

ABSTRACT
Recent worldwide outbreaks of Zika virus (ZIKV) infection and the lack of an approved vaccine raise serious concerns regarding preparedness to combat this emerging virus. We used a virus-like particle (VLP)-based approach to develop a vaccine and a microneutralization assay for ZIKV. A synthetic capsid-premembrane-envelope (C-prM-E) gene construct of ZIKV was used to generate reporter virus particles (RVPs) that package a green fluorescent protein (GFP) reporter-expressing West Nile virus (WNV) replicon. The assay was adapted to a 96-well format, similar to the plaque reduction neutralization test (PRNT), and showed high reproducibility with specific detection of ZIKV neutralizing antibodies. Furthermore, C-prM-E and prM-E VLPs were tested as vaccine candidates in mice and compared to DNA vaccination. While the ZIKV prM-E construct alone was sufficient for generating VLPs, efficient VLP production from the C-prM-E construct could be achieved in the presence of the WNV NS2B-3 protease, which cleaves C from prM, allowing virus release. Immunization studies in mice showed that VLPs generated higher neutralizing antibody titers than those with the DNA vaccines, with C-prM-E VLPs giving slightly higher titers than those with prM-E VLPs. The superiority of C-prM-E VLPs suggests that inclusion of capsid may have benefits for ZIKV and other flaviviral VLP vaccines. To facilitate the VLP platform, we generated a stable cell line expressing high levels of ZIKV prM-E proteins that constitutively produce VLPs as well as a cell line expressing ZIKV C-prM-E proteins for RVP production. While several vaccine platforms have been proposed for ZIKV, this study describes a safe, effective, and economical VLP-based vaccine against ZIKV.
IMPORTANCE To address the growing Zika virus epidemic, we undertook this study with two objectives: first, to develop a safe, effective, and economical vaccine for ZIKV, and second, to develop a rapid and versatile assay to detect the anti-ZIKV immune response. We generated a cell line stably expressing ZIKV prM-E that produces large amounts of VLPs in the supernatant and a ZIKV C-prM-E cell line that produces reporter virus particles upon transfection with a GFP replicon plasmid. The prM-E VLPs induced a strong neutralizing antibody response in mice that was better when the capsid was included. VLP-based vaccines showed significantly better neutralizing antibody responses than those with their DNA counterparts. The RVP-based microneutralization assay worked similarly to the PRNT assay, with a rapid GFP readout in a 96-well format. Our VLP-based platform provides a source for a ZIKV vaccine and diagnosis that can rapidly be adapted to current outbreaks.
KEYWORDS: C-prM-E, diagnostics, microneutralization, neutralization, PRNT, reporter, reporter virus particles, vaccine, Zika, prM-E
INTRODUCTION
Since the identification of Zika virus (ZIKV) from a rhesus monkey in Uganda in 1947 (1, 2) until 2010, the virus predominantly circulated between Aedes mosquitoes and nonhuman primates. Periodic episodes were indeed identified in the human population, which were characterized by mild self-limiting febrile disease associated with rash, headache, myalgia, and conjunctivitis (3, 4). However, the recent spread of ZIKV infections in the Western continents has caused much concern due to severe clinical outcomes in unborn fetuses (5–7), including cerebral calcifications, microcephaly, and other congenital malformations (5, 7). In adults, neurological manifestations are characterized by an autoimmune condition with symptoms of neuropathy and paralysis, also known as Guillain-Barré syndrome (8, 9). While Aedes species of mosquitos are the most common source of transmission, the virus has also been shown to be transmitted sexually both from women to men and from men to women and is capable of persisting in semen and vaginal secretions for up to 6 months after infection (10–12).
ZIKV is an enveloped RNA virus belonging to the family Flaviviridae. The 11-kb positive-sense RNA genome is translated in the cytoplasm to generate three structural and seven nonstructural proteins (13). The structural proteins C (capsid), prM/M (premembrane/membrane), and E (envelope) aid in virus assembly, which occurs predominantly in the lumen of the endoplasmic reticulum (ER). Maturation of virus particles occurs during virus egress via the secretory pathway, where furin in the Golgi apparatus cleaves prM, releasing the pr peptide as the virus reaches neutral pH upon exit from the cell (14). The E protein is the major target for neutralizing antibodies, and monoclonal antibodies against all 3 E protein domain (DI, DII, and DIII) target epitopes have been found (15–19).
The recent outbreaks of ZIKV infection have sparked efforts in the scientific community toward development of a safe and effective vaccine. These encompass the use of established approaches, such as purified inactivated virus (PIV) (20, 21), to more advanced approaches, such as DNA (prM-E) (20–22), subunit (E) (23), and recombinant adenovirus (20, 23) platforms, along with the recent development of RNA nanoparticle technology (24) or modified mRNA (prM-E) (25, 26), as vaccine candidates. Studies have demonstrated an effective neutralizing antibody response capable of protecting against ZIKV infection in both mice and nonhuman primates (20–22), leading to several clinical trials that are under way (ClinicalTrials.gov identifiers NCT02963909, NCT02840487, NCT02887482, NCT02809443, and NCT02952833). While virus-like particle (VLP)-based vaccines have been tested for nearly all flaviviruses, a VLP-based approach for ZIKV was only recently described (27) and needs further testing. One of the pros with the development of a ZIKV vaccine is that even though the virus exists as two distinct lineages (African and Asian/American) (28), the immune response generated against the virus is broadly protective (29), thus obviating the need to incorporate different serotypes in the vaccine.
The most promising measure of an effective vaccination strategy is the development of neutralizing antibodies and the availability of a rapid accurate diagnostic assay to quantitate the elicited immune response. The widely used assay for detection of neutralizing antibodies against flaviviruses is the plaque reduction neutralization test (PRNT) (30), which involves the use of live virus handled under biosafety level 2 (BSL-2) conditions. An ideal assay would be one that could be adapted to a high-throughput format with a convenient readout and eliminate the use of live virus, making it readily available to laboratories worldwide. With regard to a ZIKV vaccine, the challenge is to develop a safe, effective, and economical vaccine that can be produced easily for worldwide dissemination. All of the above criteria can be met by VLP-based vaccines, which are safe, generate an effective immune response, and can readily be scaled up for cost-effective production (31). ZIKV VLPs can be produced in cells expressing the prM-E proteins, and the particles, though noninfectious, morphologically resemble the live virus. Furthermore, addition of the capsid has been shown to promote virion stability (32) and is effective at inducing a cell-mediated immune response against flaviviruses (33, 34). VLP-based vaccines have been successful against other viral diseases, such as hepatitis B (GlaxoSmithKline's Engerix and Merck and Co., Inc.'s Recombivax HB) and human papillomavirus infection (Merck and Co., Inc.'s Gardisil), with more in the pipeline for viruses such as influenza virus, parvovirus, Norwalk virus, etc.
We studied the immune response in mice vaccinated with ZIKV VLPs generated using a prM-E or C-prM-E construct and compared it to those seen with DNA-based counterparts. To generate prM-E VLPs, we developed a cell line stably expressing the prM-E proteins, while C-prM-E VLPs were generated by cotransfection of cells with C-prM-E and the NS2B-3 protease of West Nile virus (WNV) to allow cleavage of capsid from prM-E (35). VLPs collected in culture supernatants were purified via ultracentrifugation and used to immunize mice. Concomitantly, we also developed a rapid and quantitative green fluorescent protein (GFP)-based microneutralization assay using reporter virus particles (RVPs) to measure the neutralizing immune response generated in mice. Our data demonstrate that both prM-E- and C-prM-E-based VLP vaccines are highly effective at generating neutralizing antibodies, with the latter being more potent. Interestingly, while the prM-E DNA-based vaccine was less effective than both the VLP-based vaccines, the C-prM-E DNA construct was unable to generate a significant neutralizing response, most likely due to the lack of VLP formation in the absence of NS2B-3 protease. Our RVP-based neutralization assay is safe, as it does not require use of live virus, and it yields results in <72 h, in contrast to the PRNT, which may require a week for completion. The assay was highly reproducible and effective at measuring the neutralizing antibody response against ZIKV and could be conducted in a 96-well format with simple fluorescence microscopy. Thus, this study demonstrates the feasibility of our VLP-based platform to tackle the emerging threat of ZIKV by providing a scalable source for a VLP-based vaccine and an RVP-based diagnostic assay.
RESULTS
Expression of ZIKV C-prM-E by use of a codon-optimized synthetic construct.
To develop a VLP-based platform for the ZIKV vaccine and RVP assay, we used the complete ZIKV sequence available for the current outbreak in the Americas (KU312312.1) (6) to synthesize a codon-optimized C-prM-E gene. The isolate was derived from a 52-year-old male hospitalized with symptoms of conjunctivitis and exanthema in Paramaribo (Suriname) in 2015. The synthesized gene was cloned into the pcDNA3.1 vector, which contains a cytomegalovirus (CMV) promoter (Fig. 1A). 293T cells transfected with the synthetic ZIKV C-prM-E or WNV C-prM-E construct were tested for E protein expression by immunofluorescence assay using antibodies MAB10216 (clone 4G2) and MAB8150 (clone 3.67G). As shown in Fig. 1B, MAB10216 reacted with both the ZIKV and WNV E proteins, while MAB8150 reacted only with the WNV E protein. This was expected, as MAB10216 reacts with flavivirus group-specific antigens (36) and binds to the fusion loop at the extremity of domain II of the E protein, while MAB8150 is specific for the E protein of West Nile/Kunjin virus. We also performed radioimmunoprecipitation analysis of ZIKV C-prM-E expression after transfection into 293T cells and compared it to that of WNV. As shown in Fig. 1C, both ZIKV and WNV C-prM-E proteins were immunoprecipitated with MAB10216 and also with anti-WNV mouse serum, although with different efficiencies, emphasizing the relatedness between flaviviruses (37). These data demonstrate that the ZIKV C-prM-E synthetic construct expresses the viral proteins at high levels and can be used in downstream assays requiring VLP production.
FIG 1.
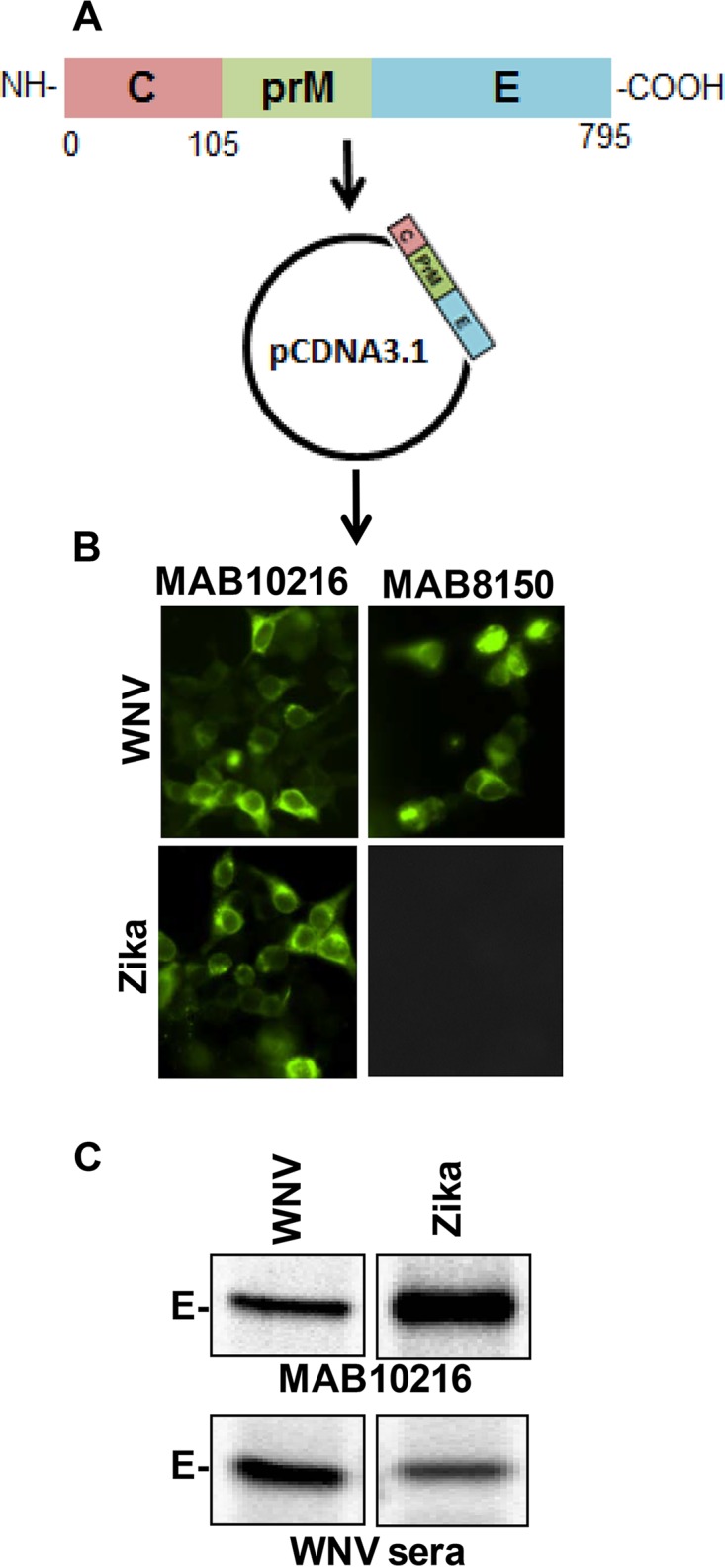
Expression of ZIKV C-prM-E by use of a codon-optimized synthetic construct. (A) A codon-optimized ZIKV C-prM-E gene was synthesized via gene synthesis technology, using the sequence available from the current outbreak in the Americas, and cloned into the pcDNA3.1 vector. (B) 293T cells were transfected with the ZIKV C-prM-E or WNV C-prM-E construct. Cells were fixed, stained with the MAB10216 and MAB8150 antibodies, and analyzed for E protein expression by fluorescence microscopy. (C) 293T cells were transfected as described above. At 48 h posttransfection, cells were radiolabeled with [35S]Met/Cys. Cell lysates were immunoprecipitated with protein A beads coated with MAB10216 or anti-WNV serum, resolved by SDS-PAGE, and visualized by PhosphorImager analysis.
RVP-based microneutralization assay for ZIKV, using a 96-well format and a GFP readout.
To make and test ZIKV RVPs, we used the method described previously by Garg et al. (38) and adapted from the work of Pierson et al. (39). 293T cells were cotransfected with the ZIKV C-prM-E construct along with the WNV replicon reporter plasmid Rep/GFP (29, 39, 40), which provided the WNV accessory proteins and the GFP reporter (Fig. 2A). RVPs generated in a similar manner, using WNV C-prM-E alongside, were used as a positive control. Both WNV and ZIKV RVPs showed robust infection of Vero and 293T cells (Fig. 2B).
FIG 2.

RVP-based microneutralization assay for ZIKV, using a 96-well plate format and a GFP readout. (A) Strategy for generation of ZIKV or WNV reporter virus particles. 293T cells were cotransfected with ZIKV/WNV C-prM-E along with the WNV subgenomic replicon construct Rep/GFP. Culture supernatants were harvested at 48 h posttransfection and used to infect 293T/Vero cells, and GFP expression was analyzed as a measure of virus infection. (B) 293T and Vero cells were infected with ZIKV or WNV RVPs, and infection was analyzed by fluorescence microscopy to detect GFP-positive cells. (C) Vero cells were infected with serial dilutions of ZIKV reporter virus-like particles in 96-well plates. Cells were fixed at 72 h postinfection, and images of whole wells were captured via fluorescence microscopy. Representative fluorescence images of whole wells infected with 6 serial dilutions (dilutions 1 to 6) of ZIKV RVPs are depicted. The top panel shows raw images acquired by fluorescence microscopy. The bottom panel shows the same images analyzed using the automated NIS Elements software, which marks GFP-positive cells by using the cell count function. The number below each well represents the number of GFP-positive cells in that well. Vero cells were infected with serial dilutions of ZIKV (D) or WNV (E) reporter virus particles in 96-well plates. The experiment was conducted with 6 technical wells infected for each dilution. The number of GFP-positive cells was determined at 72 h postinfection as described for panel C. The data show small variations between the 6 wells for each RVP dilution. (F) Quantitation of GFP-positive cells infected with ZIKV RVPs by using the automated software from Nikon versus manual counting. Representative data show a high degree of correlation between the two methods. ZIKV-117 antibody (G) or the indicated antibodies/sera (H) were serially diluted in DMEM and incubated with a predetermined amount of ZIKV RVPs for 1 h at room temperature. Subsequently, the virus-serum mixtures were added to Vero cells. The cells were incubated for 72 h, after which images were acquired and the number of GFP-positive cells quantitated as described above. The assay was conducted in technical triplicates for the ZIKV-117 antibody and ZIKV sera and in duplicates for others. Data representative of 1 of 3 independent experiment are shown.
Although ZIKV RVPs were recently used by other labs, the current method of choice is infection of Raji or Vero cells, followed by detection of GFP+ cells via flow cytometry (29, 39, 40). On the other hand, the infectious virus-based PRNT utilizes Vero cells in a 96-well format and can be tedious and time-consuming. We adapted our RVP-based assay to a 96-well plate format using Vero cells, similar to the PRNT method. Cells plated in 96-well clear-bottom black tissue culture plates were infected with serial dilutions of the RVPs. The plates were fixed 72 h later and analyzed by fluorescence microscopy, and the number of GFP-positive cells was counted using automated software (NIS Elements; Nikon). Serial dilutions (dilutions 1 to 6) of the input virus showed a dose-dependent decrease in the number of GFP-positive cells (Fig. 2C). The assay was conducted using ZIKV (Fig. 2D) and WNV (Fig. 2E) RVPs, and GFP+ cells were quantified using automated software. The assay showed a high level of reproducibility, with minimal variation between replicate wells. The linear range of the assay was 200 to 800 GFP+ cells. We also compared manual cell counting versus the automated software (NIS Elements) and found a high degree of correlation between the two methods (r = 0.9650; P < 0.0001) (Fig. 2F), suggesting that manual counting of GFP+ cells by use of a simple fluorescence microscope could also be used for the assay. Hence, this assay, much like the PRNT, can be adapted to give a reasonable number of GFP+ cells that can be counted either manually or using automated software.
We next tested whether our assay could detect neutralization of ZIKV RVPs via antibodies or polyclonal mouse sera. Experiments were performed in a manner similar to that for the standard PRNT, with serum/antibody dilutions incubated with RVPs for 1 h prior to addition to Vero cells. As shown in Fig. 2G, a human antibody against the ZIKV E protein (ZIKV-117) that is known to prevent infection via cross-linking the protein (41) potently inhibited RVP infection in a dose-dependent manner. Moreover, WNV pooled sera, polyclonal ZIKV sera, and MAB10216 also inhibited ZIKV RVP infection in a dose-dependent manner (Fig. 2H). Interestingly, and as expected, the antibody MAB8150 that failed to bind ZIKV E protein in immunofluorescence assays did not inhibit RVP infection (Fig. 2H). This demonstrates that the assay is specific and can readily be used to test for the presence of ZIKV neutralizing antibodies. Similar to PRNT, this assay can also be used to calculate the 50% effective concentration (EC50) and EC90 for serum samples. For example, the EC90 for WNV serum against ZIKV infection was determined to be a titer of 80, and the EC50 against ZIKV infection was a titer of 640 (Fig. 2H). The above data demonstrate that WNV replicon reporter construct along with ZIKV C-prM-E can yield infectious RVPs that can subsequently be used for several applications, including a microneutralization assay, while obviating the use of infectious virus and high-level biosafety containment.
Establishment of a stable cell line expressing ZIKV C-prM-E.
The sudden outbreaks of ZIKV infection caught the research community off guard, and the limited number of reagents and resources available for ZIKV studies has hampered research efforts. We hence developed a stable cell line expressing ZIKV C-prM-E to facilitate the generation of RVPs. For this purpose, the codon-optimized ZIKV C-prM-E cassette was subcloned into a lentiviral vector (pLenti6/V5) carrying a blasticidin resistance gene (Fig. 3A). Lentiviral particles generated with this vector were used to transduce 293T cells, which were then selected in the presence of 10 μg/ml blasticidin for ∼2 weeks. Bulk-selected cells were tested for expression of ZIKV E protein via fluorescence microscopy (Fig. 3B) and flow cytometry (Fig. 3C) and showed robust expression, with >90% of cells being positive for the E antigen. We also confirmed the expression of the ZIKV E protein by using Western blotting with the selected cell line and compared it to transient protein expression after transfection. As expected, high levels of E protein expression were seen for the 293T-C-prM-E-bulk cell line (Fig. 3D). As lentivirus-transduced cells can have different levels of exogenous gene expression (42, 43), we also selected single-cell clones from our parent bulk cell line. A total of 6 single clones were selected via limiting dilution cloning and characterized for ZIKV E expression by fluorescence microscopy (Fig. 3E) as well as flow cytometry (Fig. 3F). Based on our flow data, clone F6 (293T-C-prM-E-F6) showed the highest expression level and was chosen for subsequent experiments. We generated RVPs by using the 293T-C-prM-E-F6 cell line after transfecting it with the WNV Rep/GFP replicon plasmid and compared them to RVPs generated in 293T cells after transient transfection with pcDNA-C-prM-E and Rep/GFP. RVPs generated using the 293T-C-prM-E-F6 cell line had significantly higher titers than those of virus produced from 293T cells (Fig. 3G). Thus, the 293T-C-prM-E-F6 cell line stably expresses the ZIKV structural proteins and can be used for generation of high-titer RVPs.
FIG 3.

Establishment of a stable cell line expressing ZIKV C-prM-E. (A) ZIKV C-prM-E was PCR amplified by use of specific primers, using the codon-optimized construct as the template, and cloned into the lentiviral vector pLenti6/5-D-Topo. 293T cells were then transfected with the pLenti-C-prM-E construct along with the helper plasmid and VSV-G envelope, and ZIKV C-prM-E lentiviral particles were harvested at 48 h posttransfection. 293T cells were then transduced with the lentiviral particles, and cells were selected by culture in the presence of blasticidin. Bulk-selected cells were confirmed to have E protein expression via immunofluorescence assay. Subsequently, cells were plated in 96-well plates, using limiting dilution, and clones arising from single viable cells were selected. (B) 293T cells transduced with ZIKV C-prM-E lentiviral particles and bulk selected with blasticidin were stained with the MAB10216 antibody and analyzed by fluorescence microscopy. (C) The percentage of C-prM-E-positive cells was determined by flow cytometry. The “cells only” peak represents 293T cells not stained with the antibody, and the 293T peak represents 293T cells stained with MAB10216. (D) 293T cells expressing the pLenti-ZIKV-C-prM-E construct, generated as described above and bulk selected with blasticidin, were analyzed for E protein expression by Western blotting. Nontransfected 293T cells (293T) and cells transiently transfected with the ZIKV C-prM-E expression construct (transient) were used as negative and positive controls, respectively. (E and F) Single-cell clones of 293T cells expressing pLenti-ZIKV-C-prM-E were stained with the MAB10216 antibody and analyzed by fluorescence microscopy (E) or flow cytometry (F). The mean fluorescence intensity (MFI) of E protein expression for each clone is indicated on the right. FITC, fluorescein isothiocyanate. (G) pLenti-ZIKV-C-prM-E 293T cells are ideal for producing high-titer RVPs. 293T cells were transiently transfected with ZIKV C-prM-E along with the WNV Rep/GFP construct to generate reporter virus particles. Alongside, the pLenti-ZIKV-C-prM-E cell line was transfected with the WNV Rep/GFP construct. RVPs were harvested, and serial dilutions were used to infect Vero cells. The number of GFP-positive cells in each well was quantitated by fluorescence microscopy. Error bars show means ± standard deviations (SD). *, significant difference (P < 0.01) in the number of GFP+ cells in 293T versus C-prM-E-F6 cells, using the unpaired t test. Data representative of 1 of 4 independent experiments are shown.
Generation of prM-E cell line for VLP production.
Extensive studies with different flaviviruses, including ZIKV, have shown that expression of prM-E in the absence of capsid can produce subviral particles that can be used for immunization and elicit protective antibodies (44–48). These prM-E-based constructs are also the basis of DNA and mRNA nanoparticle vaccines currently in development for ZIKV (20–25). We hence developed a stable cell line that constitutively produces prM-E VLPs in the supernatant and can be used for large-scale production of VLPs for immunization studies. For this purpose, we used our original C-prM-E construct and PCR amplified and cloned the prM-E region spanning amino acids 105 to 795 (Fig. 4A) into the pcDNA3.1 vector (Fig. 4B). The last 17 amino acids of capsid, after the NS2B-3 cleavage site, were included in the prM-E construct for proper translocation into the ER lumen. Once the expression of E protein (Fig. 4C) and VLP production in the supernatant (Fig. 4D) were confirmed, the prM-E region was subcloned into the lentivirus vector, generating pLenti-prM-E (Fig. 4E). Packaged lentiviral particles were used to transduce 293T cells and to generate a bulk-selected cell line and single clones (Fig. 4E). As described above for the C-prM-E cell lines, 6 single-cell clones were selected and characterized for ZIKV E protein expression (data not shown). Clones C4 and F4 showed the highest expression levels by both immunofluorescence assay (Fig. 4F) and flow cytometry (Fig. 4G). Finally, we characterized the single-cell clones for production of VLPs in the supernatants. For this purpose, supernatants from the 293T-prM-E cell lines were ultracentrifuged through a glycerol cushion (49), the virus pellet was lysed, and the E protein was detected by Western blotting (Fig. 4H). Interestingly, the 293T-prM-E-F4 cell line showed significantly more VLP production than the 293T-prM-E-Bulk and 293T-prM-E-C4 cell lines. Thus, we have generated cell lines that constitutively produce high levels of ZIKV prM-E VLPs in the supernatant and can be used for VLP-based vaccine studies. Production of VLPs from our stable cell lines can readily be scaled up for clinical trials.
FIG 4.

Generation of prM-E cell line for VLP production. ZIKV prM-E was PCR amplified by use of specific primers, using the codon-optimized C-prM-E construct as the template (A), and cloned into the pCDNA3.1 expression vector (B). (C) E protein expression was determined by fluorescence microscopy after staining with the MAB10216 antibody. (D) Culture supernatants were harvested from ZIKV prM-E-expressing cells and ultracentrifuged. Cell and virus pellets were lysed, and E protein expression was determined by Western blotting. (E) ZIKV prM-E was PCR amplified and cloned into the lentiviral vector pLenti6/5-D-Topo. 293T cells were then transfected with the pLenti-prM-E construct along with the helper plasmid and VSV-G envelope, and ZIKV prM-E lentiviral particles were harvested at 48 h posttransfection. 293T cells were then transduced with the lentiviral particles, and cells were either bulk selected or selected as single-cell clones by culture in the presence of blasticidin. Selected cells were confirmed to have E protein expression via immunofluorescence assay after staining with the MAB10216 antibody (F) and by flow cytometry (G). The mean fluorescence intensity (MFI) of E protein expression for each prM-E clone is indicated on the right. (H) The indicated pLenti-ZIKV-prM-E cell clones were seeded in equal cell numbers, and culture supernatants were harvested and ultracentrifuged. VLP pellets were lysed and resolved by SDS-PAGE, and E protein expression was determined by Western blotting.
Immunization studies in mice.
The lack of an approved vaccine for ZIKV amid the recent outbreaks and the association of ZIKV infections with severe congenital birth defects warrant development of a safe and efficacious vaccine against the virus. For a vaccine to be available worldwide, especially in underdeveloped countries, it should be both easy to prepare and cost-effective. In this regard, stable cell lines constitutively producing ZIKV VLPs would be optimal, as they can readily be scaled up, with minimal scientific infrastructure, and can provide an economical alternative to other forms of vaccination. We hence tested the immunogenicity of the ZIKV VLPs in mice. Although prM-E VLPs are the most commonly used VLPs in flavivirus vaccine research, we also generated VLPs incorporating the capsid protein by using the C-prM-E construct. While prM-E particles can readily be generated using our stable cell lines (293T-prM-E), for generation of C-prM-E VLPs, the WNV NS2B-3 protease was needed for cleavage of C from prM-E in the ER (35, 50). As shown in Fig. 5A, and consistent with published findings (50, 51), expression of the WNV NS2B-3 protease was essential for release of C-prM-E but not prM-E VLPs. Hence, for generation of C-prM-E VLPs, cells were cotransfected with the NS2B-3 expression plasmid (52). Using these two different strategies, we generated both prM-E and C-prM-E VLPs that were purified from culture supernatants by ultracentrifugation through a glycerol cushion (49). The purified VLP pellet was analyzed for E protein expression via Western blotting and for VLP morphology by electron microscopy. As shown in Fig. 5B, there were large amounts of E protein detected in both the prM-E and C-prM-E VLP preparations. The total protein contents of the VLP preparations ranged from ∼1.7 to 2.3 mg/ml. Electron microscopy showed that the VLP morphology was consistent with that of ZIKV, with approximate particle diameters ranging from 30 to 40 nm (Fig. 5C).
FIG 5.

Expression of ZIKV prM-E alone releases VLPs into the supernatant, while C-prM-E requires the NS2B-3 protease for efficient VLP release. (A) 293T cells were transfected with the pCDNA3.1 vector expressing ZIKV prM-E or C-prM-E along with the indicated expression vectors. Cells were radiolabeled with [35S]Met/Cys, and culture supernatants were harvested and ultracentrifuged. Cell and virion samples were lysed and immunoprecipitated with MAB10216-coated protein A beads, resolved by SDS-PAGE, and visualized by PhosphorImager analysis. (B) Culture supernatants were harvested from cells expressing ZIKV C-prM-E or prM-E as indicated in Materials and Methods. Twenty-five to 30 ml of supernatant was transferred to ultracentrifuge tubes and carefully underlaid with 5 ml of 25% glycerol in TNE buffer. VLPs were pelleted by centrifugation at 110,500 × g for 3 h at 4°C. Thereafter, the supernatant was carefully removed and the VLP pellet resuspended in TNE buffer. An aliquot of concentrated VLPs was subjected to lysis with 10× RIPA buffer, and E protein in the preparations was detected by Western blotting. (C) VLPs were concentrated as described above, and images were acquired after negative staining, using a JEOL1010 transmission electron microscope with a Hamamatsu digital camera. Bars, ∼30 nm. (D and E) Immunization studies in mice. (D) BALB/c mice were divided into groups of six mice each. Mice received the primary immunization on day 0, followed by 2 boosters, at days 14 and 28, and were finally sacrificed at day 63 post-primary immunization. (E) Mice were divided into 5 groups and received immunizations with either C-prM-E/prM-E DNA or VLPs. For DNA immunization, a total of 50 μg of DNA in a volume of 100 μl PBS was injected intramuscularly (i.m.). For VLPs, the first immunization consisted of a VLP preparation mixed with TiterMax Gold adjuvant in a total volume of 100 μl injected intramuscularly. For subsequent boosters, mice received VLPs alone, without adjuvant. Control mice were injected with PBS.
The purified VLPs were subsequently used to immunize mice. The immunization schedule is shown in Fig. 5D. Mice divided into 5 groups of 6 mice each were immunized with either prM-E or C-prM-E DNA or the corresponding VLPs (Fig. 5E). Mice in the control group were injected with phosphate-buffered saline (PBS). For the first immunization, the VLPs were emulsified with a 1:1 mix of TiterMax Gold. All mice received 2 booster injections with the respective DNA or VLPs, at day 14 and day 28, and the mice were sacrificed on day 63, blood was collected by intracardiac puncture, and sera were harvested.
Anti-ZIKV immune responses in mice immunized with prM-E or C-prM-E DNA and VLPs.
We next determined the immune responses generated in mice upon immunization with the DNA- or VLP-based vaccines. Twofold serial dilutions of individual mouse sera were tested in the RVP-based microneutralization assay described for Fig. 2. Interestingly, the C-prM-E VLPs elicited the highest neutralization titers, followed by the prM-E VLPs and the prM-E DNA vaccine (Fig. 6A). Interestingly, the C-prM-E DNA vaccine worked the poorest for the generation of neutralizing antibodies, most likely because C-prM-E DNA in the absence of NS2B-3 protease failed to assemble and release VLPs, with the proteins thus being sequestered in the ER. The mean EC50 titers were >1:1,000 for both the VLP vaccines and 1:132 for prM-E DNA, while C-prM-E DNA showed titers of <1:32 (Fig. 6B). The EC90 titers followed a similar trend (Fig. 6C). Moreover, there were significant differences in EC50 (P = 0.0083) and EC90 (P = 0.0006) values between the prM-E and C-prM-E VLP-immunized mice (Fig. 6B and C). We also tested the ability of the immune sera generated in mice to neutralize the clinical ZIKV isolate PRVABC59. As evident in Fig. 6D, the mouse serum samples were able to neutralize the clinical ZIKV isolate, following a trend similar to that seen with neutralization of ZIKV RVPs in Fig. 6A. To further confirm the presence of ZIKV E-specific antibodies, we conducted an immunoprecipitation assay using pooled sera from different groups of immunized mice. For this assay, C-prM-E-expressing cells were radiolabeled with [35S]Met/Cys protein labeling mix, and cell lysates were immunoprecipitated with protein A beads coated with either 3 μl or 0.6 μl of pooled sera from the respective immunized groups. As shown in Fig. 6E, and consistent with microneutralization data, pooled sera from the C-prM-E VLP-immunized group showed the highest E protein band intensity, followed by the prM-E VLP-immunized group. Sera from the prM-E DNA group showed low levels of anti-E antibodies, followed by the C-prM-E DNA group, again consistent with the neutralization data presented in Fig. 6A. These results confirm that high levels of neutralizing antibodies against ZIKV E protein were generated using either prM-E or C-prM-E VLPs and that this approach can be used for generation of safe and effective vaccines.
FIG 6.

Anti-ZIKV immune response in mice immunized with prM-E/C-prM-E DNA or VLPs. (A) Serum samples collected from different groups of immunized mice were used in the RVP-based microneutralization assay. A serum sample from each mouse was serially diluted in DMEM and incubated with a predetermined amount of ZIKV RVPs for 1 h at room temperature. All samples were assayed in technical duplicates. Subsequently, the virus-serum mixtures were added to Vero cells in 96-well plates. The cells were incubated for 72 h, after which the plates were fixed and images acquired as described in the legend to Fig. 2. Curves were fit by using GraphPad Prism software, and neutralizing antibody EC50 (B) and EC90 (C) values were calculated. Statistical analysis was performed using the unpaired t test. Significant differences in EC50 (P = 0.0083) and EC90 (P = 0.0006) values were observed between prM-E and C-prM-E VLP-immunized mice. The dotted line denotes the limit of detection for the RVP assay (defined as the highest concentration of serum used in the neutralization experiments). Samples with titers of <20 are reported at half the limit of confidence (1:10). Neutralization data from one of two independent repeats are shown. (D) Neutralization of the clinical ZIKV isolate PRVABC59 with immune serum samples from mice. Pooled serum samples from each immunized group were serially diluted in serum-free medium as for panel A and then incubated with a predetermined amount of ZIKV for 2 h at 37°C. All samples were assayed in technical duplicates. Subsequently, the virus-serum mixtures were added to Vero cells in 96-well plates. The cells were incubated for 48 h, after which the plates were stained using MAB10216. Images were acquired as described in the legend to Fig. 2, antibody-positive cells were quantitated, and curves were fit using GraphPad Prism software. (E) Protein A beads were coated with 3 μl or 0.6 μl of pooled serum samples from each group of immunized mice. The antibody-coated beads were then incubated with radiolabeled cell lysates derived from C-prM-E-expressing cells. Cell lysates were resolved in an SDS-PAGE gel, followed by PhosphorImager analysis. The photo-stimulated luminescence (PSL) values for the bands are depicted in the graphs at bottom. (F) Pooled serum samples from each group of immunized mice were used in technical duplicates to determine the inhibition of WNV RVPs as described for panel A.
We have previously seen that immune sera from WNV-infected mice are capable of binding to the ZIKV E protein and cross-neutralizing ZIKV RVPs (Fig. 1 and 2). We hence determined if pooled sera from our ZIKV-immunized mouse groups were able to cross-neutralize WNV infection by using the RVP assay. For this assay, we prepared WNV reporter VLPs by using WNV C-prM-E and the Rep/GFP construct as described previously (38). Interestingly, sera obtained from ZIKV VLP/DNA-immunized mice were able to cross-neutralize WNV RVP infection in a trend that was similar to that of ZIKV RVP inhibition, although with a lower efficacy (Fig. 6F). Here again, sera from ZIKV C-prM-E VLP-immunized mice were most effective, followed by those from mice immunized with prM-E VLPs and prM-E DNA, with C-prM-E DNA being the least effective vaccine (Fig. 6F). These data demonstrate the efficacy of VLP-based vaccines compared to that of DNA vaccination in generating a robust neutralizing antibody response against ZIKV. Furthermore, incorporation of the capsid in VLPs gives an enhanced immune response against ZIKV, although prM-E VLPs are sufficient for inducing high level of neutralizing antibodies.
DISCUSSION
The initial outbreaks of ZIKV infection in Africa and Southeast Asia and subsequent transfer of infection to western countries, including the United States, has raised concerns regarding the lack of preparedness to combat the virus (5–7). At present, there is no approved vaccine or specific treatment for ZIKV infection. Moreover, there is limited information regarding ZIKV spread, its enhanced specificity for neural fetal tissue, and the mechanism via which it causes microcephaly. Thus, there is an urgent need not only to understand the basic virus biology but also to develop safe and efficacious vaccines to target the virus. Moreover, there is a need for development of rapid, reliable, and accurate assays that can be used to test anti-ZIKV immune responses and antiviral agents.
The ZIKV genome consists of a single-stranded positive-sense RNA and an open reading frame encoding a polyprotein (5′-C-prM-E-NS1-NS2A-NS2B-NS3-NS4A-NS4B-NS5-3′) (53, 54). This polyprotein is subsequently cleaved into capsid (C), precursor of membrane (prM), envelope (E), and seven nonstructural proteins (53, 55). The E protein (∼53 kDa) is the major virion surface protein and is involved in virus binding to the cell surface and membrane fusion (53, 55). Coexpression of the flaviviral proteins prM and E leads to secretion of VLPs that do not contain viral RNA and resemble empty particles produced during viral infection (48, 50, 56–58). These VLPs induce neutralizing antibody responses that are more potent than those induced by purified proteins (59, 60).
One of the major obstacles in flavivirus research is the requirement of high-level biosafety containment to undertake infectious virus studies, with BSL-2 or higher needed for ZIKV studies. The persistence of ZIKV in the body for prolonged periods (12) and reports of sexual transmission (10, 11) highlight the risks associated with working with infectious virus. The use of VLPs recently emerged as a powerful technology not only to study basic virus biology but also for vaccine and diagnostic assay development (59, 60). VLPs resemble viruses and are largely composed of viral structural proteins (53, 55). VLPs contain minimal or no genetic material, are nonreplicating, and may also contain a reporter gene, thus allowing for easy detection (39, 61). Moreover, the requirement of high-level biosafety containment to undertake infectious flavivirus studies (61, 62) can be overcome by using recombinant VLP-based assays. This eliminates the use of infectious virus while still allowing testing of various aspects of the pathogen, including (i) the mechanism of viral entry, (ii) neutralizing antibody sensitivity, (iii) vaccine efficacy, and (iv) compounds that inhibit viral entry (63).
In this study, a construct expressing the C-prM-E polyprotein of ZIKV was used to package a GFP reporter-containing WNV replicon to generate RVPs similar to those generated with WNV (38, 39). WNV-based RVPs have been used by us and others to study WNV E biology (29, 38, 39). ZIKV RVPs were generated using a codon-optimized synthetic ZIKV C-prM-E construct that was cotransfected with the WNV replicon construct (Rep/GFP). These RVPs were infectious in multiple cell lines and could be neutralized using anti-ZIKV antibodies or cross-neutralizing WNV immune sera. Although similar ZIKV RVPs were recently used for detection of an anti-ZIKV response, the current approach involves detection of infected cells by flow cytometry (29, 40). While flow cytometry has its benefits, especially in minimizing human error and bias and its ready use for large sample sizes, our plate-based assay may be more helpful under resource-limited conditions where flow cytometry is not available.
We adapted the RVP assay to a 96-well plate format with a GFP readout that can be quantitated using a simple fluorescence microscope. The objective was to develop an assay that can replace PRNT, requires less time to complete, does not involve use of live ZIKV, is highly reproducible, and can be used in resource-limited situations. We tested the assay extensively in 96-well format and found that both the number of GFP+ cells and the neutralization sensitivity were highly reproducible. Both automated counting using imaging software and manual counting yielded similar results, suggesting that the assay needs minimum infrastructure. For further ease and to facilitate the use of the assay, we generated a stable cell line expressing ZIKV structural proteins (293T-C-prM-E-F6) that can be used for RVP production by transfection with a GFP replicon construct, in our case a WNV replicon. However, the same cell line could also be utilized to package ZIKV reporter replicons when those become available. Besides the elimination of the use of infectious virus, our assay also provides several key advantages that make it attractive for resource-limited areas. The assay requires cell lines that are simple to grow and maintain (293T-C-prM-E-F6 and Vero), standard transfection protocols, 96-well plates, and a basic fluorescence microscope with a 4× objective. As in the case of PRNT, the plates can be fixed using formalin and saved for extended periods for quantitation of GFP-positive cells at a later time. In our hands, we saved the fixed plates for up to 6 months without a loss of GFP signal, and we quantified the signals at different time points, with comparable accuracies. With the rapid spread of ZIKV in countries with limited scientific infrastructure, this assay would be a valuable tool for assessing ZIKV neutralizing antibodies in response to vaccination drives and/or natural infections with ZIKV or related flaviviruses.
Recently, a number of studies have investigated vaccine candidates for ZIKV. These include conventional approaches, such as the use of purified inactivated virus (PIV) (20, 21), DNA (20–22), and adenovirus-based subunit vaccines incorporating the prM-E or M-E region of ZIKV (20, 23), as well as the use of lipid nanoparticle (LNP)-encapsulated RNA or modified mRNA as a vaccine candidate (24–26). These studies have demonstrated effective neutralizing antibody responses capable of protecting against ZIKV infection in various animal models. Recruitment of human subjects for testing some of these vaccine candidates is under way (ClinicalTrials.gov identifiers NCT02963909, NCT02840487, NCT02887482, NCT02809443, and NCT02952833). The geographical distribution of ZIKV is largely in developing and underdeveloped areas of the world (64). Hence, besides safety and efficacy being the top priorities for a successful vaccine, important practical aspects of a ZIKV vaccine are cost-effectiveness and ease of production. In this regard, VLP-based vaccines, especially from cell lines stably expressing and releasing ZIKV proteins, are highly attractive. Our 293T-prM-E cell lines provide a much-needed resource to take ZIKV VLP-based vaccines from the bench to the bedside. Not only do these cell lines release copious amounts of E protein into the supernatant, but VLPs produced from these cells generate a robust neutralizing antibody response in mice, making it ideal for further vaccine development.
In this study, we also found that VLP vaccines were more efficacious than their DNA counterparts at inducing a neutralizing antibody response. In contrast, other studies have reported higher antibody titers upon immunization with a single dose of prM-E DNA (22) or prM-E modified RNA (25). This may be due to use of a heterologous signal sequence from Japanese encephalitis virus (JEV) to improve expression, the stem/transmembrane region of the E protein from JEV to improve particle secretion (22), or a signal peptide from major histocompatibility complex (MHC) class II (25). Moreover, while C-prM-E VLPs worked better than prM-E VLPs, the C-prM-E DNA-based vaccine was relatively nonefficacious. This is largely because the flavivirus NS3 enzyme along with the cofactor NS2B forms an active protease that cleaves the flavivirus C protein that spans the ER membrane, producing its mature form (35, 50). Processing of the C protein by the viral protease in the ER is important for subsequent cleavage of E protein from prM by ER-resident signal peptidases (50, 65). Interestingly, our study found that inclusion of capsid in VLPs generates a better neutralizing immune response. However, it has been proposed that in the absence of an RNA genome, the nucleocapsid does not form, and hence no capsid would be released into the supernatant (66), calling into question the benefit of a C-prM-E-based vaccine. While a lack of detectable capsid protein expression in the supernatant could be due to small amounts being incorporated into VLPs, this limitation remains to be addressed due to the lack of an antibody that binds to ZIKV capsid.
Studies have suggested that T cells play an important role in generating a functional immune response in the presence of the viral capsid for hepatitis B and C viruses (67, 68). Similarly, for dengue virus 4 (DENV-4), epitopes in the capsid were shown to be recognized by cytotoxic T lymphocytes (CTLs) that were cross-reactive with other dengue virus serotypes (33). In fact, immunization with capsid alone was shown to generate a protective immune response that was independent of neutralizing antibodies and largely dependent on cell-mediated immunity (34). Moreover, CD4 T cells may also be involved in protection, as specialized subsets have been implicated in lysing flavivirus-infected cells (33, 69). Interestingly, our study shows that inclusion of capsid in VLPs requires a functional flaviviral protease, in our case the WNV NS2B-3 fusion protein. The NS2B-3 fusion protein coding sequence itself is about 2 kb long and can easily be included in VLP platforms, DNA vaccines, and modified mRNA vaccines.
In summary, we describe here the development, testing, and efficacy of a VLP-based vaccine against ZIKV and the generation of stable cell lines to facilitate this platform. We also describe the optimization of an RVP-based microneutralization assay using a ZIKV C-prM-E cell line and a WNV replicon expressing GFP. This assay recapitulates the standard PRNT routinely used by virologists, with several advantages, including ease of use, reproducibility, and elimination of the need for infectious virus. Thus, our study addresses the two most relevant aspects of ZIKV infection, i.e., a safe, effective, and economical vaccine and a neutralization assay that may be employed in the fight against the current ZIKV outbreaks.
MATERIALS AND METHODS
Cell culture and reagents.
293T and Vero cells were obtained from the ATCC and cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). All transfections were performed using Turbofect reagent (Thermo Fisher) per the manufacturer's instructions. The WNV C-prM-E and Rep/GFP plasmids have been described previously (39) and were kindly provided by Ted Pierson (NIAID). The plasmid containing the WNV NS2B-3 accessory fusion protein coding sequence and expressing the active protease has been described previously (52) and was a kind gift from Frank Scholle (NC State University). The ZIKV-117 antibody was kindly provided by James Crowe (Vanderbilt University Medical Center, Nashville, TN), and mouse polyclonal anti-ZIKV sera against isolate MR766 were kindly provided by the Centers for Disease Control and Prevention (Fort Collins, CO). The ZIKV isolate PRVABC59, derived from a human serum specimen from Puerto Rico in December 2015, was obtained from the ATCC and propagated in Vero cells by strictly following the ATCC recommendations.
Generation of vectors expressing ZIKV C-prM-E and prM-E.
A ZIKV C-prM-E construct was synthesized using the complete ZIKV sequence available from the current outbreak in the Americas (accession number KU312312.1) (6). This most current sequence was used to synthesize a codon-optimized version of the C-prM-E gene by use of gene synthesis technology by GenScript, Piscataway, NJ, USA. The synthesized gene was subcloned into the pcDNA3.1 vector (Invitrogen) by use of BamHI and XhoI restriction sites. The C-prM-E cassette was also cloned into the lentiviral vector pLenti6/V5 (Invitrogen) by use of the same restriction sites to generate plasmid pLenti-C-prM-E. The prM-E construct was generated by PCR amplification of the prM-E region spanning amino acids 105 to 795, using a Phusion high-fidelity PCR kit (New England BioLabs), and was cloned into the pcDNA3.1+ vector (Invitrogen). The prM-E cassette was also subcloned into the pLenti6/V5 vector by use of the BamHI-XhoI sites to generate plasmid pLenti-prM-E. All plasmids were sequenced to confirm their identities.
Detection of ZIKV E protein expression.
Detection of ZIKV E protein was conducted via either immunofluorescence assay or Western blotting. For immunofluorescence assay, cells were stained using the E-specific antibody MAB10216 (clone 4G2; EMD Millipore), which reacts with flavivirus group-specific antigens (36), followed by Alexa 488 (Invitrogen)-conjugated secondary antibody, and then analyzed by fluorescence microscopy. The monoclonal antibody MAB8150 (clone 3.67G; EMD Millipore) was used as a control and reacts with the E protein of West Nile/Kunjin virus. For Western blotting, lysates were resolved in an SDS-PAGE gel, transferred to polyvinylidene difluoride (PVDF) membranes, and probed with ZIKV E antibody (GTX133314; GeneTex) (1:3,000) followed by a horseradish peroxidase (HRP)-conjugated anti-mouse secondary antibody, and bands were visualized via enhanced chemiluminescence using Super Signal West Femto substrate (Pierce).
Metabolic labeling and immunoprecipitation.
The protocol for radiolabeling and immunoprecipitation of cell and virus lysates was previously described in detail (38). Briefly, transfected cells were washed with RPMI medium lacking Met and Cys. Thereafter, cells were incubated in RPMI medium supplemented with FBS and [35S]Met/Cys protein labeling mix. Culture supernatants were filtered and subjected to ultracentrifugation at 100,000 × g for 45 min. Cell and virion samples were lysed with Triton X-containing lysis buffer (0.5% Triton X-100, 300 mM NaCl, 50 mM Tris containing protease inhibitors). Thereafter, lysates were immunoprecipitated with anti-WNV serum (kindly provided by Robert Tesh, University of Texas Medical Branch, Galveston, TX) or MAB10216-coated protein A beads. Immunoprecipitated lysates were washed three times with Triton X-100 wash buffer and once with SDS-DOC wash buffer (0.1% sodium dodecyl sulfate, 300 mM NaCl, 50 mM Tris, 2.5 mM deoxycholic acid), resolved by SDS-PAGE, and visualized by PhosphorImager analysis.
Production of RVPs.
ZIKV RVPs were generated using a previously described protocol (38), with some modifications, that was originally described by Pierson et al. (39). 293T cells were cotransfected with the ZIKV C-prM-E construct and a plasmid containing a subgenomic GFP-expressing replicon derived from a lineage II strain of WNV (39). The RVPs were harvested at 48 h posttransfection, aliquoted, and stored for future use. RVPs were titrated in Vero cells plated in 96-well clear-bottom black plates at 5,000 cells per well. Thereafter, cells were infected with serial 2-fold dilutions of the RVPs and incubated for 72 h. The plates were fixed using 4% formalin-PBS, images were acquired using a Nikon EclipseTi microscope, and the number of GFP-positive cells was counted using NIS Elements software (Nikon).
RVP-based and clinical ZIKV-based microneutralization assay.
For neutralization assays, mouse sera or antibodies were serially diluted in DMEM in technical duplicates in a volume of 50 μl. Thereafter, a predetermined concentration of virus producing 200 to 500 GFP-positive cells was added to each well in a volume of 50 μl. The sera and RVPs were incubated for 1 h at room temperature. Subsequently, the virus-serum mix (100 μl) was added to Vero cells after removal of all the medium and was incubated for ∼72 h, after which the number of GFP-positive cells was quantitated. Statistical analysis was performed using the unpaired t test. The limit of detection for the RVP assay was defined as the highest concentration of serum (1:20 dilution) used in the neutralization experiments.
For neutralization assays using the ZIKV isolate PRVABC59, mouse sera were serially diluted in serum-free medium in technical duplicates in a volume of 50 μl. Thereafter, diluted ZIKV was added to each serum sample at a multiplicity of infection (MOI) of ∼5 and incubated for 2 h at 37°C. The virus-antibody mixtures were then added to Vero cells plated in 96-well plates and incubated for another 2 h at 37°C. The virus-antibody mix was then removed, and cells were incubated in DMEM containing 10% FBS for 48 h. The cells were then fixed and stained using MAB10216. Images were acquired by fluorescence microscopy, and the number of antibody-stained cells was quantitated as described above.
Generation of stable cell lines expressing ZIKV C-prM-E or prM-E.
Lentiviral vectors expressing ZIKV C-prM-E and prM-E were packaged in 293T cells by transfection with pLenti-C-prM-E or pLenti-prM-E along with the helper construct php-dl-NA (NIH AIDS reagent program) and the vesicular stomatitis virus glycoprotein (VSV-G) Env. The viral supernatants were collected at 48 h posttransfection, aliquoted, and stored. To generate stable cell lines, 293T cells were transduced with the lentiviral particles, and the cells were selected using blasticidin at a concentration of 10 μg/ml. Bulk-selected cells were passaged 8 to 10 times and stained for ZIKV E protein expression by using the monoclonal antibody MAB10216 at regular intervals to confirm selection. Subsequently, single-cell clones were generated from the bulk-selected cells by using limiting dilution cloning in 96-well plates. Up to 6 single-cell clones were selected for both the C-prM-E and prM-E constructs from wells that showed single-colony formation. Each single-cell clone was further characterized for ZIKV E protein expression by immunostaining followed by flow cytometry.
Production of DNA and VLPs for immunization.
For DNA immunizations in mice, the pcDNA-C-prM-E and pcDNA-prM-E plasmids were purified using an endotoxin-free plasmid maxikit (Qiagen) following the manufacturer's protocol.
VLPs for immunization were generated and purified as follows. For C-prM-E VLPs, 293T cells were transfected with the pcDNA3.1-C-prM-E construct along with the WNV NS2B-3 plasmid (52). The viral supernatants were harvested at 24 h and 48 h posttransfection. For production of prM-E virus particles, the 293T-Lenti-prM-E bulk cell line was cultured in the absence of blasticidin, and supernatants were harvested at 72 h and 96 h. VLPs were concentrated per the protocol of Brien et al. (49). Harvested supernatants (25 to 30 ml) were transferred to ultracentrifuge tubes and carefully underlaid with 5 ml of 25% glycerol in TNE buffer (10 mM Tris-Cl, 150 mM NaCl, 1mM EDTA). VLPs were pelleted by centrifugation at 110,500 × g for 3 h at 4°C. Thereafter, the supernatant was carefully removed and the VLP pellet resuspended in TNE buffer. The total protein content in the VLP preparations was measured using a bicinchoninic acid (BCA) microkit (Pierce), and specific E protein in the preparations was detected by Western blotting using the GTX133314 antibody.
Electron microscopy.
VLPs were concentrated as described above and imaged by electron microscopy after negative staining. Purified VLPs (3 μl) were applied to a glow-discharged carbon-coated 300-mesh grid. After ∼1 min, the grid was blotted with filter paper, and 3 μl of 2% uranyl acetate aqueous solution was added for 30 s. After blotting off excess liquid and drying, images were acquired using a JEOL1010 transmission electron microscope with a Hamamatsu digital camera and AMT Advantage image capture software, using a magnification of ×100,000.
Mouse studies.
All animal use was reviewed and approved by the Texas Tech University Health Sciences Center (TTUHSC) El Paso IACUC (study number 16024). For immunization studies, 6- to 8-week-old BALB/c mice were purchased from Jackson Laboratory and housed in a pathogen-free animal facility at TTUHSC El Paso. Mice were divided into groups of six mice each and immunized with different preparations. For DNA immunization, a total of 50 μg of DNA in a volume of 100 μl PBS was injected intramuscularly. Mice received two additional boosts, at weeks 2 and 4 of primary immunization, as described above. For VLPs, the first immunization consisted of a VLP preparation (approximate total protein content, 1.7 to 2.3 mg/ml) mixed with TiterMax Gold adjuvant (Sigma) at a 1:1 ratio in a total volume of 100 μl injected intramuscularly. Mice received two additional boosts of VLPs, at weeks 2 and 4, without adjuvant. Control mice were injected with PBS. Blood was collected at week 9 after the first immunization under terminal isoflurane anesthesia followed by intracardiac puncture. Blood samples were collected in serum separator tubes per the manufacturer's recommendations. After coagulation, the tubes were centrifuged, and sera were harvested, aliquoted, and stored at −80°C until further use.
ACKNOWLEDGMENTS
We thank Ted Pierson (NIAID) for the subgenomic GFP-expressing WNV replicon plasmids, Frank Scholle (NC State University) for the WNV NS2B-3 protease plasmid, the NIH AIDS reagent program for the php-dl-NA plasmid, Robert Tesh (University of Texas Medical Branch, Galveston, TX) for kindly providing the WNV immune mouse sera, James Crowe (Vanderbilt University Medical Center, TN) for the ZIKV-117 antibody, and Brandy Russell (CDC, Fort Collins, CO) for kindly providing the ZIKV polyclonal sera. We thank Jingchuan Sun (University of Pennsylvania) for help with electron microscopy and the TTUHSC El Paso LARC services for help with animal experiments.
This study was supported by the TTUHSC intramural seed grant program and in part by NIH grant 1R21AI131696-01 to A.J.
We declare that we have no conflicts of interest.
REFERENCES
- 1.Dick GW. 1952. Zika virus. II. Pathogenicity and physical properties. Trans R Soc Trop Med Hyg 46:521–534. doi: 10.1016/0035-9203(52)90043-6. [DOI] [PubMed] [Google Scholar]
- 2.Dick GW, Kitchen SF, Haddow AJ. 1952. Zika virus. I. Isolations and serological specificity. Trans R Soc Trop Med Hyg 46:509–520. doi: 10.1016/0035-9203(52)90042-4. [DOI] [PubMed] [Google Scholar]
- 3.Duffy MR, Chen TH, Hancock WT, Powers AM, Kool JL, Lanciotti RS, Pretrick M, Marfel M, Holzbauer S, Dubray C, Guillaumot L, Griggs A, Bel M, Lambert AJ, Laven J, Kosoy O, Panella A, Biggerstaff BJ, Fischer M, Hayes EB. 2009. Zika virus outbreak on Yap Island, Federated States of Micronesia. N Engl J Med 360:2536–2543. doi: 10.1056/NEJMoa0805715. [DOI] [PubMed] [Google Scholar]
- 4.Simpson DI. 1964. Zika virus infection in man. Trans R Soc Trop Med Hyg 58:335–338. doi: 10.1016/0035-9203(64)90200-7. [DOI] [PubMed] [Google Scholar]
- 5.Brasil P, Pereira JP Jr, Moreira ME, Ribeiro Nogueira RM, Damasceno L, Wakimoto M, Rabello RS, Valderramos SG, Halai UA, Salles TS, Zin AA, Horovitz D, Daltro P, Boechat M, Raja Gabaglia C, Carvalho de Sequeira P, Pilotto JH, Medialdea-Carrera R, Cotrim da Cunha D, Abreu de Carvalho LM, Pone M, Machado Siqueira A, Calvet GA, Rodrigues Baiao AE, Neves ES, Nassar de Carvalho PR, Hasue RH, Marschik PB, Einspieler C, Janzen C, Cherry JD, Bispo de Filippis AM, Nielsen-Saines K. 2016. Zika virus infection in pregnant women in Rio de Janeiro. N Engl J Med 375:2321–2334. doi: 10.1056/NEJMoa1602412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Enfissi A, Codrington J, Roosblad J, Kazanji M, Rousset D. 2016. Zika virus genome from the Americas. Lancet 387:227–228. doi: 10.1016/S0140-6736(16)00003-9. [DOI] [PubMed] [Google Scholar]
- 7.Rasmussen SA, Jamieson DJ, Honein MA, Petersen LR. 2016. Zika virus and birth defects—reviewing the evidence for causality. N Engl J Med 374:1981–1987. doi: 10.1056/NEJMsr1604338. [DOI] [PubMed] [Google Scholar]
- 8.Cao-Lormeau VM, Blake A, Mons S, Lastere S, Roche C, Vanhomwegen J, Dub T, Baudouin L, Teissier A, Larre P, Vial AL, Decam C, Choumet V, Halstead SK, Willison HJ, Musset L, Manuguerra JC, Despres P, Fournier E, Mallet HP, Musso D, Fontanet A, Neil J, Ghawche F. 2016. Guillain-Barre syndrome outbreak associated with Zika virus infection in French Polynesia: a case-control study. Lancet 387:1531–1539. doi: 10.1016/S0140-6736(16)00562-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oehler E, Watrin L, Larre P, Leparc-Goffart I, Lastere S, Valour F, Baudouin L, Mallet H, Musso D, Ghawche F. 2014. Zika virus infection complicated by Guillain-Barre syndrome—case report, French Polynesia, December 2013. Euro Surveill 19:20720. doi: 10.2807/1560-7917.ES2014.19.9.20720. [DOI] [PubMed] [Google Scholar]
- 10.Davidson A, Slavinski S, Komoto K, Rakeman J, Weiss D. 2016. Suspected female-to-male sexual transmission of Zika virus—New York City, 2016. MMWR Morb Mortal Wkly Rep 65:716–717. doi: 10.15585/mmwr.mm6528e2. [DOI] [PubMed] [Google Scholar]
- 11.Mansuy JM, Suberbielle E, Chapuy-Regaud S, Mengelle C, Bujan L, Marchou B, Delobel P, Gonzalez-Dunia D, Malnou CE, Izopet J, Martin-Blondel G. 2016. Zika virus in semen and spermatozoa. Lancet Infect Dis 16:1106–1107. doi: 10.1016/S1473-3099(16)30336-X. [DOI] [PubMed] [Google Scholar]
- 12.Murray KO, Gorchakov R, Carlson AR, Berry R, Lai L, Natrajan M, Garcia MN, Correa A, Patel SM, Aagaard K, Mulligan MJ. 2017. Prolonged detection of Zika virus in vaginal secretions and whole blood. Emerg Infect Dis 23:99–101. doi: 10.3201/eid2301.161394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi Y, Gao GF. 2017. Structural biology of the Zika virus. Trends Biochem Sci 42:443–456. doi: 10.1016/j.tibs.2017.02.009. [DOI] [PubMed] [Google Scholar]
- 14.Stadler K, Allison SL, Schalich J, Heinz FX. 1997. Proteolytic activation of tick-borne encephalitis virus by furin. J Virol 71:8475–8481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sapparapu G, Fernandez E, Kose N, Bin C, Fox JM, Bombardi RG, Zhao H, Nelson CA, Bryan AL, Barnes T, Davidson E, Mysorekar IU, Fremont DH, Doranz BJ, Diamond MS, Crowe JE. 2016. Neutralizing human antibodies prevent Zika virus replication and fetal disease in mice. Nature 540:443–447. doi: 10.1038/nature20564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stettler K, Beltramello M, Espinosa DA, Graham V, Cassotta A, Bianchi S, Vanzetta F, Minola A, Jaconi S, Mele F, Foglierini M, Pedotti M, Simonelli L, Dowall S, Atkinson B, Percivalle E, Simmons CP, Varani L, Blum J, Baldanti F, Cameroni E, Hewson R, Harris E, Lanzavecchia A, Sallusto F, Corti D. 2016. Specificity, cross-reactivity, and function of antibodies elicited by Zika virus infection. Science 353:823–826. doi: 10.1126/science.aaf8505. [DOI] [PubMed] [Google Scholar]
- 17.Swanstrom JA, Plante JA, Plante KS, Young EF, McGowan E, Gallichotte EN, Widman DG, Heise MT, de Silva AM, Baric RS. 2016. Dengue virus envelope dimer epitope monoclonal antibodies isolated from dengue patients are protective against Zika virus. mBio 7:e01123-16. doi: 10.1128/mBio.01123-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Q, Yang H, Liu X, Dai L, Ma T, Qi J, Wong G, Peng R, Liu S, Li J, Li S, Song J, Liu J, He J, Yuan H, Xiong Y, Liao Y, Li J, Yang J, Tong Z, Griffin BD, Bi Y, Liang M, Xu X, Qin C, Cheng G, Zhang X, Wang P, Qiu X, Kobinger G, Shi Y, Yan J, Gao GF. 2016. Molecular determinants of human neutralizing antibodies isolated from a patient infected with Zika virus. Sci Transl Med 8:369ra179. doi: 10.1126/scitranslmed.aai8336. [DOI] [PubMed] [Google Scholar]
- 19.Zhao H, Fernandez E, Dowd KA, Speer SD, Platt DJ, Gorman MJ, Govero J, Nelson CA, Pierson TC, Diamond MS, Fremont DH. 2016. Structural basis of Zika virus-specific antibody protection. Cell 166:1016–1027. doi: 10.1016/j.cell.2016.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abbink P, Larocca RA, De La Barrera RA, Bricault CA, Moseley ET, Boyd M, Kirilova M, Li Z, Ng'ang'a D, Nanayakkara O, Nityanandam R, Mercado NB, Borducchi EN, Agarwal A, Brinkman AL, Cabral C, Chandrashekar A, Giglio PB, Jetton D, Jimenez J, Lee BC, Mojta S, Molloy K, Shetty M, Neubauer GH, Stephenson KE, Peron JP, Zanotto PM, Misamore J, Finneyfrock B, Lewis MG, Alter G, Modjarrad K, Jarman RG, Eckels KH, Michael NL, Thomas SJ, Barouch DH. 2016. Protective efficacy of multiple vaccine platforms against Zika virus challenge in rhesus monkeys. Science 353:1129–1132. doi: 10.1126/science.aah6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Larocca RA, Abbink P, Peron JP, Zanotto PM, Iampietro MJ, Badamchi-Zadeh A, Boyd M, Ng'ang'a D, Kirilova M, Nityanandam R, Mercado NB, Li Z, Moseley ET, Bricault CA, Borducchi EN, Giglio PB, Jetton D, Neubauer G, Nkolola JP, Maxfield LF, De La Barrera RA, Jarman RG, Eckels KH, Michael NL, Thomas SJ, Barouch DH. 2016. Vaccine protection against Zika virus from Brazil. Nature 536:474–478. doi: 10.1038/nature18952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dowd KA, Ko SY, Morabito KM, Yang ES, Pelc RS, DeMaso CR, Castilho LR, Abbink P, Boyd M, Nityanandam R, Gordon DN, Gallagher JR, Chen X, Todd JP, Tsybovsky Y, Harris A, Huang YS, Higgs S, Vanlandingham DL, Andersen H, Lewis MG, De La Barrera R, Eckels KH, Jarman RG, Nason MC, Barouch DH, Roederer M, Kong WP, Mascola JR, Pierson TC, Graham BS. 2016. Rapid development of a DNA vaccine for Zika virus. Science 354:237–240. doi: 10.1126/science.aai9137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim E, Erdos G, Huang S, Kenniston T, Falo LD Jr, Gambotto A. 2016. Preventative vaccines for Zika virus outbreak: preliminary evaluation. EBioMedicine 13:315–320. doi: 10.1016/j.ebiom.2016.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chahal JS, Fang T, Woodham AW, Khan OF, Ling J, Anderson DG, Ploegh HL. 2017. An RNA nanoparticle vaccine against Zika virus elicits antibody and CD8+ T cell responses in a mouse model. Sci Rep 7:252. doi: 10.1038/s41598-017-00193-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pardi N, Hogan MJ, Pelc RS, Muramatsu H, Andersen H, DeMaso CR, Dowd KA, Sutherland LL, Scearce RM, Parks R, Wagner W, Granados A, Greenhouse J, Walker M, Willis E, Yu JS, McGee CE, Sempowski GD, Mui BL, Tam YK, Huang YJ, Vanlandingham D, Holmes VM, Balachandran H, Sahu S, Lifton M, Higgs S, Hensley SE, Madden TD, Hope MJ, Kariko K, Santra S, Graham BS, Lewis MG, Pierson TC, Haynes BF, Weissman D. 2017. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 543:248–251. doi: 10.1038/nature21428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Richner JM, Himansu S, Dowd KA, Butler SL, Salazar V, Fox JM, Julander JG, Tang WW, Shresta S, Pierson TC, Ciaramella G, Diamond MS. 2017. Modified mRNA vaccines protect against Zika virus infection. Cell 169:176. doi: 10.1016/j.cell.2017.03.016. [DOI] [PubMed] [Google Scholar]
- 27.Boigard H, Alimova A, Martin GR, Katz A, Gottlieb P, Galarza JM. 2017. Zika virus-like particle (VLP) based vaccine. PLoS Negl Trop Dis 11:e0005608. doi: 10.1371/journal.pntd.0005608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haddow AD, Schuh AJ, Yasuda CY, Kasper MR, Heang V, Huy R, Guzman H, Tesh RB, Weaver SC. 2012. Genetic characterization of Zika virus strains: geographic expansion of the Asian lineage. PLoS Negl Trop Dis 6:e1477. doi: 10.1371/journal.pntd.0001477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dowd KA, DeMaso CR, Pelc RS, Speer SD, Smith AR, Goo L, Platt DJ, Mascola JR, Graham BS, Mulligan MJ, Diamond MS, Ledgerwood JE, Pierson TC. 2016. Broadly neutralizing activity of Zika virus-immune sera identifies a single viral serotype. Cell Rep 16:1485–1491. doi: 10.1016/j.celrep.2016.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maeda A, Maeda J. 2013. Review of diagnostic plaque reduction neutralization tests for flavivirus infection. Vet J 195:33–40. doi: 10.1016/j.tvjl.2012.08.019. [DOI] [PubMed] [Google Scholar]
- 31.Jeong H, Seong BL. 2017. Exploiting virus-like particles as innovative vaccines against emerging viral infections. J Microbiol 55:220–230. doi: 10.1007/s12275-017-7058-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oliveira ER, Mohana-Borges R, de Alencastro RB, Horta BA. 2017. The flavivirus capsid protein: structure, function and perspectives towards drug design. Virus Res 227:115–123. doi: 10.1016/j.virusres.2016.10.005. [DOI] [PubMed] [Google Scholar]
- 33.Gagnon SJ, Zeng W, Kurane I, Ennis FA. 1996. Identification of two epitopes on the dengue 4 virus capsid protein recognized by a serotype-specific and a panel of serotype-cross-reactive human CD4+ cytotoxic T-lymphocyte clones. J Virol 70:141–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lazo L, Hermida L, Zulueta A, Sanchez J, Lopez C, Silva R, Guillen G, Guzman MG. 2007. A recombinant capsid protein from dengue-2 induces protection in mice against homologous virus. Vaccine 25:1064–1070. doi: 10.1016/j.vaccine.2006.09.068. [DOI] [PubMed] [Google Scholar]
- 35.Stocks CE, Lobigs M. 1998. Signal peptidase cleavage at the flavivirus C-prM junction: dependence on the viral NS2B-3 protease for efficient processing requires determinants in C, the signal peptide, and prM. J Virol 72:2141–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Henchal EA, Gentry MK, McCown JM, Brandt WE. 1982. Dengue virus-specific and flavivirus group determinants identified with monoclonal antibodies by indirect immunofluorescence. Am J Trop Med Hyg 31:830–836. doi: 10.4269/ajtmh.1982.31.830. [DOI] [PubMed] [Google Scholar]
- 37.Lazear HM, Diamond MS. 2016. Zika virus: new clinical syndromes and its emergence in the Western Hemisphere. J Virol 90:4864–4875. doi: 10.1128/JVI.00252-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garg H, Lee RT, Tek NO, Maurer-Stroh S, Joshi A. 2013. Identification of conserved motifs in the West Nile virus envelope essential for particle secretion. BMC Microbiol 13:197. doi: 10.1186/1471-2180-13-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pierson TC, Sanchez MD, Puffer BA, Ahmed AA, Geiss BJ, Valentine LE, Altamura LA, Diamond MS, Doms RW. 2006. A rapid and quantitative assay for measuring antibody-mediated neutralization of West Nile virus infection. Virology 346:53–65. doi: 10.1016/j.virol.2005.10.030. [DOI] [PubMed] [Google Scholar]
- 40.Dowd KA, DeMaso CR, Pierson TC. 2015. Genotypic differences in dengue virus neutralization are explained by a single amino acid mutation that modulates virus breathing. mBio 6:e01559-15. doi: 10.1128/mBio.01559-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hasan SS, Miller A, Sapparapu G, Fernandez E, Klose T, Long F, Fokine A, Porta JC, Jiang W, Diamond MS, Crowe JE Jr, Kuhn RJ, Rossmann MG. 2017. A human antibody against Zika virus crosslinks the E protein to prevent infection. Nat Commun 8:14722. doi: 10.1038/ncomms14722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garg H, Lee RT, Maurer-Stroh S, Joshi A. 2016. HIV-1 adaptation to low levels of CCR5 results in V3 and V2 loop changes that increase envelope pathogenicity, CCR5 affinity and decrease susceptibility to maraviroc. Virology 493:86–99. doi: 10.1016/j.virol.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 43.Joshi A, Nyakeriga AM, Ravi R, Garg H. 2011. HIV ENV glycoprotein-mediated bystander apoptosis depends on expression of the CCR5 co-receptor at the cell surface and ENV fusogenic activity. J Biol Chem 286:36404–36413. doi: 10.1074/jbc.M111.281659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davis BS, Chang GJ, Cropp B, Roehrig JT, Martin DA, Mitchell CJ, Bowen R, Bunning ML. 2001. West Nile virus recombinant DNA vaccine protects mouse and horse from virus challenge and expresses in vitro a noninfectious recombinant antigen that can be used in enzyme-linked immunosorbent assays. J Virol 75:4040–4047. doi: 10.1128/JVI.75.9.4040-4047.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ferlenghi I, Clarke M, Ruttan T, Allison SL, Schalich J, Heinz FX, Harrison SC, Rey FA, Fuller SD. 2001. Molecular organization of a recombinant subviral particle from tick-borne encephalitis virus. Mol Cell 7:593–602. doi: 10.1016/S1097-2765(01)00206-4. [DOI] [PubMed] [Google Scholar]
- 46.Hunt AR, Cropp CB, Chang GJ. 2001. A recombinant particulate antigen of Japanese encephalitis virus produced in stably-transformed cells is an effective noninfectious antigen and subunit immunogen. J Virol Methods 97:133–149. doi: 10.1016/S0166-0934(01)00346-9. [DOI] [PubMed] [Google Scholar]
- 47.Mason PW, Pincus S, Fournier MJ, Mason TL, Shope RE, Paoletti E. 1991. Japanese encephalitis virus-vaccinia recombinants produce particulate forms of the structural membrane proteins and induce high levels of protection against lethal JEV infection. Virology 180:294–305. doi: 10.1016/0042-6822(91)90034-9. [DOI] [PubMed] [Google Scholar]
- 48.Pincus S, Mason PW, Konishi E, Fonseca BA, Shope RE, Rice CM, Paoletti E. 1992. Recombinant vaccinia virus producing the prM and E proteins of yellow fever virus protects mice from lethal yellow fever encephalitis. Virology 187:290–297. doi: 10.1016/0042-6822(92)90317-I. [DOI] [PubMed] [Google Scholar]
- 49.Brien JD, Lazear HM, Diamond MS. 2013. Propagation, quantification, detection, and storage of West Nile virus. Curr Protoc Microbiol 31:15D.3.1–15D.3.18. doi: 10.1002/9780471729259.mc15d03s31. [DOI] [PubMed] [Google Scholar]
- 50.Lobigs M. 1993. Flavivirus premembrane protein cleavage and spike heterodimer secretion require the function of the viral proteinase NS3. Proc Natl Acad Sci U S A 90:6218–6222. doi: 10.1073/pnas.90.13.6218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bera AK, Kuhn RJ, Smith JL. 2007. Functional characterization of cis and trans activity of the flavivirus NS2B-NS3 protease. J Biol Chem 282:12883–12892. doi: 10.1074/jbc.M611318200. [DOI] [PubMed] [Google Scholar]
- 52.Wilson JR, de Sessions PF, Leon MA, Scholle F. 2008. West Nile virus nonstructural protein 1 inhibits TLR3 signal transduction. J Virol 82:8262–8271. doi: 10.1128/JVI.00226-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chambers TJ, Hahn CS, Galler R, Rice CM. 1990. Flavivirus genome organization, expression, and replication. Annu Rev Microbiol 44:649–688. doi: 10.1146/annurev.mi.44.100190.003245. [DOI] [PubMed] [Google Scholar]
- 54.Kuno G, Chang GJ. 2007. Full-length sequencing and genomic characterization of Bagaza, Kedougou, and Zika viruses. Arch Virol 152:687–696. doi: 10.1007/s00705-006-0903-z. [DOI] [PubMed] [Google Scholar]
- 55.Lindenbach BD, Rice CM. 2003. Molecular biology of flaviviruses. Adv Virus Res 59:23–61. doi: 10.1016/S0065-3527(03)59002-9. [DOI] [PubMed] [Google Scholar]
- 56.Gehrke R, Ecker M, Aberle SW, Allison SL, Heinz FX, Mandl CW. 2003. Incorporation of tick-borne encephalitis virus replicons into virus-like particles by a packaging cell line. J Virol 77:8924–8933. doi: 10.1128/JVI.77.16.8924-8933.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lorenz IC, Kartenbeck J, Mezzacasa A, Allison SL, Heinz FX, Helenius A. 2003. Intracellular assembly and secretion of recombinant subviral particles from tick-borne encephalitis virus. J Virol 77:4370–4382. doi: 10.1128/JVI.77.7.4370-4382.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pugachev KV, Mason PW, Shope RE, Frey TK. 1995. Double-subgenomic Sindbis virus recombinants expressing immunogenic proteins of Japanese encephalitis virus induce significant protection in mice against lethal JEV infection. Virology 212:587–594. doi: 10.1006/viro.1995.1516. [DOI] [PubMed] [Google Scholar]
- 59.Noad R, Roy P. 2003. Virus-like particles as immunogens. Trends Microbiol 11:438–444. doi: 10.1016/S0966-842X(03)00208-7. [DOI] [PubMed] [Google Scholar]
- 60.Pattenden LK, Middelberg AP, Niebert M, Lipin DI. 2005. Towards the preparative and large-scale precision manufacture of virus-like particles. Trends Biotechnol 23:523–529. doi: 10.1016/j.tibtech.2005.07.011. [DOI] [PubMed] [Google Scholar]
- 61.Lo MK, Tilgner M, Shi PY. 2003. Potential high-throughput assay for screening inhibitors of West Nile virus replication. J Virol 77:12901–12906. doi: 10.1128/JVI.77.23.12901-12906.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shi PY, Tilgner M, Lo MK. 2002. Construction and characterization of subgenomic replicons of New York strain of West Nile virus. Virology 296:219–233. doi: 10.1006/viro.2002.1453. [DOI] [PubMed] [Google Scholar]
- 63.Pijlman GP. 2015. Enveloped virus-like particles as vaccines against pathogenic arboviruses. Biotechnol J 10:659–670. doi: 10.1002/biot.201400427. [DOI] [PubMed] [Google Scholar]
- 64.Hayes EB. 2009. Zika virus outside Africa. Emerg Infect Dis 15:1347–1350. doi: 10.3201/eid1509.090442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yamshchikov VF, Compans RW. 1995. Formation of the flavivirus envelope: role of the viral NS2B-NS3 protease. J Virol 69:1995–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Khromykh AA, Varnavski AN, Westaway EG. 1998. Encapsidation of the flavivirus Kunjin replicon RNA by using a complementation system providing Kunjin virus structural proteins in trans. J Virol 72:5967–5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Duenas-Carrera S, Alvarez-Lajonchere L, Alvarez-Obregon JC, Herrera A, Lorenzo LJ, Pichardo D, Morales J. 2000. A truncated variant of the hepatitis C virus core induces a slow but potent immune response in mice following DNA immunization. Vaccine 19:992–997. doi: 10.1016/S0264-410X(00)00209-7. [DOI] [PubMed] [Google Scholar]
- 68.Xing YP, Huang ZH, Wang SX, Cai J, Li J, Chou TH, Lu S. 2005. Novel DNA vaccine based on hepatitis B virus core gene induces specific immune responses in Balb/c mice. World J Gastroenterol 11:4583–4586. doi: 10.3748/wjg.v11.i29.4583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Aihara H, Takasaki T, Matsutani T, Suzuki R, Kurane I. 1998. Establishment and characterization of Japanese encephalitis virus-specific, human CD4(+) T-cell clones: flavivirus cross-reactivity, protein recognition, and cytotoxic activity. J Virol 72:8032–8036. [DOI] [PMC free article] [PubMed] [Google Scholar]