

ABSTRACT
Autophagy plays important roles in maintaining cellular homeostasis. It uses double- or multiple-membrane vesicles termed autophagosomes to remove protein aggregates and damaged organelles from the cytoplasm for recycling. Hepatitis C virus (HCV) has been shown to induce autophagy to enhance its own replication. Here we describe a procedure that combines membrane flotation and affinity chromatography for the purification of autophagosomes from cells that harbor an HCV subgenomic RNA replicon. The purified autophagosomes had double- or multiple-membrane structures with a diameter ranging from 200 nm to 600 nm. The analysis of proteins associated with HCV-induced autophagosomes by proteomics led to the identification of HCV nonstructural proteins as well as proteins involved in membrane trafficking. Notably, caveolin-1, caveolin-2, and annexin A2, which are proteins associated with lipid rafts, were also identified. The association of lipid rafts with HCV-induced autophagosomes was confirmed by Western blotting, immunofluorescence microscopy, and immunoelectron microscopy. Their association with autophagosomes was also confirmed in HCV-infected cells. The association of lipid rafts with autophagosomes was specific to HCV, as it was not detected in autophagosomes induced by nutrient starvation. Further analysis indicated that the autophagosomes purified from HCV replicon cells could mediate HCV RNA replication in a lipid raft-dependent manner, as the depletion of cholesterol, a major component of lipid rafts, from autophagosomes abolished HCV RNA replication. Our studies thus demonstrated that HCV could specifically induce the association of lipid rafts with autophagosomes for its RNA replication.
IMPORTANCE HCV can cause severe liver diseases, including cirrhosis and hepatocellular carcinoma, and is one of the most important human pathogens. Infection with HCV can lead to the reorganization of membrane structures in its host cells, including the induction of autophagosomes. In this study, we developed a procedure to purify HCV-induced autophagosomes and demonstrated that HCV could induce the localization of lipid rafts to autophagosomes to mediate its RNA replication. This finding provided important information for further understanding the life cycle of HCV and its interaction with the host cells.
KEYWORDS: HCV RNA replication, autophagosomes, autophagy, hepatitis C virus, lipid rafts, proteomics
INTRODUCTION
Hepatitis C virus (HCV) is an important human pathogen that can cause severe liver diseases, including cirrhosis and hepatocellular carcinoma. It chronically infects approximately 170 million people worldwide. HCV is an enveloped positive-stranded RNA virus that belongs to the Flaviviridae family. Its 9.6-kb genome encodes a polyprotein with a length of slightly more than 3,000 amino acids. This polyprotein is processed by viral and cellular proteases into structural and nonstructural proteins (1). The structural proteins are the core, E1, and E2 proteins, which form the viral particles. The nonstructural proteins p7, NS2, and NS5A are required for viral assembly and release. NS2 and NS3 are viral proteases, and NS3, NS4A, NS4B, NS5A, and NS5B also form a complex to mediate the replication of the HCV genomic RNA (2). Many of the HCV proteins also have other regulatory functions.
Lipid rafts are membrane microdomains that are enriched in sphingolipids and cholesterol (3, 4). They can serve as “rafts” for the transport of selected molecules, signal transduction (5), or protein sorting in cells (5, 6). They may also be involved in the attachment, entry, assembly, or egress of a variety of pathogens, including enveloped viruses (7–11). Lipid rafts have also been shown to be important for HCV RNA replication (12–14), and NA255, a lipophilic long-chain base compound that disrupts the de novo synthesis of sphingolipid, a major component of lipid rafts, prevents the association of HCV nonstructural proteins with lipid rafts and inhibits HCV RNA replication (15).
Autophagy (i.e., macroautophagy) is a catabolic process that is important for maintaining cellular homeostasis. This process involves the formation of double- or multiple-membrane structures, called autophagosomes, to sequester part of the cytoplasm that may contain damaged organelles and protein aggregates. Autophagosomes mature by fusing with lysosomes, in which the cargos of autophagosomes are degraded for recycling (16). It has been demonstrated that HCV can induce autophagy and transiently suppresses the maturation of autophagosomes in its host cells to enhance its replication (17).
To understand the nature of the autophagosomes induced by HCV, we developed a procedure to purify autophagosomes from Huh7 hepatoma cells that harbored an HCV subgenomic RNA replicon. Our subsequent proteomics studies revealed the association of HCV nonstructural proteins and lipid raft proteins with HCV-induced autophagosomes. Our further analysis indicated that HCV could specifically induce the association of lipid rafts with autophagosomes to mediate its RNA replication.
RESULTS
Purification of autophagosomes.
The microtubule-associated protein light chain 3 (LC3) is a cytosolic protein, but it is lipidated and becomes associated with autophagosomal membranes during autophagy (18). Hence, it is often used as a marker for autophagosomes. We had previously established an Huh7 hepatoma cell line that stably expressed the green fluorescent protein (GFP)-LC3 fusion protein (i.e., Huh7-GFP-LC3 cells) (19) and used this cell line to produce another stable cell line that contained the HCV genotype 1b subgenomic RNA replicon. This GFP-LC3-replicon cell line was termed GLR cells (20). To confirm the induction of autophagy by HCV, we performed fluorescence microscopy. In agreement with the previous report (20), few GFP-LC3 puncta (i.e., autophagosomes) could be detected in the parental Huh7-GFP-LC3 cells, but their number was significantly increased in GLR cells (Fig. 1A). This result was confirmed by Western blot analysis. As shown in Fig. 1B, GLR cells had increased levels of LC3-II (i.e., lipidated LC3) and GFP-LC3-II. Similarly, a stable HCV subgenomic RNA replicon cell line that did not express GFP-LC3 also had elevated levels of LC3-II. To purify autophagosomes, we enriched cellular membranes by membrane flotation using a discontinuous sucrose gradient, followed by affinity purification of autophagosomes using the an anti-GFP antibody. The HCV subgenomic RNA replicon cell that did not express GFP-LC3 was used as the negative control. The membrane flotation analysis indicated that the autophagosome-associated LC3-II and GFP-LC3-II (fraction 3) could be efficiently separated from their respective nonlipidated forms, LC3-I and GFP-LC3-I (Fig. 1C). NS5A also cofractionated mostly with the membrane fractions, as previously reported (13), whereas GAPDH (glyceraldehyde-3-phosphate dehydrogenase), a cytosolic protein, was detected predominantly in the cytosolic fractions (fractions 7 to 10) at the bottom of the sucrose gradient (Fig. 1C).
FIG 1.

Induction of autophagosomes by HCV and the membrane flotation assay. (A) Stable Huh7-GFP-LC3 cells with (right panel) or without (left panel) the HCV subgenomic RNA replicon were analyzed by fluorescence microscopy. Blue, DAPI staining of nuclei; green, GFP-LC3 puncta. Scale bar, 20 μm. (B) Huh7-GFP-LC3 cells (Huh7), GLR cells, and HCV subgenomic replicon cells without the expression of GFP-LC3 (Replicon) were lysed for Western blot analysis. The locations of LC3-I/II and GFP-LC3-I/II are marked. Actin served as the loading control. (C) Cell lysates of Huh7-GFP-LC3, GLR, and replicon cells were fractionated by membrane flotation in a discontinuous sucrose gradient. Individual fractions were analyzed by Western blotting.
Cellular membranes enriched in fraction 3 were isolated and subjected to affinity purification using the anti-GFP antibody. The purified autophagosomes were then negatively stained with uranyl acetate and examined by electron microscopy. As shown in Fig. 2A to C, double- or multiple-layered membrane vesicles with diameters of approximately 200 to 600 nm were detected when GLR cells were analyzed. These membrane vesicles, which were consistent with the morphology of autophagosomes, were not detected when Huh7-GFP-LC3 cells were analyzed, due to the low basal level of autophagosomes in these cells. They were also not detected in replicon cells that did not express GFP-LC3, ruling out the possibility that they were nonspecific membrane contaminants. These autophagosomes were also subjected to immunoelectron microscopy for the staining of HCV NS5A. The staining for NS5A was difficult, likely due to the protection of NS5A by membranes. However, NS5A could be detected if the membrane vesicles were broken (Fig. 2D). We also analyzed autophagosomes purified from Huh7-GFP-LC3 cells that were nutrient starved. The autophagosomes induced by nutrient starvation were larger and more uniform in size, with most of them having a diameter of >500 nm, and no multiple-membrane structures were detected (Fig. 2E and F).
FIG 2.

Negative staining of autophagosomes purified from cells. (A to C) Autophagosomes purified from GLR cells by membrane flotation followed by affinity purification were negatively stained using uranyl acetate for electron microscopy. (D) Representative example of autophagosomes that were purified from GLR cells and stained for NS5A by immunogold staining. Arrows highlight the gold particles that are located on the rim of the broken membrane. (E and F) Negative staining of autophagosomes purified from nutrient-starved Huh7-GFP-LC3 cells.
Proteomic analysis of autophagosomes reveals the association of HCV nonstructural proteins and lipid raft proteins.
The autophagosomes purified from GLR cells and Huh7-GFP-LC3 cells were solubilized and subjected to proteomics analysis for the characterization of their associated proteins. Replicon cells that did not express GFP-LC3 were also analyzed to serve as a negative control for the exclusion of nonspecific protein species that might be isolated by the purification procedures. As shown in Table S1 in the supplemental material, a total of 78 protein species and 103 protein species were identified for GLR cells and Huh7-GFP-LC3 cells, respectively, and among those proteins, 19 of them were unique to GLR cells, 58 of them were shared between GLR cells and Huh7-GFP-LC3 cells, and 45 of them were unique to Huh7-GFP-LC3 cells. The most notable proteins that were unique to GLR cells were HCV nonstructural proteins (Table 1). These proteins were not detected when replicon cells that did not express GFP-LC3 were used for the purification procedures, and thus it is unlikely that they were nonspecific contaminants. Other notable proteins were annexin A2, caveolin-1 (Cav-1), and caveolin-2 (Cav-2), which are proteins associated with lipid rafts and often used as the markers for lipid rafts. The detection of these lipid raft proteins suggested the possible association of lipid rafts with HCV-induced autophagosomes. The association of the HCV NS5A protein and Cav-1 with autophagosomes was confirmed by Western blot analysis of the purified autophagosomes (Fig. 3A). Interestingly, Cav-1 was not detected when autophagosomes purified from nutrient-starved Huh7-GFP-LC3 cells were analyzed by Western blotting (Fig. 3B) (see below).
TABLE 1.
Proteins associated with autophagosomes purified from GLR cellsa
| Gene | Protein | No. of: |
|
|---|---|---|---|
| Peptides | Unique peptides | ||
| HCV polyprotein gene | HCV polyprotein | 12 | 5 |
| ANXA2 | Annexin A2 | 5 | 5 |
| CAV1 | Caveolin-1 | 1 | 1 |
| CAV2 | Caveolin-2 | 1 | 1 |
The complete list of proteins that were identified by proteomics is shown in Table S1 in the supplemental material.
FIG 3.
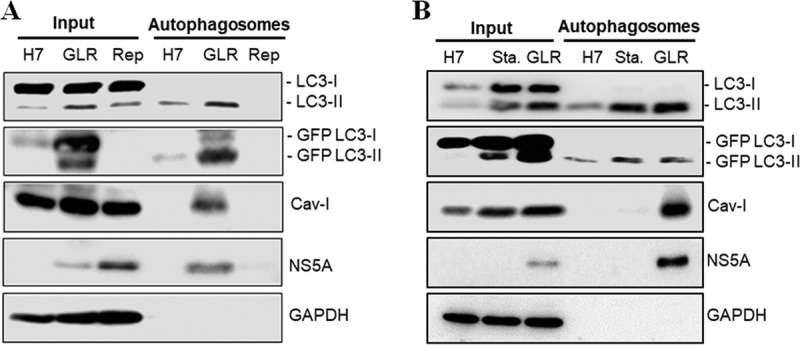
Western blot analysis of proteins associated with autophagosomes. (A) A fraction of total lysates of Huh7-GFP-LC3, GLR, and replicon cells was used as the input control for Western blot analysis. Autophagosomes purified by membrane flotation and the anti-GFP antibody were also analyzed. (B) The experiment was conducted as for panel A, with the exception that replicon cells were replaced by nutrient-starved Huh7-GFP-LC3 cells (Sta.) in the analysis.
Specific association of lipid rafts with HCV-induced autophagosomes.
To confirm the possible association of lipid rafts with HCV-induced autophagosomes, we performed immunofluorescence microscopy to analyze the subcellular localization of Cav-1 in Huh7-GFP-LC3 cells and GLR cells. As shown in Fig. 4A, significant amounts of Cav-1 (red) were associated with the plasma membranes of Huh7-GFP-LC3 cells, in agreement with a previous report (21). In contrast, in GLR cells, Cav-1 was detected primarily in the cytoplasm, and almost all of the GFP-LC3 puncta (i.e., autophagosomes) were also positive for Cav-1. Immunogold double-staining analysis using autophagosomes purified from GLR cells confirmed the colocalization of Cav-1 and GFP-LC3 on autophagosomes (Fig. 4B). To further confirm the association of lipid rafts with autophagosomes in GLR cells, we stained these cells with filipin, a fluorescent dye that binds to cholesterol, which is a major component of lipid rafts. As shown in Fig. 4C, cholesterol was localized primarily to plasma membranes in control Huh7-GFP-LC3 cells, but it was localized to the cytoplasm in GLR cells with a significant fraction of it associated with autophagic puncta, in agreement with the Cav-1 staining results.
FIG 4.

HCV-specific association of lipid rafts with autophagosomes. (A) Immunofluorescence microscopy of autophagosomes (GFP-LC3), Cav-1 (red), and nuclei (blue). White arrowheads denote the plasma membranes. (B) Immunogold staining of autophagosomes purified from HCV replicon cells. Red arrows denote Cav-1, which was stained with 10-nm gold particles, and blue arrows denote GFP-LC3, which was stained with 20-nm gold particles. (C) Staining of cholesterol with filipin in stable Huh7-GFP-LC3 cells and GLR cells. (D) Fluorescence imaging of GFP-LC3 and Cav-1 in nutrient-starved cells. Stable Huh7 cells that expressed GFP-LC3 without (upper panels) or with (lower panels) nutrient starvation were stained for Cav-1 (red). (E) Percentages of GFP-LC3 puncta that were positive for Cav-1 in starved Huh7 cells or GLR cells (i.e., yellow/green ratio). The results represent the average for >50 cells.
To determine whether the association of lipid rafts with autophagosomes was specific to HCV, we nutrient starved Huh7-GFP-LC3 cells for 1 h to induce autophagy. As shown in Fig. 4D, this nutrient starvation induced autophagic puncta. However, it had no significant effect on the subcellular localization of Cav-1, which remained largely on plasma membranes and did not colocalize with autophagosomes. The quantitative analysis of GFP-LC3 puncta that were positive for Cav-1 in starved cells and GLR cells revealed that fewer than 10% of GFP-LC3 puncta were positive for Cav-1 in starved cells, but this percentage was increased to approximately 90% in GLR cells (Fig. 4E). We also repeated the same experiment by treating Huh7-GFP-LC3 cells with rapamycin, which induces autophagy via the inhibition of mTOR. Cells were then stained with filipin for the identification of lipid rafts. As shown in Fig. 5, rapamycin indeed induced autophagy, as evidenced by the appearance of GFP-LC3 puncta. However, it had no effect on lipid rafts, which remained largely associated with plasma membranes and did not colocalize with autophagic puncta. These results indicated the specific association of lipid rafts with HCV-induced autophagosomes.
FIG 5.
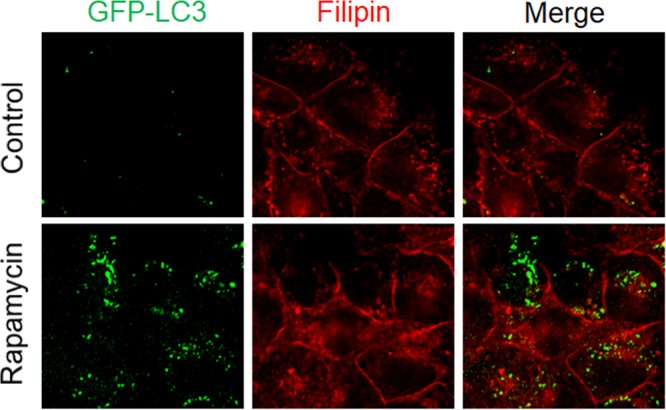
Lack of association of lipid rafts with autophagosomes induced by rapamycin. The experiments were conducted on Huh7-GFP-LC3 cells without (top panels) or with (bottom panels) treatment with 100 nM rapamycin for 12 h. Lipid rafts were stained with filipin, which binds to cholesterol.
Colocalization of lipid rafts with autophagosomes in HCV-infected cells.
To determine whether lipid rafts could also be associated with autophagosomes in HCV-infected cells, we infected stable GFP-LC3 cells with a cell culture-adapted HCV JFH1 variant (22). Cells were fixed at 24 and 48 h postinfection for fluorescence microscopy. As shown in Fig. 6A, the association of Cav-1 with the plasma membranes was apparent in mock-infected cells, in agreement with the results shown in Fig. 4A and D. However, Cav-1 became colocalized with GFP-LC3 puncta at 24 and 48 h postinfection. The quantitative analysis indicated that approximately 80% of the GFP-LC3 puncta were positive for Cav-1 at both 24 and 48 h postinfection (Fig. 6B). These data confirmed that lipid rafts were also associated with autophagosomes induced by HCV infection. As HCV JFH1 is a genotype 2a virus and GLR cells were established using the HCV genotype 1b RNA, these results also indicated that the association of lipid rafts with autophagosomes was independent of HCV genotypes. In agreement with the proteomics results, part of the HCV NS5A protein was also found to colocalize with GFP-LC3 puncta that were positive for Cav-1 (Fig. 6A).
FIG 6.

Colocalization of lipid rafts with autophagosomes in HCV-infected cells. (A) Fluorescence imaging of GFP-LC3, Cav-1, and HCV NS5A in mock-infected and HCV-infected cells. Stable Huh7 cells that expressed GFP-LC3 were infected with HCV (multiplicity of infection, 1), fixed at the time points indicated, and stained for Cav-1 (red) and NS5A (blue). (B) Percentages of GFP-LC3 puncta that were positive for Cav-1 (i.e., yellow/green ratio). The results represent the average from >50 cells that were analyzed.
Autophagosomes purified from HCV replicon cells mediate HCV RNA replication in vitro.
As lipid rafts are important for HCV RNA replication, the observation that lipid rafts and HCV nonstructural proteins were associated with autophagosomes would be consistent with our previous report that autophagosomes could mediate HCV RNA replication (20). To confirm this possibility, we performed the cell-free HCV RNA replication assay using whole-cell lysates and autophagosomes that were purified. As shown in Fig. 7A, the whole-cell lysates of GLR cells and replicon cells, but not those of the control Huh7-GFP-LC3 cells, could mediate the replication of HCV RNA, which was labeled with [α-32P]UTP. Autophagosomes purified from GLR cells could also mediate the replication of HCV RNA (Fig. 7B), and this replication was abolished by the HCV NS5B inhibitor filibuvir (Fig. 7C) (23). As expected, this HCV RNA replication was not detected when autophagosomes purified from Huh7-GFP-LC3 cells were used for the replication assay (Fig. 7B). It was also not detected when the replicon cells that did not express GFP-LC3 were used for the studies, ruling out the possibility of contamination of other cellular membranes.
FIG 7.
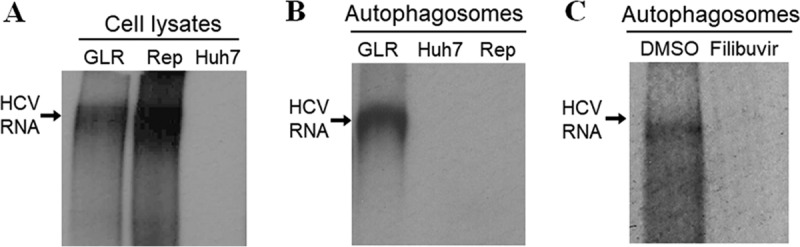
Autophagosomes purified from HCV replicon cells mediate HCV RNA replication in vitro. The in vitro HCV RNA replication assay was conducted using whole-cell lysates of GLR, replicon (Rep), and Huh7-GFP-LC3 (Huh7) cells (A), purified autophagosomes (B), or purified autophagosomes treated with the HCV NS5B inhibitor filibuvir (C). The location of the replicated HCV RNA is indicated with an arrow.
Depletion of cholesterol from autophagosomes abolishes HCV RNA replication.
To test whether the HCV RNA replication on autophagosomes required lipid rafts, we treated autophagosomes purified from GLR cells with methyl-β-cyclodextrin (MβCD), which removes cholesterol from membranes and disrupts lipid rafts. For a control, we treated autophagosomes with MβCD plus cholesterol. The inclusion of cholesterol, which forms a complex with MβCD, reduced the ability of MβCD to extract cholesterol from autophagosomes. As shown in Fig. 8A, MβCD efficiently depleted cholesterol from autophagosomes, and this depletion was inhibited if cholesterol was included in the treatment. MβCD had no significant effects on LC3-II, GFP-LC3-II, and NS5A that were associated with autophagosomes (Fig. 8B). It seemed to only slightly, if at all, reduce the level of Cav-1 (Fig. 8B). Similarly, the inclusion of cholesterol together with MβCD in the treatment also had little effect on LC3-II, GFP-LC3-II, NS5A, and Cav-1. We then performed another in vitro HCV RNA replication assay. As shown in Fig. 8C, the deletion of cholesterol from autophagosomes with MβCD abolished HCV RNA replication, and this effect of MβCD on HCV RNA replication was largely suppressed in the presence of free cholesterol. These results demonstrated that HCV-induced autophagosomes mediated HCV RNA replication in vitro in a cholesterol-dependent manner.
FIG 8.

Lipid rafts associated with autophagosomes are important for HCV RNA replication. Autophagosomes purified from GLR cells were untreated (Cont.) or treated with MβCD or MβCD plus cholesterol for 30 min at 37°C and then washed several times with buffer (see Materials and Methods for details). (A) Quantification of total cholesterol in autophagosomes by ELISA. (B) Western blot analysis for proteins that were associated with autophagosomes. (C) In vitro HCV RNA replication assay.
DISCUSSION
Previous studies indicated that HCV induced autophagy to enhance its replication (24). To further understand the relationship between HCV and autophagy, we developed a two-step procedure, which included a sucrose gradient centrifugation step to enrich cellular membranes (Fig. 1) and an affinity purification step to purify autophagosomes induced by HCV. Autophagosomes thus purified had double- or multiple-membrane structures with a diameter ranging from 200 nm to 600 nm (Fig. 2), resembling those of autophagosomes. Their nature was further confirmed by Western blot and immunogold analyses (Fig. 3 and 4B). We also conducted proteomics analysis on the proteins associated with HCV-induced autophagosomes and found that the autophagosomes induced by HCV shared many proteins with autophagosomes purified from the parental Huh7 cells. Many of these proteins, such as syntaxin-7, syntaxin-12, Rab5B, and N-ethylmaleimide-sensitive factor (NSF), are unique to HCV-induced autophagosomes and are known to be involved in membrane trafficking (see Table S1 in the supplemental material). It is interesting that nucleoporin NUP188 was found to be associated with autophagosomes in both Huh7-GFP-LC3 cells and GLR cells. Nucleoporins had previously been shown to be important for HCV RNA replication. Thus, the detection of NUP188 in purified autophagosomes would be consistent with its role in HCV RNA replication. It should be noted that many proteins were identified by our proteomics analysis, and although some of them had been confirmed to be important for HCV replication in previous reports (Table S1), additional studies will be required to confirm the roles of the other proteins in the biogenesis of autophagosomes and/or the life cycle of HCV.
By conducting Western blot and fluorescence microscopy analyses, we confirmed that HCV nonstructural proteins and the lipid raft protein Cav-1 were indeed associated with autophagosomes (Fig. 3 and 6). We also confirmed the association of lipid rafts with autophagosomes by using the fluorescence dye filipin to stain cholesterol, a critical component of lipid rafts (Fig. 4C). The association of lipid rafts with autophagosomes was specific to HCV, as this association was not detected when autophagosomes induced by nutrient starvation or rapamycin were analyzed (Fig. 4D and 5). How HCV induced the association of lipid rafts with autophagosomes is unclear. However, as the synthesis of cholesterol and sphingolipids initiates in the endoplasmic reticulum (ER), which can serve as the membrane source of autophagosomes (25, 26), it is conceivable that HCV-induced autophagosomes may directly recruit cholesterol and sphingolipids from the ER when these autophagosomes are being formed. Alternatively, as cholesterol and sphingolipids are enriched in plasma membranes, it is also possible that HCV-induced autophagosomes may acquire lipid rafts via the endocytic pathway followed by the fusion of endocytic membrane vesicles with autophagosomes (27). The latter possibility is supported by the previous observations that early endocytic compartments could fuse with endosomes and autophagosomes via a process mediated by Rab proteins such as Rab5 and soluble NSF attachment protein receptors (SNAREs), which include syntaxin-7 (28–30). In any case, this association of lipid rafts with autophagosomes led to their loss from the cell surface (Fig. 4A and C), which will likely have a significant impact on cellular physiology, as lipid rafts are known to mediate molecular transport, cellular signaling, and protein sorting (5, 6).
Lipid rafts have been shown to be important for HCV RNA replication (12–14), and their disruption in cell cultures abolished HCV RNA replication (31). Our finding that lipid rafts as well as HCV nonstructural proteins were associated with HCV-induced autophagosomes is in support of the previous finding that HCV RNA replication can take place on autophagosomes (20). Indeed, we demonstrated that the autophagosomes purified from GLR cells could mediate HCV RNA replication in vitro (Fig. 7) and that this RNA replication was abolished if lipid rafts associated with purified autophagosomes were disrupted with MβCD (Fig. 8C), which removed cholesterol from autophagosomes (Fig. 8A), and was largely restored if the activity of MβCD was suppressed by free cholesterol (Fig. 8C). These results indicate that lipid rafts play an essential role in HCV RNA replication. How lipid rafts affect HCV RNA replication is unclear. It does not appear likely that their loss leads to the dissociation of HCV nonstructural proteins from autophagosomes, as the depletion of cholesterol from autophagosomes had little effect on the association of HCV NS5A and LC3-II with these membrane vesicles (Fig. 8B). As lipid rafts can invaginate to form caveolae, it is possible that this structure may be important for protecting the integrity of the HCV RNA template, as previously suggested (13).
Previous studies had also suggested that the HCV RNA replication took place on membranous webs, which are clusters of poorly defined membrane structures (32). It was subsequently demonstrated that HCV RNA replication took place primarily on double-membrane vesicles (DMVs) approximately 200 nm in diameter (33–35). The morphology of DMVs resembled that of small autophagosomes (36), although the relationship between DMVs and autophagosomes remains unclear. It has been suggested that autophagosomes could be the source of DMVs (37). In this regard, it is interesting to note that Prentice et al. reported that the knockdown of ATG5, a key factor of autophagy, reduced the number of DMVs in coronavirus-infected cells and suppressed viral replication, indicating that autophagy was required for the formation of DMVs in coronavirus-infected cells (38). We had previously demonstrated that HCV temporally regulated the maturation of autophagosomes by inducing the expression of Rubicon, which suppressed the fusion between autophagosomes and lysosomes, at the early time points of infection (17). Huh7 cells containing HCV subgenomic RNA replicon also had an increased expression level of Rubicon and inefficient maturation of autophagosomes (17). Since HCV could use autophagosomes for its RNA replication, the inefficient maturation of autophagosomes during the early stage of viral infection would be beneficial to viral RNA replication. In the later stage of infection, the maturation of autophagosomes was facilitated due to the expression of UVRAG, which antagonizes the activity of Rubicon (17). It is possible that at this stage, either efficient viral RNA replication is no longer important for HCV or the locations of viral RNA replication had transitioned from autophagosomes to DMVs. It is interesting to note that although autophagic puncta in replicon cells and HCV-infected cells were mostly positive for Cav-1 (Fig. 4 and 6), Cav-1 did not always colocalize with autophagic puncta. It will be interesting to determine whether Cav-1 that is not associated with autophagosomes is associated with DMVs.
MATERIALS AND METHODS
Cell lines, nutrient starvation, and rapamycin treatment.
Huh7 cells are a human hepatoma cell line. They were maintained at 37°C in Dulbecco's modified essential medium (DMEM) supplemented with 10% fetal bovine serum (FBS). Huh7 cells that stably expressed the GFP-LC3 fusion protein have been described before (19). HCV subgenomic RNA replicon cells (i.e., GLR cells) that were established from Huh7-GFP-LC3 cells have also been described previously (20). This replicon contained the HCV NS3-NS5B sequence but not the preceding HCV protein-coding sequence. GLR cells were maintained in DMEM containing 150 μg/ml hygromycin B (20). Replicon cells were identical to GLR cells with the exception that they did not express the GFP-LC3 fusion protein. For nutrient starvation, cells were incubated in Hanks' balanced salt solution for 1 h prior to fixation in 4% formaldehyde for fluorescence imaging analysis. For rapamycin treatment, cells were treated with 100 nmol/liter rapamycin for 12 h for the induction of autophagy.
Membrane flotation and affinity purification of autophagosomes.
The membrane flotation assay was performed as previously described (38). Briefly, cells were seeded in three 100-mm plates, lysed with 1 ml hypotonic buffer (10 mM Tris-HCl [pH 7.5], 10 mM KCl, 5 mM MgCl2), and passed through a 26-guage needle 20 times. Nuclei and other cell debris were removed by centrifugation at 1,000 × g for 5 min at 4°C. Cell lysates were pooled and mixed with 4 ml of 80% sucrose solution in low-salt buffer (LSB) (50 mM Tris-HCl [pH 7.5], 25 mM KCl, and 5 mM MgCl2) and overlaid with 4 ml of 55% sucrose solution, followed by the addition of 1.5 ml 10% sucrose solution in LSB. The sucrose gradient was centrifuged at 38,000 rpm in a Beckman SW40Ti rotor for 15 h at 4°C. After centrifugation, 1-ml fractions were collected from the top of the gradient, and 20 μl of each fraction was subjected to Western blot analysis. For the affinity purification, the membrane fraction was incubated with 50 μl anti-GFP monoclonal antibody-conjugated magnetic beads (MBL) for 2 h at 4°C. The immune complex was then isolated with a magnetic separator and subjected to proteomics analysis, in vitro RNA replication assay, Western blot analysis, or electron microscopy.
Antibodies.
The primary antibodies used in this study included the rabbit anti-caveolin-1 antibody (Abcam), rabbit anti-LC3 antibody (Sigma), and mouse anti-HCV NS5A monoclonal antibody 9E10 (a gift from Charles Rice, Rockefeller University).
Western blot analysis.
Depending on the experiments, cells were lysed with M-PER mammalian protein extraction reagent (Thermo) or with a hypotonic buffer. After centrifugation to remove cell debris, cell lysates were subjected to SDS-PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane. The membrane was blocked with 5% skim milk for 1 h and incubated with the primary antibody overnight at 4°C. After three washes with Tris-buffered saline (TBS) (10 mM Tris-HCl [pH 7.5], 150 mM NaCl) containing 1% Tween 20 (TBST), the membrane was incubated with horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h. After further washes with TBST, chemiluminescent substrates (Pierce) were added to the membrane, and the image was captured using the LAS-4000 imaging system (Fujifilm).
Proteomics analysis.
After the enrichment of membranes by membrane flotation and subsequent affinity purification of autophagosomes, the protein concentration of the purified autophagosomes was determined by the Bradford method (Bio-Rad Laboratories). Proteins (15 μg) were then loaded on a 10% SDS-polyacrylamide gel for electrophoresis and stained with the SYPRO Ruby protein gel stain (Thermo Fisher). Each lane was cut into six slices to separate proteins of different sizes, and proteins in different gel slices were then subjected to proteomics analysis by mass spectrometry in the USC Proteomics Core facility.
In vitro HCV RNA replication assay.
Autophagosomes that were affinity purified using the anti-GFP-conjugated magnetic beads as described above were resuspended in a reaction mixture containing 50 mM HEPES, 5 mM MgCl2, 0.5 mM MnCl2, 10 mM KCl, 1 mM ATP, 1 mM GTP, 1 mM UTP, 10 mM CTP, 5 μg/ml actinomycin D, and 30 μCi of [α-32P]CTP while they were still associated with the beads. For the inhibition of HCV RNA replication, filibuvir at a final concentration of 100 nM was added to the reaction mixture. The reaction mixture was then incubated at 30°C for 90 min. The HCV RNA synthesized was then isolated using TRIzol (Invitrogen) following the manufacturer's instructions and analyzed by agarose gel electrophoresis and autoradiography.
Immunoelectron microscopy.
Autophagosomes that were affinity purified were eluted from the anti-GFP beads using 3 M guanidine-HCl and fixed with 2% paraformaldehyde. For negative staining, the fixed autophagosomes were mounted on a copper grid, followed by incubation with 5% uranyl acetate for 5 min. For immunogold staining, fixed autophagosomes were mounted on a nickel grid and incubated with rabbit anti-Cav-1 (Abcam) and mouse anti-GFP primary antibodies (Santa Cruz), followed by incubation with 10-nm gold-conjugated goat anti-rabbit and 20-nm-gold-conjugated goat anti-mouse secondary antibodies. For the staining of HCV NS5A, the anti-NS5A antibody (a gift of Charles Rice, Rockefeller University) was used, followed by staining with the 10-nm-gold-conjugated secondary antibody.
Immunofluorescence staining and microscopy.
Cells were rinsed with phosphate-buffered saline (PBS), fixed with 3.7% formaldehyde, permeabilized with PBS containing 0.1% saponin, 1% bovine serum albumin (BSA), and 0.05% sodium azide for 5 min, and then incubated with the rabbit anti-caveolin-1 antibody and then with the rhodamine-conjugated goat anti-rabbit antibody for immunofluorescence microscopy. Coverslips were mounted in Vector Shield (Vector) containing 4,6-diamidino-2-phenylindole (DAPI), which stained the nucleus. The staining of cholesterol was conducted using a cholesterol assay kit (Abcam 133116), which contained the fluorescence dye filipin, following the manufacturer's instructions. Images were acquired with a Keyence All-in-One fluorescence microscope. The colocalization coefficient, which measured the fraction of green fluorescent protein (GFP) pixels that were also positive for Cav-1 (red), was determined from randomly selected cells (>50) using the Keyence All-in-One software.
Depletion of cholesterol with MβCD.
For the depletion of cholesterol, autophagosomes affinity purified with the anti-GFP beads were treated with LSB, 15 mM MβCD (Sigma-Aldrich) in LSB, or 15 mM MβCD saturated with cholesterol (Sigma C4951) (8:1, mol/mol) at 37°C for 30 min (39). After several washes with LSB, autophagosomes were subjected to Western blotting, in vitro RNA replication, or cholesterol quantification.
Cholesterol quantification.
Total cholesterol in untreated, MβCD-treated, or MβCD-plus-cholesterol-treated autophagosomes was measured using the cholesterol/cholesteryl ester quantitation assay kit (Abcam 65359) following the manufacturer's instructions.
Supplementary Material
ACKNOWLEDGMENTS
We thank Ebrahim Zandi and Yu Zhou of the USC Proteomics Core Facility for help with the proteomics analysis and the USC Tissue Imaging Core Facility for help with electron microscopy.
This work was supported by NIH grant DK094652.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.00541-17.
REFERENCES
- 1.Moradpour D, Penin F, Rice CM. 2007. Replication of hepatitis C virus. Nat Rev Microbiol 5:453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 2.Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 3.van der Goot FG, Harder T. 2001. Raft membrane domains: from a liquid-ordered membrane phase to a site of pathogen attack. Semin Immunol 13:89–97. doi: 10.1006/smim.2000.0300. [DOI] [PubMed] [Google Scholar]
- 4.Harder T, Simons K. 1997. Caveolae, DIGs, and the dynamics of sphingolipid-cholesterol microdomains. Curr Opin Cell Biol 9:534–542. doi: 10.1016/S0955-0674(97)80030-0. [DOI] [PubMed] [Google Scholar]
- 5.Smart EJ, Graf GA, McNiven MA, Sessa WC, Engelman JA, Scherer PE, Okamoto T, Lisanti MP. 1999. Caveolins, liquid-ordered domains, and signal transduction. Mol Cell Biol 19:7289–7304. doi: 10.1128/MCB.19.11.7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lafont F, Verkade P, Galli T, Wimmer C, Louvard D, Simons K. 1999. Raft association of SNAP receptors acting in apical trafficking in Madin-Darby canine kidney cells. Proc Natl Acad Sci U S A 96:3734–3738. doi: 10.1073/pnas.96.7.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bavari S, Bosio CM, Wiegand E, Ruthel G, Will AB, Geisbert TW, Hevey M, Schmaljohn C, Schmaljohn A, Aman MJ. 2002. Lipid raft microdomains: a gateway for compartmentalized trafficking of Ebola and Marburg viruses. J Exp Med 195:593–602. doi: 10.1084/jem.20011500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manié SN, de Breyne S, Debreyne S, Vincent S, Gerlier D. 2000. Measles virus structural components are enriched into lipid raft microdomains: a potential cellular location for virus assembly. J Virol 74:305–311. doi: 10.1128/JVI.74.1.305-311.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nguyen DH, Hildreth JE. 2000. Evidence for budding of human immunodeficiency virus type 1 selectively from glycolipid-enriched membrane lipid rafts. J Virol 74:3264–3272. doi: 10.1128/JVI.74.7.3264-3272.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barman S, Nayak DP. 2007. Lipid raft disruption by cholesterol depletion enhances influenza A virus budding from MDCK cells. J Virol 81:12169–12178. doi: 10.1128/JVI.00835-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noisakran S, Dechtawewat T, Avirutnan P, Kinoshita T, Siripanyaphinyo U, Puttikhunt C, Kasinrerk W, Malasit P, Sittisombut N. 2008. Association of dengue virus NS1 protein with lipid rafts. J Gen Virol 89:2492–2500. doi: 10.1099/vir.0.83620-0. [DOI] [PubMed] [Google Scholar]
- 12.Shi ST, Lee KJ, Aizaki H, Hwang SB, Lai MM. 2003. Hepatitis C virus RNA replication occurs on a detergent-resistant membrane that cofractionates with caveolin-2. J Virol 77:4160–4168. doi: 10.1128/JVI.77.7.4160-4168.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aizaki H, Lee KJ, Sung VM, Ishiko H, Lai MM. 2004. Characterization of the hepatitis C virus RNA replication complex associated with lipid rafts. Virology 324:450–461. doi: 10.1016/j.virol.2004.03.034. [DOI] [PubMed] [Google Scholar]
- 14.Shanmugam S, Saravanabalaji D, Yi M. 2015. Detergent-resistant membrane association of NS2 and E2 during hepatitis C virus replication. J Virol 89:4562–4574. doi: 10.1128/JVI.00123-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sakamoto H, Okamoto K, Aoki M, Kato H, Katsume A, Ohta A, Tsukuda T, Shimma N, Aoki Y, Arisawa M, Kohara M, Sudoh M. 2005. Host sphingolipid biosynthesis as a target for hepatitis C virus therapy. Nat Chem Biol 1:333–337. doi: 10.1038/nchembio742. [DOI] [PubMed] [Google Scholar]
- 16.He C, Klionsky DJ. 2009. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang L, Tian Y, Ou JH. 2015. HCV induces the expression of Rubicon and UVRAG to temporally regulate the maturation of autophagosomes and viral replication. PLoS Pathog 11:e1004764. doi: 10.1371/journal.ppat.1004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, Noda T, Ohsumi Y. 2000. A ubiquitin-like system mediates protein lipidation. Nature 408:488–492. doi: 10.1038/35044114. [DOI] [PubMed] [Google Scholar]
- 19.Sir D, Chen WL, Choi J, Wakita T, Yen TS, Ou JH. 2008. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 48:1054–1061. doi: 10.1002/hep.22464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sir D, Kuo CF, Tian Y, Liu HM, Huang EJ, Jung JU, Machida K, Ou JH. 2012. Replication of hepatitis C virus RNA on autophagosomal membranes. J Biol Chem 287:18036–18043. doi: 10.1074/jbc.M111.320085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scherer PE, Okamoto T, Chun M, Nishimoto I, Lodish HF, Lisanti MP. 1996. Identification, sequence, and expression of caveolin-2 defines a caveolin gene family. Proc Natl Acad Sci U S A 93:131–135. doi: 10.1073/pnas.93.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang J, Luo G. 2012. Cell culture-adaptive mutations promote viral protein-protein interactions and morphogenesis of infectious hepatitis C virus. J Virol 86:8987–8997. doi: 10.1128/JVI.00004-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Troke PJ, Lewis M, Simpson P, Gore K, Hammond J, Craig C, Westby M. 2012. Characterization of resistance to the nonnucleoside NS5B inhibitor filibuvir in hepatitis C virus-infected patients. Antimicrob Agents Chemother 56:1331–1341. doi: 10.1128/AAC.05611-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang L, Ou JH. 2015. Hepatitis C virus and autophagy. Biol Chem 396:1215–1222. doi: 10.1515/hsz-2015-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, Akira S, Noda T, Yoshimori T. 2009. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 11:385–396. doi: 10.1038/ncb1846. [DOI] [PubMed] [Google Scholar]
- 26.Matsunaga K, Morita E, Saitoh T, Akira S, Ktistakis NT, Izumi T, Noda T, Yoshimori T. 2010. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol 190:511–521. doi: 10.1083/jcb.200911141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lamb CA, Dooley HC, Tooze SA. 2013. Endocytosis and autophagy: shared machinery for degradation. Bioessays 35:34–45. doi: 10.1002/bies.201200130. [DOI] [PubMed] [Google Scholar]
- 28.Risselada HJ, Grubmüller H. 2012. How SNARE molecules mediate membrane fusion: recent insights from molecular simulations. Curr Opin Struct Biol 22:187–196. doi: 10.1016/j.sbi.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 29.Zeigerer A, Gilleron J, Bogorad RL, Marsico G, Nonaka H, Seifert S, Epstein-Barash H, Kuchimanchi S, Peng CG, Ruda VM, Del Conte-Zerial P, Hengstler JG, Kalaidzidis Y, Koteliansky V, Zerial M. 2012. Rab5 is necessary for the biogenesis of the endolysosomal system in vivo. Nature 485:465–470. doi: 10.1038/nature11133. [DOI] [PubMed] [Google Scholar]
- 30.Ao X, Zou L, Wu Y. 2014. Regulation of autophagy by the Rab GTPase network. Cell Death Differ 21:348–358. doi: 10.1038/cdd.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang JT, Tseng CP, Liao MH, Lu SC, Yeh WZ, Sakamoto N, Chen CM, Cheng JC. 2013. Hepatitis C virus replication is modulated by the interaction of nonstructural protein NS5B and fatty acid synthase. J Virol 87:4994–5004. doi: 10.1128/JVI.02526-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gosert R, Egger D, Lohmann V, Bartenschlager R, Blum HE, Bienz K, Moradpour D. 2003. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J Virol 77:5487–5492. doi: 10.1128/JVI.77.9.5487-5492.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferraris P, Blanchard E, Roingeard P. 2010. Ultrastructural and biochemical analyses of hepatitis C virus-associated host cell membranes. J Gen Virol 91:2230–2237. doi: 10.1099/vir.0.022186-0. [DOI] [PubMed] [Google Scholar]
- 34.Romero-Brey I, Merz A, Chiramel A, Lee JY, Chlanda P, Haselman U, Santarella-Mellwig R, Habermann A, Hoppe S, Kallis S, Walther P, Antony C, Krijnse-Locker J, Bartenschlager R. 2012. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog 8:e1003056. doi: 10.1371/journal.ppat.1003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paul D, Romero-Brey I, Gouttenoire J, Stoitsova S, Krijnse-Locker J, Moradpour D, Bartenschlager R. 2011. NS4B self-interaction through conserved C-terminal elements is required for the establishment of functional hepatitis C virus replication complexes. J Virol 85:6963–6976. doi: 10.1128/JVI.00502-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paul D, Hoppe S, Saher G, Krijnse-Locker J, Bartenschlager R. 2013. Morphological and biochemical characterization of the membranous hepatitis C virus replication compartment. J Virol 87:10612–10627. doi: 10.1128/JVI.01370-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blanchard E, Roingeard P. 2015. Virus-induced double-membrane vesicles. Cell Microbiol 17:45–50. doi: 10.1111/cmi.12372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prentice E, Jerome WG, Yoshimori T, Mizushima N, Denison MR. 2004. Coronavirus replication complex formation utilizes components of cellular autophagy. J Biol Chem 279:10136–10141. doi: 10.1074/jbc.M306124200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Christian AE, Haynes MP, Phillips MC, Rothblat GH. 1997. Use of cyclodextrins for manipulating cellular cholesterol content. J Lipid Res 38:2264–2272. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.