

ABSTRACT
The papillomavirus (PV) E2 protein is a DNA binding, protein interaction platform that recruits viral and host factors necessary for transcription and replication. We recently discovered phosphorylation of a tyrosine (Y102) in bovine PV (BPV) E2. To identify the responsible factor, we tested several candidate tyrosine kinases that are highly expressed in keratinocytes for binding to BPV-1 E2. Fibroblast growth factor receptor 3 (FGFR3) coimmunoprecipitated with the BPV-1 E2 protein, as did human papillomavirus 31 (HPV-31) E2, which also colocalized with FGFR3 within the nucleus. A constitutively active mutant form of FGFR3 decreased BPV-1 and HPV-31 transient replication although this result also occurred in a BPV-1 E2 mutant lacking a previously identified phosphorylation site of interest (Y102). Furthermore, FGFR3 depletion in cell lines that maintain HPV-31 episomes increased viral copy number. These results suggest that FGFR3 kinase activity may regulate the PV reproductive program through phosphorylation of the E2 protein although this is unlikely to occur through the Y102 residue of HPV E2.
IMPORTANCE The papillomavirus (PV) is a double-stranded DNA tumor virus infecting cervix, mouth, and throat tissues. The viral protein E2 is responsible for the replication of the virus. Understanding the mechanisms of the replicative life cycle of the virus may bring to light direct targets and treatments against viral infection. We recently found that the fibroblast growth factor receptor 3 (FGFR3) interacts with and mediates PV E2 function through phosphorylation of the E2 protein. Our study suggests that the function of the E2 protein may be regulated through a direct FGFR3 target during the maintenance stage of the PV life cycle.
KEYWORDS: BPV, FGFR3, HPV, kinase, papillomavirus, protein phosphorylation, viral replication
INTRODUCTION
Replication of the 8-kb circular, double-stranded DNA genome of the papillomavirus (PV) occurs in three distinct phases that are orchestrated by the epithelial cell environment (1, 2). In the initial infection of basal keratinocytes, the entering viral genome replicates and persists as low-copy-number episomes. As these cells divide, viral episomes duplicate and partition to the progeny cells, thereby maintaining copy number. As the infected cells are pushed into the upper epithelial strata, their usual differentiation program is subverted by viral proteins that induce a prolonged S/G2 phase (3). During this stage, viral episomes amplify to hundreds of copies that can be packaged into virion particles. PVs infect virtually all mammals and cause both benign and malignant epithelial tumors in their natural host, and medical intervention to abort their triphasic replicative program will require detailed insights (4).
The E2 protein is encoded by all PVs and is composed of an amino-terminal activation domain (TAD), a nonconserved hinge extension, and the sequence-specific DNA binding carboxy-terminal domain (CTD) (5, 6). The CTD binds with high affinity to specific palindromes within the viral genome and thereby attracts the host factors and enzymes necessary for both transcription and DNA synthesis. E2 also binds to the viral helicase E1, which initiates replication of the PV episome. Posttranslational modifications have been found to regulate the functions of the E2 protein (7–10). Our lab recently discovered and characterized a novel tyrosine phosphorylation site at position 102 (Y102) within the TAD of bovine papillomavirus type 1 (BPV-1) E2 (8). Additional mass spectroscopy studies of purified E2 from human papillomavirus 31 (HPV-31), which causes a small percentage of cervical cancers and is therefore included in the high-risk clade, have revealed the presence of other phosphorylated tyrosine residues (unpublished data). The responsible protein tyrosine kinases (PTKs) have not been elucidated. In this study, we tested a series of PTKs expressed in skin and found fibroblast growth factor receptor 3 (FGFR3) in complexes with the BPV-1 and HPV-31 E2 proteins.
The FGF receptors are a family of tyrosine kinases; the FGFR3 kinase is expressed in the cervix and skin (11, 12). FGFR3 point mutations and elevated expression are hallmarks of benign overgrowths of keratinocytes reported in seborrheic keratoses (13). Constitutively active kinase mutations of FGFR3 drive epidermal hyperplasia but are insufficient to induce malignant skin tumors (12). Consistent with FGFR3's lack of oncogenic properties, its expression is nearly undetectable in squamous cell carcinomas (SCCs) (11).
Using phosphotyrosine-specific antibodies, we demonstrate here that FGFR3 induces E2 phosphorylation. Expression of a constitutively activated FGFR3 mutant led to suppression of viral replication, consistent with our published results in which a phosphomimetic tyrosine-to-glutamate mutant (BPV-1 E2 Y102E) was unable to stimulate E1-dependent viral replication. However, further studies revealed that the effect of FGFR3 is unlikely to occur by targeting Y102, implying action through another tyrosine residue(s) of E2.
RESULTS
Characterization of FGFR3 interaction with E2.
In a search for tyrosine kinases responsible for phosphorylation of E2 proteins, including the tyrosine (Y) at position 102 that we previously identified (Y102 in BPV-1 and HPV-31), we used online predictive software based on the peptide regions flanking each tyrosine; however, these programs yielded inconsistent results. Analysis of a protein atlas database indicated that the fibroblast growth factor receptor (FGFR) family of proteins was a lead candidate, which was noteworthy as FGFR proteins are expressed in keratinocytes (14–16). Specifically, FGFR3 and FGFR2 are abundantly expressed in keratinocytes compared to levels of FGFR1 and FGFR4 (Table 1). Immunofluorescence studies were first conducted with a commercial FGFR3 antibody (Sigma-Aldrich) or FLAG antibodies in C33A cells transfected with wild-type (WT) FLAG-FGFR3 or the constitutively active mutant form FLAG-FGFR3 K650E as endogenous FGFR3 expression could not be detected. WT and K650E FGFR3 were present throughout the cytoplasm, near the nuclear membrane, and within the nucleus with both FGFR3 and FLAG antibodies (Fig. 1A to C). FGFR3 protein was observed throughout the cytoplasm and at the nuclear membrane in the HPV-negative cell lines HaCat and NIKS, normal human foreskin keratinocytes (HFK), and the HPV-positive cell lines W12 (HPV-16), HPV-BP (HPV-16), and CIN612-9E (HPV-31) (Fig. 1D).
TABLE 1.
Expression of FGFR in skin cells in the Human Protein Atlas database
| HGNC gene namea | Ensembl gene no. | Tissue sample no. | Cell typeb | Relative expression level |
|---|---|---|---|---|
| FGFR2 | ENSG00000066468 | Skin 1 | Keratinocytes | High |
| Skin 2 | Epidermal cells | High | ||
| FGFR3 | ENSG00000068078 | Skin 1 | Keratinocytes | High |
| Skin 2 | Epidermal cells | High | ||
| FGFR4 | ENSG00000160867 | Skin 1 | Keratinocytes | Medium |
| Skin 2 | Epidermal cells | Medium | ||
| FGFR1 | ENSG00000077782 | Skin 1 | Keratinocytes | Low |
| Skin 2 | Epidermal cells | Low |
HGNC, Human Gene Nomenclature Committee.
Langerhans, fibroblasts, and melanocytes were excluded.
FIG 1.

FGFR3 is expressed in keratinocytes with and without HPV genomes. C33A cells were transfected with FLAG-FGFR3 WT (A) or FLAG-FGFR3 K650E (B) expression plasmids. Cells were stained for immunofluorescence with FLAG (M2) and FGFR3 antibodies (ab). Red, FLAG or FGFR3 antibodies; blue, DAPI. Magnification, ×100. (C) C33A cells were transfected with a FLAG-FGFR3 WT expression plasmid and stained with FLAG (M2; red) and FGFR3 (green) antibodies. Blue, DAPI. Cells were viewed under a magnification of ×60 using confocal microscopy. (D) HaCat, NIKS, W12, HFK, CIN612-9E, and HPV-BP cells were stained with FGFR3 antibodies (red) and DAPI (blue) for immunofluorescence. Magnifications, ×100 (left panels) and ×40 (right panels).
We therefore investigated association of BPV-1 E2 and HPV-31 E2 with FGFR3 by cotransfection of HEK293TT cells with constructs expressing WT FGFR3 or mutations that are constitutively active (K650E) or kinase inactive (K508R) (17). Immunoprecipitations of BPV-1 and HPV-31 E2 proteins contained these FGFR3 proteins (Fig. 2A and B). In contrast, we did not observe coimmunoprecipitation of the epidermal growth factor receptor (EGFR) or the ephrin B2 receptor (EphB2) with BPV-1 E2 (Fig. 2C and D). FGFR3 K650E appeared to interact with the greatest affinity, followed by the WT and then the K508R form of FGFR3. We noted that FGFR3 K650E expression increased the levels of the BPV-1 and HPV-31 E2 proteins (Fig. 2). Interestingly, FGFR3 K650E did not alter the levels of FLAG-HPV-31 E1 driven by the same cytomegalovirus (CMV) promoter, suggesting that HPV-31 E2 levels are stabilized by expression of this activated kinase, FGFR3 K650E (Fig. 3).
FIG 2.

FGFR3 interacts with PV E2 but not with EGFR or EphB2. (A) HEK293TT cells were transfected with FGFR3 WT, K650E, and K508R constructs along with a BPV-1 E2 expression plasmid. BPV-1 E2 pulldown was completed with B201/B202 antibodies and blotted with FGFR3, B201, and β-actin antibodies. (B) HEK293TT cells were transfected with FGFR3 WT, K650E, and K508R constructs along with a FLAG-HPV-31 E2 expression plasmid. HPV-31 E2 pulldown was completed with M2 antibodies and blotted with FGFR3, M2, and β-actin antibodies. C33A cells were transfected with wild-type BPV-1 E2, E2R, and EGFR (C) or EphB2 (D) expression plasmids. Twenty-four hours later, cells were lysed, and BPV-1 E2 was immunoprecipitated with B201 antibodies. Immunoblotting was completed with EGFR, EphB2, and B201 antibodies. IP, immunoprecipitation.
FIG 3.
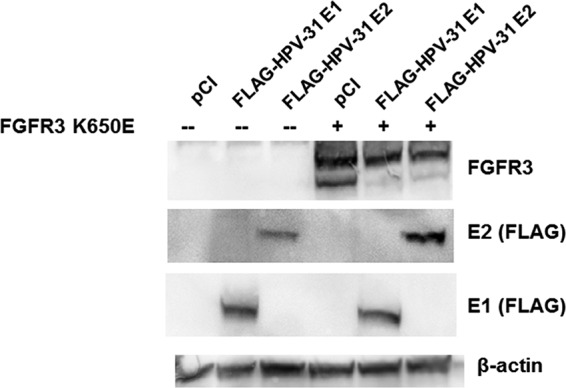
Activated FGFR3 increases HPV-31 E2 levels. HEK293TT cells were transfected with pCI or FLAG-FGFR3 K650E and FLAG-HPV-31 E1 or FLAG-HPV-31 E2 constructs. Forty-eight hours later, cells were lysed in 2% SDS, 150 mM NaCl, 10 mM Tris-HCl, pH 8.0. Immunoblotting was completed with FGFR3, M2 (FLAG), and β-actin antibodies.
To identify the region of E2 that associates with FGFR3, C33A cells were transfected with vectors expressing full-length BPV-1 E2, the transactivation domain (TAD; amino acids [aa] 1 to 216), or the E2 repressor form, E2R (aa 162 to 410), along with FLAG-FGFR3 WT. FGFR3 coimmunoprecipitated with the full-length and TAD domains of BPV-1 E2 but not with E2R (Fig. 4A). To determine whether E2 protein expressed in W12 cells, which stably maintain episomal HPV-16 genomes, was in complex with endogenous levels of FGFR3, we used a sheep anti-HPV-16 E2 antibody (Fig. 4B), with HPV-16 E2 transfected C33A cells serving as a positive control for the antibody (18). These experiments demonstrated that HPV-16 E2 in W12 lysates coimmunoprecipitated with FGFR3 (Fig. 4C).
FIG 4.
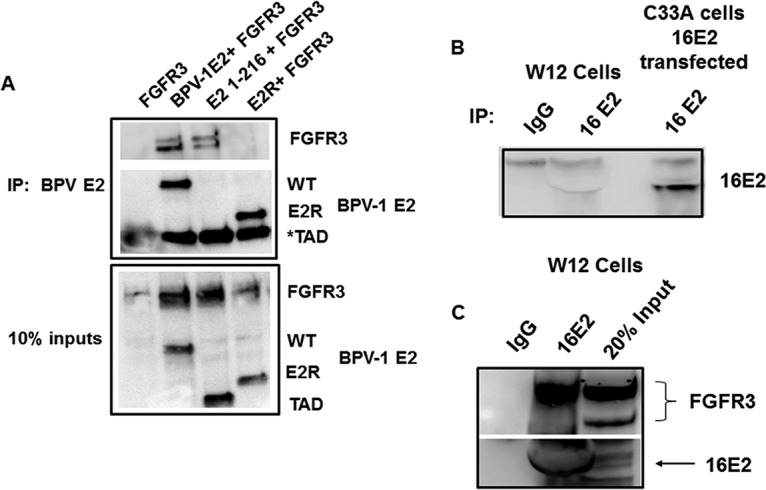
FGFR3 interacts with TAD of BPV-1 E2 and endogenous HPV-16 E2. (A) C33A cells were transfected with FLAG-FGFR3 WT and BPV-1 full-length, TAD (aa 1 to 216), and E2R (aa 162 to 410) constructs. BPV-1 E2 protein pulldown was completed with B201 antibodies and blotted with B201 and M2 antibodies. TAD (*) is the same size as light-chain IgG in immunoprecipitation groups. (B) Endogenous levels of HPV-16 E2 (16E2) in W12 cells were confirmed by immunoprecipitation using sheep anti-HPV-16 E2 polyclonal serum. C33A cells transfected with FLAG-HPV-16 E2 served as the positive control for the detection. HPV-16 E2 was immunoblotted with TVG-261 antibody. (C) The immune complexes of HPV-16 E2 from the W12 cells were analyzed for the presence of endogenous FGFR3 using specific antibodies. Normal sheep IgG served as the negative control.
Papillomavirus E2 proteins primarily reside in the nucleus (19, 20). Previous studies have shown that FGFR2 and FGFR3 may be present in the nucleus of different cell types (21–23). We surveyed the localization of FGFR3 and E2 using confocal microscopy. These experiments revealed colocalization of FGFR3 WT, K650E, and K508R with HPV-31 E2 in CV-1 cells within the cytoplasm and nucleus and at the nuclear membrane (Fig. 5).
FIG 5.

FGFR3 is expressed in the nucleus where it colocalizes with E2. CV-1 cells were transfected with FGFR3 and FLAG-HPV-31 E2 constructs. Immunofluorescence staining for FGFR3, M2 for FLAG-HPV-31 E2, and DAPI (blue) was completed. Cells were visualized under a magnification of ×60 using confocal microscopy. FGFR3 KE, FGFR3 K650E; FGFR3 KR, FGFR3 K508R.
A proximal ligation assay (PLA) was utilized to address whether endogenous FGFR3 and E2 proteins are within close proximity in live cells and to determine the cellular location at which this interaction occurs (Fig. 6). The PLA disclosed nuclear foci in cells expressing HPV-31 E2 in contrast to results in cells without HPV-31 E2, with more than 80% of the E2 transfected cells showing two or more positive signals in C33A cells (Fig. 6B). To rule out the possibility that this observation was exclusive to this cell line, we completed this experiment in a physiologically relevant model, CIN612-9E cells. While these cells have a much lower transfection efficiency than C33A cells, FLAG-HPV-31 E2 interaction foci with the endogenous FGFR3 were detected (Fig. 6C). These results illustrated the juxtaposition of a subpopulation of FGFR3 with E2 in nuclei but not the cytoplasm.
FIG 6.
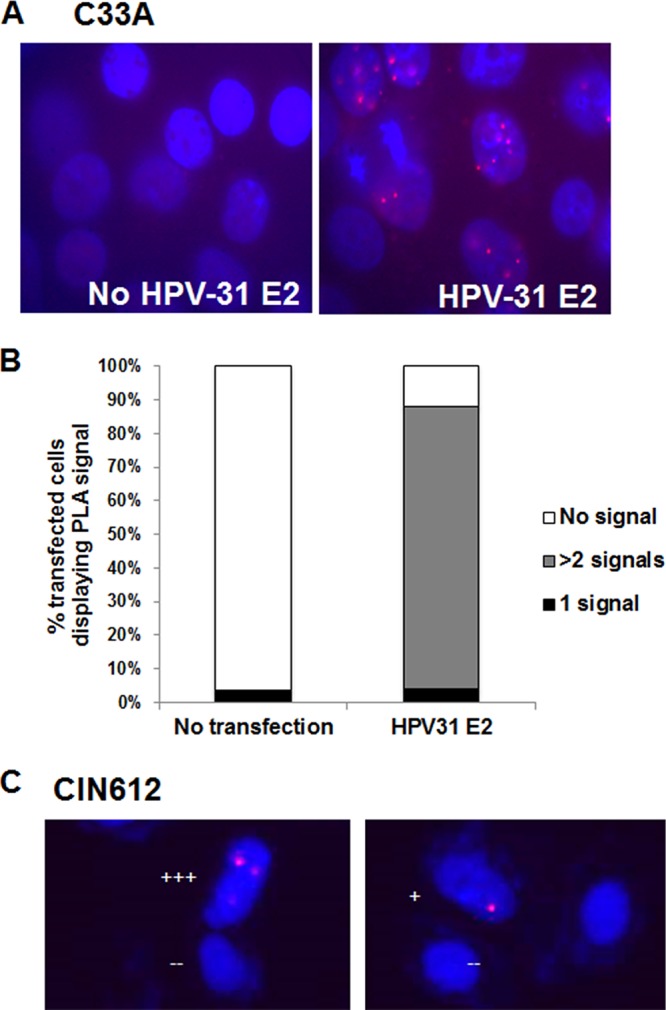
FGFR3 interacts with E2 within the nucleus. (A) C33A cells were transfected with and without FLAG-HPV-31 E2. PLA was completed with M2 (mouse) and FGFR3 (rabbit) antibodies. Red foci are an indication that the FLAG and FGFR3 proteins are within close proximity of each other. Red, focus formation; blue, DAPI. (B) FGFR3 and FLAG-HPV-31 E2 PLA foci were counted without and with FLAG-HPV-31 E2 expression. The percentages of cells displaying 0, 1, or more than 2 signals are shown. (C) CIN612-9E cells were transfected with and without FLAG-HPV-31 E2. PLA was completed with M2 (mouse) and FGFR3 (rabbit) antibodies. Red foci are an indication that the FLAG and FGFR3 proteins are within close proximity of each other. Red, focus formation; blue, DAPI. Each plus sign indicates the number of foci.
Activated FGFR3 increases levels of phosphotyrosine E2 and decreases PV replication.
While our intent was to identify the tyrosine kinase that modifies Y102, antibodies to this specific phosphopeptide in BPV or HPV E2 are not available. As a surrogate, we performed immunoblotting with the phosphotyrosine-specific PY-99 antibody to assess overall tyrosine phosphorylation. HEK293TT cells were transfected with HPV-31 E2 and WT, K650E, and K508R forms of FGFR3. First, we showed that HPV-31 E2 coimmunoprecipitated with each of these forms of FGFR3 (Fig. 7). As expected, only the FGFR3 K650E protein showed elevated auto-phosphorylation whereas WT FGFR3 and the kinase-dead form K508R were not auto-phosphorylated. Consistently, there appeared to be increased phosphorylation of HPV-31 E2 with coexpression of FGFR3 K650E but not of the WT and K508R forms.
FIG 7.
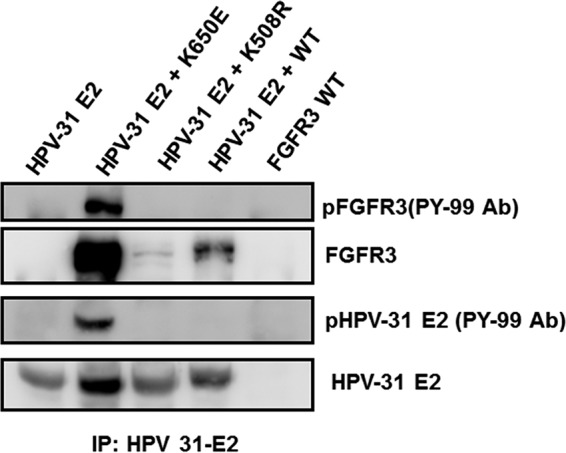
Activated FGFR3 complexes with and phosphorylates E2. HEK293TT cells were transfected with FLAG-HPV-31 E2 and FGFR3 WT, K650E, and K508R constructs. HPV-31 E2 pulldown was completed with M2 antibodies and blotted with FGFR3, M2, and phospho-tyrosine (PY-99) antibodies.
The effect of FGFR3 kinase activity on E2-stimulated viral DNA replication was subsequently evaluated using C33A cells transfected with BPV-1 E1 and E2 expression vectors, along with a BPV-1 luciferase-linked reporter in the presence of FGFR3 WT, K650E, and K508R expression constructs. In this assay, luciferase expression is proportional to episomal copy number and is sensitive to DNA replication inhibitors (24–26). Firefly luciferase readout was normalized to Renilla luciferase levels as a transcription and transfection control (Fluc/Rluc). First we tested the effect of the FGFR3 constructs on the BPV-1 luciferase-linked reporter in the absence of any HPV gene. The WT FGFR3 construct had a small (−20%) effect on basal firefly/Renilla luciferase levels of the BPV-1 origin of DNA replication (ori) (Fig. 8A). Next, the experiment was completed with E1 and E2 transfections with the BPV-1 ori-linked luciferase constructs. E1 and E2 Fluc/Rluc levels were normalized to those with E2 alone to account for BPV-1 E2 transcriptional activation and stabilization since there was elevated E2 protein with K650E coexpression (Fig. 2). Luciferase levels were unaltered by coexpression of the WT and K508R FGFR3 proteins, but these were significantly reduced with coexpression of the constitutively active FGFR3 mutant K650E (Fig. 8B).
FIG 8.

Activated FGFR3 decreases HPV replication. (A) C33A cells were transfected with pFLORI-BPV-1 (Fluc) and pRL (Rluc) constructs in the presence of FGFR3 WT, K650E, and K508R expression plasmids. Twenty-four hours later, cells were lysed, and firefly and Renilla luciferase levels were measured using Dual-Glo luciferase reagent. Firefly luciferase levels were normalized to Renilla luciferase levels. Number of samples per group, 4. (B) C33A cells were transfected with BPV-1 E1, E2, pFLORI-BPV-1 (Fluc), and pRL (Rluc) constructs in the presence of FGFR3 WT, K650E, and K508R expression plasmids. Seventy-two hours later, cells were lysed, and firefly and Renilla luciferase levels were measured using Dual-Glo luciferase reagent. Firefly luciferase levels were normalized to Renilla luciferase levels. E1 and E2 luciferase-normalized values were normalized to E2-normalized luciferase values. Values are expressed as means ± SEM (n = 4). *, P < 0.05. (C) CIN612-9E wells were transfected with pFLORI-HPV-31 (Fluc), pRL (Rluc), and WT, K650E, and K508R constructs. Seventy-two hours later, cells were lysed, and firefly and Renilla luciferase levels were measured using Dual-Glo luciferase reagent. Firefly luciferase levels were normalized to Renilla luciferase levels. Values are expressed as means ± SEM (n = 8). *, P < 0.05.
The reporter-based system offers strong evidence for the effects of FGFR3; we sought to confirm the consequences of FGFR3 kinase activity on HPV-31 replication without using heterologous expression of E2 and E1. The FGFR3 constructs were cotransfected with an HPV-31 luciferase-linked replicon analogous to that employed to measure BPV-1 transient replication into the CIN612-9E cell line, which maintains HPV-31 episomes mediated by constitutive expression of the E2 and E1 proteins. As observed with BPV-1, HPV-31 replication decreased in the presence of FGFR3 K650E but was unaffected by the WT and the kinase-dead K508R FGFR3 forms (Fig. 8C). To support these observations, we studied the effects of FGFR3 depletion on the resident HPV-31 DNA content in CIN612-9E cells. As a control for these silencing experiments, an EGFR small interfering RNA (siRNA) was utilized since EGFR did not complex with E2 (Fig. 2C). A fluorescein siRNA revealed a transfection efficiency of >90%. FGFR3 and EGFR siRNAs successfully reduced levels of their corresponding proteins (Fig. 9A). FGFR3 depletion resulted in increased HPV-31 DNA content by at least 1.5-fold, which was not changed following EGFR depletion (Fig. 9B). Finally, we questioned whether Y102 was the target of FGFR3 activation. We previously reported that a phospho-deficient BPV-1 E2 mutant with phenylalanine in place of Y102 (Y102F) retained full capability to stimulate E1-dependent viral replication, whereas the phosphomimetic glutamate mutant (Y102E) is inactive. Initially we found that both BPV-1 Y102F and Y102E E2 were each capable of binding to FGFR3 (Fig. 10A). As seen in Fig. 10B, replication stimulated by Y102F was significantly reduced following cotransfection of FGFR3 K650E, implying that the FGFR3-mediated decrease in replication does not depend on phosphorylation of Y102. Consistent with this observation, we found that the amount of tyrosine phosphorylation for the BPV-1 E2 Y102F mutant did not decrease compared to that of the WT with FGFR3 activation (Fig. 10C).
FIG 9.

FGFR3 siRNA increases HPV-31 DNA content in CIN612-9E cells. (A) CIN612-9E cells were transfected with a control, FGFR3, or EGFR siRNA. After 48 h cells were lysed (0.5% NP-40), and immunoblotting was completed with FGFR3, EGFR, and β-actin antibodies. (B) CIN612-9E cells were transfected with a control, FGFR3, or EGFR siRNA. After 48 h cells were lysed, and DNA was isolated for reverse transcription-PCR against the HPV-31 LCR and normalized to the level with the β-actin primer set. Values are expressed as means ± SEM (n = 14). *, P < 0.1.
FIG 10.

FGFR3-mediated phosphorylation is not mediated through Y102. (A) HEK293TT cells were transfected with FLAG-FGFR3 or a BPV-1 E2 WT, Y102F, or Y102E construct. After 48 h, cells were lysed, and BPV-1 E2 was immunoprecipitated with BPV-1 E2 II-I antibody. Immunocomplexes were blotted with FLAG and B201 antibodies. (B) C33A cells were transfected with BPV-1 E1, E2, pFLORI-BPV-1 (Fluc), and pRL (Rluc) constructs in the presence of pCI or FLAG-FGFR3 K650E expression plasmids. Seventy-two hours later, cells were lysed, and firefly and Renilla luciferase levels were measured using Dual-Glo luciferase reagent. Firefly luciferase levels were normalized to Renilla luciferase levels. Values are expressed as means ± SEM (n = 4). (C) HEK293TT cells were transfected with a FLAG-FGFR3 K650E (KE), BPV-1 E2 WT, Y102F, or Y102E construct. Cells were lysed, and BPV-1 E2 was immunoprecipitated with B201 antibodies. Immunocomplexes were blotted with PY-99, B201, and M2 antibodies.
DISCUSSION
The papillomavirus E2 protein has multiple functions including binding to its consensus DNA motif at the viral origin and the E1 protein along with an undefined set of cellular factors that enable viral transcription and replication. These activities must be coordinated within the differentiating host epithelium. Regulation of E2 activities by posttranslational modifications during the viral life cycle has not been adequately characterized. A few serine-threonine kinases targeting E2 have been identified. Casein kinase II (CKII or CK2) phosphorylates BPV-1 E2 serine (S) 301 to destabilize the protein (27). Protein kinase A (PKA) phosphorylates HPV-8 E2 on S253 to increase E2 stability and chromatin binding (28). Polo-like kinase 1 (PLK1) interacts with HPV-5 E2, which was proposed to be mediated by the chromatin recognition protein Brd4 (29). Using a proteomics approach, the tyrosine kinase PTK2B was reported to be in complex with HPV-18 E2 (30). However, how these kinases modulate E2 actions during viral replication has yet to be elucidated. We recently described the role of lysine 111 acetylation and tyrosine 102 phosphorylation in modulating BPV-1 E2 function (7, 8).
Our intention was to identify the tyrosine kinase responsible for phosphorylation of E2 at position 102. During this screen, a member of the FGFR family, FGFR3, was discovered to coimmunoprecipitate with PV E2. The FGFR family of proteins is composed of four members (FGFR 1 to 4), each with several isoforms (31). The FGFR family is a subset of receptor tyrosine kinases (RTKs) and is responsible for regulating many of the cell's biological activities, including proliferation, differentiation, and angiogenesis (32). FGFR proteins consist of three extracellular immunoglobulin-like domains, a hydrophobic transmembrane domain, and a cytoplasmic kinase domain (33). FGFR3 is highly expressed in skin and in HFK cell lines with and without HPV (Fig. 1). Ligand binding results in receptor dimerization within the plasma membrane and consequent auto-tyrosine phosphorylation of the kinase domain (33). Sequentially, STAT, phospholipase Cγ (PLCγ), RAS–mitogen-activated protein kinase (MAPK), and AKT signaling pathways may become activated.
The FGFR3 interaction with E2 proteins represents an interesting partnership that may modulate viral replication within keratinocytes. FGFR3 expression was found throughout the cell, including in the nucleus. It is possible that the variations of FGFR3 localization depend on the cell cycle. Colocalization of FGFR3 and E2 in the nucleus was confirmed by direct immunofluorescence and PLA.
The potential impact of FGFR3-mediated phosphorylation on the replicative properties of PV E2 was the obvious enquiry. This is a technically complicated question since FGFR3 is responsible for inducing multiple downstream kinase effectors. We used kinase-active and kinase-dead forms of FGFR3 to provide suitable comparisons for such experiments. The phospho-activated mutant of FGFR3, K650E, had robust association with E2 and mapped to the N-terminal TAD. The literature shows that overexpression of wild-type FGFR3 itself has minimal basal kinase activity (17), as confirmed in our studies. Heterologous expression of FGFR3 K650E enhanced tyrosine phosphorylation of E2.
We initially examined transient replication of BPV-1 by transfection of PV ori-linked luciferase constructs into C33A cells. Heterologous expression of the active form of FGFR3 K650E significantly decreased BPV-1 replication. While heterologous expression of E2 and E1 in the transient PV replication reporter assay has advantages, it may have limitations due to the influence of relatively elevated levels of these proteins. We used CIN612-9E cells to drive endogenous expression of E2 since we also observed that FGFR3 K650E increased E2 protein levels in transfected cells. FGFR3 K650E expression impaired E2/E1-dependent transient replication in CIN612-9E cells cotransfected with the HPV-31 ori-linked luciferase construct. Importantly, HPV-31 genome content in CIN612-9E cells increased after FGFR3 depletion using an siRNA. EGFR silencing did not affect HPV-31 DNA content after 48 h. We used an EGFR siRNA as a control for these experiments since it did not complex with BPV-1 E2 (Fig. 2C). This observation is consistent with a previous study showing that another EGFR tyrosine kinase family member, ErbB3, did not bind to HPV-16 E2 (34).
Our experiments demonstrate that FGFR3 phosphorylates E2 and thereby restricts PV replication. Which tyrosine residue(s) of E2 is targeted by FGFR3 remains to be determined. We speculated that this might be the Y102 residue as we reported that the phosphomimetic form of this residue, Y102E, prohibited BPV replication (8). Consistent with this observation, the kinase-active form of FGFR3 mediated phosphorylation of E2 and inhibited HPV replication. However, the Y102F BPV E2 mutant was still tyrosine phosphorylated and displayed reduced E1-dependent transient replication in the presence of activated FGFR3. These observations imply that the suppression of replication by FGFR3 is not mediated through Y102. Experiments with the phosphotyrosine antibody PY-99 persuasively indicate that other tyrosine residues within E2 become phosphorylated by FGFR3. There are six highly conserved tyrosine residues in E2 (amino acids 32, 131, 138, 158, 159, and 319) among the BPV-1, HPV-16, HPV-18, and HPV-31 genotypes. Further investigations are designed to determine the precise amino acid substrates for FGFR3 and other tyrosine kinases that may mediate phosphorylation of E2.
Our data provide evidence for an inhibitory FGFR3-mediated phosphorylation of E2. During the initial and maintenance stages, E2 activity is low. Amplification of viral genomes and licensing of the viral origin require robust E2 activity for cooperation with the E2 binding partner E1. Gene expression data sets from several studies have shown that during keratinocyte differentiation, FGFR3 mRNA expression decreased (35–37). Prolonged FGFR3 activity may also promote differentiation. For example, increased FGFR3 activity promoted FOXN1-induced differentiation in benign epithelial tumors called seborrheic keratoses (11). We envision that FGFR3 phosphorylates E2 during the early stages of the viral replicative program within dividing basal keratinocytes to restrict E2 activity. During differentiation, FGFR3 expression decreases, and the inhibitory phosphorylation of E2 can be removed to permit PV genome amplification. Experiments are under way to fully characterize the phosphopeptides in HPV E2 and the repertoire of tyrosine kinases and phosphatases that modify these residues and thereby regulate viral transcription and genome replication throughout E2's course in the epithelium.
MATERIALS AND METHODS
Plasmids and antibodies.
The following plasmids were used: codon-optimized triple-FLAG HPV-31 E1 (24); pSG5-HA-HPV-31 E2 (where HA is hemagglutinin) (38); pGEM-EGFR (M. Korc, Indiana University); pCG-BPV-1 E1 Eag123 (39); ori-luciferase reporter plasmids for the PV transient replication assay (24); FLAG-FGFR3 WT and K650E from L. Thompson (UC Irvine); pCDNA-FGFR3 WT, K650E, and K508R from D. Donoghue (UC San Diego); EphB2 from E. Pasquale (Sanford Burnham Prebys Medical Discovery Institute); and pCG-BPV-1 E2, pCG-BPV-1 E2 TAD, and pCG-BPV-1 E2R as described previously (40). Codon-optimized FLAG HPV-31 E2 (41) was cloned between the BamHI and HindIII sites of pcDNA3.
The following antibodies were used: mouse anti-FLAG M2, anti-β-actin (Sigma), PY-99 (Santa Cruz), rabbit anti-EGFR (Cell Signaling), mouse anti-EphB2 (Acris Antibodies), and rabbit anti-FGFR3 (Sigma). BPV-1 E2 was identified with B201 (a mouse monoclonal antibody with an epitope between aa 160 and 220) and B202 (a mouse monoclonal antibody with an epitope between aa 286 and 310 [40, 42]). Mouse-anti HPV-16 E2 (TVG-261) and HPV-16 E2 sheep antiserum (18) were used to identify HPV-16 E2 proteins.
Cell culture.
All cell lines were maintained at 37°C and 5% CO2. HEK293TT (from J. Schiller and C. Buck), CV-1, and C33A (from D. Lowy) cells were cultured in Dulbecco's modified Eagle medium (Life Technologies) with 10% fetal bovine serum (Perk Serum) and penicillin-streptomycin (100 U/ml; Life Technologies). HaCat cells (from N. Fusenig) were cultured in Dulbecco's modified Eagle medium with 10% fetal bovine serum with 0.2 mM calcium. CIN612-9E (from L. Laimins) was grown in E medium with J23T3 fibroblast feeders (from Howard Green). W12 (from M. Stanley and P. Lambert) and normal immortalized keratinocytes (NIKS) were grown in F medium with J23T3 fibroblast feeders. HFK cells were maintained in keratinocyte serum-free medium (SFM) containing human recombinant epidermal growth factor (EGF; aa 1 to 53), bovine pituitary extract (BPE), and penicillin-streptomycin (100 U/ml; Life Technologies).
Immunofluorescence.
C33A cells were transfected with 2 μg of either FLAG-FGFR3 WT or FLAG-FGFR3 K650E using Lipofectamine 2000. CV-1 cells were transfected with 1 μg of FLAG-HPV-31 E2 and pCDNA3-FGFR3 constructs. These cell lines after transfection and the HFK cell lines were seeded into six-well dishes with coverslips. After 24 to 48 h, coverslips were washed twice in phosphate-buffered saline (PBS), fixed in 4% paraformaldehyde for 15 min, and washed three times in PBS. Coverslips were placed in 10% goat serum in PBS-Triton X (PBS-T; 0.25%) for 30 min, followed by primary antibody in PBS-T overnight; they were then washed three times with PBS, incubated in a 1:5,000 dilution of Alexa Fluor goat anti-mouse/rabbit 594 or 488 (Invitrogen) for 1 h, washed three times in PBS, and mounted onto slides with Vectashield mounting medium with 4′,6′-diamidino-2-phenylindole (DAPI). Cells were analyzed with a Nikon microscope or with an Olympus FV1000 MPE confocal microscope.
In situ PLA.
An in situ proximity ligation assay (PLA) was performed using a PLA Red kit (Olink Biosciences). C33A or CIN612-9E cells were transfected with FLAG-HPV-31 E2. Twenty-four hours later, cells were fixed in 4% paraformaldehyde for 10 min, permeabilized for 15 min in 0.5% Triton X-100–PBS, washed in PBS, blocked with 5% goat serum in 0.2% Triton X-100–PBS, and then incubated overnight with primary antibody combinations (E2-M2 FLAG or rabbit FGFR3) at 4°C. The PLA followed the manufacturer's protocol. Briefly, coverslips were washed in buffer A, incubated with PLA probe Plus and Minus for 1 h at 37°C, and washed twice, and the probes were ligated for 30 min. Amplification was performed for 100 min at 37°C and washed twice in buffers A and B provided in the kit. Coverslips were mounted in mounting medium provided in the PLA kit.
Coimmunoprecipitations and immunoblotting.
Cells were transfected using Lipofectamine 2000 (Life Technologies) or polyethyleneimine (PEI; 2 mg/ml) according to the manufacturer's instructions. After 24 to 48 h, cells were lysed in 0.5% NP-40 containing 150 mM NaCl, 20 mM HEPES, pH 7.4, 1 mM pervanadate, and protease inhibitor cocktail (Sigma). To each reaction mixture, 30 μl of a 50% protein A/G slurry (Invitrogen) and 1 μg of antibody was added and rotated overnight at 4°C. Beads were washed five times in lysis buffer. Proteins were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE). Gels were transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore), blocked in 3% Tris-buffered saline (TBS)–Tween–0.1% bovine serum albumin (BSA), and probed with antibodies. Chemiluminescence substrates (Thermo Scientific) were used to detect antibody signal. For endogenous HPV-16 E2 coimmunoprecipitation, W12 cells were seeded and grown to 80% confluence. Cells lysed in 50 mM HEPES [pH 7.4], 150 mM NaCl, 1 mM Na3VO4, 0.5% NP-40, 1 mM dithiothreitol (DTT), and 1× protease inhibitor cocktail. Ten microliters of sheep anti-HPV-16 E2 polyclonal serum or 1 μg of sheep nonspecific IgG (Santa Cruz) was added to the lysates and rotated overnight at 4°C. The immunoprecipitates were collected using a protein A/G slurry for 3 h at 4°C. Beads were washed four times with lysis buffer and once with high-salt buffer (50 mM HEPES [pH 7.4], 500 mM NaCl, 1 mM Na3VO4, 0.5% NP-40, 1 mM DTT, and 1× protease inhibitor cocktail). Proteins were removed from the beads with 2× SDS lysis buffer, separated by 4 to 12% SDS-PAGE, and detected by Western blotting using anti-FGFR3 and anti-HPV-16 E2 (TVG-261) antibodies.
Luciferase DNA replication assays.
C33A and CIN612-9E cells were transfected with 3 μg of FGFR3 constructs, 15 ng of pRL (Rluc), 75 ng of pFLORI31 or pFLORIBPV-1, 0.3 μg of pE1 (only for C33A cells), and 0.3 μg of pE2 (only for C33A cells) using Lipofectamine 2000 in a six-well dish. For the BPV replication assays, pCG-BPV-1 E1 Eag123 was used for E1 expression. Seventy-two hours later cells were lysed, and luciferase activity was measured using a Dual-Glo kit (Promega). Firefly luciferase levels were normalized to Renilla luciferase levels.
PCR DNA replication assays.
CIN612-9E cells, without feeders, were seeded in 6-cm plates with the addition of the transfection reaction mixture. The transfection reaction mixture contained Lipofectamine 2000 (Invitrogen) and either a control siRNA (sc-37007; Santa Cruz), FGFR3 siRNA duplexes (sc-29314; Santa Cruz), or EGFR siRNA (sc-29301; Santa Cruz) at a final concentration of 15 nM in E-medium. Forty-eight hours later, cells were lysed in Tris-EDTA (TE) medium with 0.1% SDS and 10 ng/μl RNase. DNA was isolated with phenol-chloroform-isoamyl alcohol (25:24:1; Fisher Scientific), and real-time PCR was performed as described above. HPV-31 DNA content was measured using primers to the HPV-31 long control region (LCR) (5′-GTTCTGCGGTTTTTGGTTTC-3′ and 5′-TGTTGGCAAGGTGTGTTAGG-3′). HPV-31 DNA content was normalized to that of β-actin DNA (using primers 5′-GAGGCACTCTTCCAGCCTTC-3′ and 5′-CGGATGTCCACGTCACACTT-3′) (43).
Statistical analysis.
A two-way or one-way t test was used for analysis. Means are expressed ± standard errors of the means (SEM).
ACKNOWLEDGMENTS
We appreciate the generosity of the following for plasmids: Alison McBride (NIAID/NIH) for the codon-optimized HPV-31 E2, Jacques Archambault (McGill University) for HPV-31 and BPV-1 luciferase replicons, Leslie Thompson (University of California, Irvine) for FLAG-FGFR3, Daniel Donoghue (University of California, San Diego) for pCDNA-FGFR3, Murray Korc (Indiana University School of Medicine) for pGEM-EGFR, and Elena Pasquale (Sanford Burnham Prebys Medical Discovery Institute) for EphB2 expression plasmid. Jo Parish (Birmingham University) kindly provided the HPV-16 E2 antiserum.
This work was supported by National Cancer Institute/National Institutes of Health R01CA58376 (E.J.A.), T32AI060519 (M.D.), T32AI060519 and F30AI114284 (S.P.C.), and T32AI007637 and T32AR062495 (T.G.) and a Beijing Natural Science Foundation grant 7174347 (F.X.).
REFERENCES
- 1.McKinney CC, Hussmann KL, McBride AA. 2015. The Role of the DNA damage response throughout the papillomavirus life cycle. Viruses 7:2450–2469. doi: 10.3390/v7052450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hong S, Laimins LA. 2013. Regulation of the life cycle of HPVs by differentiation and the DNA damage response. Future Microbiol 8:1547–1557. doi: 10.2217/fmb.13.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Banerjee NS, Wang HK, Broker TR, Chow LT. 2011. Human papillomavirus (HPV) E7 induces prolonged G2 following S phase reentry in differentiated human keratinocytes. J Biol Chem 286:15473–15482. doi: 10.1074/jbc.M110.197574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bravo IG, Felez-Sanchez M. 2015. Papillomaviruses: viral evolution, cancer and evolutionary medicine. Evol Med Public Health 2015:32–51. doi: 10.1093/emph/eov003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McBride AA, Romanczuk H, Howley PM. 1991. The papillomavirus E2 regulatory proteins. J Biol Chem 266:18411–18414. [PubMed] [Google Scholar]
- 6.McBride AA. 2013. The papillomavirus E2 proteins. Virology 445:57–79. doi: 10.1016/j.virol.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Quinlan EJ, Culleton SP, Wu SY, Chiang CM, Androphy EJ. 2013. Acetylation of conserved lysines in bovine papillomavirus E2 by p300. J Virol 87:1497–1507. doi: 10.1128/JVI.02771-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Culleton SP, Kanginakudru S, DeSmet M, Gilson T, Xie F, Wu SY, Chiang CM, Qi G, Wang M, Androphy EJ. 2017. Phosphorylation of the bovine papillomavirus E2 protein on tyrosine regulates its transcription and replication functions. J Virol 91:e01854-16. doi: 10.1128/JVI.01854-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang SW, Liu WC, Liao KY, Tsao YP, Hsu PH, Chen SL. 2014. Phosphorylation of HPV-16 E2 at serine 243 enables binding to Brd4 and mitotic chromosomes. PLoS One 9:e110882. doi: 10.1371/journal.pone.0110882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schuck S, Ruse C, Stenlund A. 2013. CK2 phosphorylation inactivates DNA binding by the papillomavirus E1 and E2 proteins. J Virol 87:7668–7679. doi: 10.1128/JVI.00345-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mandinova A, Kolev V, Neel V, Hu B, Stonely W, Lieb J, Wu X, Colli C, Han R, Pazin MJ, Ostano P, Dummer R, Brissette JL, Dotto GP. 2009. A positive FGFR3/FOXN1 feedback loop underlies benign skin keratosis versus squamous cell carcinoma formation in humans. J Clin Invest 119:3127–3137. doi: 10.1172/JCI38543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duperret EK, Oh SJ, McNeal A, Prouty SM, Ridky TW. 2014. Activating FGFR3 mutations cause mild hyperplasia in human skin, but are insufficient to drive benign or malignant skin tumors. Cell Cycle 13:1551–1559. doi: 10.4161/cc.28492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hafner C, van Oers JM, Hartmann A, Landthaler M, Stoehr R, Blaszyk H, Hofstaedter F, Zwarthoff EC, Vogt T. 2006. High frequency of FGFR3 mutations in adenoid seborrheic keratoses. J Invest Dermatol 126:2404–2407. [DOI] [PubMed] [Google Scholar]
- 14.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Ponten F. 2015. Proteomics Tissue-based map of the human proteome. Science 347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 15.Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, Zwahlen M, Kampf C, Wester K, Hober S, Wernerus H, Bjorling L, Ponten F. 2010. Towards a knowledge-based Human Protein Atlas. Nat Biotechnol 28:1248–1250. doi: 10.1038/nbt1210-1248. [DOI] [PubMed] [Google Scholar]
- 16.Uhlen M, Bjorling E, Agaton C, Szigyarto CA, Amini B, Andersen E, Andersson AC, Angelidou P, Asplund A, Asplund C, Berglund L, Bergstrom K, Brumer H, Cerjan D, Ekstrom M, Elobeid A, Eriksson C, Fagerberg L, Falk R, Fall J, Forsberg M, Bjorklund MG, Gumbel K, Halimi A, Hallin I, Hamsten C, Hansson M, Hedhammar M, Hercules G, Kampf C, Larsson K, Lindskog M, Lodewyckx W, Lund J, Lundeberg J, Magnusson K, Malm E, Nilsson P, Odling J, Oksvold P, Olsson I, Oster E, Ottosson J, Paavilainen L, Persson A, Rimini R, Rockberg J, Runeson M, Sivertsson A, Skollermo A, et al. . 2005. A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol Cell Proteomics 4:1920–1932. doi: 10.1074/mcp.M500279-MCP200. [DOI] [PubMed] [Google Scholar]
- 17.Nelson KN, Meyer AN, Siari A, Campos AR, Motamedchaboki K, Donoghue DJ. 2016. Oncogenic gene fusion FGFR3-TACC3 is regulated by tyrosine phosphorylation. Mol Cancer Res 14:458–469. doi: 10.1158/1541-7786.MCR-15-0497. [DOI] [PubMed] [Google Scholar]
- 18.Siddiqa A, Leon KC, James CD, Bhatti MF, Roberts S, Parish JL. 2015. The human papillomavirus type 16 L1 protein directly interacts with E2 and enhances E2-dependent replication and transcription activation. J Gen Virol 96:2274–2285. doi: 10.1099/vir.0.000162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klucevsek K, Wertz M, Lucchi J, Leszczynski A, Moroianu J. 2007. Characterization of the nuclear localization signal of high risk HPV16 E2 protein. Virology 360:191–198. doi: 10.1016/j.virol.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hubbert NL, Schiller JT, Lowy DR, Androphy EJ. 1988. Bovine papilloma virus-transformed cells contain multiple E2 proteins. Proc Natl Acad Sci U S A 85:5864–5868. doi: 10.1073/pnas.85.16.5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou L, Yao LT, Liang ZY, Zhou WX, You L, Shao QQ, Huang S, Guo JC, Zhao YP. 2015. Nuclear translocation of fibroblast growth factor receptor 3 and its significance in pancreatic cancer. Int J Clin Exp Pathol 8:14640–14648. [PMC free article] [PubMed] [Google Scholar]
- 22.Schmahl J, Kim Y, Colvin JS, Ornitz DM, Capel B. 2004. Fgf9 induces proliferation and nuclear localization of FGFR2 in Sertoli precursors during male sex determination. Development 131:3627–3636. doi: 10.1242/dev.01239. [DOI] [PubMed] [Google Scholar]
- 23.Neben CL, Idoni B, Salva JE, Tuzon CT, Rice JC, Krakow D, Merrill AE. 2014. Bent bone dysplasia syndrome reveals nucleolar activity for FGFR2 in ribosomal DNA transcription. Hum Mol Genet 23:5659–5671. doi: 10.1093/hmg/ddu282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fradet-Turcotte A, Morin G, Lehoux M, Bullock PA, Archambault J. 2010. Development of quantitative and high-throughput assays of polyomavirus and papillomavirus DNA replication. Virology 399:65–76. doi: 10.1016/j.virol.2009.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lehoux M, Gagnon D, Archambault J. 2014. E1-mediated recruitment of a UAF1-USP deubiquitinase complex facilitates human papillomavirus DNA replication. J Virol 88:8545–8555. doi: 10.1128/JVI.00379-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gagnon D, Lehoux M, Archambault J. 2015. Artificial recruitment of UAF1-USP complexes by a PHLPP1-E1 chimeric helicase enhances human papillomavirus DNA replication. J Virol 89:6227–6239. doi: 10.1128/JVI.00560-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Penrose KJ, Garcia-Alai M, de Prat-Gay G, McBride AA. 2004. Casein Kinase II phosphorylation-induced conformational switch triggers degradation of the papillomavirus E2 protein. J Biol Chem 279:22430–22439. doi: 10.1074/jbc.M314340200. [DOI] [PubMed] [Google Scholar]
- 28.Sekhar V, McBride AA. 2012. Phosphorylation regulates binding of the human papillomavirus type 8 E2 protein to host chromosomes. J Virol 86:10047–10058. doi: 10.1128/JVI.01140-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang WS, Lee MS, Tseng CE, Liao IH, Huang SP, Lin RI, Li C. 2009. Interaction between human papillomavirus type 5 E2 and polo-like kinase 1. J Med Virol 81:536–544. doi: 10.1002/jmv.21404. [DOI] [PubMed] [Google Scholar]
- 30.Muller M, Jacob Y, Jones L, Weiss A, Brino L, Chantier T, Lotteau V, Favre M, Demeret C. 2012. Large scale genotype comparison of human papillomavirus E2-host interaction networks provides new insights for e2 molecular functions. PLoS Pathog 8:e1002761. doi: 10.1371/journal.ppat.1002761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Logie A, Dunois-Larde C, Rosty C, Levrel O, Blanche M, Ribeiro A, Gasc JM, Jorcano J, Werner S, Sastre-Garau X, Thiery JP, Radvanyi F. 2005. Activating mutations of the tyrosine kinase receptor FGFR3 are associated with benign skin tumors in mice and humans. Hum Mol Genet 14:1153–1160. doi: 10.1093/hmg/ddi127. [DOI] [PubMed] [Google Scholar]
- 32.Wesche JHK, Haugsten EM. 2011. Fibroblast growth factors and their receptors in cancer. Biochem J 15:199–213. doi: 10.1042/BJ20101603. [DOI] [PubMed] [Google Scholar]
- 33.Gallo LH, Nelson KN, Meyer AN, Donoghue DJ. 2015. Functions of fibroblast growth factor receptors in cancer defined by novel translocations and mutations. Cytokine Growth Factor Rev 26:425–449. doi: 10.1016/j.cytogfr.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 34.Paolini F, Curzio G, Melucci E, Terrenato I, Antoniani B, Carosi M, Mottolese M, Vici P, Mariani L, Venuti A. 2016. Human papillomavirus 16 E2 interacts with neuregulin receptor degradation protein 1 affecting ErbB-3 expression in vitro and in clinical samples of cervical lesions. Eur J Cancer 58:52–61. doi: 10.1016/j.ejca.2016.02.001. [DOI] [PubMed] [Google Scholar]
- 35.Cavazza A, Miccio A, Romano O, Petiti L, Malagoli Tagliazucchi G, Peano C, Severgnini M, Rizzi E, De Bellis G, Bicciato S, Mavilio F. 2016. Dynamic transcriptional and epigenetic regulation of human epidermal keratinocyte differentiation. Stem Cell Reports 6:618–632. doi: 10.1016/j.stemcr.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cohen I, Birnbaum RY, Leibson K, Taube R, Sivan S, Birk OS. 2012. ZNF750 is expressed in differentiated keratinocytes and regulates epidermal late differentiation genes. PLoS One 7:e42628. doi: 10.1371/journal.pone.0042628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sen GL, Boxer LD, Webster DE, Bussat RT, Qu K, Zarnegar BJ, Johnston D, Siprashvili Z, Khavari PA. 2012. ZNF750 is a p63 target gene that induces KLF4 to drive terminal epidermal differentiation. Dev Cell 22:669–677. doi: 10.1016/j.devcel.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Powell ML, Smith JA, Sowa ME, Harper JW, Iftner T, Stubenrauch F, Howley PM. 2010. NCoR1 mediates papillomavirus E8;E2C transcriptional repression. J Virol 84:4451–4460. doi: 10.1128/JVI.02390-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ustav M, Ustav E, Szymanski P, Stenlund A. 1991. Identification of the origin of replication of bovine papillomavirus and characterization of the viral origin recognition factor E1. EMBO J 10:4321–4329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Breiding DE, Grossel MJ, Androphy EJ. 1996. Genetic analysis of the bovine papillomavirus E2 transcriptional activation domain. Virology 221:34–43. doi: 10.1006/viro.1996.0350. [DOI] [PubMed] [Google Scholar]
- 41.Sakakibara N, Mitra R, McBride AA. 2011. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J Virol 85:8981–8995. doi: 10.1128/JVI.00541-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Androphy EJ, Lowy DR, Schiller JT. 1987. Bovine papillomavirus E2 trans-activating gene product binds to specific sites in papillomavirus DNA. Nature 325:70–73. doi: 10.1038/325070a0. [DOI] [PubMed] [Google Scholar]
- 43.DeSmet M, Kanginakudru S, Rietz A, Wu WH, Roden R, Androphy EJ. 2016. The replicative consequences of papillomavirus E2 protein binding to the origin replication factor ORC2. PLoS Pathog 12:e1005934. doi: 10.1371/journal.ppat.1005934. [DOI] [PMC free article] [PubMed] [Google Scholar]