

ABSTRACT
Viral manipulation of cellular proteins allows viruses to suppress host defenses and generate infectious progeny. Due to the linear double-stranded DNA nature of the adenovirus genome, the cellular DNA damage response (DDR) is considered a barrier to successful infection. The adenovirus genome is packaged with protein VII, a virally encoded histone-like core protein that is suggested to protect incoming viral genomes from detection by the cellular DNA damage machinery. We showed that protein VII localizes to host chromatin during infection, leading us to hypothesize that protein VII may affect DNA damage responses on the cellular genome. Here we show that protein VII at cellular chromatin results in a significant decrease in accumulation of phosphorylated H2AX (γH2AX) following irradiation, indicating that protein VII inhibits DDR signaling. The oncoprotein SET was recently suggested to modulate the DDR by affecting access of repair proteins to chromatin. Since protein VII binds SET, we investigated a role for SET in DDR inhibition by protein VII. We show that knockdown of SET partially rescues the protein VII-induced decrease in γH2AX accumulation on the host genome, suggesting that SET is required for inhibition. Finally, we show that knockdown of SET also allows ATM to localize to incoming viral genomes bound by protein VII during infection with a mutant lacking early region E4. Together, our data suggest that the protein VII-SET interaction contributes to DDR evasion by adenovirus. Our results provide an additional example of a strategy used by adenovirus to abrogate the host DDR and show how viruses can modify cellular processes through manipulation of host chromatin.
IMPORTANCE The DNA damage response (DDR) is a cellular network that is crucial for maintaining genome integrity. DNA viruses replicating in the nucleus challenge the resident genome and must overcome cellular responses, including the DDR. Adenoviruses are prevalent human pathogens that can cause a multitude of diseases, such as respiratory infections and conjunctivitis. Here we describe how a small adenovirus core protein that localizes to host chromatin during infection can globally downregulate the DDR. Our study focuses on key players in the damage signaling pathway and highlights how viral manipulation of chromatin may influence access of DDR proteins to the host genome.
KEYWORDS: adenovirus, DNA damage, chromatin, histone-like proteins
INTRODUCTION
The double-stranded linear DNA genome of adenovirus (Ad) is packaged with several core proteins, the most abundant of which is the histone-like protein VII (1–3). Infection and replication of the viral DNA genome have been suggested to elicit a damage response through recognition by host cell proteins (4–7). Host DNA repair proteins and damage signaling pathways can inhibit viral DNA replication (6, 8–11). Thus, adenovirus has evolved numerous mechanisms to manipulate the cellular DNA damage response (DDR) during infection (4, 5, 7, 10, 12–16). The DDR is a complex signal transduction network that senses DNA damage and transduces this information to a myriad of downstream proteins which then effect repair (17, 18). Depending on the type of damage incurred, different members of the phosphatidylinositol 3-kinase-like protein kinase (PIKK) family of damage transducers (ATM, ATR, and DNA-PK) are activated and then phosphorylate numerous protein targets (18, 19). Damage sensors include the MRN (Mre11, Rad50, and Nbs1) complex, which can recognize double-strand DNA breaks and promote ATM activation (20–22). ATM becomes activated by autophosphorylation, and the histone variant H2AX is one of the earliest substrates to be phosphorylated (23, 24). Together, the H2AX phosphorylation mark at serine 139 (γH2AX) and activated ATM phosphorylated at serine 1981 (pATM) are considered classic markers of double-strand breaks and DDR activation (23, 24). Spreading of phosphorylated H2AX (γH2AX) around the damage site leads to recruitment of mediators, such as MDC1 (25, 26). Mediators further amplify the signaling cascade, leading to activation of downstream effectors (17, 27–29). The γH2AX mark can extend up to 1 Mb around the break, and this spreading is thought to facilitate amplification of the damage signal and recruitment of further downstream proteins (27). This cascade leads to activation of cell cycle checkpoints and arrest until the damage is repaired, or it may signal apoptosis.
Chromatin compaction of the cellular genome has recently come to light as a factor influencing the efficiency of the DNA damage response (30–35). At sites of heterochromatin, compaction serves to protect DNA from damage, but this compaction can restrict access of repair proteins when lesions do occur (30, 36, 37). The oncoprotein SET was recently implicated in mediating heterochromatin formation at double-strand breaks through recruitment of factors such as HP1γ (38). In addition, SET was shown to localize to incoming adenovirus genomes, was suggested to facilitate deposition of histones to promote transcription on viral genomes, and binds viral protein VII (39–43). However, the ability of SET to modulate heterochromatin and the DDR on viral genomes has not yet been investigated. It is important to note that the oncoprotein SET, also known as TAF-1 (for template activating factor) (44), does not contain a histone methyltransferase domain (45) and should not be confused with this family of enzymes.
Protein VII coats packaged viral genomes in what has been described as a “beads on a string” assembly (3, 46–48). Although structural details remain unclear, protein VII is not required for virion formation (49). Protein VII is a small basic protein rich in arginine and lysine residues (50), but does not appear to contain a histone fold domain. Due to its high positive charge, protein VII binds DNA (3, 51) and has been well described to bind the acidic C terminus of SET (41–43, 52). We recently reported that protein VII also binds the nuclear factor and immune danger signal HMGB1 (51), which similarly contains an acidic C terminus (53). It has been suggested that protein VII is removed from viral genomes and replaced by histones, allowing effective transcription to begin (40, 54, 55). Once early viral proteins are expressed, adenovirus (Ad) E1b55K and E4orf6 form an E3 ubiquitin ligase complex that targets several host proteins for degradation, including MRN and DNA ligase IV (4, 12, 56–60). To study the DDR during infection, viral mutants deficient in the E4 region have been employed (6–8, 15, 16, 61). By use of a mutant virus deficient in E4 proteins, protein VII was suggested to protect incoming viral genomes from recognition by the DDR (61). However, the relationship between the DDR and the removal of protein VII from viral genomes is currently unclear.
In light of the above-mentioned studies, we investigated whether protein VII in concert with SET can modulate the DDR on both viral and host genomes. We recently reported that protein VII localizes to the host genome during infection, where it directly binds nucleosomes and leads to changes in the appearance and composition of cellular chromatin (51). Here we show that protein VII is sufficient to block accumulation of γH2AX upon irradiation of cellular DNA. We found that this block occurs after the autophosphorylation of ATM but before MDC1 recruitment and is dependent on chromatin localization of protein VII. We further investigated whether binding partners of protein VII are involved in limiting the DDR and found that SET, but not HMGB1, contributes to the DDR block. Finally, we show that SET is also required to block pATM association with viral genomes early during infection. Taken together, our data suggest that protein VII limits the DDR through recruitment of SET to both the viral and host genomes. Our results highlight how viral proteins can manipulate cellular processes through chromatin and reveal a previously unknown mechanism of DDR inhibition by adenovirus.
RESULTS
Protein VII downregulates accumulation of γH2AX foci on cellular chromatin.
Protein VII on incoming viral genomes is thought to block activation of the DDR (61). We therefore hypothesized that protein VII may also affect the damage response on the cellular genome. To investigate the role of protein VII on the host genome, we utilized an inducible cell line that expresses hemagglutinin (HA)-tagged protein VII upon treatment with doxycycline. Expression of protein VII results in a characteristic increase in nuclear size and a punctate pattern of DAPI (4′,6-diamidino-2-phenylindole)-stained DNA (51). To investigate the impact of protein VII on the DDR, we expressed protein VII in our inducible cell line and then irradiated cells to cause double-strand DNA breaks. We used Western blot analysis to investigate the levels of the classic markers of double-strand breaks, i.e., H2AX phosphorylated at serine 139 (γH2AX) and activated ATM phosphorylated at serine 1981 (pATM) (17, 23, 24, 62). We observed a dramatic decrease in accumulation of γH2AX and pATM in the presence of protein VII at 2 h postirradiation (Fig. 1A). We then visualized γH2AX by immunofluorescence assay and observed significant decreases in the number and intensity of foci at 1, 5, and 10 Gy of irradiation upon protein VII expression (Fig. 1B). At 20 Gy, the quantification of γH2AX immunofluorescence showed more individual foci in the presence of protein VII, although each focus was smaller and less intense than those in control cells (Fig. 1B), consistent with the total decrease in γH2AX observed by immunoblotting. To represent the robust difference, we therefore chose to use 10 Gy of irradiation for the rest of this study. We also confirmed by pulsed-field gel electrophoresis that the presence of protein VII did not affect the amount of fragmented DNA under each irradiated condition (data not shown). To determine whether the lack of γH2AX in protein VII-expressing cells was dependent on the response time, we carried out a time course assay with 10 Gy of irradiation. In the presence of protein VII, we did not detect accumulation of γH2AX at time points up to 4 h postirradiation (Fig. 1C). These data indicate that expression of protein VII results in decreased accumulation of γH2AX and pATM upon DNA damage, suggesting that protein VII limits a robust DDR.
FIG 1.

Protein VII expression results in downregulation of the DDR postirradiation. (A) Western blot analysis after irradiation (IR) for A549 cells with inducible protein VII-HA. Cells were treated with doxycycline (dox) for 4 days to induce protein VII expression prior to treatment (0, 1, 5, 10, or 20 Gy). Samples were collected 2 h after irradiation and probed for proteins or phospho-specific marks as indicated. Decreases in γH2AX and pATM were observed in the presence of protein VII-HA. Actin is shown as a loading control. (B) Immunofluorescence assay and quantification under the same conditions as those for panel A. γH2AX is shown in red and DAPI staining in gray to indicate the change in chromatin structure characteristic of protein VII expression. Bar, 10 μm. Quantification of the number of γH2AX foci per nucleus was carried out for each condition, as indicated. Significantly fewer foci were observed in cells expressing protein VII-HA (orange dots) than in no-dox control cells (gray dots). Each dot represents one cell (n = 25 per condition). The differences are statistically significant within each dose (**, P < 0.0001 by two-tailed t test). We also employed a Poisson-based regression which demonstrated that the trend across the doses was significantly different (P < 2e−16). (C) Western blot analysis over the time course after irradiation with 10 Gy, with or without dox treatment to express protein VII-HA. The γH2AX signal displayed a steady increase for up to 1 h and then a decrease toward 4 h in control cells (no dox), whereas no γH2AX signal was observed in the presence of protein VII-HA. Total H2AX and tubulin are shown as loading controls.
Protein VII blocks the DDR after ATM phosphorylation but before MDC1 accumulation.
We hypothesized that protein VII may inhibit signaling required for recruitment of downstream DDR proteins or may block access to sites of damaged DNA. We investigated whether downstream DDR proteins accumulate at the few remaining foci that are present in protein VII-expressing cells. We observed that pATM colocalizes with γH2AX foci both in control cells and in cells expressing protein VII (Fig. 2A). We quantified the colocalization coefficient for γH2AX and pATM and found that despite the smaller number of total foci, the colocalization was not significantly different in the presence of protein VII. To investigate the recruitment of downstream DDR proteins, we next examined colocalization of the mediator MDC1 with γH2AX foci. MDC1 functions to amplify DDR signaling by recruiting additional proteins to damage sites (26–29). We found that in the presence of protein VII, the colocalization coefficient for γH2AX and MDC1 was significantly reduced compared to that for control cells (Fig. 2B). These data indicate that protein VII blocks recruitment of DDR proteins downstream of pATM, suggesting an impairment of repair signaling.
FIG 2.

Protein VII blocks the DDR after ATM autophosphorylation but before MDC1 recruitment. (A) Immunofluorescence assay of pATM (green) and γH2AX (red) 30 min and 4 h after irradiation with 10 Gy in the absence (no dox) or presence (+ dox) of protein VII-HA expression. DAPI staining is shown in blue. The top panels show multiple cells, while the bottom zoom shows one nucleus, as indicated by the arrowhead in the 4-h γH2AX image. Yellow in the merge image indicates colocalization of the red and green channels. Quantification of the colocalization coefficient indicated no significant difference between γH2AX and pATM colocalization in the presence or absence of protein VII. (B) Same as for panel A, but for γH2AX and MDC1. Quantification indicated a significant decrease in the colocalization coefficient of γH2AX and MDC1 upon protein VII induction. Each dot represents one cell (n = 25 per condition), **, P < 0.0001 by two-tailed t test; n.s., not significant. Bars, 10 μm.
Protein VII localization to chromatin contributes to downregulation of the DDR.
To determine whether the decrease in damage signaling upon protein VII expression was caused by protein VII accumulation at chromatin, we examined whether the chromatin distortion caused by protein VII correlates with downregulation of the cellular response. We induced protein VII expression by doxycycline treatment over the course of 4 days, irradiated cells, and visualized γH2AX foci by immunofluorescence assay. We compared equivalent doxycycline treatment, irradiation, and γH2AX immunofluorescence conditions in parental A549 cells as a control (Fig. 3A). We observed an inverse correlation between the protein VII effect on chromatin and γH2AX accumulation: as the effect of protein VII on host chromatin increased, the γH2AX signal upon damage decreased, which did not occur in the control cells. We also observed the protein VII-induced decrease in total γH2AX upon irradiation by Western blotting (Fig. 3B). To determine whether localization of protein VII to chromatin contributes to downregulation of the DDR, we utilized point mutants previously shown to affect protein VII localization (51). Alanine substitution of specific posttranslationally modified (PTM) residues results in nucleolar localization of protein VII (51). We used an immunofluorescence assay to analyze whether these nucleolar mutants affect γH2AX accumulation similarly to wild-type protein VII. We found that mutating all five PTM sites resulted in a protein VII mutant (ΔPTM) that did not affect the appearance of chromatin or the γH2AX level (Fig. 3C and D). Two lysines found to be acetylated contribute significantly to chromatin localization of protein VII (51). We found that mutation of these lysines (K2AK3A) resulted in a partial effect on chromatin and normal levels of γH2AX after damage. Replacement of the K3 residue with the acetyl mimic glutamine generated a mutant (K3Q) that affected the chromatin appearance similarly to wild-type protein VII and resulted in a decrease in γH2AX (Fig. 3C and D). Taken together, these data suggest that chromatin localization of protein VII is required for downregulation of the DDR.
FIG 3.

Downregulation of the DDR by protein VII correlates with chromatin localization. (A) Immunofluorescence assay showing γH2AX (red) and DAPI (gray) staining of either parental A549 cells or cells with inducible protein VII-HA. All cells were treated with doxycycline for the indicated number of days (d = days with dox), irradiated with 10 Gy, and fixed at 2 h postirradiation (IR). DAPI staining (gray) is shown to indicate the change in chromatin structure observed after 3 days of protein VII expression. Bar, 10 μm. (B) Western blot analysis of the samples from panel A, showing that the increase in protein VII-HA levels over the course of doxycycline treatment correlates with the γH2AX level decrease. (C) Immunofluorescence assay of irradiated (10 Gy) cells expressing wild-type (WT) protein VII or the indicated mutants with C-terminal HA tags, showing γH2AX (red), HA (green), and DAPI (gray) staining. γH2AX was evident only in cells that did not have chromatin-localized protein VII. Bar, 10 μm. DAPI is shown in blue in the merged images. (D) Western blot analysis of the samples from panel C, probed with the indicated antibodies.
The protein VII interactor HMGB1 is not required for the protein VII-mediated block to the DDR.
In our previous report, we found that protein VII significantly altered the association of SET and the HMGB family proteins with chromatin (51). To investigate the mechanism of protein VII's effect on the DDR, we examined the roles of these known binding partners of protein VII. Since the protein VII interaction partner HMGB1 has a reported role in the DDR (63–65), we asked whether HMGB1 is required for protein VII to downregulate the DDR on host DNA. We used clustered regularly interspaced short palindromic repeat-Cas9 (CRISPR-Cas9) technology to knock out HMGB1 in A549 cells, expressed protein VII by using a recombinant vector (66), and investigated the levels of γH2AX postirradiation. We found that despite the loss of HMGB1, protein VII still decreased γH2AX levels at all doses of irradiation, most notably with 10 Gy, in each cell line (Fig. 4A). We visualized the transduction efficiency of our protein VII-green fluorescent protein (protein VII-GFP) vector and observed that not all cells expressed the fluorescent protein due to variability in transduction efficiency. Nevertheless, we found that in cells that expressed protein VII-GFP, γH2AX foci were markedly reduced after 10 Gy of irradiation. Conversely, in cells within the same population that did not express protein VII-GFP, the γH2AX foci accumulated normally (Fig. 4B). Thus, the loss of HMGB1 appears to have no effect on the downregulation of γH2AX caused by protein VII.
FIG 4.

HMGB1 is not required for DDR downregulation by protein VII. (A) Western blot analysis of the indicated proteins upon irradiation for A549 cells expressing protein VII-GFP and with HMGB1 deleted. Cells were harvested at 2 h postirradiation. Deletion of HMGB1 did not affect the ability of protein VII-GFP to decrease γH2AX accumulation postirradiation. (B) Immunofluorescence assay of the 10-Gy-treated samples from panel A, showing γH2AX (red), protein VII-GFP (green), and DAPI (blue in merged images) staining. Cells that expressed protein VII-GFP did not accumulate γH2AX irrespective of the HMGB1 level, whereas those that did not express protein VII-GFP show a normal γH2AX signal. Dotted circles indicate nuclei that are present. Bar, 10 μm.
The protein VII interactor SET contributes to downregulation of the DDR.
The oncoprotein SET (also known as TAF-1) was shown to interact with protein VII (42, 52) and to localize to incoming viral genomes (39, 41). SET was also recently reported to modulate the damage response on the cellular genome by affecting access of DDR proteins to damage sites (38). We therefore investigated whether protein VII is able to downregulate the DDR in the absence of SET. Knockdown of SET led to an increase in γH2AX levels in the absence of any treatment (Fig. 5, compare lanes 1 and 4), as reported by Kalousi et al. (38). Upon irradiation, we found that the levels of γH2AX were higher in the absence of SET than in irradiated control cells (Fig. 5, compare lanes 2 and 3 with lanes 5 and 6). In cells with SET, induction of protein VII blunted the response to irradiation, as observed by a reduced γH2AX level (Fig. 5, compare lanes 3 and 9). The degree of block achieved by protein VII for γH2AX in irradiated cells was lower in the absence of SET (Fig. 5, compare lanes 8 and 9 with lanes 11 and 12). It should be noted that the baseline levels of γH2AX were increased in the absence of irradiation upon SET knockdown alone, regardless of the presence of protein VII (Fig. 5, compare lanes 1 and 7 with lanes 4 and 10). Since protein VII did not prevent the increase in γH2AX when SET was depleted, this result implies that SET is required for protein VII to block the signaling event. Taken together, these results suggest that SET plays a role in downregulation of the DDR by protein VII.
FIG 5.
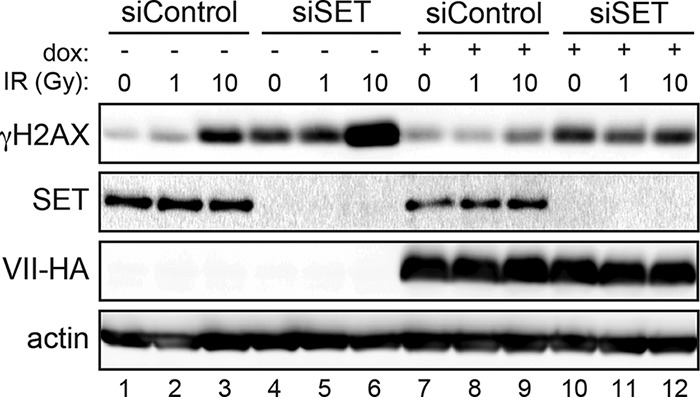
SET contributes to protein VII downregulation of the DDR. The Western blot shows γH2AX levels upon SET knockdown in the presence or absence of protein VII-HA, with or without irradiation (0, 1, or 10 Gy), as indicated. A549 cells were transfected with siRNAs, incubated for 3 days, and then induced with doxycycline for 4 days. On the first day of induction, cells were transfected a second time to ensure efficient knockdown. On the fourth day, cells were irradiated at the doses indicated and harvested at 2 h postirradiation. Loss of SET led to higher γH2AX levels under all conditions (compare lane 3 with lane 10 and lane 9 with lane 12). Actin is shown as a loading control.
SET is required for protein VII to prevent ATM recognition of incoming viral genomes.
Protein VII was reported to protect incoming viral genomes from association with the DDR kinase ATM (61). This was determined using an adenovirus deficient in E4 proteins such that it cannot inactivate the damage response through degradation or mislocalization of MRN (61, 67). Karen and Hearing showed by immunofluorescence assay that pATM and protein VII are mutually exclusive on early viral genomes (61). They confirmed that both protein VII and pATM foci colocalized with 5-ethynyl-2′-deoxyuridine (EdU)-labeled viral genomes but not with each other. Therefore, they concluded that protein VII prevents pATM from binding to viral genomes. Based on our results and the finding that SET also localizes to incoming viral genomes (39, 41), we hypothesized that protein VII may recruit SET to incoming viral genomes to prevent recognition by ATM. To test this hypothesis, we first titrated the multiplicity of infection (MOI) to visualize incoming protein VII from an E4-deleted virus at 10 h postinfection (Fig. 6A and B). Similar to the experiment first described by Karen and Hearing, we found that increasing the MOI resulted in more protein VII foci, confirming that foci represent incoming viral genomes bound by protein VII. We chose to use an MOI of 100 to test the role of SET in pATM colocalization with these discrete foci. It is important that the antibody and virus used here differ from those used by Karen and Hearing, and the precise staining patterns are slightly different. However, the images shown here are comparable to those previously reported in that pATM and protein VII do not overlap spatially in siControl-infected cells (Fig. 6B). We used small interfering RNA (siRNA) to knock down SET, infected cells with an E4-deleted virus, and visualized pATM and protein VII at 10 h postinfection. We showed mutually exclusive protein VII and pATM staining on incoming viral genomes in our control cells. Conversely, we found that protein VII and pATM colocalized in the absence of SET under the same conditions (Fig. 6B). We quantified the colocalization coefficient and found significantly more colocalization of protein VII and pATM in the absence of SET (Fig. 6B). Taken together, our data suggest that protein VII recruits SET to incoming viral genomes, where it limits activation of the DDR.
FIG 6.

Protein VII requires SET on incoming viral genomes to disrupt detection by pATM. (A) Immunofluorescence assay of protein VII (red) visualized on incoming viral genomes at increasing MOIs. The higher the MOI, the more discrete foci are visible. (Right) Infection with the E4-deleted mutant dl1004 was fixed at 10 h postinfection, and the intensity across four separate images was quantified using ImageJ. (B) Immunofluorescence assay of pATM (green) and protein VII (red) on incoming viral genomes (E4-deleted mutant dl1004) upon knockdown of SET. Quantification of the colocalization coefficient showed a significant increase in colocalization of protein VII with pATM in the absence of SET. Each dot represents one cell (n = 29 per condition). **, P < 0.0001 by two-tailed t test. Bars, 10 μm.
DISCUSSION
Adenovirus infection systematically inactivates the host DDR signaling and repair pathways (7). This inactivation is considered to block the activity of DNA repair proteins on viral genomes, thereby facilitating efficient viral replication (6–8, 10, 11). Incoming viral genomes are “chromatinized” to incorporate histones, and it is thought that repressive protein VII-only regions are removed (40, 54, 55). The resulting “viral chromatin” is not well understood but is highly active and likely represents open or easily accessible DNA. The host damage response is more effective when DNA is easily accessible (31–33), making the viral genome a prime trigger for damage signaling. Thus, the virus has evolved multiple mechanisms for manipulating the host DDR (7). In this study, we have demonstrated that the histone-like protein VII can blunt DDR activation on both viral and host DNA genomes. Our investigation of the molecular mechanism suggests a role for the nuclear oncoprotein SET, which is recruited to DNA through binding to protein VII (Fig. 7).
FIG 7.

Model of protein VII and SET on host and viral genomes. (A) Model of protein VII blocking ATM recognition of viral genomes through SET. Protein VII on incoming viral genomes forms “beads on a string”-like structures that are replaced by histones. The protein VII-bound genome recruits SET and blocks ATM from associating with the viral genome. Upon knockdown of SET, ATM is now able to phosphorylate H2AX on nucleosomes assembled on the viral genome. (B) Model of protein VII blocking pATM spread on the host genome. pATM colocalizes with γH2AX on the host genome upon irradiation, but protein VII binding nucleosomes with SET blocks the spread of γH2AX and the recruitment of downstream DDR proteins. Upon knockdown of SET, pATM is able to access neighboring H2AX histones and leads to a more robust DDR.
It has been observed that removal of protein VII from incoming viral genomes correlates with the beginning of viral transcription (54, 68). On early viral genomes, SET has been reported both to act as a histone chaperone to deposit histones and to activate transcription (44, 69). SET binds directly to protein VII, presumably on incoming viral genomes, but the mechanism of protein VII replacement by histones is unclear. Chromatin immunoprecipitation followed by PCR (ChIP-PCR) showed that viral genomes contain both histones and protein VII (55), suggesting that viral chromatin may contain regions exclusively comprised of either protein VII or histones. However, ChIP experiments cannot distinguish between multiple copies of the viral genome, and it is therefore possible that some genomes have protein VII only, whereas others have only histones. After viral transcription begins, protein VII has also been suggested to bring E1A to the viral genome and to promote transcription activation, since protein VII and E1A directly interact on DNA (54). These data have not been reconciled with protein VII removal from viral genomes or SET-facilitated deposition of histones. Regardless of the order of events, it is clear from the previous work of Karen and Hearing (61) and the data reported here that the protein VII-SET complex on the viral genome is key to preventing pATM activation and accumulation of γH2AX (Fig. 7A). These observations come from mutant viruses that lack the E4 region and therefore have a crippled ability to suppress the damage response through the known interactions of E4 proteins (E4orf3 and E4orf6) with host repair pathways. Although γH2AX cannot spread to the length of megabases on the 36-kb viral genome, as noted by Shah and O'Shea (6), blocking recruitment of downstream DDR proteins may still be crucial to prevent inhibition of viral DNA replication, and potentially processing of viral genomes into end-to-end joined concatemers (4, 12, 70).
In addition to their impact on proteins that associate with viral genomes, adenovirus proteins can also affect histones and chromatin-associated proteins on the host genome (51, 71–74). For example, the viral E1A transactivator can affect specific chromatin marks, such as decreasing global histone H3 lysine 18 acetylation (H3K18ac) and increasing nucleosome density to promote transcriptional repression at specific antiviral gene promoters (72, 75). In addition, mass spectrometry studies have identified significant changes to global histone modification patterns during adenovirus infection (76). Host chromatin contains regions with differing degrees of chromatin accessibility, including “open” or euchromatin that is highly transcriptionally active and multiple states of closed or repressed heterochromatin. Expression of protein VII results in a characteristic change to host chromatin appearance that may result from directly binding to nucleosomes (51), but it is not yet clear how protein VII affects the extent of chromatin accessibility. We recently reported that high levels of protein VII expressed late during infection accumulate in the host chromatin, retaining the host proteins SET and HMGB family members (51). Here we have shown that protein VII on the host genome is sufficient to block accumulation of γH2AX and recruitment of MDC1. By blocking the spread of γH2AX, protein VII robustly inhibits the damage response on the host genome (Fig. 7). It is clear from our results that loss of SET, but not HMGB1, results in higher levels of γH2AX in the presence of protein VII. This result can be interpreted in two ways: (i) protein VII may be unable to block γH2AX spread in the absence of SET, or (ii) loss of SET leads to an increase in γH2AX that is still somewhat suppressed by protein VII. We are unable to distinguish between these two possibilities. The second possible explanation is consistent with the result observed by Kalousi et al. (38) showing that knockdown of SET results in an increase in γH2AX. But since protein VII does not prevent the increase in γH2AX when SET is depleted, we interpret this to mean that SET is required for protein VII to block the signaling event. Therefore, given the well-established binding of protein VII to SET, we conclude that the accumulation of γH2AX observed upon knockdown of SET indicates a role for SET in DDR inhibition by protein VII on the host genome.
The abundance of protein VII on the host genome during adenovirus infection can lead to several perturbations within host nuclei. The impacts of protein VII on host responses and gene expression patterns are only beginning to be understood. We have suggested that protein VII alters the composition of host chromatin to prevent downstream inflammation through HMGB1 (51), and here we show that retention of SET affects DDR signaling. Our model, building upon previous findings, proposes that protein VII blocks the spread of γH2AX on the host genome (Fig. 7B). Although adenovirus has been suggested to activate global γH2AX on the host genome (77), we suggest that abundant protein VII on the host genome may block the spread of γH2AX during infection to prevent downstream events that could inhibit viral replication. Since protein VII is produced in the late phase of infection, it accumulates in the host chromatin only after replication has begun, suggesting that the functional relevance of downregulating the DDR on the host genome may be important for late stages of infection. In addition, it should be noted that protein VII is expressed as a larger precursor protein that is proteolytically cleaved after the first 24 amino acids by a viral protease (50, 78), and only the mature version localizes to host chromatin (51, 66). Future studies will determine the roles of other proteins, such as linker histones, in chromatin manipulation by adenovirus and will investigate the impact of protein VII on other functions of SET and HMGB1. This study advances our understanding of protein VII's impact on host genome processes while highlighting the importance of chromatin on both the viral and host genomes.
MATERIALS AND METHODS
Cell culture.
A549 and 293T cells were purchased from the American Type Culture Collection (ATCC) and cultured in Kaighn's modification of Ham's F-12 medium (F-12K) containing 100 U/ml of penicillin and 100 μg/ml of streptomycin and supplemented with 10% fetal bovine serum (FBS) as recommended by the provider. Protein VII-inducible cells were generated and cultured as previously described (51, 79). Doxycycline was administered at 0.2 μg/ml and replenished every 24 h for 4 days unless otherwise indicated.
Construction of HMGB1 knockout A549 cells.
Small guide RNAs (sgRNAs) for HMGB1 knockout were chosen from a public sgRNA database (80). A total of 3 sgRNA sequence pairs were used for HMGB1, one of which resulted in HMGB1 knockout (forward primer, CACCGGGCGATACTCAGAGCAGAAG; and reverse primer, AAACTTCTGCTCTGAGTATCGCCC). These primers (IDT) were annealed and inserted into the pLentiCRISPR plasmid (Addgene) according to a protocol available from the Gecko lentiviral CRISPR toolbox (http://sanjanalab.org/library/protocol_lentiOligo.pdf). HMGB1 was knocked out either by stable transduction of A549 cells with a lentivirus containing the HMGB1-LentiCRISPR construct [HMGB1 (−/−) 1] or by transfection of the plasmid [HMGB1 (−/−) 2]. To generate the lentivirus, 1.2 × 106 293T cells were transfected with 4 μg of the HMGB1-LentiCRISPR plasmid together with 2.5 μg pMDL, 1.5 μg pVSVg, and 1 μg pREV in 250 μl of Opti-MEM (Gibco) and 13.5 μl of Lipofectamine 2000 (Invitrogen) in 250 μl of Opti-MEM. Supernatant containing the lentivirus was collected at 24 and 48 h posttransfection, combined, and passed through a 0.45-μm filter (Life Technologies). Lentivirus was used to transduce A549 cells (1.2 × 106 cells) by addition of 750 μl of virus combined with 750 μl of medium and 1.5 μl 10-mg/ml Polybrene (Santa Cruz), followed by spinoculation for 30 min at 1,500 relative centrifugal force (RCF) and 37°C. At 48 h posttransduction, cells were cultured in medium containing 1 μg/ml of puromycin (Clontech) for 7 days. Puromycin-selected cells were serially diluted into 96-well plates to achieve single-cell clones. For the transfection strategy, A549 cells (0.8 × 106 cells) were transfected with 3 μg of the HMGB1-LentiCRISPR plasmid in 250 μl of Opti-MEM and 7.5 μl of Lipofectamine 2000 in 250 μl of Opti-MEM. At 24 h posttransfection, cells were cultured in medium containing 1 μg/ml of puromycin for 3 days. Puromycin-selected cells were serially diluted into 96-well plates to achieve single-cell clones. HMGB1 knockout in the cell lines was confirmed using immunoblotting, sequencing of the genomic locus (forward primer, GGACAAGGCCCGTTATGAAAG; and reverse primer, CAGCTTCGCAGCCTTCTTTTC), and RT-qPCR for the HMGB1 mRNA (forward primer, TAACTAAACATGGGCAAAGGAG; and reverse primer, TAGCAGACATGGTCTTCCAC).
Immunofluorescence assay.
Cells were seeded on glass coverslips and treated as indicated. Protein VII was induced by doxycycline treatment at 0.2 μg/ml. Cells were irradiated at the indicated dose. Cells were harvested for immunofluorescence assay at the indicated times following irradiation and prepared by standard methods (81). Briefly, coverslips were washed in Dulbecco's phosphate-buffered saline (PBS) and fixed in cold 4% paraformaldehyde for 15 min. The coverslips were permeabilized with 0.5% Triton-X in PBS, blocked in 3% bovine serum albumin (BSA) in PBS, incubated with primary antibodies (see “Antibodies”) for 1 h in 3% BSA in PBS, washed three times in blocking buffer, incubated with secondary antibodies (Life Technologies) and 4′,6-diamidino-2-phenylindole (DAPI) for 1 h, and washed again with PBS. The coverslips were then mounted using ProLong Gold antifade reagent (Life Technologies) and visualized with a Zeiss LSM 710 confocal microscope (Cell and Developmental Microscopy Core at the University of Pennsylvania). Images were processed using ImageJ and Adobe CS6.
Antibodies.
Commercially available antibodies were purchased through Abcam (ATM [ab32420], GFP [ab290], phosphorylated S139 H2AX [ab111174], HMGB1 [ab18256], MDC1 [ab11169], pATM [ab81292], and SET [ab1183]), BioLegend (HA [901514]), Cell Signaling (H2AX [2595S]), Millipore (phosphorylated S139 H2AX [05-636]), Santa Cruz (HA [sc-805] and tubulin [sc-69969]), and Sigma (actin [A5441]). The protein VII antibody was a generous gift from H. Wodrich (39). Secondary antibodies used for immunoblotting were purchased from Jackson ImmunoResearch, and secondary antibodies used for immunofluorescence assay were purchased from Life Technologies.
Immunoblotting.
Western blot analysis was carried out using standard methods. Briefly, samples were prepared with lithium dodecyl sulfate (LDS) sample buffer (NuPage) and boiled for 10 min. Total protein lysates of equal concentrations were separated by SDS-PAGE and transferred to a nitrocellulose membrane (Amersham) at 30 V for a minimum of 40 min. Even protein loading was confirmed by Ponceau staining, and the membrane was blocked in 5% milk in Tris-buffered saline with Tween (TBST) containing 0.1% azide for 30 min. Membranes were incubated overnight with primary antibodies, washed with TBST for 30 min, and incubated with secondary antibodies conjugated to horseradish peroxidase (Jackson ImmunoResearch) for 1 h. The membranes were then washed in TBST, and the proteins were detected with enhanced chemiluminescence (ECL) Western blotting substrate (Thermo Scientific) on a Syngene G-Box.
Transduction and infection.
For expression of protein VII-GFP, A549 cells and HMGB1 knockout cells were transduced with a recombinant adenovirus vector according to standard protocols, as previously described (51, 66). A multiplicity of infection (MOI) was used that resulted in approximately 50 to 60% of the cells expressing protein VII-GFP at 24 h posttransduction. For infection using the E4-deleted adenovirus dl1004 (82), A549 cells were seeded on glass coverslips, infected at an MOI of 100, adsorbed for 1 h before addition of complete medium, and fixed using 4% paraformaldehyde at 10 h postinfection to recapitulate the previously reported conditions (61).
Quantification of immunofluorescence.
For colocalization analysis, representative cells were quantified using ImageJ on the red and green channels, using the standard method (83). Each representative cell was selected individually, and the colocalization coefficient (Pearson's correlation coefficient) was calculated. For each condition analyzed, the minimum number of cells analyzed was 25 (n ≥ 25), and the results were plotted using GraphPad Prism. To score the total number of γH2AX foci per cell, images were quantified using ImageJ on the blue and red channels, using standard methods (84). The background was removed, a threshold was set, and any cytoplasmic pixels were set to zero, using a region of interest based on the DAPI channel. The number of foci per cell was counted for at least 25 cells (n ≥ 25) for each condition and was plotted using GraphPad Prism.
Small interfering RNA knockdown of SET.
Our previously described A549 cells that inducibly express protein VII-HA were transfected with 20 nM siRNA for 1 × 106 cells (SMARTpool siGENOME SET or siControl) by use of Lipofectamine RNAiMAX (Life Technologies) diluted in Opti-MEM medium. At 3 days posttransfection, the cells were split, and half were induced to express protein VII by use of 0.2 μg/ml of doxycycline. The next day, cells were transfected a second time and harvested 72 h later, when doxycycline treatment reached 4 days. The commercially available siRNAs siSET (M-019586-01-0005) and siControl (D-001206-13-20) were purchased through Dharmacon GE Healthcare.
ACKNOWLEDGMENTS
We thank the members of the Weitzman lab for helpful discussions and input, especially R. Dilley, B. Simpson, and R. Fernandez for valuable technical help. We thank P. Hearing for helpful comments on experiments and a careful reading of the manuscript. We are grateful to D. Curiel for sharing recombinant protein VII-GFP vectors, H. Wodrich for generously sharing anti-protein VII antibodies, G. Ketner for the dl1004 mutant virus, and the University of Pennsylvania CDB Microscopy Core for imaging assistance.
D.C.A. was supported in part by grants T32 CA115299 and F32 GM112414. N.J.P. was supported in part by grant T32 NS007180. Research was supported by a grant from the National Institutes of Health (grant CA097093 to M.D.W.) and by funds from the Children's Hospital of Philadelphia (M.D.W.).
REFERENCES
- 1.Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed). 2013. Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Alqahtani A, Heesom K, Bramson JL, Curiel D, Ugai H, Matthews DA. 2014. Analysis of purified wild type and mutant adenovirus particles by SILAC based quantitative proteomics. J Gen Virol 95:2504–2511. doi: 10.1099/vir.0.068221-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vayda ME, Rogers AE, Flint SJ. 1983. The structure of nucleoprotein cores released from adenovirions. Nucleic Acids Res 11:441–460. doi: 10.1093/nar/11.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stracker TH, Carson CT, Weitzman MD. 2002. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature 418:348–352. doi: 10.1038/nature00863. [DOI] [PubMed] [Google Scholar]
- 5.Carson CT, Schwartz RA, Stracker TH, Lilley CE, Lee DV, Weitzman MD. 2003. The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J 22:6610–6620. doi: 10.1093/emboj/cdg630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shah GA, O'Shea CC. 2015. Viral and cellular genomes activate distinct DNA damage responses. Cell 162:987–1002. doi: 10.1016/j.cell.2015.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turnell AS, Grand RJ. 2012. DNA viruses and the cellular DNA-damage response. J Gen Virol 93:2076–2097. doi: 10.1099/vir.0.044412-0. [DOI] [PubMed] [Google Scholar]
- 8.Lakdawala SS, Schwartz RA, Ferenchak K, Carson CT, McSharry BP, Wilkinson GW, Weitzman MD. 2008. Differential requirements of the C terminus of Nbs1 in suppressing adenovirus DNA replication and promoting concatemer formation. J Virol 82:8362–8372. doi: 10.1128/JVI.00900-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mathew SS, Bridge E. 2007. The cellular Mre11 protein interferes with adenovirus E4 mutant DNA replication. Virology 365:346–355. doi: 10.1016/j.virol.2007.03.049. [DOI] [PubMed] [Google Scholar]
- 10.Evans JD, Hearing P. 2005. Relocalization of the Mre11-Rad50-Nbs1 complex by the adenovirus E4 ORF3 protein is required for viral replication. J Virol 79:6207–6215. doi: 10.1128/JVI.79.10.6207-6215.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gautam D, Bridge E. 2013. The kinase activity of ataxia-telangiectasia mutated interferes with adenovirus E4 mutant DNA replication. J Virol 87:8687–8696. doi: 10.1128/JVI.00376-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baker A, Rohleder KJ, Hanakahi LA, Ketner G. 2007. Adenovirus E4 34k and E1b 55k oncoproteins target host DNA ligase IV for proteasomal degradation. J Virol 81:7034–7040. doi: 10.1128/JVI.00029-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boyer J, Rohleder K, Ketner G. 1999. Adenovirus E4 34k and E4 11k inhibit double strand break repair and are physically associated with the cellular DNA-dependent protein kinase. Virology 263:307–312. doi: 10.1006/viro.1999.9866. [DOI] [PubMed] [Google Scholar]
- 14.Araujo FD, Stracker TH, Carson CT, Lee DV, Weitzman MD. 2005. Adenovirus type 5 E4orf3 protein targets the Mre11 complex to cytoplasmic aggresomes. J Virol 79:11382–11391. doi: 10.1128/JVI.79.17.11382-11391.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carson CT, Orazio NI, Lee DV, Suh J, Bekker-Jensen S, Araujo FD, Lakdawala SS, Lilley CE, Bartek J, Lukas J, Weitzman MD. 2009. Mislocalization of the MRN complex prevents ATR signaling during adenovirus infection. EMBO J 28:652–662. doi: 10.1038/emboj.2009.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Evans JD, Hearing P. 2003. Distinct roles of the adenovirus E4 ORF3 protein in viral DNA replication and inhibition of genome concatenation. J Virol 77:5295–5304. doi: 10.1128/JVI.77.9.5295-5304.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ciccia A, Elledge SJ. 2010. The DNA damage response: making it safe to play with knives. Mol Cell 40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shiloh Y, Ziv Y. 2013. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 14:197–210. doi: 10.1038/nrm3546. [DOI] [PubMed] [Google Scholar]
- 19.Blackford AN, Jackson SP. 2017. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol Cell 66:801–817. doi: 10.1016/j.molcel.2017.05.015. [DOI] [PubMed] [Google Scholar]
- 20.Paull TT, Lee J-H. 2005. The Mre11/Rad50/Nbs1 complex and its role as a DNA double-strand break sensor for ATM. Cell Cycle 4:737–740. doi: 10.4161/cc.4.6.1715. [DOI] [PubMed] [Google Scholar]
- 21.Lamarche BJ, Orazio NI, Weitzman MD. 2010. The MRN complex in double-strand break repair and telomere maintenance. FEBS Lett 584:3682–3695. doi: 10.1016/j.febslet.2010.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stracker TH, Petrini JHJ. 2011. The MRE11 complex: starting from the ends. Nat Rev Mol Cell Biol 12:90–103. doi: 10.1038/nrm3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bakkenist CJ, Kastan MB. 2003. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- 24.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. 1998. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 25.Lou Z, Minter-Dykhouse K, Franco S, Gostissa M, Rivera MA, Celeste A, Manis JP, van Deursen J, Nussenzweig A, Paull TT, Alt FW, Chen J. 2006. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol Cell 21:187–200. doi: 10.1016/j.molcel.2005.11.025. [DOI] [PubMed] [Google Scholar]
- 26.Stewart GS, Wang B, Bignell CR, Taylor AMR, Elledge SJ. 2003. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 421:961–966. doi: 10.1038/nature01446. [DOI] [PubMed] [Google Scholar]
- 27.Stucki M, Jackson SP. 2006. gammaH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair (Amst) 5:534–543. doi: 10.1016/j.dnarep.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 28.Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. 2005. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 123:1213–1226. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 29.Coster G, Goldberg M. 2010. The cellular response to DNA damage: a focus on MDC1 and its interacting proteins. Nucleus 1:166–178. doi: 10.4161/nucl.1.2.11176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goodarzi AA, Noon AT, Deckbar D, Ziv Y, Shiloh Y, Löbrich M, Jeggo PA. 2008. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol Cell 31:167–177. doi: 10.1016/j.molcel.2008.05.017. [DOI] [PubMed] [Google Scholar]
- 31.Falk M, Lukásová E, Kozubek S. 2008. Chromatin structure influences the sensitivity of DNA to gamma-radiation. Biochim Biophys Acta 1783:2398–2414. doi: 10.1016/j.bbamcr.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 32.Vasireddy RS, Karagiannis TC, El-Osta A. 2010. Gamma-radiation-induced gammaH2AX formation occurs preferentially in actively transcribing euchromatic loci. Cell Mol Life Sci 67:291–294. doi: 10.1007/s00018-009-0181-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cann KL, Dellaire G. 2011. Heterochromatin and the DNA damage response: the need to relax. Biochem Cell Biol 89:45–60. doi: 10.1139/O10-113. [DOI] [PubMed] [Google Scholar]
- 34.Polo SE, Jackson SP. 2011. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev 25:409–433. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bakkenist CJ, Kastan MB. 2015. Chromatin perturbations during the DNA damage response in higher eukaryotes. DNA Repair (Amst) 36:8–12. doi: 10.1016/j.dnarep.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goodarzi AA, Noon AT, Jeggo PA. 2009. The impact of heterochromatin on DSB repair. Biochem Soc Trans 37:569–576. doi: 10.1042/BST0370569. [DOI] [PubMed] [Google Scholar]
- 37.Noon AT, Shibata A, Rief N, Löbrich M, Stewart GS, Jeggo PA, Goodarzi AA. 2010. 53BP1-dependent robust localized KAP-1 phosphorylation is essential for heterochromatic DNA double-strand break repair. Nat Cell Biol 12:177–184. doi: 10.1038/ncb2017. [DOI] [PubMed] [Google Scholar]
- 38.Kalousi A, Hoffbeck A-S, Selemenakis PN, Pinder J, Savage KI, Khanna KK, Brino L, Dellaire G, Gorgoulis VG, Soutoglou E. 2015. The nuclear oncogene SET controls DNA repair by KAP1 and HP1 retention to chromatin. Cell Rep 11:149–163. doi: 10.1016/j.celrep.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 39.Komatsu T, Dacheux D, Kreppel F, Nagata K, Wodrich H. 2015. A method for visualization of incoming adenovirus chromatin complexes in fixed and living cells. PLoS One 10:e0137102. doi: 10.1371/journal.pone.0137102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giberson AN, Davidson AR, Parks RJ. 2012. Chromatin structure of adenovirus DNA throughout infection. Nucleic Acids Res 40:2369–2376. doi: 10.1093/nar/gkr1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haruki H, Okuwaki M, Miyagishi M, Taira K, Nagata K. 2006. Involvement of template-activating factor I/SET in transcription of adenovirus early genes as a positive-acting factor. J Virol 80:794–801. doi: 10.1128/JVI.80.2.794-801.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haruki H, Gyurcsik B, Okuwaki M, Nagata K. 2003. Ternary complex formation between DNA-adenovirus core protein VII and TAF-Ibeta/SET, an acidic molecular chaperone. FEBS Lett 555:521–527. doi: 10.1016/S0014-5793(03)01336-X. [DOI] [PubMed] [Google Scholar]
- 43.Xue Y, Johnson JS, Ornelles DA, Lieberman J, Engel DA. 2005. Adenovirus protein VII functions throughout early phase and interacts with cellular proteins SET and pp32. J Virol 79:2474–2483. doi: 10.1128/JVI.79.4.2474-2483.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matsumoto K, Nagata K, Ui M, Hanaoka F. 1993. Template activating factor I, a novel host factor required to stimulate the adenovirus core DNA replication. J Biol Chem 268:10582–10587. [PubMed] [Google Scholar]
- 45.Yeates TO. 2002. Structures of SET domain proteins: protein lysine methyltransferases make their mark. Cell 111:5–7. doi: 10.1016/S0092-8674(02)01010-3. [DOI] [PubMed] [Google Scholar]
- 46.Mirza MA, Weber JM. 1982. Structure of adenovirus chromatin. Biochim Biophys Acta 696:76–86. doi: 10.1016/0167-4781(82)90012-4. [DOI] [PubMed] [Google Scholar]
- 47.Chatterjee PK, Vayda ME, Flint SJ. 1985. Interactions among the three adenovirus core proteins. J Virol 55:379–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chatterjee PK, Vayda ME, Flint SJ. 1986. Identification of proteins and protein domains that contact DNA within adenovirus nucleoprotein cores by ultraviolet light crosslinking of oligonucleotides 32P-labelled in vivo. J Mol Biol 188:23–37. doi: 10.1016/0022-2836(86)90477-8. [DOI] [PubMed] [Google Scholar]
- 49.Ostapchuk P, Suomalainen M, Zheng Y, Boucke K, Greber UF, Hearing P. 2017. The adenovirus major core protein VII is dispensable for virion assembly but is essential for lytic infection. PLoS Pathog 13:e1006455. doi: 10.1371/journal.ppat.1006455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lischwe MA, Sung MT. 1977. A histone-like protein from adenovirus chromatin. Nature 267:552–554. doi: 10.1038/267552a0. [DOI] [PubMed] [Google Scholar]
- 51.Avgousti DC, Herrmann C, Kulej K, Pancholi NJ, Sekulic N, Petrescu J, Molden RC, Blumenthal D, Paris AJ, Reyes ED, Ostapchuk P, Hearing P, Seeholzer SH, Worthen GS, Black BE, Garcia BA, Weitzman MD. 2016. A core viral protein binds host nucleosomes to sequester immune danger signals. Nature 535:173–177. doi: 10.1038/nature18317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gyurcsik B, Haruki H, Takahashi T, Mihara H, Nagata K. 2006. Binding modes of the precursor of adenovirus major core protein VII to DNA and template activating factor I: implication for the mechanism of remodeling of the adenovirus chromatin. Biochemistry 45:303–313. doi: 10.1021/bi051248+. [DOI] [PubMed] [Google Scholar]
- 53.Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao L, Huang J, Yu Y, Fan X-G, Yan Z, Sun X, Wang H, Wang Q, Tsung A, Billiar TR, Zeh HJ, Lotze MT, Tang D. 2014. HMGB1 in health and disease. Mol Aspects Med 40:1–116. doi: 10.1016/j.mam.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Johnson JS, Osheim YN, Xue Y, Emanuel MR, Lewis PW, Bankovich A, Beyer AL, Engel DA. 2004. Adenovirus protein VII condenses DNA, represses transcription, and associates with transcriptional activator E1A. J Virol 78:6459–6468. doi: 10.1128/JVI.78.12.6459-6468.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Komatsu T, Haruki H, Nagata K. 2011. Cellular and viral chromatin proteins are positive factors in the regulation of adenovirus gene expression. Nucleic Acids Res 39:889–901. doi: 10.1093/nar/gkq783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harada JN, Shevchenko A, Shevchenko A, Pallas DC, Berk AJ. 2002. Analysis of the adenovirus E1B-55K-anchored proteome reveals its link to ubiquitination machinery. J Virol 76:9194–9206. doi: 10.1128/JVI.76.18.9194-9206.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dallaire F, Blanchette P, Groitl P, Dobner T, Branton PE. 2009. Identification of integrin alpha3 as a new substrate of the adenovirus E4orf6/E1B 55-kilodalton E3 ubiquitin ligase complex. J Virol 83:5329–5338. doi: 10.1128/JVI.00089-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Forrester NA, Sedgwick GG, Thomas A, Blackford AN, Speiseder T, Dobner T, Byrd PJ, Stewart GS, Turnell AS, Grand RJ. 2011. Serotype-specific inactivation of the cellular DNA damage response during adenovirus infection. J Virol 85:2201–2211. doi: 10.1128/JVI.01748-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Täuber B, Dobner T. 2001. Molecular regulation and biological function of adenovirus early genes: the E4 ORFs. Gene 278:1–23. doi: 10.1016/S0378-1119(01)00722-3. [DOI] [PubMed] [Google Scholar]
- 60.Querido E, Blanchette P, Yan Q, Kamura T, Morrison M, Boivin D, Kaelin WG, Conaway RC, Conaway JW, Branton PE. 2001. Degradation of p53 by adenovirus E4orf6 and E1B55K proteins occurs via a novel mechanism involving a Cullin-containing complex. Genes Dev 15:3104–3117. doi: 10.1101/gad.926401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Karen KA, Hearing P. 2011. Adenovirus core protein VII protects the viral genome from a DNA damage response at early times after infection. J Virol 85:4135–4142. doi: 10.1128/JVI.02540-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. 2001. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem 276:42462–42467. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 63.Liu Y, Prasad R, Wilson SH. 2010. HMGB1: roles in base excision repair and related function. Biochim Biophys Acta 1799:119–130. doi: 10.1016/j.bbagrm.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lange SS, Mitchell DL, Vasquez KM. 2008. High mobility group protein B1 enhances DNA repair and chromatin modification after DNA damage. Proc Natl Acad Sci U S A 105:10320–10325. doi: 10.1073/pnas.0803181105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lange SS, Vasquez KM. 2009. HMGB1: the jack-of-all-trades protein is a master DNA repair mechanic. Mol Carcinog 48:571–580. doi: 10.1002/mc.20544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Le LP, Le HN, Nelson AR, Matthews DA, Yamamoto M, Curiel DT. 2006. Core labeling of adenovirus with EGFP. Virology 351:291–302. doi: 10.1016/j.virol.2006.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang MM, Hearing P. 1989. Adenovirus early region 4 encodes two gene products with redundant effects in lytic infection. J Virol 63:2605–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen J, Morral N, Engel DA. 2007. Transcription releases protein VII from adenovirus chromatin. Virology 369:411–422. doi: 10.1016/j.virol.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 69.Okuwaki M, Nagata K. 1998. Template activating factor-I remodels the chromatin structure and stimulates transcription from the chromatin template. J Biol Chem 273:34511–34518. doi: 10.1074/jbc.273.51.34511. [DOI] [PubMed] [Google Scholar]
- 70.Weiden MD, Ginsberg HS. 1994. Deletion of the E4 region of the genome produces adenovirus DNA concatemers. Proc Natl Acad Sci U S A 91:153–157. doi: 10.1073/pnas.91.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Horwitz GA, Zhang K, McBrian MA, Grunstein M, Kurdistani SK, Berk AJ. 2008. Adenovirus small e1a alters global patterns of histone modification. Science 321:1084–1085. doi: 10.1126/science.1155544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ferrari R, Pellegrini M, Horwitz GA, Xie W, Berk AJ, Kurdistani SK. 2008. Epigenetic reprogramming by adenovirus e1a. Science 321:1086–1088. doi: 10.1126/science.1155546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ferrari R, Su T, Li B, Bonora G, Oberai A, Chan Y, Sasidharan R, Berk AJ, Pellegrini M, Kurdistani SK. 2012. Reorganization of the host epigenome by a viral oncogene. Genome Res 22:1212–1221. doi: 10.1101/gr.132308.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fonseca GJ, Thillainadesan G, Yousef AF, Ablack JN, Mossman KL, Torchia J, Mymryk JS. 2012. Adenovirus evasion of interferon-mediated innate immunity by direct antagonism of a cellular histone posttranslational modification. Cell Host Microbe 11:597–606. doi: 10.1016/j.chom.2012.05.005. [DOI] [PubMed] [Google Scholar]
- 75.Ferrari R, Gou D, Jawdekar G, Johnson SA, Nava M, Su T, Yousef AF, Zemke NR, Pellegrini M, Kurdistani SK, Berk AJ. 2014. Adenovirus small E1A employs the lysine acetylases p300/CBP and tumor suppressor Rb to repress select host genes and promote productive virus infection. Cell Host Microbe 16:663–676. doi: 10.1016/j.chom.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kulej K, Avgousti DC, Weitzman MD, Garcia BA. 2015. Characterization of histone post-translational modifications during virus infection using mass spectrometry-based proteomics. Methods 90:8–20. doi: 10.1016/j.ymeth.2015.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nichols GJ, Schaack J, Ornelles DA. 2009. Widespread phosphorylation of histone H2AX by species C adenovirus infection requires viral DNA replication. J Virol 83:5987–5998. doi: 10.1128/JVI.00091-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ruzindana-Umunyana A, Imbeault L, Weber JM. 2002. Substrate specificity of adenovirus protease. Virus Res 89:41–52. doi: 10.1016/S0168-1702(02)00111-9. [DOI] [PubMed] [Google Scholar]
- 79.Khandelia P, Yap K, Makeyev EV. 2011. Streamlined platform for short hairpin RNA interference and transgenesis in cultured mammalian cells. Proc Natl Acad Sci U S A 108:12799–12804. doi: 10.1073/pnas.1103532108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F. 2014. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lilley CE, Carson CT, Muotri AR, Gage FH, Weitzman MD. 2005. DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc Natl Acad Sci U S A 102:5844–5849. doi: 10.1073/pnas.0501916102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bridge E, Ketner G. 1989. Redundant control of adenovirus late gene expression by early region 4. J Virol 63:631–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Solovjeva L, Firsanov D, Vasilishina A, Chagin V, Pleskach N, Kropotov A, Svetlova M. 2015. DNA double-strand break repair is impaired in presenescent Syrian hamster fibroblasts. BMC Mol Biol 16:18. doi: 10.1186/s12867-015-0046-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Leatherbarrow EL, Harper JV, Cucinotta FA, O'Neill P. 2006. Induction and quantification of gamma-H2AX foci following low and high LET-irradiation. Int J Radiat Biol 82:111–118. doi: 10.1080/09553000600599783. [DOI] [PubMed] [Google Scholar]