, Elliger C, Podsakoff G, Villarreal L, Kurtzman GJ, Iwaki Y, Colosi P. 1998. Adeno-associated virus vectors can be efficiently produced without helper virus. Gene Ther 5:938–945. doi: 10.1038/sj.gt.3300680. [DOI] [PubMed] [Google Scholar]
- 47.Grimm D, Streetz KL, Jopling CL, Storm TA, Pandey K, Davis CR, Marion P, Salazar F, Kay MA. 2006. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 441:537–541. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 48.D'Costa S, Blouin V, Broucque F, Penaud-Budloo M, Francois A, Perez IC, Le Bec C, Moullier P, Snyder RO, Ayuso E. 2016. Practical utilization of recombinant AAV vector reference standards: focus on vector genomes titration by free ITR qPCR. Mol Ther Methods Clin Dev 5:16019. doi: 10.1038/mtm.2016.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wistuba A, Weger S, Kern A, Kleinschmidt JA. 1995. Intermediates of adeno-associated virus type 2 assembly: identification of soluble complexes containing Rep and Cap proteins. J Virol 69:5311–5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Borner K, Niopek D, Cotugno G, Kaldenbach M, Pankert T, Willemsen J, Zhang X, Schurmann N, Mockenhaupt S, Serva A, Hiet MS, Wiedtke E, Castoldi M, Starkuviene V, Erfle H, Gilbert DF, Bartenschlager R, Boutros M, Binder M, Streetz K, Krausslich HG, Grimm D. 2013. Robust RNAi enhancement via human Argonaute-2 overexpression from plasmids, viral vectors and cell lines. Nucleic Acids Res 41:e199. doi: 10.1093/nar/gkt836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Borner K, Hermle J, Sommer C, Brown NP, Knapp B, Glass B, Kunkel J, Torralba G, Reymann J, Beil N, Beneke J, Pepperkok R, Schneider R, Ludwig T, Hausmann M, Hamprecht F, Erfle H, Kaderali L, Krausslich HG, Lehmann MJ. 2010. From experimental setup to bioinformatics: an RNAi screening platform to identify host factors involved in HIV-1 replication. Biotechnol J 5:39–49. doi: 10.1002/biot.200900226. [DOI] [PubMed] [Google Scholar]
- 52.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.