

ABSTRACT
Progressive multifocal leukoencephalopathy (PML) is an often-fatal demyelinating disease of the central nervous system. PML results when oligodendrocytes within immunocompromised individuals are infected with the human JC virus (JCV). We have identified an oligodendrocyte precursor cell line, termed G144, that supports robust levels of JCV DNA replication, a central part of the JCV life cycle. In addition, we have determined that JC virus readily infects G144 cells. Furthermore, we have determined that JCV DNA replication in G144 cells is stimulated by myristoylated (i.e., constitutively active) Akt and reduced by the Akt-specific inhibitor MK2206. Thus, this oligodendrocyte-based model system will be useful for a number of purposes, such as studies of JCV infection, establishing key pathways needed for the regulation of JCV DNA replication, and identifying inhibitors of this process.
IMPORTANCE The disease progressive multifocal leukoencephalopathy (PML) is caused by the infection of particular brain cells, termed oligodendrocytes, by the JC virus. Studies of PML, however, have been hampered by the lack of an immortalized human cell line derived from oligodendrocytes. Here, we report that the G144 oligodendrocyte cell line supports both infection by JC virus and robust levels of JCV DNA replication. Moreover, we have established that the Akt pathway regulates JCV DNA replication and that JCV DNA replication can be inhibited by MK2206, a compound that is specific for Akt. These and related findings suggest that we have established a powerful oligodendrocyte-based model system for studies of JCV-dependent PML.
KEYWORDS: AKT, JC virus, oligodendrocytes
INTRODUCTION
JC polyomavirus (JCV) is the causative agent of progressive multifocal leukoencephalopathy (PML), an often fatal demyelinating disease of the central nervous system (CNS) in immunocompromised individuals (reviewed in references 1–5). In addition, JCV-dependent PML is a not uncommon side effect of immunomodulatory therapies, such as treatment of multiple sclerosis patients with natalizumab (Tysabri) (6, 7), and it is also associated with infections by human immunodeficiency virus (8). Studies have also suggested a possible link between JCV and human brain and non-central nervous system tumors (9, 10).
The principal CNS targets for JCV infection are oligodendrocytes (OGs) (11; reviewed in references 2, 12, and 13). JCV also infects neurons, where it causes the novel neurological disorders JC virus granule cell neuronopathy and JC virus encephalopathy (reviewed in references 1 and 14). In vitro cell types that support, to varying extents, the JCV life cycle include human embryonic stem cell-derived oligodendrocyte progenitor cells (15), human fetal glial cells (16–19), human embryonic kidney cells (20), progenitor-derived astrocytes (21–23), astrocytes (24), and glial progenitor cells (24). Nevertheless, a significant limitation in the JCV field has been the lack of a rapidly proliferating oligodendrocyte cell line that can be differentiated and that also supports critical steps in the JCV life cycle, such as viral infection (25) and DNA replication.
Given the need for a tractable oligodendrocyte cell line that supports the JCV life cycle, it was interesting that a glioma-derived stem cell line, termed G144, has an oligodendrocyte precursor (OPC)-like phenotype that is stable through passaging (26). Moreover, it has a doubling time of 3 to 5 days, and upon withdrawal of the growth factors epidermal growth factor (EGF) and fibroblast growth factor 2 (FGF-2), the G144 cells differentiated into mature OGs (26, 27). In view of these properties, we obtained the G144 cell line and conducted experiments designed to determine if this oligodendrocyte precursor cell line could be used as a model system for studies of the JCV life cycle, with initial emphasis on JCV DNA replication. The results of these studies are presented here.
RESULTS
Characterization of the G144 oligodendrocyte precursor cell line.
Images of undifferentiated G144 cells are presented in Fig. 1A (top). Upon growth factor withdrawal, G144 cells extend the radial processes characteristic of mature oligodendrocytes (26) (Fig. 1A, bottom). This finding replicates the in vivo behavior of OPCs, which undergo maturation after cell cycle exit (28). To continue the characterization of the G144 cell line, we screened for both surface and stage-specific markers for oligodendrocytes.
FIG 1.

Characterization of the G144 cell line. (A) Bright-field images of undifferentiated G144 cells (top) and cells differentiated by 7 days of growth factor withdrawal (bottom). In contrast to the bipolar morphology of undifferentiated cells, differentiated cells exhibited standard oligodendrocyte morphology, including radial processes. (B) Staining of G144 cells for proteins selectively made in oligodendrocytes. Shown are representative images of undifferentiated and differentiated G144 cells stained for the sulfatide surface antigen O4, a marker of late immature OGs (top), sulfatide surface antigen O1, a marker of immature OLs, and MBP, a marker of mature OLs.
(i) G144 cells express oligodendrocyte surface markers.
To demonstrate that undifferentiated and differentiated G144 cells grown under our experimental conditions express well-known oligodendrocyte-specific surface antigens (29), we stained for the premyelinating stage O4 protein (30). It is apparent from the immunofluorescence (IF) studies presented in Fig. 1B (top) that both undifferentiated and differentiated G144 cells are O4 positive. In addition, we established that G144 cells express the myelinating stage O1 protein (Fig. 1B, middle). Moreover, we analyzed whether G144 cells express myelin basic protein (MBP), a protein selectively expressed in mature oligodendrocytes (31). It is clear from Fig. 1B (bottom) that MBP is expressed in both undifferentiated and differentiated G144 cells. Of interest, colocalization studies with DAPI (4′,6-diamidino-2-phenylindole) indicated that MBP is concentrated in the nuclei of G144 cells (data not shown), a finding supported by previous studies of the subcellular localization of MBP (32). Collectively, the results from these IF experiments support the conclusion that G144 cells are oligodendrocyte progenitors (26, 27). We note, however, that despite dramatic differences in morphology, noticeable differences in the levels of these proteins between undifferentiated and differentiated G144 cells were not detected. This might simply reflect a more advanced degree of differentiation in our stock of G144 precursors than previously reported (26) or the inherently nonquantitative nature of IF experiments.
(ii) qRT-PCR studies of stage-specific oligodendrocyte mRNA levels in G144 cells.
In addition to protein markers, a number of stage-specific mRNA markers for OPCs and OGs have been identified (28). Therefore, to further characterize the differentiation of G144 cells following growth factor withdrawal, quantitative reverse transcription (qRT)-PCR was used to analyze the expression levels of representative markers in both undifferentiated and differentiated G144 cells. Examination of Fig. 2 establishes that upon removal of the growth factors EGF and FGF-2, mRNA levels for neuraminidase 1 (Neu1) and chondroitin sulfate proteoglycan 4 (CSPG4) (also termed NG2) declined. (The legend to Fig. 2 provides brief descriptions of the functions of the proteins encoded by these mRNAs). In contrast, mRNA levels for myelin and lymphocyte protein (Mal) and Forkhead box protein O4 (Foxo4) increased. Moreover, the mRNA levels for MBP and Forkhead box protein O1 (Foxo1) also increased. Collectively, these studies provide further evidence that G144 cells maintained in the presence of growth factors are OPCs, which are induced to mature into OGs by growth factor withdrawal.
FIG 2.

G144 cells express stage-specific oligodendrocyte mRNAs during in vitro differentiation. qRT-PCR analysis of oligodendrocyte lineage markers following 7 days of growth factor withdrawal revealed reduced expression of the OPC markers Neu1 and CSPG4 (NG2) compared with undifferentiated controls. (Neu1 makes a significant contribution to B-series ganglioside degradation in the brain [103], while NG2 is characteristically used to identify OPCs [reference 104 and references therein]). This reduction is accompanied by an increase in expression of the OG-associated Mal, Foxo4, MBP, and Foxo1 genes. As described by Goldman and Kuypers (28), the OG mRNAs were subdivided into those associated with premyelinating oligodendrocytes and myelinating oligodendrocytes. (Mal is predominantly localized in compact myelin [105], while Foxo4 is a transcription factor that is involved in growth and differentiation [106].) MBP, which is expressed in the nuclei of G144 cells, maintains the correct structure of myelin (107, 108), while the transcription factor Foxo1 has multiple roles, including the promotion of oligodendrocyte regeneration (109) and the regulation of apoptosis (reviewed in reference 110). The y axis presents the log2-fold change between the ΔCT values for the mRNAs collected from the undifferentiated and differentiated samples. Means (n = 3) are reported, and the error bars indicate standard deviations from the mean. The asterisks indicate significant differences between the undifferentiated and differentiated cells (P < 0.05).
The G144 oligodendrocyte precursor cell line supports robust levels of T-ag-dependent JCV DNA replication. (i) JCV T-ag accumulates in the nuclei of G144 cells.
The major gene product encoded by the early region of the JCV genome is large T antigen (T-ag), a 688-residue modular protein that has an N-terminal J domain, an origin binding domain, and C-terminal helicase domain (reviewed in references 33–37); the structures of the JCV T-ag origin binding domain (38, 39) and helicase domains (40) have been recently solved. JCV T-ag accumulates in the nuclei of glial cells derived from PML patients (41). Moreover, localization of T-ag to the nuclei of cells is essential for polyomavirus DNA replication (33, 34, 36). Therefore, following nucleofection of a plasmid encoding JCV T-ag (i.e., pCMV-JCVT-ag) (38, 42) into undifferentiated G144 cells, we determined the subcellular localization of JCV T-ag. Examination of the representative immunofluorescence image presented in Fig. 3A establishes that JCV T-ag readily accumulates in the nuclei of undifferentiated G144 cells (T-ag also accumulates in the nuclei of differentiated G144 cells [data not shown]). In contrast, JCV T-ag does not accumulate in the nuclei of Hs683 cells (Fig. 3B), a glial cell line that does not support JCV DNA replication (42).
FIG 3.

Efficient expression of JCV T-ag in the nuclei of G144 cells. (A) Immunofluorescence analysis revealed localization of T antigen (left) in the nucleus (indicated by DAPI [right]) of undifferentiated G144 cells following nucleofection of pCMV-JCT-ag. (B) In contrast, the results of a representative control experiment demonstrated that T-ag (left) does not accumulate in the nuclei of the Hs683 glioma cell line (indicated by DAPI [right]).
(ii) The G144 cell line supports the firefly luciferase (Fluc)-based JCV DNA replication assay.
Given that G144 cells are oligodendrocyte precursors (Fig. 1 and 2) and that JCV T-ag accumulates in the nuclei of these cells (Fig. 3A), we elected to determine if they support T-ag-dependent JCV DNA replication. Therefore, the JCV DNA replication plasmids (42, 43) were nucleofected into both undifferentiated and differentiated G144 cells, and the levels of JCV DNA replication were determined. It is apparent from Fig. 4A that G144 cells support robust levels of T-ag-dependent JCV DNA replication and that replication is elevated in the differentiated cells. Moreover, the data presented in the accompanying Western blot (Fig. 4B) establish that in the differentiated cells, increased levels of JCV T-ag do not account for the elevated levels of JCV DNA replication. Finally, the T-ag gene in plasmid pCMV-JCVT-ag is based on the cDNA sequence of large T-ag; thus, levels of JCV DNA replication do not depend on the expression of small T antigen.
FIG 4.
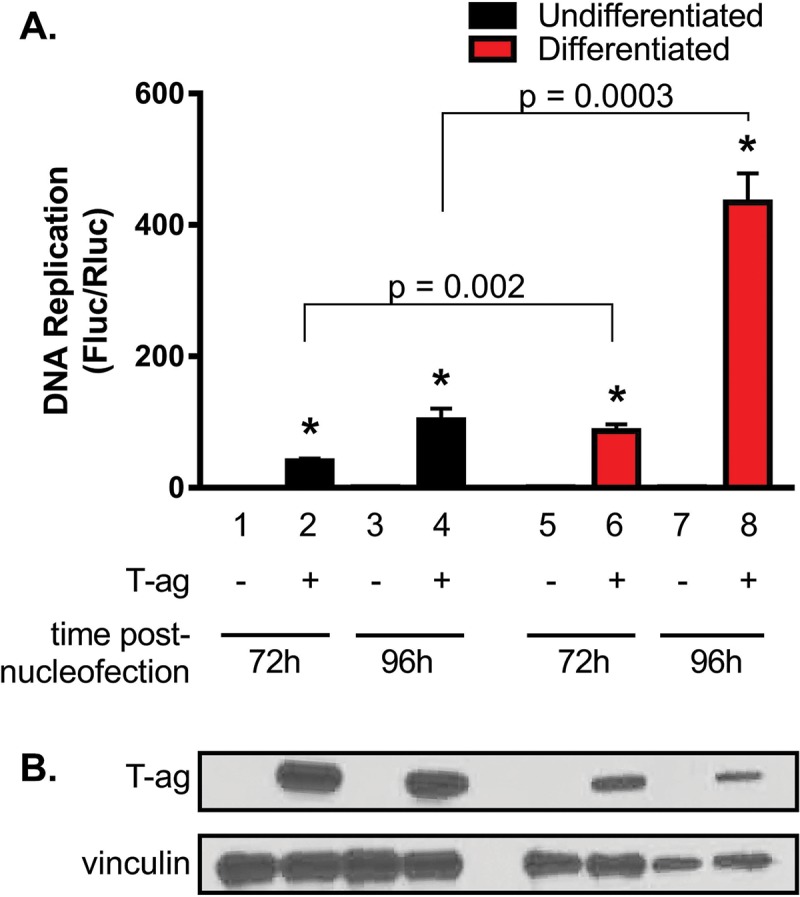
G144 cells support robust levels of JCV DNA replication, particularly when differentiated. (A) Levels of replication in both undifferentiated and differentiated G144 cells were determined by luciferase assay after 72 h (bars 2 and 6) and 96 h (bars 4 and 8). Reactions conducted in the absence of T-ag at the indicated times are represented by the odd-numbered bars. Means (n = 3) are reported, and the error bars indicate standard deviations from the mean. The asterisks indicate significant increases in replication (P < 0.05) compared with T-ag-minus (−) controls. (B) Results of a Western blot used to determine the amounts of JCV T-ag in G144 cells after 72 h (lanes 2 and 6) and 96 h (lanes 4 and 8) following nucleofection of the replication plasmids. As a loading control, an additional Western blot was performed to determine vinculin levels.
(iii) Using a T-ag mutant to validate JCV DNA replication in the G144 cell line.
To confirm that the Fluc signal is dependent upon JCV DNA replication, we measured the amount of viral DNA replication catalyzed by the JCV T-ag H514A mutant. T-ag residue H514 is at the tip of the beta-hairpin (Fig. 5A). In simian virus 40 (SV40), the corresponding residue (i.e., H513) is known to play critical roles during both the melting of the viral origin (44–47) and the subsequent pumping of single-stranded DNA (ssDNA) through the center of the helicase domain (48, 49). Examination of Fig. 5B establishes that in differentiated G144 cells, the JCV T-ag H514A mutant does not support JCV DNA replication. Furthermore, the Western blot presented in Fig. 5C demonstrates that the failure of the H514A T-ag mutant to support JCV DNA replication is not due to increased protein lability caused by this surface mutation. Of interest, SV40 T-ag containing mutations at the tip of the beta-hairpin is not impaired in terms of its ability to bind site specifically to DNA (45). Thus, the JCV H514A T-ag mutant is likely to be defective for replication at a stage subsequent to origin binding (reviewed in references 35–37 and 50).
FIG 5.
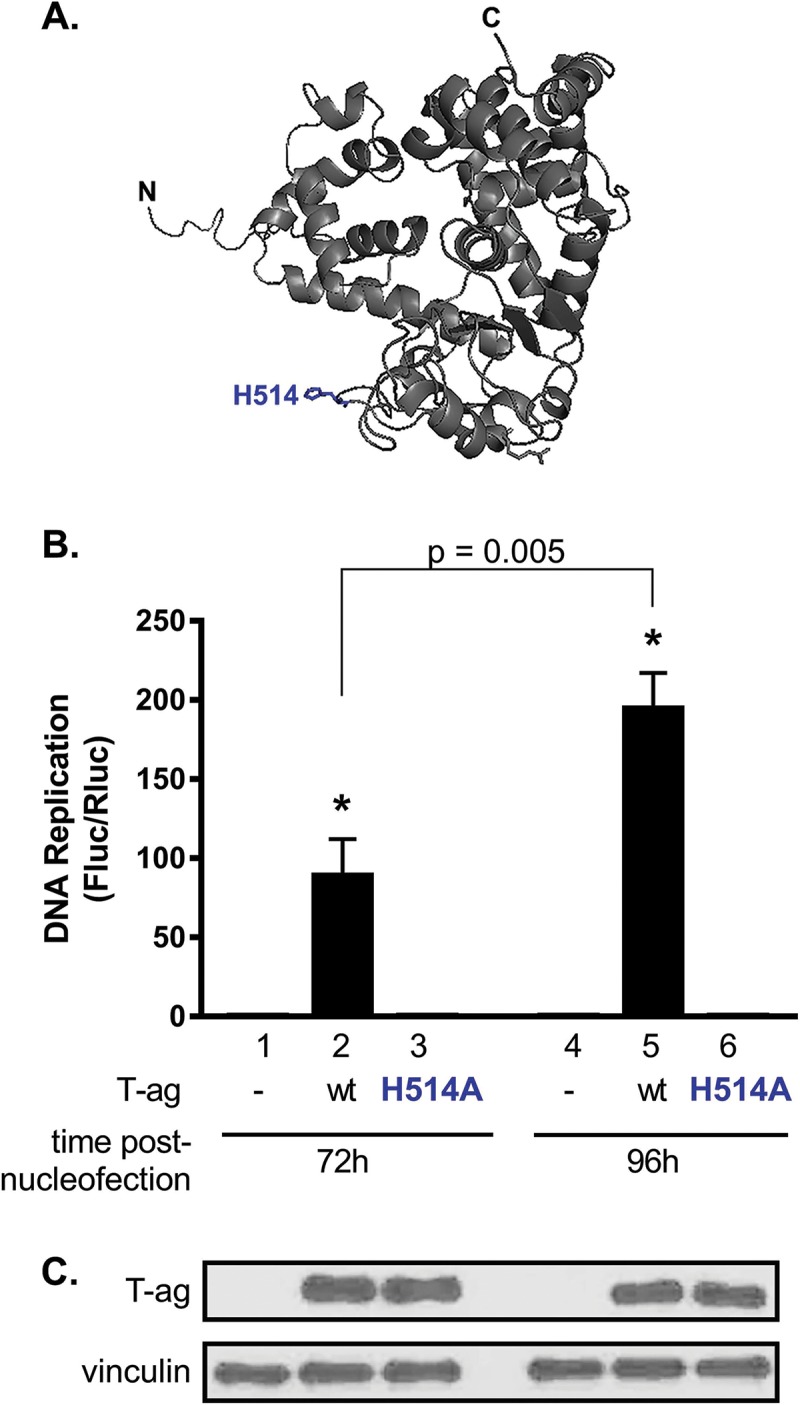
JCV T-ag containing a point mutation at the tip of the beta-hairpin does not support JCV DNA replication. (A) Residue H514, situated at the tip of the beta-hairpin within the helicase domain of JCV T-ag and critical for viral DNA replication (45, 46, 48), is shown in blue. (B) Following nucleofection of both wt JCV T-ag and the H514A mutant into undifferentiated G144 cells, levels of JCV DNA replication were measured after incubation for 72 and 96 h. Cells containing an H514 mutation did not exhibit significant replication compared with T-ag-minus (−) controls. Means (n = 3) are reported, and the error bars indicate standard deviations from the mean. *, P < 0.05. (C) Results of a Western blot used to determine the amounts of JCV T-ag in G144 cells 72 h and 96 h following nucleofection of the replication plasmids, including wt T-ag (lanes 2 and 5) or the H514A mutant (lanes 3 and 6). As a loading control, a Western blot was performed to determine vinculin levels.
(iv) Confirming JCV DNA replication in G144 cells by qPCR.
To provide further evidence that the increase in luciferase activity results from an increase in the copy number of origin-containing plasmid DNA, quantitative PCR (qPCR) experiments were used to measure the amounts of Fluc-containing plasmid DNA present in undifferentiated G144 cells as a function of time. Examination of Fig. 6 demonstrates that in the absence of the plasmid encoding JCV T-ag, the firefly luciferase-specific primers amplified relatively low levels of plasmid DNA (gray line). In contrast, during a 4-day time course in the presence JCV T-ag, there was a marked increase in the amount of amplified Fluc DNA derived from the pFL-JCV ori plasmid (green line).
FIG 6.

qPCR-based demonstration of the T-antigen-dependent amplification of JCV DNA in G144 cells. The results of a qPCR-based time course, showing levels of JCV DNA replication obtained in assays conducted with the full complement of replication plasmids, is presented in green. Shown in gray is a similar replication time course conducted with the replication plasmids, but in the absence of the plasmid encoding JCV T-ag. The y axis presents the fold change in the FLuc (replicating)/RLuc (nonreplicating) plasmid DNA ratios. Means (n = 3) are reported, and the error bars indicate standard deviations from the mean.
JCV DNA replication in the G144 cell line is enhancer dependent.
Tissue-dependent gene expression of JCV is directed by the enhancer-containing noncoding control region (NCCR) (51, 52). Studies conducted in the cervical carcinoma cell line C33A also established that JCV enhancers play a direct role in the replication of viral DNA (42). Therefore, to test the hypothesis that enhancers are also directly required for JCV DNA replication in the G144 cell line, the following experiments were conducted.
(i) In G144 cells, JCV DNA replication is inactivated by mutations in the enhancer at the consensus binding sites for NFI family members.
Members of the Nuclear Factor I (NFI) transcription family (reviewed in reference 53) were previously reported to have multiple roles in JCV transcription and replication (42, 54–58). Extending these studies, we elected to determine if the two most origin-proximal nfi binding sites in the tandemly duplicated Mad-1 enhancer (i.e., nfi a and b) are required for JCV DNA replication in G144 cells. The locations of the nfi a and b sites within the Mad-1 enhancer are depicted in Fig. 7A (magenta). When JCV replication assays were conducted with plasmids containing mutations at the nfi a and b binding sites, viral DNA replication in both undifferentiated and differentiated G144 cells was significantly reduced (Fig. 7B and D, respectively). (For the sequence of the nfi mutations, see Materials and Methods.) This finding confirms the hypothesis that in G144 cells, intact enhancers are essential for JCV DNA replication and further indicates that interactions between the NCCR and an NFI family member(s) are critically important for viral replication.
FIG 7.

JCV DNA replication in G144 cells is reduced by a derivative of pFL-JCVori containing mutations at the nfi a and b binding sites. (A) Depiction of the Mad-1 enhancer showing the relative locations of the nfi a and b binding sites (magenta). (B) Luciferase-based replication assays conducted in undifferentiated G144 cells for 72 and 96 h comparing levels of JCV DNA replication obtained with the wt Mad-1 enhancer and a Mad-1 derivative containing mutant nfi a and b binding sites. Means (n = 3) are reported, and the error bars indicate standard deviations from the mean. The asterisks indicate significant increases in JC replication (P < 0.05) compared with reactions performed in the absence of T-ag (−). (C) Western blots establishing the relative levels of the NFI family members in undifferentiated G144 cells. Vinculin levels were determined as loading controls. (D) Results of JCV DNA replication assays conducted in differentiated G144 cells for 72 and 96 h. As in panel B, levels of JCV DNA replication obtained with the Mad-1 enhancer containing mutant nfi a and b binding sites were greatly reduced relative to the wt Mad-1 enhancer. (E) Western blots establishing the relative levels of the NFI family members in differentiated G144 cells; vinculin levels were determined as loading controls.
(ii) G144 cells express NFI family members NFI-B and NFI-C.
The NFI family contains four members termed NFI-A, NFI-B, NFI-C, and NFI-X (59–61). In light of the finding that the nfi a and b binding sites are needed for JCV DNA replication in G144 cells, we determined which NFI family member(s) was present in G144 cells by Western blotting. Examination of Fig. 7C and E establishes that the 47-kDa NFI-B and 56-kDa NFI-C proteins were detected in both undifferentiated and differentiated G144 cells but the 56-kDa NFI-A and the 55-kDa NFI-X were not. This finding suggests that NFI-B and NFI-C, perhaps acting together, are necessary to promote JCV DNA replication in G144 cells.
The PI3K-Akt pathway promotes JCV DNA replication in the G144 oligodendrocyte cell line.
Little is known about the processes that regulate the JCV life cycle in oligodendrocytes. One candidate mechanism for controlling JCV replication is the phosphatidylinositol 3-kinase (PI3K)-Akt pathway. It is regulated by both EGF (reviewed in reference 62) and FGF (63), the two factors required to maintain G144 cells in the undifferentiated state. Furthermore, the PI3K-Akt pathway is known to play critical roles in the survival of many viruses (reviewed in reference 64). In addition, SV40 T-ag activates Akt by inducing the phosphorylation of Akt at both T308 and S473 (65, 66), and infection with BK polyomavirus (BKPyV) causes an increase in Akt phosphorylation (67). In view of these studies and the structural and functional similarities between SV40 and JCV T-ags (38, 40), we elected to determine if Akt plays a role in the replication of JCV DNA in the G144 cell line.
(i) In G144 cells, JCV DNA replication is stimulated by the myristoylated Akt-1 isoform.
The JCV replication assay was modified by conucleofection of the pcDNA Myr-HA-Akt1 plasmid, which encodes a myristoylated (myr), and thus constitutively activated, Akt-1 isoform (68). In undifferentiated G144 cells, the inclusion of 100 ng of plasmid pcDNA Myr-HA-Akt1 activated JCV DNA replication (Fig. 8A). Corresponding Western blots demonstrated the expression of myr-Akt1 in undifferentiated G144 cells as a function of time (Fig. 8B). From these data, it is apparent that increased levels of JCV DNA replication correlate with increased levels of myr-Akt1. Finally, it is also apparent from Fig. 8B (middle row) that expression of myr-Akt1 has little or no effect on levels of T-ag expression.
FIG 8.

JCV DNA replication in G144 cells is stimulated by a plasmid encoding myristolated Akt1. (A) Time course showing levels of JCV DNA replication in undifferentiated G144 cells, used as a control (red). The levels of JCV DNA replication obtained in identical reactions but in the presence of 100 ng of plasmid pcDNA Myr-HA-Akt1 are shown in blue. Additional control reactions, conducted in the absence of JCV T-ag, are shown in black. Means (n = 3) are reported, and the error bars indicate standard deviations from the mean. The asterisks indicate increased JCV replication in myr-Akt samples compared with wt controls (*, P < 0.05). (B) Western blot used to detect the expression of HA-tagged myr-Akt1 in undifferentiated G144 cells during the replication time course presented in panel A. The anti-HA MAb used in these studies (Santa Cruz; SC-7392) was raised against the canonical HA sequence YPYDVPDYA. However, the HA sequence in pcDNA Myr-HA-Akt1 has an Ala in place of the N-terminal Pro (underlined); this may help to explain the relatively weak binding to the Myr-HA-Akt1 substrates. The additional Western blots demonstrate that expression of Myr-Akt1 does not alter expression levels of JCV T-ag. Finally, as loading controls, vinculin levels were also determined.
(ii) JCV DNA replication in G144 cells is inhibited by the Akt-specific inhibitor MK2206.
Given the stimulation of JCV DNA replication by constitutive activation of Akt, we also tested the inverse prediction that JCV DNA replication in G144 cells is inhibited by the Akt-specific inhibitor MK2206 (69). In undifferentiated G144 cells, MK2206 significantly inhibits JCV DNA replication (Fig. 9A) without altering expression of JCV T-ag, even at the highest levels of MK2206 (Fig. 9B). Collectively, these studies provide further support for the conclusion that in G144 cells, JCV DNA replication is dependent upon the PI3K-Akt pathway.
FIG 9.
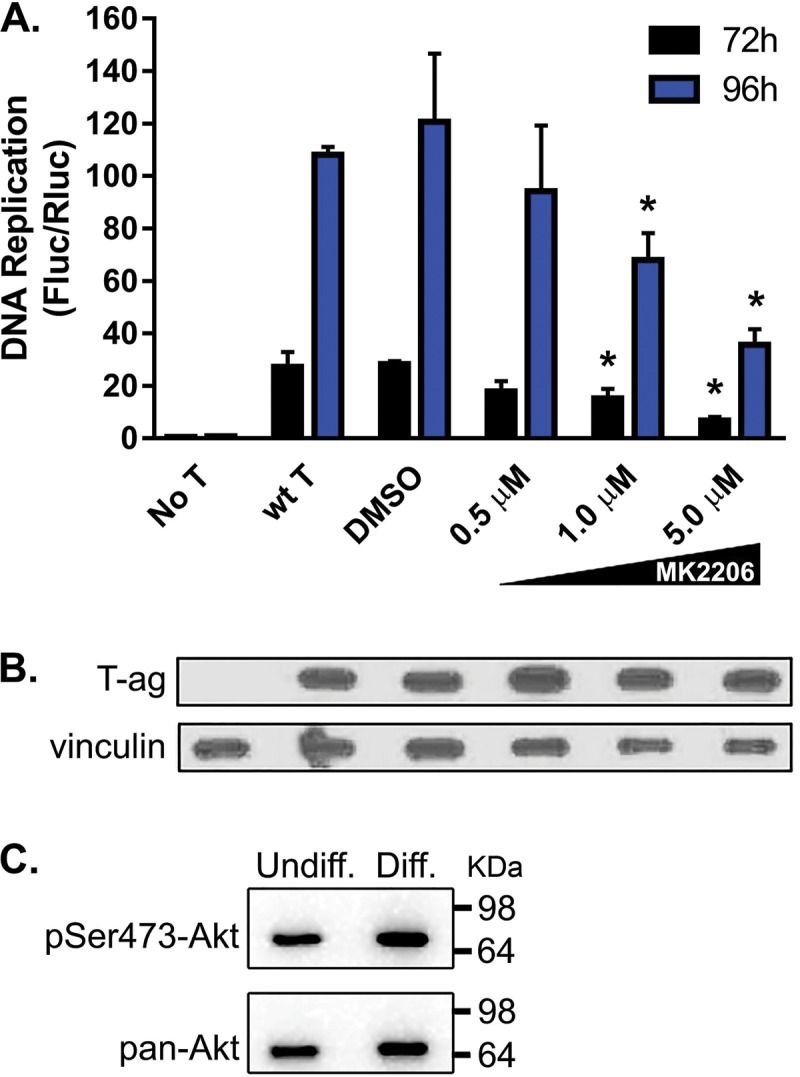
Inhibition of JCV DNA replication in nondifferentiated G144 cells by the Akt inhibitor MK2206. (A) Following nucleofection of the replication plasmids, the cells were allowed to adhere to a 96-well plate for 12 h. The indicated amounts of MK2206 were then added, and levels of DNA replication were subsequently determined (after either 72 or 96 h). Means (n = 3) are reported, and the error bars indicate standard deviations from the mean. The asterisks indicate differences from wt T-ag controls at equivalent time points (*, P < 0.05). DMSO, dimethyl sulfoxide. (B) Western blot-based evidence that addition of MK2206 to G144 cells at the indicated concentrations does not alter T-ag levels. (Note that in an additional series of IF experiments, we also determined that the subcellular distribution of JCV T-ag within G144 cells does not change upon the addition of 5 μM MK2206 [data not shown].) (C) An additional Western blot was used to confirm that the transformed G144 cell line contains activated Akt. (Top) G144 cell extracts, either undifferentiated (left) or differentiated (right), were probed using the phospho-Akt (Ser 473) MAb. (Bottom) The same G144 extracts were probed with a pan-Akt MAb.
In view of the above-mentioned results linking Akt to JCV DNA replication, we determined the status of Akt in both undifferentiated and differentiated G144 cells (grown in the presence and absence of EGF and FGF2, respectively). A Western blot probed with a phospho-Akt (Ser473) monoclonal antibody (MAb) demonstrated that Akt is activated in both undifferentiated and differentiated G144 cells (Fig. 9C, top). This result is consistent with previous studies of Akt activation in transformed cells (70). As a control, an additional Western blot was performed with a pan-Akt MAb (Fig. 9C, bottom). Thus, in the transformed G144 cell line, Akt activation is not appreciably altered by growth factor removal and subsequent differentiation.
Do G144 cells support infection by JC virus?
Previous studies established the plasmid-based JCV replication assay as a powerful tool for addressing a wide range of issues pertaining to JCV DNA replication in an oligodendrocyte cell line. They also raised a question, namely, do G144 cells also support infection by JC virus? To address this issue, differentiated G144 cells were infected with the Mad-4 strain of JC virus (see Materials and Methods). The infected cells were then analyzed for the expression of proteins encoded by the viral early and late genes. Examination of the representative IF image presented in Fig. 10A establishes that 48 h postinfection, JCV T-ag is readily detected within G144 cells. Similarly, it is apparent from Fig. 10B that following infection with the Mad-4 strain for 48 h, the “late” viral protein VP1 is also produced.
FIG 10.

Evidence that differentiated G144 cells support infection by JC virus. (A) (Right) Results of immunofluorescence-based experiments demonstrating that differentiated G144 cells infected with JC virus for 48 h express JCV T-ag. (Left) As a control, additional immunofluorescence studies of T-ag expression were conducted in uninfected G144 cells. (B) (Right) Additional immunofluorescence-based experiments establishing that after infection with the Mad-4 strain of JC virus for 48 h, differentiated G144 cells express the late viral protein VP1 (probed with the anti-VP1 MAb Pab 597). (Left) Results of identical immunofluorescence-based studies of uninfected G144 cells. (C) Results of a qPCR-based study demonstrating that the JCV genome is amplified following infection of differentiated G144 cells with the Mad-4 strain of JC virus for 6 days. The y axis presents the log2-fold change between the ΔCT values of the input and JCV DNA samples collected at the indicated times. Means (n = 3) are reported, and the error bars indicate standard deviations from the mean. *, P < 0.05.
In polyomaviruses, the switch from early- to late-mRNA synthesis is dependent upon viral DNA replication (reviewed in reference 71). Since late mRNA encodes VP1, the detection of VP1 (Fig. 10B) implies that G144 cells also support the amplification of the JCV genome. To test this theory, differentiated G144 cells were infected with the Mad-4 JCV strain (see Materials and Methods). Following incubation for the indicated periods, the medium was removed and the infected cells were washed with phosphate-buffered saline (PBS). The JCV genomic DNA present in the G144 cells was then purified via modified Hirt extractions (72). The relative amount of JCV DNA present in the G144 cells was determined by additional qPCR studies (Fig. 10C). Examination of Fig. 10C confirms that amplification of the JCV genome occurs within G144 cells. In summary, the studies presented in Fig. 10A to C establish that G144 cells support infection by the Mad-4 strain of JC virus.
DISCUSSION
The JCV life cycle takes place predominantly in oligodendrocytes (reviewed in references 2, 5, and 13). The data presented in this study extend previous observations (26) indicating that G144 cells are an oligodendrocyte precursor cell line and demonstrate that upon growth factor withdrawal they exhibit features characteristic of differentiating oligodendrocytes. Accordingly, we elected to determine whether G144 cells support JCV DNA replication, a central stage of the viral life cycle. Here, we demonstrate that JCV T-ag accumulates in the nuclei of these cells and that the G144 oligodendrocyte cell line supports robust levels of JCV DNA replication. We also demonstrate that JC virus readily infects differentiated G144 cells. Therefore, G144 cells can serve as an integral part of the first oligodendrocyte-based model system for studies of major events during the JCV life cycle.
The selective targeting of JCV to oligodendrocytes is due primarily to interactions between the viral NCCR and oligodendrocyte-specific transcription factors (reviewed in references 2 and 13). It was noted that a novel feature of the plasmid-based JCV replication assay is that T-ag is expressed from a separate plasmid than the origin-containing Fluc plasmid; as a result, the regulation of T-ag transcription and viral replication are uncoupled (42, 43). Therefore, it is of interest that in G144 cells a derivative of the Mad-1 enhancer, containing mutations at the nfi a and b sites within the NCCR, supported levels of JCV DNA replication that were much lower than wild type (wt). Thus, the diminished ability of the nfi a and b mutants to support JCV DNA replication indicates that within G144 cells, NFI family members are directly required for JCV DNA replication, rather than the more indirect stimulation of T-ag expression. Related experiments indicated that the NFI family member(s) that is likely to bind to the nfi sites within G144 cells is NFI-B or NFI-C (or perhaps both). However, T-ag is a transcriptional activator (73); thus, it is possible that the levels of the NFI family members will change following viral infection and T-ag expression. Therefore, establishing the role(s) of the NFI family members during JCV DNA replication will require additional biochemical and structural studies. Nevertheless, continued studies of the NFI family member(s) bound to the JCV enhancer will be broadly informative, since the regulatory regions of many other DNA viruses contain binding sites for NFI family members (i.e., polyomavirus [74], BKPyV [75], human papillomavirus 16 [HPV16] [76], cytomegalovirus [77, 78], hepatitis B virus [79], and adenovirus [reviewed in reference 80]). Furthermore, NFI family members regulate the transcription of a large number of neuronal genes (81, 82), including oligodendrocyte-specific expression of MBP (83, 84), and they are essential for multiple steps in development (reviewed in reference 53). In the long term, similar detailed investigations of the other G144-specific factors that bind to the NCCR are warranted.
The experiments presented here indicate that Akt plays a role in the regulation of JCV DNA replication in G144 cells. For instance, JCV DNA replication was promoted by myr-Akt1, a molecule that is known to be associated with the plasma membrane. These data raise the possibility that the elevated levels of JCV DNA replication observed in the differentiated G144 cells relative to the undifferentiated cells may reflect, in part, differences in the subcellular distribution of Akt isoforms. However, there are many other potential links between Akt and JCV replication. Akt and T-ag have regulators in common; for example, Akt is activated by Cdk2/cyclin A (85), a known regulator of T-ag (reviewed in reference 86). In addition, cellular proteins bound to the NCCR (reviewed in reference 2), such as NFI family members, may also be regulated by Akt. However, it is unlikely that Akt directly phosphorylates JCV T-ag, since consensus recognition sites for Akt (87) are not present in JCV T-ag (data not shown). Conversely, previous SV40-based studies (65, 66) suggested that JCV T-ag might also regulate Akt. Complex modes of Akt-dependent regulation of JCV DNA replication are also suggested by Akt-specific modulation of proteins in many additional replication-relevant pathways, including cell cycle progression (e.g., p21 and p27) (reviewed in reference 88), maintenance of translation (reviewed in reference 89), and apoptosis (reviewed in reference 90). However, G144 cells contain a mutant p53 protein (Pro152Leu) (S. Pollard, personal communication). This may abrogate or alter the usual interconnections between the p53 and Akt pathways (reviewed in reference 91). Finally, in addition to the Akt pathway, other key cellular signaling pathways are likely to be involved in the regulation of JCV DNA replication and other central stages during the JCV life cycle. Among the candidate pathways are those that were shown to change expression levels when G144 and other glioblastoma-derived neural stem cells were compared to normal neural stem cells (92). These pathways include factors having roles in adhesion and/or migration, differentiation, and apoptosis.
Future experiments with G144 cells will also address more practical issues. For example, since G144 cells support JCV DNA replication in 96-well plates (Fig. 4A), the cells may be suitable for high-throughput screening and may be able to substitute for the cervical carcinoma cell line C33A that was previously used to study JCV DNA replication (42). Finding an inhibitor that is selective for T-ag-dependent JCV DNA replication will be a significant step toward developing a treatment for PML and related diseases (40, 93–95).
In summary, we have identified an oligodendrocyte cell line that supports both JCV DNA replication and infection by JC virus. We also demonstrated that the G144 cell line can be used for studies of signaling pathways within oligodendrocytes that promote JCV DNA replication. In addition, JCV DNA replication in the G144 cell line is inhibited by MK2206, a selective inhibitor of Akt. Therefore, it is likely that the G144 cell line will be used to identify additional inhibitors of JCV DNA replication and viral infection. Finally, glioblastoma-derived neural stem cells, such as G144 cells, retain a diploid karyotype and can be genetically modified (96). Thus, in addition to studies of the JCV life cycle, there will be many additional applications for the G144 cell line, such as continued investigations of the molecular underpinnings of glioblastomas (92).
MATERIALS AND METHODS
Plasmids.
Several of the plasmids used in these studies were previously described (38, 42, 43). They include (i) a plasmid that expresses JCV T-ag (pCMV-JCT-ag), (ii) a plasmid that contains both the firefly luciferase (FL) gene and the noncoding control region (which includes the tandemly duplicated enhancer [enh]) from the Mad-1 JCV strain (97) (pFL-JCVori-Mad1enh), (iii) a plasmid that expresses Renilla luciferase (Rluc) (pRL), and (iv) a plasmid that is used as a carrier (pCI). For newly generated plasmids, the QuikChange II kit (Agilent Technologies) was used to introduce DNA mutations into the Mad-1 enhancer at identical nfi a and b sites in pFL-JCVori-Mad1enh. The oligonucleotide used to mutate the nfi-a and b sites (98) was 5′-CCCTAGGTATGAGCTCATGCTCAGCTGGCAGTAATCCCTTCCC-3′ (the complementary oligonucleotide is not listed); the mutated bases in the nfi-a and b sites are underlined (54). Finally, upon purification using a Qiagen maxiprep kit, plasmid DNA sequences were confirmed by DNA sequencing.
Cell culture.
G144 cells were maintained in Corning T75 flasks at 37°C and 5% CO2. Serum-free Dulbecco's modified Eagle's medium (DMEM), which was previously described (26), contained laminin (1.5 μg/ml) and EGF and FGF2 (PeproTech; both used at a final concentration of 20 pg/μl). When passaging the cells, accutase (Gibco; StemPro) was used at 1.5 ml/flask. To differentiate the G144 cells, EGF and FGF-2 were withheld from the growth medium for 7 days (26). Differentiated and undifferentiated G144 cells were also grown in chamber slides coated with laminin (Corning; 354688), using the same medium and growth conditions. The human glioma cell line Hs683 (99) was grown in DMEM in the presence of 10% fetal bovine serum (FBS).
Infection of G144 cells with JC virus.
Differentiated G144 cells were grown on chamber slides until ∼75% confluent. The cells were then infected with the Mad-4 strain of JCV (ATCC VR-1583) using the protocol provided by the ATCC. In brief, the medium was removed and replaced for 2 h with 125 μl of infection medium (i.e., growth factor- and laminin-free medium) containing JC virus (∼0.1 × 106 tissue culture infectious doses). In the control reactions, the medium was replaced with infection medium lacking JC virus. During the incubation period, the cells were kept at 37°C in a 5% CO2 atmosphere and rocked every 20 min to redistribute the inoculum. The incubation was ended upon the addition of 250 μl of differentiation medium.
Characterization of stage-specific mRNA molecules via qRT-PCR.
RNA was isolated from G144 cells using a Zymo Quick-RNA Microprep kit, following the manufacturer's protocol, and reverse transcribed using Clontech PrimeScript RT master mix. Real-time PCRs were performed using the RT2 SYBR green qPCR kit from Qiagen and the following primer pairs: Neu1, Fwd (GGACCGCTGAGCTATTGGG) and Rev (CGGGATGCGGAAAGTGTCTA); CSPG4, Fwd (GGGCTGTGCTGTCTGTTGA) and Rev (TGATTCCCTTCAGGTAAGGCA); Mal, Fwd (TTTGTGAGTTTGTCTTTGGAGGC) and Rev (CCGCCATGAGTACCAATTATGT); Foxo4, Fwd (CTTCCTCGACCAGACCTCG) and Rev (ACAGGATCGGTTCGGAGTGT); MBP, Fwd (GGCGGTGACAGACTCCAAG) and Rev (GAAGCTCGTCGGACTCTGAG); and finally, Foxo1, Fwd (CCCAGGCCGGAGTTTAACC) and Rev (GTTGCTCATAAAGTCGGTGCT).
Fluc-based DNA replication assays.
The Fluc-based JCV DNA replication assays were conducted largely as described previously (42, 43). However, with G144 cells, the plasmids needed for DNA replication were introduced by nucleofection (using the Lonza mouse neural stem cell nucleofector kit and an Amaxa Nucleofector II [set at X005]). Firefly and Renilla luciferase activities were measured at the indicated times using the Dual-Glo luciferase assay system and a Glomax Multi microplate reader (Promega). After normalizing to the 72-h wt-T-ag-containing sample, levels of JCV DNA replication were measured by determining the ratio of Fluc to Rluc.
qPCR-based measurements of DNA amplification. (i) Plasmid-based DNA replication.
Following nucleofection of the replication plasmids into G144 cells, levels of JCV DNA replication were also measured by SYBR green-based qPCR. To conduct these experiments, plasmid DNA was isolated at the indicated time points using a Zymo Zyppy Plasmid MiniPrep kit. Input plasmid DNA was removed by DpnI and exonuclease III treatment, as previously described (43). The qPCRs were performed using the RT2 SYBR green qPCR kit from Qiagen. The primer pairs used in the assay were as follows: Fluc, Fwd (TCAAAGAGGCGAACTGTGTG) and Rev (GGTGTTGGAGCAAGATGGAT), and Rluc, Fwd (GATAACTGGTCCGCAGTGGT) and Rev (ACCAGATTTGCCTGATTTGC).
(ii) Amplification of the JCV genome following infection with JC virus.
Following infection with the JCV Mad-4 strain (see above) for the indicated lengths of time, the JCV genome was purified using a modified Hirt extraction protocol (72) and the Zymo Zyppy Plasmid MiniPrep kit. Amplification of the JCV genome was subsequently detected by additional SYBR green-based qPCRs. The JCV-specific primers, designed using the NCBI Primer-BLAST program, target the region of the viral genome encoding VP1. The sequences of the VP1-specific primer pair are Fwd, CAGAGCACAAGGCGTACCTA, and Rev, TTTGCAAAGTGGCCCAACAC.
Cell lysis and Western blotting.
G144 cells were washed with PBS and then lysed in Triton X-100 lysis buffer (1× PBS, 1% Triton X-100, 2 mM EDTA, 1 mM phenylmethylsulfonyl fluoride [PMSF] supplemented with a protease inhibitor cocktail [Sigma]). Cellular extracts (15 μg/lane) were separated by SDS-PAGE, employing 10% acrylamide gels. The separated proteins were then transferred to polyvinylidene difluoride (PVDF) membranes (Millipore). After overnight blocking with 5% dry milk in TBST (20 mM Tris [pH 7.5], 137 mM NaCl, and 0.1% Tween 20), the membranes were incubated with the appropriate primary antibodies. JCV T-ag was detected using the Pab 416 monoclonal antibody (1:1,000; Santa Cruz Biotechnology); the blot was developed using a horseradish peroxidase (HRP)-conjugated rat anti-mouse secondary antibody (BD Pharmingen) and an enhanced chemiluminescence detection kit (Millipore). Members of the NFI family (reviewed in reference 53) of transcription factors (i.e., NFI-A, -B, -C, and -X) were detected using rabbit polyclonal antibodies from Abcam (ab104967, ab11989, ab86570, and ab126899, respectively), an HRP-conjugated goat anti-rabbit secondary Ab (Santa Cruz), and the enhanced chemiluminescence detection kit. As a loading control, vinculin levels were determined using the HVIN-1 MAb (Sigma) and an HRP-conjugated anti-mouse secondary antibody. Activated Akt was detected using a phospho-Akt (Ser473) rabbit MAb from Cell Signaling Technology (CST) (no. 4060). When using this MAb, 5% bovine serum albumin (BSA) was used as the blocking agent and the PhosStop phosphatase inhibitor cocktail (Roche) was added to the G144 lysates. The pan-Akt MAb was also obtained from CST (no. 4691). Finally, the construct used to express the myristoylated-Akt (68) had an N-terminal hemagglutinin (HA) tag; therefore, it was detected using an anti-HA antibody from Santa Cruz Biotechnology (sc-7392).
Immunofluorescence.
The protocols used to detect cell surface markers were based on the manufacturers' instructions (R&D). In summary, G144 cells grown on chamber slides were fixed with 4% paraformaldehyde, washed with PBS, permeabilized with ice-cold methanol, washed again, and blocked with normal donkey block (NDB) (100) for 1 h before overnight incubation in primary antibody (anti-O1 [1:100; R&D Systems; MAb 1327], anti-O4 [1:250; R&D Systems; MAb 1326], and anti-MBP [1:250; Novus; MAb 42282]) at 4°C. After removal of the primary Ab and washing with PBS, the cells were incubated with an anti-mouse secondary Ab conjugated to either Cy3 or Alexa 488 for 1 h at room temperature. After counterstaining with DAPI and coverslipping with n-propyl gallate, the samples were visualized with a Nikon E800 epifluorescence microscope or a Zeiss LSM800 confocal microscope.
The subcellular localization within G144 cells of JCV-encoded T-ag and VP1 were determined by standard immunofluorescence techniques (references 42 and 101 and references therein). JCV T-ag was detected using the Pab 416 MAb (1:1,000; Santa Cruz Biotechnology; SC-53448) and an anti-mouse secondary Ab conjugated with Cy3 or Alexa 488. The JCV coat protein VP1 was detected using the Pab 597 anti-VP1 MAb (a gift from W. Atwood) and an anti-mouse secondary Ab conjugated to Cy3. The cells were visualized using a Nikon E800 epifluorescence microscope.
Molecular modeling.
An image of the JCV T-ag helicase domain based on a threaded model of the SV40 helicase domain (Protein Data Bank [PDB] code 4GDF) was generated using the program PyMOL (102) (note that the recently solved structure of the JCV helicase domain [40] was not used because it did not include electron density for residue H514).
Statistical analyses.
Data were analyzed and graphed using GraphPad Prism software with appropriate statistical tests applied as described in the figure legends. Statistical significance was determined by two-way analysis of variance (ANOVA), followed by Tukey's multiple-comparison test or Student's t test.
ACKNOWLEDGMENTS
This work was supported by funding from Tufts University to P.A.B. and by NIH grants R01 DC002167 (J.E.S.) and F31 DC014637 (B.L.).
We thank Steve Pollard for providing the G144 cell line, Vivien Grant for advice on its growth, and Walter Atwood for the Pab 597 MAb. We also thank Jacques Archambault for the Abcam antibodies directed against the NFI family members and Ole Gjoerup for plasmid pcDNA Myr-HA-Akt1 and for helpful discussions.
REFERENCES
- 1.Tan CS, Koralnik IJ. 2010. Beyond progressive multifocal leukoencephalopathy: expanded pathogenesis of JC virus infection in the central nervous system. Lancet Neurol 9:425–437. doi: 10.1016/S1474-4422(10)70040-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ferenczy MW, Marshall LJ, Nelson CDS, Atwood WJ, Nath A, Khalili K, Major EO. 2012. Molecular biology, epidemiology, and pathogenesis of progressive multifocal leukoencephalopathy, the JC virus-induced deymelinating disease of the human brain. Clin Microbiol Rev 25:471–506. doi: 10.1128/CMR.05031-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brew BJ, Davies NWS, Cinque P, Clifford DB, Nath A. 2010. Progressive multifocal leukoencephalopathy and other forms of JC virus disease. Nat Rev Neurol 6:667–679. doi: 10.1038/nrneurol.2010.164. [DOI] [PubMed] [Google Scholar]
- 4.Bellizzi A, Anzivino E, Rodio DM, Palamara AT, Nencioni L, Pietropaolo V. 2013. New insights on human polyomavirus JC and pathogenesis of progressive multifocal leukoencephalopathy. Clin Dev Immunol 2013:839719. doi: 10.1155/2013/839719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gheuens S, Wuthrich C, Koralnik IJ. 2013. Progressive multifocal leukoencephalopathy: why gray and white matter. Annu Rev Pathol 8:189–215. doi: 10.1146/annurev-pathol-020712-164018. [DOI] [PubMed] [Google Scholar]
- 6.Langer-Gould A, Atlas SW, Green AJ, Bollen AW, Pelletier D. 2005. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N Engl J Med 353:375–381. doi: 10.1056/NEJMoa051847. [DOI] [PubMed] [Google Scholar]
- 7.Kleinschmidt-DeMasters BK, Tyler KL. 2005. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. N Engl J Med 353:369–374. doi: 10.1056/NEJMoa051782. [DOI] [PubMed] [Google Scholar]
- 8.Berger JR, Kaszovitz B, Post MJ, Dickinson G. 1987. Progressive multifocal leukoencephalopathy associated with human immunodeficiency virus infection. A review of the literature with a report of sixteen cases. Ann Intern Med 107:78–87. [DOI] [PubMed] [Google Scholar]
- 9.Maginnis MS, Atwood WJ. 2009. JC virus: an oncogenic virus in animals and humans? Semin Cancer Biol 19:261–269. doi: 10.1016/j.semcancer.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Del Valle L, White MK, Khalili K. 2008. Potential mechanisms of the human polyomavirus JC in neural oncogenesis. J Neuropathol Exp Neurol 67:729–740. doi: 10.1097/NEN.0b013e318180e631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Valeo T. 2014. A new model of the pathology of the JC virus. Neurol Today 18:1–6. [Google Scholar]
- 12.Barth H, Solis M, Kack W, Soulier E, Velay A, Fafi-Kremer S. 2016. In vitro and in vivo models for the study of human polyomavirus infection. Viruses 8:E292. doi: 10.3390/v8100292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Imperiale MJ, Major EO. 2007. Polyomaviruses, In Knipe DM, Howley PM (ed), Fields virology, 5th ed, vol 2, p 2263–2298. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 14.Miskin DP, Koralnik IJ. 2015. Novel syndromes associated with JC virus infection of neurons and meningeal cells: no longer a gray area. Curr Opin Neurol 28:288–294. doi: 10.1097/WCO.0000000000000201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schaumburg C, O'Hara BA, Lane TE, Atwood WJ. 2008. Human embryonic stem cell-derived oligodendrocyte progenitor cells express the Serotonin receptor and are susceptible to JC virus infection. J Virol 82:8896–8899. doi: 10.1128/JVI.00406-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Padgett BL, Zurhein GM, Walker DL, Eckroade RJ, Dessel BH. 1971. Cultivation of papova-like virus from human brain with progressive multifocal leukoencephalopathy. Lancet 297:1257–1260. doi: 10.1016/S0140-6736(71)91777-6. [DOI] [PubMed] [Google Scholar]
- 17.Padgett BL, Rogers CM, Walker DL. 1977. JC virus, a human polyomavirus associated with progressive multifocal leukoencephalopathy: additional biological characteristics and antigenic relationships. Infect Immunol 15:656–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Major EO, Miller AE, Mourrain P, Traub RG, de Widt E, Sever J. 1985. Establishment of a line of human fetal glial cells that supports JC virus multiplication. Proc Natl Acad Sci U S A 82:1257–1261. doi: 10.1073/pnas.82.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mandl C, Walker DL, Frisque RJ. 1987. Derivation and characterization of POJ cells, transformed human fetal glial cells that retain their permissivity for JC virus. J Virol 61:755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miyamura T, Yoshiike K, Takemoto KK. 1980. Characterization of JC papovavirus adapted to growth in human embryonic kidney cells. J Virol 35:498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferenczy MW, Johnson KR, Marshall LJ, Monaco MC, Major EO. 2013. Differentiation of human fetal multipotential neural progenitor cells to astrocytes reveals susceptibility factors for JC virus. J Virol 87:6221–6231. doi: 10.1128/JVI.00396-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Messam CA, Hou J, Gronostajski RM, Major EO. 2003. Lineage pathway of human brain progenitor cells identified by JC virus susceptibility. Ann Neurol 53:636–646. doi: 10.1002/ana.10523. [DOI] [PubMed] [Google Scholar]
- 23.Major EO, Vacante DA. 1989. Human fetal astrocytes in culture support the growth of the neurotropic human polyomavirus, JCV. Exp Neurol 48:425–436. [DOI] [PubMed] [Google Scholar]
- 24.Kondo Y, Windrem MS, Zou L, Chandler-Militello D, Schanz SJ, Auvergne RM, Betstadt SJ, Harrington AR, Johnson M, Kazarov A, Gorelik L, Goldman SA. 2014. Human glial chimeric mice reveal astrocytic dependence of JC virus infection. J Clin Invest 124:5323–5336. doi: 10.1172/JCI76629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seth P, Diaz F, Ta Cheng JH, Major EO. 2004. JC virus induces nonapoptotic cell death of human central nervous system progenitor cell-derived astrocytes. J Virol 78:4884–4891. doi: 10.1128/JVI.78.9.4884-4891.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pollard SM, Yoshikawa K, Clarke ID, Danovi D, Stricker S, Russell R, Bayani J, Head R, Lee M, Bernstein M, Squire JA, Smith A, Dirks P. 2009. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 4:568–580. doi: 10.1016/j.stem.2009.03.014. [DOI] [PubMed] [Google Scholar]
- 27.Caren H, Stricker SH, Bulstrode H, Gagrica S, Johnstone E, Bartlett TE, Feber A, Wilson G, Teschendorff AE, Bertone P, Beck S, Pollard SM. 2015. Glioblastoma stem cells respond to differentiation cues but fail to undergo commitment and terminal cell-cycle arrest. Stem Cell Reports 5:829–842. doi: 10.1016/j.stemcr.2015.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goldman SA, Kuypers NJ. 2015. How to make an oligodendrocyte. Development 142:3983–3995. doi: 10.1242/dev.126409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang SC. 2001. Defining glial cells during CNS development. Nat Rev Neurosci 2:840–843. doi: 10.1038/35097593. [DOI] [PubMed] [Google Scholar]
- 30.Sommer I, Schachner M. 1981. Monoclonal antibodies (O1 to O4) to oligodendrocyte cell surfaces: an immunocytological study in the central nervous system. Dev Biol 83:311–327. doi: 10.1016/0012-1606(81)90477-2. [DOI] [PubMed] [Google Scholar]
- 31.Kamholz J, DeFerra F, Puckett C, Lazzarini RA. 1986. Identification of three forms of human myelin basic protein by cDNA cloning. Proc Natl Acad Sci U S A 83:4962–4966. doi: 10.1073/pnas.83.13.4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pedraza L, Fidler L, Staugaitis SM, Colman DR. 1997. The active transport of myelin basic protein into the nucleus suggests a regulatory role in myelination. Neuron 18:579–589. doi: 10.1016/S0896-6273(00)80299-8. [DOI] [PubMed] [Google Scholar]
- 33.An P, Saenz Robles MT, Pipas JM. 2012. Large T antigens of polyomaviruses: amazing molecular machines. Annu Rev Microbiol 66:213–236. doi: 10.1146/annurev-micro-092611-150154. [DOI] [PubMed] [Google Scholar]
- 34.Topalis D, Andrei G, Snoeck R. 2013. The large tumor antigen: a “Swiss army knife” protein possessing the functions required for the polyomavirus life cycle. Antiviral Res 97:122–136. doi: 10.1016/j.antiviral.2012.11.007. [DOI] [PubMed] [Google Scholar]
- 35.Bullock PA. 1997. The Initiation of simian virus 40 DNA replication in vitro. Crit Rev Biochem Mol Biol 32:503–568. doi: 10.3109/10409239709082001. [DOI] [PubMed] [Google Scholar]
- 36.Borowiec JA, Dean FB, Bullock PA, Hurwitz J. 1990. Binding and unwinding—how T antigen engages the SV40 origin of DNA replication. Cell 60:181–184. doi: 10.1016/0092-8674(90)90730-3. [DOI] [PubMed] [Google Scholar]
- 37.Fanning E, Zhao K. 2009. SV40 DNA replication: from the A gene to a nanomachine. Virology 384:352–359. doi: 10.1016/j.virol.2008.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meinke G, Phelan PJ, Kalekar R, Shin J, Bohm A, Bullock PA. 2014. Insights into the initiation of JC virus DNA replication derived from the crystal structure of the T-antigen origin binding domain. PLoS Pathog 10:e1003966. doi: 10.1371/journal.ppat.1003966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meinke G, Phelan PJ, Shin J, Gagnon D, Archambault J, Bohm A, Bullock PA. 2016. Structural based analyses of the JCV T-antigen F258L mutant provides evidence for DNA dependent conformational changes in the C-termini of polyomavirus origin binding domains. PLoS Pathog 12:e1005362. doi: 10.1371/journal.ppat.1005362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bonafoux D, Nanthakumar S, Bandarage UK, Memmott C, Lowe D, Aronov AM, Bhisetti GR, Bonanno KC, Coll J, Leeman J, Lepre CA, Lu F, Perola E, Rijnbrand R, Taylor WP, Wilson D, Zhou Y, Zwahlen J, ter Haar E. 2016. Fragment-based discovery of dual JC virus and BK virus helicase inhibitors. J Med Chem 59:7138–7151. doi: 10.1021/acs.jmedchem.6b00486. [DOI] [PubMed] [Google Scholar]
- 41.Stoner GL, Ryschkewitsch CF, Walker DL, Webster HD. 1986. JC papovavirus large tumor (T)-antigen expression in brain tissue of acquired immune deficiency syndrome (AIDS) and non-AIDS patients with progressive multifocal leukoencephalopathy. Proc Natl Acad Sci U S A 83:2271–2275. doi: 10.1073/pnas.83.7.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shin J, Phelan PJ, Chhum P, Bashkenova N, Yim S, Parker R, Gagnon D, Gjoerup O, Archambault J, Bullock PA. 2014. Analysis of JC virus DNA replication using a quantitative and high-throughput assay. Virology 468–470:113–125. doi: 10.1016/j.virol.2014.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fradet-Turcotte A, Morin G, Lehoux M, Bullock PA, Archambault J. 2010. Development of quantitative and high-throughput assays of polyomavirus and papillomavirus DNA replication. Virology 399:65–76. doi: 10.1016/j.virol.2009.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reese DK, Sreekumar KR, Bullock PA. 2004. Interactions required for binding of simian virus 40 T antigen to the viral origin and molecular modeling of initial assembly events. J Virol 78:2921–2934. doi: 10.1128/JVI.78.6.2921-2934.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumar A, Meinke G, Reese DK, Moine S, Phelan PJ, Fradet-Turcotte A, Archambault J, Bohm A, Bullock PA. 2007. Model for T-antigen-dependent melting of the simian virus 40 core origin based on studies of the interaction of the beta-hairpin with DNA. J Virol 81:4808–4818. doi: 10.1128/JVI.02451-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shen J, Gai D, Patrick A, Greenleaf WB, Chen XS. 2005. The roles of the residues on the channel β-hairpin and loop structures of simian virus 40 hexameric helicase. Proc Natl Acad Sci U S A 102:11248–11253. doi: 10.1073/pnas.0409646102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gai D, Wang D, Li S-X, Chen XS. 2016. The structure of SV40 large T hexameric helicase in complex with AT-rich origin DNA. eLife 5:e18129. doi: 10.7554/eLife.18129. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48.Gai D, Zhao R, Li D, Finkielstein CV, Chen XS. 2004. Mechanisms of conformational change for a replicative hexameric helicase of SV40 large tumor antigen. Cell 119:47–60. doi: 10.1016/j.cell.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 49.Li D, Zhao R, Lilyestrom W, Gai D, Zhang R, DeCaprio JA, Fanning E, Jochimiak A, Szakonyi G, Chen XS. 2003. Structure of the replicative helicase of the oncoprotein SV40 large tumour antigen. Nature 423:512–518. doi: 10.1038/nature01691. [DOI] [PubMed] [Google Scholar]
- 50.Meinke G, Bullock PA. 2012. Structural “snap-shots” of the initiation of SV40 replication, p 195–215. In Gaston K. (ed), Small DNA tumor viruses. Horizon Scientific Press, Norwich, United Kingdom. [Google Scholar]
- 51.Kenny S, Natarajan V, Strike D, Khoury G, Salzman NP. 1984. JC virus enhancer-promoter active in human brain cells. Science 226:1337–1339. doi: 10.1126/science.6095453. [DOI] [PubMed] [Google Scholar]
- 52.Trapp BD, Small JA, Pulley M, Khoury G, Scangos GA. 1988. Dysmyelination in transgenic mice containing JC virus early region. Ann Neurol 23:38–48. doi: 10.1002/ana.410230108. [DOI] [PubMed] [Google Scholar]
- 53.Gronostajski RM. 2000. Roles of the NFI/CTF gene family in transcription and development. Gene 249:31–45. doi: 10.1016/S0378-1119(00)00140-2. [DOI] [PubMed] [Google Scholar]
- 54.Ravichandran V, Sabath BF, Jensen PN, Houff SA, Major EO. 2006. Interactions between c-Jun, nuclear factor 1, and JC virus promoter sequences: implications for viral tropism. J Virol 80:10506–10513. doi: 10.1128/JVI.01355-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Amemiya K, Traub RG, Durham L, Major EO. 1989. Interaction of a nuclear factor-1-like protein with the regulatory region of the human polyomavirus JC virus. J Biol Chem 264:7025–7032. [PubMed] [Google Scholar]
- 56.Marshall LJ, Dunham L, Major EO. 2010. Transcription factor Spi-B binds unique sequences present in the tandem repeat promoter/enhancer of JC virus and supports viral activity. J Gen Virol 91:3042–3052. doi: 10.1099/vir.0.023184-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Monaco MC, Sabath BF, Durham LC, Major EO. 2001. JC virus multiplication in human hematopoietic progenitor cells requires the NF-1 class D transcription factor. J Virol 75:9687–9695. doi: 10.1128/JVI.75.20.9687-9695.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sock E, Wegner M, Grummt F. 1993. Large T-antigen and sequences within the regulatory region of JC virus both contribute to the features of JC virus DNA replication. Virology 197:537–548. doi: 10.1006/viro.1993.1627. [DOI] [PubMed] [Google Scholar]
- 59.Gil G, Smith JR, Goldstein JL, Slaugher CA, Orth K, Brown MS, Osborne TF. 1988. Multiple genes encode nuclear factor 1-like proteins that bind to the promoter for 3-hydroxy-3-methylglutaryl-coenzyme A reductase. Proc Natl Acad Sci U S A 85:8963–8967. doi: 10.1073/pnas.85.23.8963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Paonessa G, Gounari F, Frank R, Cortese R. 1988. Purification of a NF1-like DNA-binding protein from rat liver and cloning of the corresponding cDNA. EMBO J 7:3115–3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Santoro C, Mermod N, Andrews PC, Tjian R. 1988. A family of human CCAAT-box-binding proteins active in transcription and DNA replication: cloning and expression of multiple cDNAs. Nature 334:218–224. doi: 10.1038/334218a0. [DOI] [PubMed] [Google Scholar]
- 62.Engelman JA, Luo J, Cantley L. 2006. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet 7:606–619. [DOI] [PubMed] [Google Scholar]
- 63.Chen Y, Li X, Eswarakumar VP, Seger R, Lonai P. 2000. Fibroblast growth factor (FGF) signaling through PI 3-kinase and Akt/PKB is required for embryoid body differentiation. Oncogene 19:3750–3756. doi: 10.1038/sj.onc.1203726. [DOI] [PubMed] [Google Scholar]
- 64.Cooray S. 2004. The pivotal role of phosphatidylinositol 3-kinse-Akt signal transduction in virus survival. J Gen Virol 85:1065–1076. doi: 10.1099/vir.0.19771-0. [DOI] [PubMed] [Google Scholar]
- 65.Yu Y, Alwine JC. 2002. Human cytomegalovirus major immediate-early proteins and simian virus 40 large T antigen can inhibit apoptosis through activation of the phosphatidylinositide 3′-OH kinase pathway and the cellular kinase Akt. J Virol 76:3731–3738. doi: 10.1128/JVI.76.8.3731-3738.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yu Y, Alwine JC. 2008. Interactions between simian virus 40 large T antigen and insulin receptor substrate 1 is disrupted by the K1 mutation, resulting in the loss of large T antigen-mediated phosphorylation of Akt. J Virol 82:4521–4526. doi: 10.1128/JVI.02365-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hirsch HH, Yakhontova ML, Manzetti J. 2016. BK polyomavirus replication in renal tubular epithelial cells is inhibited by sirolimus, but activated by tacrolimus through a pathway involving FKBP-12. Am J Transplant 16:821–832. doi: 10.1111/ajt.13541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ramaswamy S, Nakamura N, Vazques F, Batt DB, Perera S, Roberts TM, Sellers WR. 1999. Regulation of G1 progression by the PtEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc Natl Acad Sci U S A 96:2110–2115. doi: 10.1073/pnas.96.5.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hirai H, Sootome H, Nakatauru Y, Miyama K, Taguchi S, Tsujioka K, Ueno Y, Hatch H, Majumder PK, Pan B-S, Kotani H. 2010. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther 9:1956–1967. doi: 10.1158/1535-7163.MCT-09-1012. [DOI] [PubMed] [Google Scholar]
- 70.Nicholson KM, Anderson NG. 2002. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal 14:381–395. doi: 10.1016/S0898-6568(01)00271-6. [DOI] [PubMed] [Google Scholar]
- 71.Carmichael GG. 2016. Gene regulation and quality control in murine polyomavirus infection. Viruses 8:E284. doi: 10.3390/v8100284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Arad U. 1998. Modified Hirt procedure for rapid purification of extrachromosomal DNA from mammalian cells. Biotechniques 24:760–762. [DOI] [PubMed] [Google Scholar]
- 73.Gilinger G, Alwine JC. 1993. Transcriptional activation by simian virus 40 large T antigen: requirements for simple promoter structures containing either TATA or initiator elements with variable upstream factor binding sites. J Virol 67:6682–6688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shivakumar CV, Das GC. 1994. Biochemical and mutational analysis of the polyomavirus core promoter: involvement of nuclear factor-1 in early promoter function. J Gen Virol 75:1281–1290. doi: 10.1099/0022-1317-75-6-1281. [DOI] [PubMed] [Google Scholar]
- 75.Liang B, Tikhanovich I, Nasheuer HP, Folk WR. 2012. Stimulation of BK virus DNA replication by NF1 family transcription factors. J Virol 86:3264–3275. doi: 10.1128/JVI.06369-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chong T, Apt D, Gloss B, Isa M, Bernard H. 1991. The enhancer of human papillomavirus type 16: binding sites for the ubiquitous transcription factors Oct 1, NFA, TEF-2, NF-1 and AP-1 participate in epithelial cell-specific expression. J Virol 65:5933–5943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hennighausen L, Fleckenstein B. 1986. Nuclear factor 1 interacts with five DNA elements in the promoter region of the human cytomegalovirus major immediate early gene. EMBO J 5:1367–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jeang KT, Rawlins DR, Rosenfeld PJ, Shero JH, Kelly TJ, Hayward GS. 1987. Multiple tandemly repeated binding sites for cellular nuclear factor 1 that surround the major immediate-early promoters of simian and human cytomegalovirus. J Virol 61:1559–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shaul Y, Ben-Levy R, De Medina T. 1986. The high affinity binding site for nuclear factor I next to the hepatitis B virus S gene promoter. EMBO J 5:1967–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.de Jong D, van der Vliet PC. 1999. Mechanism of DNA replication in eukaryotic cells: cellular host factors stimulating adenovirus DNA replication. Gene 236:1–12. doi: 10.1016/S0378-1119(99)00249-8. [DOI] [PubMed] [Google Scholar]
- 81.Lee LT, Tan-Un KC, Lin MC, Chow BK. 2005. Retinoic acid activates human secretin gene expression by Sp proteins and nuclear factor I in neuronal SH-SY5Y cells. J Neurochem 93:339–350. doi: 10.1111/j.1471-4159.2005.03018.x. [DOI] [PubMed] [Google Scholar]
- 82.Wong YW, Schulze C, Streichert T, Gronostajski RM, Schachner M, Tilling T. 2007. Gene expression analysis of nuclear factor I-A deficient mice indicates delayed brain maturation. Genome Biol 8:R72. doi: 10.1186/gb-2007-8-5-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Aoyama A, Tamura TA, Mikoshiba K. 1990. Regulation of brain-specific transcription of the mouse myelin basic protein gene: function of the NFI-binding site in the distal promoter. Biochem Biophys Res Commun 167:648–653. doi: 10.1016/0006-291X(90)92074-A. [DOI] [PubMed] [Google Scholar]
- 84.Tamura TA, Miura M, Ikenaka K, Mikoshiba K. 1988. Analysis of transcription control elements of the mouse myelin basic protein gene in HeLa cell extracts: demonstration of a strong NFI-binding motif in the upstream region. Nucleic Acids Res 16:11441–11459. doi: 10.1093/nar/16.24.11441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liu P, Begley M, Michowski W, Inuzuka H, Ginzberg M, Gao D, Tsou P, Gan W, Papa A, Kim BM, Wan L, Singh A, Zhai B, Yuan M, Wang Z, Gygi SP, Lee TH, Lu KP, Toker A, Pandolfi PP, Asara JM, Kirschner MW, Sicinski P, Cantley L, Wei W. 2014. Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature 508:541–545. doi: 10.1038/nature13079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fanning E. 1994. Control of SV40 DNA replication by protein phosphorylation: a model for cellular DNA replication? Trends Cell Biol 4:250–255. doi: 10.1016/0962-8924(94)90123-6. [DOI] [PubMed] [Google Scholar]
- 87.Rust HL, Thompson PR. 2011. Kinase consensus sequences: a breeding ground for crosstalk. ACS Chem Biol 6:881–892. doi: 10.1021/cb200171d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Manning BD, Cantley L. 2007. Akt/PKB signaling: navigating downstream. Cell 129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Buchkovich NJ, Yu Y, Zampieri CA, Alwine JC. 2008. The TORrid affairs of viruses: effects of mammalian DNA viruses on the PI3K-Akt-mTOR signalling pathway. Nat Rev Microbiol 6:266–275. doi: 10.1038/nrmicro1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chang F, Lee JT, Navolanic PM, Steelman LS, Shelton JG, Blalock WL, Franklin RA, McCrubrey JA. 2003. Involvement of PI3K/Akt pathway in cell cycle progression, apoptosis, and neoplastic transformation: a target for cancer chemotherapy. Leukemia 17:590–603. doi: 10.1038/sj.leu.2402824. [DOI] [PubMed] [Google Scholar]
- 91.Levine AJ, Feng Z, Mak TW, You H, Jin S. 2006. Coordination and communication between the p53 and IGF-1-Akt-TOR signal transduction pathways. Genes Dev 20:267–275. doi: 10.1101/gad.1363206. [DOI] [PubMed] [Google Scholar]
- 92.Engstrom PG, Tommei D, Stricker SH, Ender C, Pollard SM, Bertone P. 2012. Digital transcriptome profiling of normal and glioblastoma-derived neural stem cells identifies genes associated with patient survival. Genome Med 4:76–95. doi: 10.1186/gm377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pavlovic D, Patera AC, Nyberg F, Gerber M, Liu M. 2015. Progressive multifocal leukoencephalopathy: current treatment options and future perspectives. Ther Adv Neurol Disord 8:255–273. doi: 10.1177/1756285615602832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Brickelmaier M, Lugovskoy A, Kartikeyan R, Reviriego-Mendoza MM, Allaire N, Simon K, Frisque RJ, Gorelik L. 2009. Identification and characterization of mefloquine efficacy against JC virus in vitro. Antimicrob Agents Chemother 53:1840–1849. doi: 10.1128/AAC.01614-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Danovi D, Folarin AA, Baranowski B, Pollard SM. 2012. High content screening of defined chemical libraries using normal and glioma-derived neural stem cell lines. Methods Enzymol 506:311–329. doi: 10.1016/B978-0-12-391856-7.00040-8. [DOI] [PubMed] [Google Scholar]
- 96.Sun Y, Pollard S, Conti L, Toselli M, Biella G, Parkin G, Willatt L, Falk A, Cattaneo E, Smith A. 2008. Long-term tripotent differentiation capacitiy of human neural stem (NS) cells in adherent culture. Mol Cell Neurosci 38:245–258. doi: 10.1016/j.mcn.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 97.Frisque RJ, Bream GL, Cannella MT. 1984. Human polyomavirus JC virus genome. J Virol 51:458–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Amemiya K, Traub RG, Durham L, Major EO. 1992. Adjacent nuclear factor-1 and activator protein binding sites in the enhancer of the neurotropic JC virus. J Biol Chem 267:14204–14211. [PubMed] [Google Scholar]
- 99.Owens RB, Smith HS, Nelson-Rees WA, Springer EL. 1976. Epithelial cell cultures from normal and cancerous human tissues. J Natl Cancer Inst 56:843–849. doi: 10.1093/jnci/56.4.843. [DOI] [PubMed] [Google Scholar]
- 100.Schnittke N, Herrick DB, Lin B, Peterson J, Coleman JH, Packard AI, Jang W, Schwob JE. 2015. Transcription factor p63 controls the reserve status but not the stemness of horizontal basal cells in the olfactory epithelium. Proc Natl Acad Sci U S A 112:E5068–E5077. doi: 10.1073/pnas.1512272112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Boichuk S, Hu L, Hein J, Gjoerup OV. 2010. Multiple DNA damage signaling and repair pathways deregulated by simian virus 40 large T antigen. J Virol 84:8007–8020. doi: 10.1128/JVI.00334-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.DeLano WL. 2002. The PyMOL molecular graphics system. Delano Scientific, Palo Alto, CA. [Google Scholar]
- 103.Timur ZK, Demir SA, Marsching C, Sandhoff R, Seyrantepe V. 2015. Neuraminidase-1 contributes significantly to the degradation of neuronal B-series gangliosides but not to the bypass of the catabolic block in Tay-Sachs mouse models. Mol Genet Metab Rep 4:72–82. doi: 10.1016/j.ymgmr.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sakry D, Yigit H, Dimou L, Trotter J. 2015. Oligodendrocyte precursor cells synthesize neuromodulatory factors. PLoS One 10:e0127222. doi: 10.1371/journal.pone.0127222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Frank M. 2000. MAL, a proteolipid in glycosphingolipid enriched domains: functional implications in myelin and beyond. Prog Neurobiol 60:531–544. doi: 10.1016/S0301-0082(99)00039-8. [DOI] [PubMed] [Google Scholar]
- 106.Yang H, Zhao R, Yang H-Y, Lee M-H. 2005. Constitutively active FOXO4 inhibits Akt activity, regulates p27 Kip1 stability, and suppresses HER2-mediated tumorigenicity. Oncogene 24:1924–1935. doi: 10.1038/sj.onc.1208352. [DOI] [PubMed] [Google Scholar]
- 107.Deber CM, Reynolds SJ. 1991. Central nervous system myelin: structure, function and pathology. Clin Biochem 24:113–134. doi: 10.1016/0009-9120(91)90421-A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Inouye H, Kirschner DA. 1991. Folding and function of the myelin proteins from primary sequence data. J Neurosci Res 28:1–17. doi: 10.1002/jnr.490280102. [DOI] [PubMed] [Google Scholar]
- 109.Jablonska B, Scafidi J, Aguirre A, Vaccarino F, Nguyen V, Borok E, Horvath TL, Rowitch DH, Gallo V. 2012. Oligodendrocyte regeneration after neonatal hypoxia requires Fox1-Mediated p27 Kip1 expression. J Neurosci 32:14775–14793. doi: 10.1523/JNEUROSCI.2060-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fu Z, Tindall DJ. 2008. FOXOs, cancer and regulation of apoptosis. Oncogene 27:2312–2319. doi: 10.1038/onc.2008.24. [DOI] [PMC free article] [PubMed] [Google Scholar]