

ABSTRACT
Human metapneumovirus (HMPV) has the ability to inhibit Toll-like receptor 7 (TLR7)- and TLR9-dependent alpha interferon (IFN-α) production by plasmacytoid dendritic cells (pDCs). However, the inhibition mechanism remains largely unknown. To identify viral proteins responsible for this inhibition, we performed a screening of HMPV open reading frames (ORFs) for the ability to block TLR7/9-dependent signaling reconstituted in HEK293T cells by transfection with myeloid differentiation factor 88 (MyD88), tumor necrosis factor receptor-associated factor 6 (TRAF6), IKKα, and IFN regulatory factor 7 (IRF7). This screening demonstrated that the M2-2 protein was the most potent inhibitor of TLR7/9-dependent IFN-α induction. A recombinant HMPV in which the M2-2 ORF was silenced indeed induced greater IFN-α production by human pDCs than wild-type HMPV did. Immunoprecipitation experiments showed direct physical association of the M2-2 protein with the inhibitory domain (ID) of IRF7. As a natural consequence of this, transfection of IRF7 lacking the ID, a constitutively active mutant, resulted in activation of the IFN-α promoter even in the presence of M2-2. Bioluminescence resonance energy transfer assays and split Renilla luciferase complementation assays revealed that M2-2 inhibited MyD88/TRAF6/IKKα-induced homodimerization of IRF7. In contrast, expression of the M2-2 protein did not result in inhibition of IPS-1-induced homodimerization and resultant activation of IRF7. This indicates that inhibition of MyD88/TRAF6/IKKα-induced IRF7 homodimerization does not result from a steric effect of M2-2 binding. Instead, it was found that M2-2 inhibited MyD88/TRAF6/IKKα-induced phosphorylation of IRF7 on Ser477. These results suggest that M2-2 blocks TLR7/9-dependent IFN-α induction by preventing IRF7 homodimerization, possibly through its effects on the phosphorylation status of IRF7.
IMPORTANCE The family Paramyxoviridae is divided into two subfamilies, the Paramyxovirinae and the Pneumovirinae. Members of the subfamily Paramyxovirinae have the ability to inhibit TLR7/9-dependent IFN-α production, and the underlying inhibition mechanism has been intensively studied. In contrast, little is known about how members of the subfamily Pneumovirinae regulate IFN-α production by pDCs. We identified the M2-2 protein of HMPV, a member of the subfamily Pneumovirinae, as a negative regulator of IFN-α production by pDCs and uncovered the underlying mechanism. This study explains in part why the M2-2 knockout recombinant HMPV is attenuated and further suggests that M2-2 is a potential target for HMPV therapy.
KEYWORDS: IRF7, TLR7, human metapneumovirus, immune evasion, interferon, plasmacytoid dendritic cell
INTRODUCTION
Human metapneumovirus (HMPV) is a member of the subfamily Pneumovirinae in the family Paramyxoviridae and is a causal agent of respiratory diseases in all age groups. HMPV infection is clinically important, especially in the field of pediatric medicine, because HMPV, like human respiratory syncytial virus (HRSV) in the same subfamily, potentially causes severe lower respiratory tract infection in young children, such as infants with congenital heart disease and low-birth-weight babies. Pathogenesis of HMPV involves complicated processes that are affected by multiple factors, including viral evasion strategies for the host immune system. In general, silencing viral genes responsible for immune evasion resulted in attenuation of the virus. Therefore, a full understanding of viral immune evasion mechanisms contributes to not only elucidation of viral pathogenesis, but also development of effective vaccines and antiviral agents.
Interferons (IFNs) play a central role in innate immunity, constituting the first line of host defense. IFNs not only induce an antiviral state in cells via the JAK-STAT signaling cascade, but also contribute to activation of macrophages and T lymphocytes. A variety of cells produce IFNs, but plasmacytoid dendritic cells (pDCs) are unique in producing enormous amounts of alpha IFN (IFN-α). pDCs recognize pathogen-associated molecular patterns (PAMPs) derived from invading pathogens by using their endosomal Toll-like receptor 7 (TLR7) and TLR9 (TLR7/9). This recognition initiates signaling cascades, one of which leads to the ultimate activation of latent IFN regulatory factor 7 (IRF7). IRF7 is phosphorylated by the serine/threonine kinases, interleukin 1 (IL-1) receptor-associated kinase 1 (IRAK1) and/or IκB kinase alpha (IKKα), forms a homodimer, and translocates into the nucleus to activate the IFN-α genes (1–4). The core of the TLR7/9-dependent signaling cascade consists of myeloid differentiation factor 88 (MyD88), IRAK4, tumor necrosis factor receptor-associated factor 6 (TRAF6), TRAF3, IRAK1, IKKα, viperin, and osteopontin (5–12).
To maintain efficient virus growth in the body, viruses have independently developed strategies by which they minimize host IFN production. Indeed, it was found that members of the subfamily Paramyxovirinae encode V and C proteins that have the ability to block the TLR7/9-dependent signaling cascade leading to IFN-α production in pDCs (13–16). This activity is shared by all the C and V proteins tested in the subfamily Paramyxovirinae, suggesting its importance for survival of members of the Paramyxovirinae throughout evolution. We investigated their underlying molecular mechanisms and have presented evidence to indicate that the V protein of human parainfluenza virus type 2 (PIV2) targets TRAF6 for inhibition of K63-linked polyubiquitination and the C protein of Sendai virus (SeV) binds to IKKα to inhibit phosphorylation of IRF7 (13–15). On the other hand, Pfaller and Conzelmann reported that the V protein of measles virus inhibited phosphorylation of IRF7 by serving as a decoy substrate for IKKα (16). In contrast, little is known about the mechanisms by which members of the subfamily Pneumovirinae inhibit IFN-α production by pDCs, although it has been suggested that HRSV and HMPV have strategies for suppressing IFN-α production by pDCs (17–19). Guerrero-Plata et al. revealed that infection with HRSV or HMPV in vitro inhibited IFN-α production by pDCs in response to a TLR9 ligand, synthetic CpG oligodeoxynucleotide (CpG-ODN), and that viral replication was necessary to elicit this effect (20). They also found that lung pDCs isolated from mice infected with HRSV or HMPV in vivo exhibited reduced ability to produce IFN-α in response to the synthetic CpG-ODN (19).
Considering these circumstances, we designed this study to identify HMPV proteins responsible for the inhibition of TLR7/9-dependent signaling and to elucidate the underlying molecular mechanisms. Screening of the HMPV open reading frames (ORFs) for the ability to inhibit TLR7/9-dependent IFN-α induction revealed that the M2-2 protein was the most potent inhibitor. We show that the M2-2 protein directly binds to IRF7 and specifically prevents MyD88/TRAF6/IKKα-stimulated homodimerization of IRF7, possibly through inhibition of IRF7 phosphorylation.
RESULTS
M2-2 serves as an inhibitor of IFN-α production by pDCs.
HMPV infection of mice in vivo and pDCs in vitro resulted in reduction in the pDCs' ability to produce IFN-α in response to TLR9 stimulation (18, 19). This finding suggests that HMPV encodes proteins that inhibit the TLR7/9-dependent signaling cascade leading to IFN-α production. To identify viral proteins responsible for this inhibition, we carried out a screening of HMPV ORFs for the ability to block TLR7/9-dependent signaling reconstituted in HEK293T cells. In this reconstitution system, a set of upstream signaling components (human MyD88, TRAF6, and IKKα), in addition to human IRF7, were transfected into HEK293T cells, along with the IFN-α6 promoter-driven firefly luciferase (Fluc) reporter plasmid and an internal control, pRL-TK, according to the procedure described by Pfaller and Conzelmann (16). Signaling components of human origin were used in this and subsequent experiments unless otherwise noted. As shown in Fig. 1A, transfection of MyD88, TRAF6, and IKKα, along with IRF7, resulted in striking enhancement of the IFN-α promoter activation compared with transfection of IRF7 alone. This enhancement was suppressed when M2-2, M, SH, P, N, or F was cotransfected. The most potent suppression was observed in M2-2, which exhibited dose-dependent inhibition, like human PIV2 V (Fig. 1B). This inhibition was further confirmed by measuring the IFN-α levels of culture media (Fig. 1C). Activation of TLR7/9-dependent signaling leads to not only IRF7-mediated IFN production, but also NF-κB-mediated cytokine production. Thus, a similar experiment was performed using an NF-κB-responsive Fluc plasmid. However, MyD88-induced NF-κB activation was not inhibited by M2-2 (Fig. 1D). TRAF6 C70A, a dominant-negative mutant in which the cysteine residue at position 70 important for ubiquitin-conjugating activity is mutated to alanine, was used as a positive control (21). We next created recombinant HMPV (rHMPV) expressing green fluorescent protein (GFP) (rHMPV-GFP) and rHMPV-GFPΔM2-2, in which the M2-2 ORF is silenced, and infected human pDCs isolated from human peripheral blood mononuclear cells (PBMCs). At 36 h postinfection, the levels of IFN-α secreted by pDCs were determined. As shown in Fig. 1E, rHMPV-GFPΔM2-2 induced IFN-α production about 17 times more than rHMPV-GFP did, demonstrating that the M2-2 protein actually functions as a negative regulator of IFN-α production by pDCs.
FIG 1.

Effects of HMPV proteins on the TLR7/9-dependent signaling pathway. (A, B, and D) An IFN-α6 promoter-driven (A and B) or NF-κB-dependent (D) Fluc reporter plasmid was transfected into HEK293T cells, along with the internal control, pRL-TK, and the indicated plasmids. At 24 h posttransfection, Fluc and Rluc activities were measured. Relative luciferase activity was calculated as the ratio of Fluc activity to Rluc activity. (C and E) The indicated plasmids were transfected into HEK293T cells without the reporter plasmids and pRL-TK (C) or human pDCs (∼104 cells/well) were infected with wild-type (wt) rHMPV-GFP or rHMPV-GFPΔM2-2 at a multiplicity of infection (MOI) of 3 (E). At 36 h posttransfection (C) or postinfection (E), levels of IFN-α in the culture media were measured by ELISA. The mean values from three independent experiments are shown, with standard deviations. C70A, human TRAF6 C70A mutant.
Activation of the TLR7/9-dependent signaling pathway induces production of IFN-α, which in turn activates transcription of the IRF7 gene, one of the IFN-stimulated genes, via the JAK-STAT pathway. This upregulation of IRF7 serves as positive feedback and consequently augments IFN-α production. Assuming that the M2-2 protein has the ability to block the JAK-STAT pathway, this assumptive blockade suppresses upregulation of IRF7, thereby reducing the activation level of the IFN-α promoter. To exclude this possibility, we examined the effect of the M2-2 protein on IFN-α-mediated activation of the JAK-STAT pathway. HEK293T cells were transfected with an IFN-sensitive response element (ISRE)-containing promoter-driven Fluc plasmid and pRL-TK and then treated with IFN-α (Fig. 2A). When SeV C protein, a well-known inhibitor of the JAK-STAT pathway, was transfected along with the reporter plasmids, IFN-α-mediated activation of the ISRE-containing promoter was clearly inhibited. In contrast, none of the HMPV proteins, including M2-2, exhibited an inhibitory effect (Fig. 2A). As expected from this result, the inhibitory effect of M2-2 on MyD88/TRAF6/IKKα-induced IFN-α promoter activation was seen even in Vero cells that had a defect in the type I IFN genes (Fig. 2B). These results ensured the authenticity of the ability of M2-2 to block TLR7/9-dependent signaling leading to IFN-α production.
FIG 2.
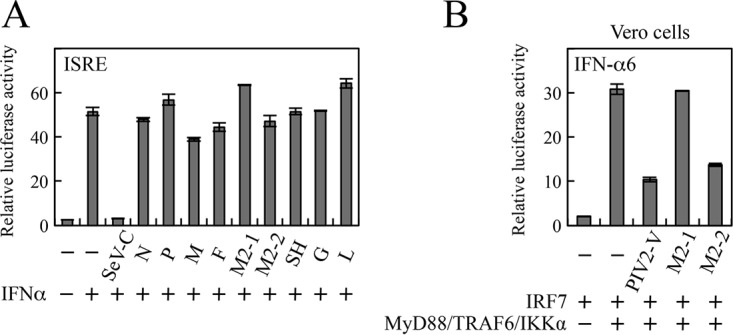
Effects of HMPV proteins on the JAK-STAT signaling pathway. (A) HEK293T cells were transfected with the indicated plasmids, pRL-T, and an ISRE Fluc reporter plasmid. At 24 h posttransfection, the cells were treated with IFN-α2b (1,000 U/ml). (B) Vero cells were transfected with the indicated plasmids, pRL-TK, and an IFN-α6 promoter-driven Fluc reporter plasmid. After 6 h of IFN treatment (A) or at 24 h posttransfection (B), Fluc and Rluc activities were measured. Relative luciferase activity was calculated as the ratio of Fluc activity to Rluc activity. The mean values from three independent experiments are shown, with standard deviations.
IRF7 is a potential target of the M2-2 protein.
To find a molecular target of M2-2, we investigated the interaction between the M2-2 protein and signaling components of the TLR7/9-dependent signaling pathway by immunoprecipitation experiments. FLAG-M2-2 was transfected into HEK293T cells, along with one of the V5-tagged signaling components, and then the transfected cells were subjected to immunoprecipitation. As shown in Fig. 3A, FLAG-M2-2 was coimmunoprecipitated with anti-V5 antibody in cells transfected with V5-tagged TRAF6, IKKα, and IRF7. Conversely, V5-tagged TRAF6, IKKα, and IRF7 were coimmunoprecipitated with anti-FLAG antibody (Fig. 3B). To determine whether these interactions were direct, similar experiments were carried out for mixtures of myc-M2-2 and V5-tagged signaling components, which were synthesized by using the wheat germ cell-free expression system. As shown in Fig. 3C, only V5-tagged IRF7 was coimmunoprecipitated with anti-myc antibody, indicating that the M2-2–IRF7 interaction was direct. Further immunoprecipitation experiments for cells transfected with myc-IRF7 and various FLAG-tagged HMPV proteins showed that none of the HMPV proteins other than M2-2 interacted with IRF7 (Fig. 3D), suggesting specificity of the interaction between M2-2 and IRF7.
FIG 3.

Interaction of the M2-2 protein with IRF7. (A, B, and D) HEK293T cells were transfected with the indicated plasmids. At 24 h posttransfection, the cells were lysed in lysis buffer and subjected to immunoprecipitation (IP) with anti-V5 (A) or anti-FLAG (B and D) antibody, followed by immunoblot analysis (WB) with anti-FLAG, anti-V5, or anti-myc antibody. (C) V5-TRAF6, V5-IKKα, V5-IRF7, and myc-M2-2 were synthesized in the wheat germ cell-free expression system. Then, the in vitro transcription/translation products were mixed in various combinations and subjected to IP with anti-myc antibody followed by WB with anti-V5 and anti-myc antibodies. A portion of the whole-cell lysates (A, B, and D) or the in vitro transcription/translation products (Input) (C) prepared for IP was also subjected to WB. The asterisks indicate the positions of the antibody heavy chain (AbHC).
The M2-2 protein binds to the inhibitory domain of IRF7.
IRF7 comprises four domains; the DNA-binding domain, transactivation domain, inhibitory domain (ID), and regulatory domain (22, 23) (Fig. 4A). To identify the domains interacting with M2-2, we first used mouse IRF7 (mIRF7) deletion mutants previously created (13) for immunoprecipitation experiments. These FLAG-tagged IRF7 deletion mutants, such as C1 and C2, were transfected into HEK293T cells, together with myc-M2-2. As shown in Fig. 4B, myc-M2-2 was coimmunoprecipitated with anti-FLAG antibody in cells transfected with FLAG-tagged C2, C3, and N3, but not C1, N1, and N2, suggesting that the ID of mIRF7 interacted with M2-2. We then created expression plasmids that carried FLAG-IRF7 of human origin and its deletion mutants, FLAG-ID and FLAG-ΔID (IRF7 lacking the ID). As expected, myc-M2-2 was coimmunoprecipitated with anti-FLAG antibody in cells transfected with FLAG-ID, but not FLAG-ΔID (Fig. 4C). A similar result was obtained for the ID and ΔID mutants of mIRF7 (data not shown). Since the ΔID mutant is a constitutively active mutant, transfection of the ΔID mutant alone resulted in significant activation of the IFN-α6 promoter without transfection of upstream signaling components (Fig. 4D). This activation was not suppressed by M2-2 at all, even if the dose of the M2-2 plasmid was increased, suggesting the importance of the M2-2–IRF7 interaction.
FIG 4.

Interaction of the M2-2 protein with IRF7 deletion mutants. (A) Schematic diagram of FLAG-tagged deletion mutants of mIRF7. (B and C) HEK293T cells were transfected with the indicated plasmids and lysed at 24 h posttransfection. The lysates were subjected to IP with anti-FLAG antibody followed by WB with anti-myc or anti-FLAG antibody. A portion of the whole-cell lysates prepared for IP was also subjected to WB. (D) HEK293T cells were transfected with the indicated plasmids, an IFN-α6 promoter-driven reporter plasmid, and pRL-TK. At 24 h posttransfection, Fluc and Rluc activities were measured. Relative luciferase activity was calculated as the ratio of Fluc activity to Rluc activity. The mean values from three independent experiments are shown, with standard deviations. The IRF7 deletion mutants used in panel B were of mouse origin, whereas those in panels C and D were of human origin.
M2-2 inhibits MyD88/TRAF6/IKKα-induced homodimerization of IRF7.
IRF7 is activated through its phosphorylation and subsequent homodimerization. Since homodimerization involves the ID-ID association (24), it raises the possibility that M2-2 inhibits IRF7 homodimerization. To monitor IRF7 homodimerization, we tried immunoprecipitation experiments using extracts from cells transfected with FLAG-IRF7 and myc-IRF7. However, this method was not suitable for analysis of IRF7 homodimerization, because a large proportion of phosphorylated IRF7 after stimulation of the TLR7/9-dependent pathway was found to become insoluble in lysis buffer while unphosphorylated IRF7 remained soluble (data not shown). Therefore, we performed a bioluminescence resonance energy transfer (BRET) assay instead (Fig. 5A). The BRET assay is useful for analyzing protein-protein interactions in living cells. We constructed plasmids encoding IRF7-YFP (IRF7 fused to the N terminus of yellow fluorescent protein [YFP]) and IRF7-Nluc (IRF7 fused to the N terminus of NanoLuc [Nluc]; Promega). HEK293T cells were transfected with various combinations of IRF7-YFP, IRF7-Nluc, YFP, and Nluc. At 24 h posttransfection, the YFP emission and the Nluc emission were measured immediately after addition of furimazine, a substrate for Nluc. The Nluc emission resulting from catalytic degradation of furimazine provides energy to YFP, resulting in YFP emission only when YFP and Nluc are in close proximity. The BRET ratio is defined by the YFP emission (530 to 570 nm) relative to the Nluc emission (370 to 450 nm). As shown in Fig. 5A, the BRET signal was less than 0.1 in cells transfected with YFP/Nluc, YFP/IRF7-Nluc, IRF7-YFP/Nluc, IRF7-YFP/IRF7-Nluc, or Nluc alone. Cotransfection of MyD88, TRAF6, and IKKα strikingly enhanced the BRET signal in cells transfected with IRF7-YFP/IRF7-Nluc but not in cells transfected with YFP/IRF7-Nluc or IRF7-YFP/Nluc. This enhancement was suppressed by cotransfection of M2-2 or PIV2 V, but not M2-1 (Fig. 5B). PIV2 V was tested as a positive control that targets TRAF6-mediated K63-linked polyubiquitination, a prerequisite for phosphorylation and homodimerization of IRF7 (14).
FIG 5.

Effect of the M2-2 protein on MyD88-TRAF6-IKKα-induced homodimerization of IRF7. (A and B) HEK293T cells were transfected with the indicated plasmids. At 24 h posttransfection, the cells were suspended in Dulbecco's PBS. After addition of furimazine, luminescence and fluorescence signals were immediately detected. The BRET ratio was calculated as described in Materials and Methods. (C and D) HEK293T cells were transfected with the indicated plasmids and an internal control, pCA7-Fluc. At 36 h posttransfection, Rluc and Fluc activities were measured. Relative luciferase activity was calculated as the ratio of Fluc activity to Rluc activity. The mean values from three independent experiments are shown, with standard deviations.
To further confirm the authenticity of this inhibitory effect, a split Renilla luciferase (Rluc) complementation (SRC) assay was carried out (25, 26). We constructed plasmids that encode the N-terminal portion of Rluc (RlucN) attached to the N terminus of IRF7 through an intervening linker GGGGSG peptide (RlucN-IRF7) and the C-terminal portion of Rluc (RlucC) connected to the C terminus of IRF7 through the same intervening linker peptide (IRF7-RlucC). HEK293T cells were transfected with various combinations of these constructs, and at 24 h posttransfection, Rluc activity was measured. Homodimerization of IRF7 between RlucN-IRF7 and IRF7-RlucC facilitates interaction between the RlucN and RlucC halves, which results in restoration of Rluc activity. Transfection of either RlucN-IRF7 or IRF7-RlucC alone exhibited little Rluc activity, but transfection of both showed distinct Rluc activity (Fig. 5C). This activity was significantly enhanced by cotransfection of MyD88, TRAF6, and IKKα, indicating restoration of the Rluc activity by facilitation of homodimerization of IRF7. This enhancement was suppressed by coexpression of M2-2 or IRF7, but not M2-1 (Fig. 5D). The magnitude of the inhibition by M2-2 was comparable to that by intact IRF7 acting as a competitive inhibitor. From these results, we conclude that M2-2 inhibits MyD88/TRAF6/IKKα-induced homodimerization of IRF7.
Inhibition of MyD88/TRAF6/IKKα-induced IRF7 homodimerization does not result from a steric effect of M2-2 binding.
It is possible that M2-2 binding to the ID sterically hinders dimer formation of IRF7. To evaluate this possibility, we determined whether the M2-2 protein similarly affected IRF7 activation mediated by IFN-β promoter stimulator 1 (IPS-1), which activates the serine/threonine kinases, TANK-binding kinase 1 (TBK1), and IKKi, for IRF7, in the context of the retinoic acid-inducible gene I (RIG-I)/melanoma differentiation-associated gene 5 (MDA5)-dependent signaling pathway. As shown in Fig. 6A, transfection of IPS-1 and IRF7 resulted in striking enhancement of IFN-α promoter activation. However, this enhancement was not inhibited by M2-2. Hepatitis C virus (HCV) NS3/4A, whose expression leads to degradation of IPS-1, was tested as a positive control (27, 28). Consistent with this result, the SRC assay showed that M2-2 never inhibited IPS-1-induced homodimerization of IRF7 (Fig. 6B), suggesting that inhibition of MyD88/TRAF6/IKKα-induced IRF7 homodimerization does not result from a steric effect of M2-2 binding.
FIG 6.
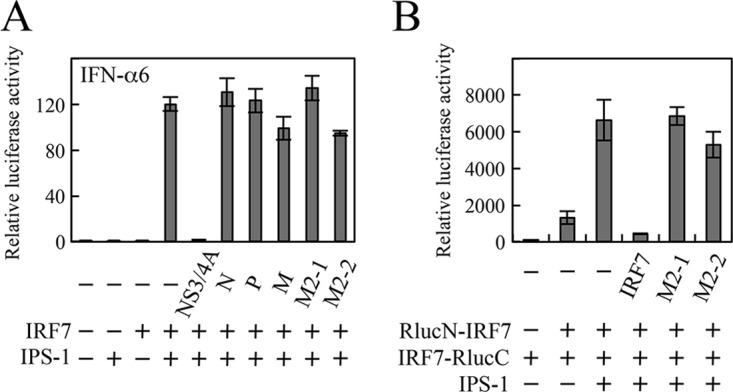
Effect of the M2-2 protein on IPS-1-induced activation and homodimerization of IRF7. (A) HEK293T cells were transfected with the indicated plasmids, pRL-TK, and an IFN-α6 promoter-driven Fluc reporter plasmid. At 24 h posttransfection, Fluc and Rluc activities were measured. Relative luciferase activity was calculated as the ratio of Fluc activity to Rluc activity. (B) HEK293T cells were transfected with the indicated plasmids and pCA7-Fluc. At 36 h posttransfection, Fluc and Rluc activities were measured. Relative luciferase activity was calculated as the ratio of Rluc activity to Fluc activity. The mean values from three independent experiments are shown, with standard deviations. NS3/4A, HCV NS3/4A protein.
M2-2 inhibits phosphorylation of IRF7 on Ser477.
Activation of IRF7 requires phosphorylation of Ser residues in its C-terminal regulatory domain by upstream kinases, IKKα and/or IRAK1. Phosphorylation of IRF7 induces homodimerization of IRF7, which unmasks a DNA binding domain and a bipartite transactivation domain (23). The phosphorylation status of IRF7 was therefore investigated by immunoblot analysis (Fig. 7A). When myc-IRF7 was transfected alone, phosphorylation of myc-IRF7 was not observed. Additional expression of MyD88, TRAF6, and IKKα resulted in phosphorylation of IRF7 on Ser477, Ser471, and Ser472 (the numbering is that for IRF7 isotype a). The PIV2 V protein was tested as a positive control and inhibited phosphorylation of all the Ser residues. In contrast, M2-2 inhibited phosphorylation on Ser477, but not on Ser471 and Ser472. On the other hand, M2-2 did not inhibit IPS-1-mediated phosphorylation on all the Ser residues examined (Fig. 7B). These results suggest that the M2-2 protein prevents IKKα/IRAK1, but not TBK1/IKKi, from gaining access to and phosphorylating Ser477 of IRF7.
FIG 7.

Effect of the M2-2 protein on MyD88-TRAF6-IKKα- or IPS-1-induced phosphorylation of IRF7. (A and B) HEK293T cells were transfected with the indicated plasmids. At 24 h posttransfection, the cells were lysed in Laemmli sample buffer, and the lysates were subjected to WB with the indicated antibodies.
M2-2 does not inhibit nuclear translocation of IRF7.
Subcellular localization of IRF7 in the presence of M2-2 was investigated by the immunostaining method. HEK293T cells were transfected with myc-IRF7 and then treated with anti-myc antibody and anti-mouse IgG antibody conjugated to Alexa Fluor 488. It was found that myc-IRF7, when expressed alone, was localized predominantly in the cytoplasm with uneven distribution (Fig. 8A). Cotransfection of V5-MyD88, V5-TRAF6, and V5-IKKα resulted in accumulation of myc-IRF7 in the nucleus, indicating MyD88/TRAF6/IKKα-induced nuclear translocation of myc-IRF7. However, this nuclear translocation was suppressed by coexpression with PIV2 V, but not M2-1 and M2-2. To confirm this finding, transfected cells were fractionated into the nuclear and cytoplasmic fractions, and then the level of IRF7 in the nuclear fraction was examined by immunoblot analysis (Fig. 8B). Transfection of MyD88, TRAF6, and IKKα, along with IRF7, resulted in elevation of the IRF7 level in the nuclear fraction compared with transfection of IRF7 alone. This elevation was inhibited by transfection of PIV2 V, whereas it was not inhibited by transfection of M2-2 and M2-1. Taken together, these results demonstrate that M2-2 does not inhibit nuclear translocation of IRF7.
FIG 8.
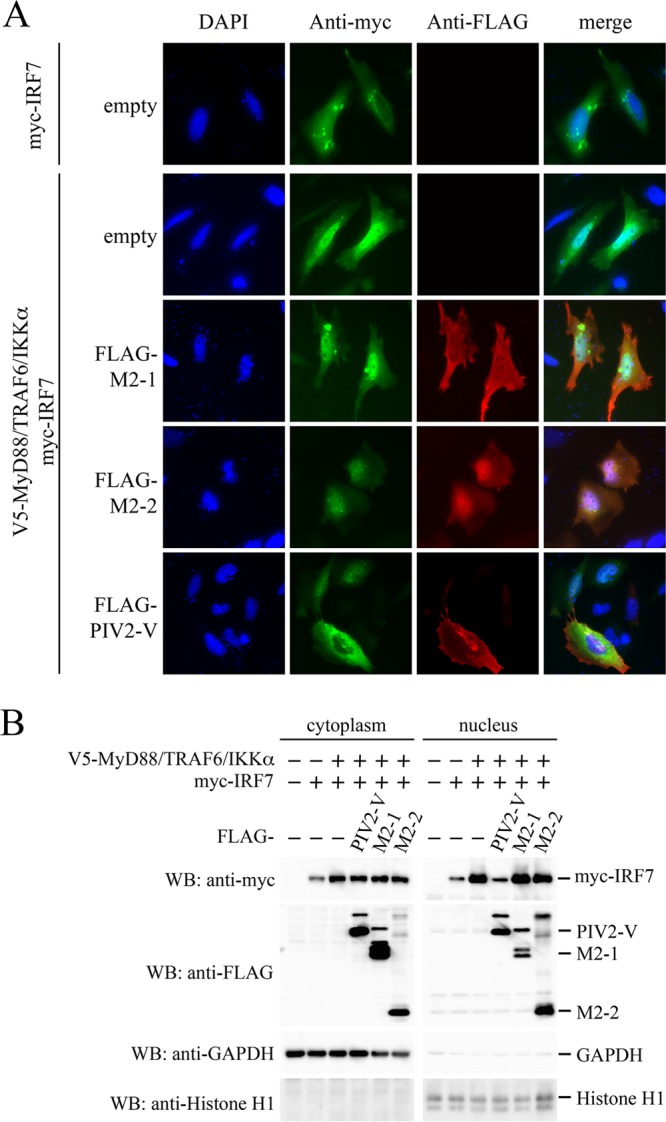
Effect of the M2-2 protein on MyD88-TRAF6-IKKα-induced nuclear translocation of IRF7. (A) HeLa cells cultured in 8-well glass chamber slides were transfected with the indicated plasmids. At 24 h posttransfection, the cells were fixed and permeabilized. Then, the cells were incubated with anti-myc mouse MAb or anti-FLAG rabbit polyclonal antibody as the primary antibody, followed by anti-mouse IgG antibody conjugated to Alexa Fluor 488 (green) or anti-rabbit IgG antibody conjugated to Alexa Fluor 647 (red) as the secondary antibody. The stained cells were mounted with ProLong Gold reagent with DAPI (blue). (B) HEK293T cells were transfected with the indicated plasmids. At 24 h posttransfection, nuclear and cytoplasmic extracts were prepared and then subjected to WB with the indicated antibodies.
DISCUSSION
In this study, we identified the M2-2 protein as a negative regulator of IFN-α production by pDCs. Recombinant ΔM2-2 virus, in which expression of M2-2 is abrogated, indeed induced a high level of IFN-α in human pDCs compared with wild-type HMPV (Fig. 1E). This enhancement of IFN-α production seems to be causally related to two kinds of independent activities of M2-2 that appear to explain in part why the ΔM2-2 virus is attenuated (29). One is the inhibitory activity for TLR7/9-dependent IFN-α induction. We demonstrated, by using TLR7/9-dependent signaling reconstituted in HEK293T cells, that M2-2 was the most potent inhibitor of all the proteins encoded by the HMPV genome (Fig. 1A). The other is an inhibitory activity for viral RNA synthesis. ΔM2-2 virus produces high levels of viral mRNAs in Vero cells compared with wild-type virus (30). The inhibitory effect of M2-2 on viral RNA synthesis was also confirmed by experiments with an HMPV minigenome construct carrying the luciferase reporter gene (31). Possible enhancement of viral RNA synthesis in pDCs infected with ΔM2-2 virus may cause elevation of the PAMP level, which stimulates IFN-α production. However, it is difficult to determine the degree to which each activity contributes to suppression of IFN-α production.
How does the M2-2 protein block TLR7/9-dependent signaling? BRET and SRC assays revealed that the M2-2 protein inhibits MyD88/TRAF6/IKKα-induced homodimerization of IRF7 (Fig. 5). This inhibition seems to be critical, since the homodimerization derepresses transactivation and allows specific DNA binding (23). We initially assessed the IRF7-IRF7 interaction by immunoprecipitation with extracts from cells cotransfected with two kinds of tagged IRF7 in the absence of stimulation with upstream signaling components, according to the standard method described in other studies (22, 32). This experiment showed that expression of M2-2 had no appreciable effect on IRF7-IRF7 interaction (data not shown). This result was also confirmed by the SRC assay. Since in these experiments we could assess only the interaction between static unphosphorylated IRF7 molecules, we next attempted to examine interaction between phosphorylated IRF7 molecules after stimulation of the TLR7/9-dependent signaling pathway using extracts from cells cotransfected with MyD88, TRAF6, and IKKα. Unexpectedly, we found that phosphorylated IRF7 became insoluble in a lysis buffer containing Triton X-100 (even in radioimmunoprecipitation assay [RIPA] buffer) and was therefore removed from cell extracts by centrifugation before immunoprecipitation. It remains unclear why phosphorylated IRF7 becomes insoluble, although it may be related to TLR9 stimulation-dependent colocalization of IRF7 with viperin, which recruits IRAK1 and TRAF6 to lipid droplets after TLR9 stimulation (10). We therefore used a BRET assay and an SRC assay in place of the immunoprecipitation method and succeeded in showing inhibition of the IRF7-IRF7 interaction by M2-2. To our knowledge, few reports on examining the IRF7-IRF7 interaction after TLR7/9 stimulation are available. BRET and SRC assays may be useful to study interaction of other viral proteins with TLR7/9 signaling components after TLR7/9 stimulation.
The M2-2 protein binds to the ID of IRF7 (Fig. 4). Since the ID-ID interaction seems to be essential for IRF7 homodimerization (24), we assumed that the IRF7-IRF7 interaction was interrupted by a steric effect of M2-2. However, it is unlikely, because the M2-2 protein inhibits neither IPS-1-induced homodimerization of IRF7 (Fig. 6B) nor homodimerization of static unphosphorylated IRF7. IRF7 is subjected to two kinds of modification: K63-linked polyubiquitination by TRAF6 and subsequent serine phosphorylation by IKKα/IRAK1. We examined the effect of M2-2 on K63-linked polyubiquitination of IRF7 according to the procedure previously described (14), but K63-linked polyubiquitination was not affected by M2-2 (data not shown). Instead, M2-2 was found to inhibit phosphorylation of IRF7 on Ser477, but not Ser471/Ser472 (Fig. 7A). When an IRF7 mutant in which only Ser477 and Ser479 are replaced with the phosphomimetic Asp is transfected into cells, the mutant is homodimerized and activates the IFN-α promoter without transfection of upstream signaling components (22). It is thus possible that inhibition of phosphorylation of IRF7 on Ser residues, including Ser477, is involved in inhibition of IRF7 homodimerization. To prove this hypothesis, it is necessary to examine the effect of M2-2 on phosphorylation of all the Ser residues in the regulatory domain and, further, to analyze the role of each phosphorylated Ser residue in IRF7 homodimerization.
It should be noted that inhibition of phosphorylation of IRF7 by M2-2 is partial. This indicates that M2-2 never inhibits the kinase activity per se of the responsible kinases, IKKα and/or IRAK1, and does not target upstream components of the TLR7/9-dependent signaling pathway. This is in good agreement with the fact that M2-2 has no ability to inhibit MyD88-induced activation of NF-κB (Fig. 1D). It is possible that the M2-2 protein masks Ser477, but not Ser471 and Ser472, by binding to the ID near the regulatory domain and thereby prevents IKKα and/or IRAK1 from gaining access to Ser residues near Ser477. The masking effect of the M2-2 protein might be insufficient to prevent IKKi/TBK1 from gaining access to Ser477.
In spite of the inhibition of IRF7 homodimerization, M2-2 did not inhibit nuclear translocation of IRF7. Similar observations have been reported for other viruses. LF2 of Epstein-Barr virus interacts with the ID of IRF7, and this interaction results in inhibition of IRF7 homodimerization (33). However, nuclear translocation of IRF7 is not affected by the LF2 protein. The ML protein of Thogoto virus interacts with IRF7 and prevents homodimerization of IRF7 and interaction between IRF7 and TRAF6 (32). However, it never inhibits nuclear translocation of IRF7. These findings, including ours, suggest that homodimerization is not a prerequisite for nuclear translocation of IRF7.
Although our present study defined the function of M2-2 in immune evasion, conflicting results have been reported (34). Ren et al. showed that the M2-2 protein targeted MyD88 for inhibition of the activation of NF-κB and activation of the IFN-β promoter. They did not examine the effect of the M2-2 protein on IRF7 activation, but it is likely that activation of IRF7 is also inhibited, because MyD88 is an upstream component of the TLR7/9-dependent signaling pathway. However, we could confirm neither significant interaction between M2-2 and MyD88 (Fig. 3) nor an inhibitory effect on MyD88-induced activation of NF-κB (Fig. 1D). Also, not obtained were positive results supporting the previous finding reported by Ren et al. and Chen et al. regarding inhibition of MAVS (IPS-1)-dependent gene transcription by M2-2 (Fig. 6 and 7B) (35, 36). The reasons for these conflicting results are unclear. Compared with the CAN97/83 strain used in the other studies, the Jpn03-1 strain used in our study has a single amino acid difference at position 58 in M2-2 (I and L at this position in Jpn03-1 and CAN97-83, respectively). Position 58 is located in the C-terminal half of the M2-2 protein (72 amino acids [aa]). As the C-terminal half is responsible for binding to MyD88 (34), it will be of interest to investigate whether the amino acid at position 58 determines the target of the M2-2 protein in immune evasion. In this regard, it should be noted that there is a study pointing out strain-dependent difference in HMPV immune evasion. Goutagny et al. reported that the P protein of strain B1 specifically impaired RIG-I-mediated sensing of viral 5′-triphosphate RNA derived from HMPV, but this activity was not seen in the P protein of strain A1 (37).
In summary, our study reveals that HMPV employs M2-2 as an IFN antagonist that suppresses IFN-α production by pDCs and that the M2-2 protein blocks the TLR7/9-dependent signaling pathway by inhibiting IRF7 homodimerization, possibly through partial inhibition of IRF7 phosphorylation. Infection with RSV, a closely related virus, in vivo or in vitro, also results in reduction of the pDCs' ability to produce IFN-α in response to TLR7 or TLR9 stimulation (17, 19, 20). Unlike HMPV, RSV carries additional viral genes, NS1 and NS2, and uses them as IFN antagonists that block the IFN-responsive JAK-STAT signaling pathway or inhibit activation of IRF3 (38–41). What proteins does RSV use for regulation of IFN-α production by pDCs? An investigation of the underlying mechanism for RSV is now in progress.
MATERIALS AND METHODS
Cells.
HEK293T, Vero, LLC-MK2, and HeLa cells were maintained in Dulbecco's modified Eagle's medium supplemented with 2 mM l-glutamine, penicillin (100 IU/ml), streptomycin (100 μg/ml), and 10% fetal bovine serum (FBS). pDCs were isolated from human PBMCs by magnetically activated cell sorting with a Diamond plasmacytoid dendritic cell isolation kit II (Miltenyi Biotec, Auburn, CA) according to the manufacturer's instructions. The isolated pDCs were resuspended in RPMI medium (Nacalai Tesque) supplemented with 10% FBS, human IL-3 (10 ng/ml; Peprotech), 2 mM l-glutamine, penicillin (100 IU/ml), and streptomycin (100 μg/ml) (14).
Plasmids.
Mammalian expression plasmids encoding viral or cellular protein were created by insertion of a cDNA fragment containing each ORF into the multicloning site downstream of the cytomegalovirus enhancer chicken β-actin hybrid promoter of pCA7. The cDNAs to be inserted were created by PCR or reverse transcription (RT)-PCR. Mutations, including deletion and protein fusion, were introduced by PCR-based overlap mutagenesis as described previously (13). Newly created were cDNAs for N, P, M, F, M2-1, M2-2, SH, G, L (derived from HMPV strain Jpn03-1), SeV C (derived from strain Z), human MyD88, TRAF6, IKKα, IRF7 (isoform d; aa 1 to 516), the ID (aa 276 to 469) of human IRF7, human IRF7 lacking the ID (IRF7ΔID), TRAF6 C70A, IRF7-YFP, IRF7-Nluc, RlucN-IRF7 (IRF7 fused to the C terminus of RlucN [aa 1 to 229] with an intervening linker peptide [GGGGSG]), and IRF7-RlucC (IRF7 fused to the N terminus of RlucC [aa 230 to 331] with the same linker peptide) (25, 42). The pNL1.1 vector was purchased from Promega Corp. and was used as a template for Nluc in PCR. Construction of pCA7 encoding Fluc, PIV2 V (derived from strain Toshiba), HCV NS3/4A, or mIRF7 deletion mutants (with or without an N-terminal 3× FLAG, V5, or myc tag), and pEFneo-IPS-1 were described previously (13, 43). The sequence fidelity of all the plasmids was confirmed by sequence analysis.
Recovery of rHMPV-GFPΔM2-2.
The genome construction of rHMPV-GFP derived from strain Jpn03-1 was described previously (44). To create rHMPV-GFPΔM2-2, in which the M2-2 ORF is silenced, two putative start codons of M2-2 at positions 5236 and 5248 were mutagenized from AUG to ACG. In addition, UUA at position 5272 was mutagenized to UAA to introduce a stop codon, and the major part of the M2-2 ORF (from positions 5288 to 5445), the sequence downstream of the stop codon of the M2-1 ORF, was deleted according to the method of Buchholz et al. (30). These nucleotide changes do not affect the amino acid sequence of the M2-1 protein. rHMPV-GFPΔM2-2 was recovered by transfecting the M2-2-silenced antigenome plasmid into BSR T7/5 cells (a gift from K. K. Conzelmann), which constitutively express T7 polymerase, as described previously (44). Sequence fidelity was confirmed by sequence analysis of the genome of the recovered virus. The recovered rHMPV was propagated in LLC-MK2 cells in the presence of 4 μg/ml of N-acetyl trypsin. Trypsin was added to culture medium at a final concentration of 4 μg/ml every 3 days to promote multistep replication.
Reporter assay.
An IFN-α6 promoter-driven Fluc reporter (13) (80 ng/well), an NF-κB Fluc reporter (Clontech) (100 ng/well), or an ISRE-driven Fluc reporter (Clontech) (80 ng/well) plasmid was transfected into HEK293T (∼1.0 × 105) or Vero (∼2.0 × 104) cells cultured in a 24-well plate in triplicate, together with pRL-TK (10 ng/well; Promega Corp.) and various combinations of plasmids that express MyD88 (25 ng/well), TRAF6 (25 ng/well), IKKα (25 ng/well), IPS-1 (50 ng/well), IRF7 (15 ng/well), IRF7ΔID (15 ng/well), N (100 ng/well), P (100 ng/well), M (100 ng/well), F (100 ng/well), M2-1 (100 ng/well), M2-2 (100 ng/well), SH (100 ng/well), G (100 ng/well), L (100 ng/well), PIV2 V (100 ng/well), TRAF6 C70A (100 ng/well), SeV C (100 ng/well), or HCV NS3/4A (100 ng/well), using polyethyleneimine (PEI) (Polysciences) (13). The total mass of transfected DNA was held constant in all the experiments by adding an appropriate amount of pCA7 empty plasmid. The cells were lysed at 24 h posttransfection, and the relative luciferase activity was determined with the dual-luciferase reporter assay system (Promega). In certain experiments, transfected cells were treated with recombinant human IFN-α2b (1,000 IU/ml; Schering-Plough) for 6 h at 24 h posttransfection.
Measurement of IFN-α.
Levels of human IFN-α in culture media were measured by enzyme-linked immunosorbent assay (ELISA) with a human IFN alpha ELISA kit (PBL Interferon Source, Piscataway, NJ) according to the manufacturer's instructions.
Immunoprecipitation.
HEK293T cells (∼5.0 × 105/well) in a 6-well plate were transfected with various combinations of plasmids (500 ng/well each), using PEI. At 24 h posttransfection, the cells were lysed in 400 μl of lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, and protease inhibitor cocktail). Then, the cell lysates were incubated with anti-V5 mouse monoclonal antibody (MAb) (SV5-Pk1; Invitrogen), anti-FLAG mouse MAb (1E6; Wako), or anti-myc mouse MAb (9B11; Cell Signaling Technology), together with SureBeads protein G (Bio-Rad) at 4°C for 2 h. In certain experiments, cell-free protein synthesis was performed using the TNT SP6 high-yield wheat germ protein expression system (Promega). Mixtures of the products synthesized in vitro were incubated in place of cell lysates. After washing the beads five times with the lysis buffer, proteins were eluted from the beads by boiling with Laemmli sample buffer (50 mM Tris-HCl, pH 6.8, 2% sodium dodecyl sulfate [SDS], 0.1% bromophenol blue, 10% glycerol, and 5% 2-mercaptoethanol), and then the samples were subjected to immunoblot analysis.
Immunoblot analysis.
Samples were resolved by SDS-10 to 15% polyacrylamide gel electrophoresis and then electroblotted onto a membrane filter (Immobilon-P; Millipore). The membrane was blocked in phosphate-buffered saline (PBS) containing 5% skim milk and 0.05% Tween 20, and was incubated at 4°C overnight with anti-FLAG mouse MAb (1E6), anti-V5 mouse MAb (SV5-Pk1), anti-myc mouse MAb (9E10; Wako), anti-phospho-IRF7 (anti-pIRF7) (S471/472) rabbit polyclonal antibody (Cell Signaling Technology), anti-pIRF7 (S477) rabbit MAb (D7E1W; Cell Signaling Technology), anti-IRF7 mouse MAb (F-1; Santa Cruz), anti-actin mouse MAb (AC-74; Sigma), anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) rabbit MAb (D16H11; Cell Signaling Technology), or anti-histone H1 mouse MAb (AE-4; Santa Cruz). The membrane was then incubated at room temperature for 2 h with horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG antibody (GE Healthcare Bio-Science). Immunoreactive bands were visualized by using the ECL select substrate (GE Healthcare Bio-Science).
BRET assay.
HEK293T cells (∼1.0 × 105) in a 24-well plate were transfected with various combinations of plasmids that express MyD88 (25 ng), TRAF6 (25 ng), IKKα (25 ng), YFP (85 ng), IRF7-YFP (85 ng), Nluc (15 ng), IRF7-Nluc (15 ng), or viral proteins (100 ng). At 24 h posttransfection, the cells were suspended in Dulbecco's PBS and transferred to a 96-well microplate. After the addition of furimazine, a substrate for Nluc from the Nano-Glo luciferase assay system (Promega), luminescence and fluorescence signals were immediately detected using an Infinite F500 microplate reader (Tecan). The BRET ratio is defined as the light signal emitted by YFP (530 to 570 nm) relative to the light signal emitted by Nluc (370 to 450 nm) (45, 46). The actual BRET ratio was calculated by subtracting a background BRET ratio, which was obtained for cells expressing Nluc-IRF7 alone, from the directly measured BRET ratio of each sample.
SRC assay.
HEK293T cells (∼1.0 × 105) in a 24-well plate were transfected with various combinations of plasmids that expressed MyD88 (25 ng), TRAF6 (25 ng), IKKα (25 ng), IPS-1 (50 ng), RlucN-IRF7 (15 ng), IRF7-RlucC (15 ng), or viral proteins (100 ng), along with an internal control, pCA7-Fluc (10 ng). The cells were lysed at 36 h posttransfection. Rluc and Fluc activities were measured with an Rluc assay system (Promega) and an Fluc assay system (Promega), respectively. The relative activity was defined as the ratio of Rluc activity to Fluc activity.
Immunofluorescence staining.
HeLa cells (∼1.0 × 104/well) cultured in an 8-well glass chamber slide (Matsunami Glass) were transfected with various combinations of plasmids that expressed V5-MyD88 (25 ng), V5-TRAF6 (25 ng), V5-IKKα (25 ng), myc-IRF7 (15 ng), FLAG-M2-1 (100 ng), FLAG-M2-2 (100 ng), or FLAG-PIV2 V (100 ng). At 24 h posttransfection, the cells were washed twice with PBS, fixed with 4% paraformaldehyde for 20 min, and permeabilized with 0.2% Triton X-100 for 5 min, followed by three washes with PBS. The cells were incubated with anti-myc mouse MAb (9B11) or anti-FLAG rabbit polyclonal antibody (Cell Signaling Technology) as the primary antibody, followed by anti-mouse IgG(H+L) antibody conjugated to Alexa Fluor 488 or anti-rabbit IgG(H+L) antibody conjugated to Alexa Fluor 647 as the secondary antibody. After each incubation step, the cells were washed with PBS three times. The stained cells were mounted with ProLong Gold reagent with DAPI (4′,6-diamidino-2-phenylindole) (Thermo Fisher Scientific) and then visualized under a BX-61 fluorescence microscope (Olympus).
Preparation of cytoplasmic and nuclear fractions.
HEK293T cells (∼5.0 × 105/well) in a 6-well plate were transfected with various combinations of plasmids that expressed MyD88 (125 ng), TRAF6 (125 ng), IKKα (125 ng), IRF7 (150 ng), or viral proteins (500 ng). At 24 h posttransfection, nuclear and cytoplasmic protein extracts were prepared by using nuclear and cytoplasmic extraction reagents (Thermo Fisher) according to the manufacturer's instructions.
ACKNOWLEDGMENTS
We thank K. K. Conzelmann (Munich, Germany) for providing BSR T7/5 cells and T. Komatsu (Aichi, Japan) for helpful discussions. Sequence analysis was performed using an ABI Prism 3130xl genetic analyzer in the Central Research Laboratory, Shiga University of Medical Science.
This work was supported by JSPS Kakenhi grants (no. 22590414, no. 25460563, and no. 26460553) and by grants from the Shiga University of Medical Science, Wajinkai, and the Yakult Foundation, Japan.
REFERENCES
- 1.Kumar H, Kawai T, Akira S. 2009. Toll-like receptors and innate immunity. Biochem Biophys Res Commun 388:621–625. doi: 10.1016/j.bbrc.2009.08.062. [DOI] [PubMed] [Google Scholar]
- 2.Kawai T, Akira S. 2009. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol 21:317–337. doi: 10.1093/intimm/dxp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blasius AL, Beutler B. 2010. Intracellular Toll-like receptors. Immunity 32:305–315. doi: 10.1016/j.immuni.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 4.Ning S, Pagano JS, Barber GN. 2011. IRF7: activation, regulation, modification and function. Genes Immun 12:399–414. doi: 10.1038/gene.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawai T, Sato S, Ishii KJ, Coban C, Hemmi H, Yamamoto M, Terai K, Matsuda M, Inoue J, Uematsu S, Takeuchi O, Akira S. 2004. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol 5:1061–1068. doi: 10.1038/ni1118. [DOI] [PubMed] [Google Scholar]
- 6.Honda K, Yanai H, Mizutani T, Negishi H, Shimada N, Suzuki N, Ohba Y, Takaoka A, Yeh WC, Taniguchi T. 2004. Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in Toll-like receptor signaling. Proc Natl Acad Sci U S A 101:15416–15421. doi: 10.1073/pnas.0406933101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hacker H, Redecke V, Blagoev B, Kratchmarova I, Hsu LC, Wang GG, Kamps MP, Raz E, Wagner H, Hacker G, Mann M, Karin M. 2006. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 439:204–207. doi: 10.1038/nature04369. [DOI] [PubMed] [Google Scholar]
- 8.Hoshino K, Sugiyama T, Matsumoto M, Tanaka T, Saito M, Hemmi H, Ohara O, Akira S, Kaisho T. 2006. IkappaB kinase-alpha is critical for interferon-alpha production induced by Toll-like receptors 7 and 9. Nature 440:949–953. doi: 10.1038/nature04641. [DOI] [PubMed] [Google Scholar]
- 9.Oganesyan G, Saha SK, Guo B, He JQ, Shahangian A, Zarnegar B, Perry A, Cheng G. 2006. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature 439:208–211. doi: 10.1038/nature04374. [DOI] [PubMed] [Google Scholar]
- 10.Saitoh T, Satoh T, Yamamoto N, Uematsu S, Takeuchi O, Kawai T, Akira S. 2011. Antiviral protein Viperin promotes Toll-like receptor 7- and Toll-like receptor 9-mediated type I interferon production in plasmacytoid dendritic cells. Immunity 34:352–363. doi: 10.1016/j.immuni.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 11.Shinohara ML, Lu L, Bu J, Werneck MB, Kobayashi KS, Glimcher LH, Cantor H. 2006. Osteopontin expression is essential for interferon-alpha production by plasmacytoid dendritic cells. Nat Immunol 7:498–506. doi: 10.1038/ni1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uematsu S, Sato S, Yamamoto M, Hirotani T, Kato H, Takeshita F, Matsuda M, Coban C, Ishii KJ, Kawai T, Takeuchi O, Akira S. 2005. Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7- and TLR9-mediated interferon-α induction. J Exp Med 201:915–923. doi: 10.1084/jem.20042372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kitagawa Y, Yamaguchi M, Zhou M, Komatsu T, Nishio M, Sugiyama T, Takeuchi K, Itoh M, Gotoh B. 2011. A tryptophan-rich motif in the human parainfluenza virus type 2 V protein is critical for the blockade of Toll-like receptor 7 (TLR7)- and TLR9-dependent signaling. J Virol 85:4606–4611. doi: 10.1128/JVI.02012-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kitagawa Y, Yamaguchi M, Zhou M, Nishio M, Itoh M, Gotoh B. 2013. Human parainfluenza virus type 2 V protein inhibits TRAF6-mediated ubiquitination of IRF7 to prevent TLR7- and TLR9-dependent interferon induction. J Virol 87:7966–7976. doi: 10.1128/JVI.03525-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamaguchi M, Kitagawa Y, Zhou M, Itoh M, Gotoh B. 2014. An anti-interferon activity shared by paramyxovirus C proteins: inhibition of Toll-like receptor 7/9-dependent alpha interferon induction. FEBS Lett 588:28–34. doi: 10.1016/j.febslet.2013.11.015. [DOI] [PubMed] [Google Scholar]
- 16.Pfaller CK, Conzelmann KK. 2008. Measles virus V protein is a decoy substrate for IkappaB kinase alpha and prevents Toll-like receptor 7/9-mediated interferon induction. J Virol 82:12365–12373. doi: 10.1128/JVI.01321-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schlender J, Hornung V, Finke S, Gunthner-Biller M, Marozin S, Brzozka K, Moghim S, Endres S, Hartmann G, Conzelmann KK. 2005. Inhibition of Toll-like receptor 7- and 9-mediated alpha/beta interferon production in human plasmacytoid dendritic cells by respiratory syncytial virus and measles virus. J Virol 79:5507–5515. doi: 10.1128/JVI.79.9.5507-5515.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guerrero-Plata A, Baron S, Poast JS, Adegboyega PA, Casola A, Garofalo RP. 2005. Activity and regulation of alpha interferon in respiratory syncytial virus and human metapneumovirus experimental infections. J Virol 79:10190–10199. doi: 10.1128/JVI.79.16.10190-10199.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guerrero-Plata A, Kolli D, Hong C, Casola A, Garofalo RP. 2009. Subversion of pulmonary dendritic cell function by paramyxovirus infections. J Immunol 182:3072–3083. doi: 10.4049/jimmunol.0802262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guerrero-Plata A, Casola A, Suarez G, Yu X, Spetch L, Peeples ME, Garofalo RP. 2006. Differential response of dendritic cells to human metapneumovirus and respiratory syncytial virus. Am J Respir Cell Mol Biol 34:320–329. doi: 10.1165/rcmb.2005-0287OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ. 2000. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 103:351–361. doi: 10.1016/S0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 22.Lin R, Mamane Y, Hiscott J. 2000. Multiple regulatory domains control IRF-7 activity in response to virus infection. J Biol Chem 275:34320–34327. doi: 10.1074/jbc.M002814200. [DOI] [PubMed] [Google Scholar]
- 23.Marie I, Smith E, Prakash A, Levy DE. 2000. Phosphorylation-induced dimerization of interferon regulatory factor 7 unmasks DNA binding and a bipartite transactivation domain. Mol Cell Biol 20:8803–8814. doi: 10.1128/MCB.20.23.8803-8814.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee KJ, Ye JS, Choe H, Nam YR, Kim N, Lee U, Joo CH. 2014. Serine cluster phosphorylation liberates the C-terminal helix of IFN regulatory factor 7 to bind histone acetyltransferase p300. J Immunol 193:4137–4148. doi: 10.4049/jimmunol.1401290. [DOI] [PubMed] [Google Scholar]
- 25.Paulmurugan R, Umezawa Y, Gambhir SS. 2002. Noninvasive imaging of protein-protein interactions in living subjects by using reporter protein complementation and reconstitution strategies. Proc Natl Acad Sci U S A 99:15608–15613. doi: 10.1073/pnas.242594299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paulmurugan R, Gambhir SS. 2003. Monitoring protein-protein interactions using split synthetic renilla luciferase protein-fragment-assisted complementation. Anal Chem 75:1584–1589. doi: 10.1021/ac020731c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. 2005. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci U S A 102:17717–17722. doi: 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, Tschopp J. 2005. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437:1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- 29.Biacchesi S, Pham QN, Skiadopoulos MH, Murphy BR, Collins PL, Buchholz UJ. 2005. Infection of nonhuman primates with recombinant human metapneumovirus lacking the SH, G, or M2-2 protein categorizes each as a nonessential accessory protein and identifies vaccine candidates. J Virol 79:12608–12613. doi: 10.1128/JVI.79.19.12608-12613.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buchholz UJ, Biacchesi S, Pham QN, Tran KC, Yang L, Luongo CL, Skiadopoulos MH, Murphy BR, Collins PL. 2005. Deletion of M2 gene open reading frames 1 and 2 of human metapneumovirus: effects on RNA synthesis, attenuation, and immunogenicity. J Virol 79:6588–6597. doi: 10.1128/JVI.79.11.6588-6597.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kitagawa Y, Zhou M, Yamaguchi M, Komatsu T, Takeuchi K, Itoh M, Gotoh B. 2010. Human metapneumovirus M2-2 protein inhibits viral transcription and replication. Microbes Infect 12:135–145. doi: 10.1016/j.micinf.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 32.Buettner N, Vogt C, Martinez-Sobrido L, Weber F, Waibler Z, Kochs G. 2010. Thogoto virus ML protein is a potent inhibitor of the interferon regulatory factor-7 transcription factor. J Gen Virol 91:220–227. doi: 10.1099/vir.0.015172-0. [DOI] [PubMed] [Google Scholar]
- 33.Wu L, Fossum E, Joo CH, Inn KS, Shin YC, Johannsen E, Hutt-Fletcher LM, Hass J, Jung JU. 2009. Epstein-Barr virus LF2: an antagonist to type I interferon. J Virol 83:1140–1146. doi: 10.1128/JVI.00602-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ren J, Liu G, Go J, Kolli D, Zhang G, Bao X. 2014. Human metapneumovirus M2-2 protein inhibits innate immune response in monocyte-derived dendritic cells. PLoS One 9:e91865. doi: 10.1371/journal.pone.0091865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ren J, Wang Q, Kolli D, Prusak DJ, Tseng CT, Chen ZJ, Li K, Wood TG, Bao X. 2012. Human metapneumovirus M2-2 protein inhibits innate cellular signaling by targeting MAVS. J Virol 86:13049–13061. doi: 10.1128/JVI.01248-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen Y, Deng X, Deng J, Zhou J, Ren Y, Liu S, Prusak DJ, Wood TG, Bao X. 2016. Functional motifs responsible for human metapneumovirus M2-2-mediated innate immune evasion. Virology 499:361–368. doi: 10.1016/j.virol.2016.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goutagny N, Jiang Z, Tian J, Parroche P, Schickli J, Monks BG, Ulbrandt N, Ji H, Kiener PA, Coyle AJ, Fitzgerald KA. 2010. Cell type-specific recognition of human metapneumoviruses (HMPVs) by retinoic acid-inducible gene I (RIG-I) and TLR7 and viral interference of RIG-I ligand recognition by HMPV-B1 phosphoprotein. J Immunol 184:1168–1179. doi: 10.4049/jimmunol.0902750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramaswamy M, Shi L, Varga SM, Barik S, Behlke MA, Look DC. 2006. Respiratory syncytial virus nonstructural protein 2 specifically inhibits type I interferon signal transduction. Virology 344:328–339. doi: 10.1016/j.virol.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 39.Schlender J, Bossert B, Buchholz U, Conzelmann KK. 2000. Bovine respiratory syncytial virus nonstructural proteins NS1 and NS2 cooperatively antagonize alpha/beta interferon-induced antiviral response. J Virol 74:8234–8242. doi: 10.1128/JVI.74.18.8234-8242.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu X, Zheng J, Zheng K, Hou Y, Zhao F, Zhao D. 2014. Respiratory syncytial virus NS1 protein degrades STAT2 by inducing SOCS1 expression. Intervirology 57:65–73. doi: 10.1159/000357327. [DOI] [PubMed] [Google Scholar]
- 41.Bossert B, Marozin S, Conzelmann KK. 2003. Nonstructural proteins NS1 and NS2 of bovine respiratory syncytial virus block activation of interferon regulatory factor 3. J Virol 77:8661–8668. doi: 10.1128/JVI.77.16.8661-8668.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim SB, Ozawa T, Watanabe S, Umezawa Y. 2004. High-throughput sensing and noninvasive imaging of protein nuclear transport by using reconstitution of split Renilla luciferase. Proc Natl Acad Sci U S A 101:11542–11547. doi: 10.1073/pnas.0401722101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kitagawa Y, Tani H, Limn CK, Matsunaga TM, Moriishi K, Matsuura Y. 2005. Ligand-directed gene targeting to mammalian cells by pseudotype baculoviruses. J Virol 79:3639–3652. doi: 10.1128/JVI.79.6.3639-3652.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou M, Kitagawa Y, Yamaguchi M, Uchiyama C, Itoh M, Gotoh B. 2013. Expeditious neutralization assay for human metapneumovirus based on a recombinant virus expressing Renilla luciferase. J Clin Virol 56:31–36. doi: 10.1016/j.jcv.2012.09.014. [DOI] [PubMed] [Google Scholar]
- 45.Sun S, Yang X, Wang Y, Shen X. 2016. In vivo analysis of protein-protein interactions with bioluminescence resonance energy transfer (BRET): progress and prospects. Int J Mol Sci 17:E1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Borroto-Escuela DO, Flajolet M, Agnati LF, Greengard P, Fuxe K. 2013. Bioluminescence resonance energy transfer methods to study G protein-coupled receptor-receptor tyrosine kinase heteroreceptor complexes. Methods Cell Biol 117:141–164. doi: 10.1016/B978-0-12-408143-7.00008-6. [DOI] [PMC free article] [PubMed] [Google Scholar]