

Abstract
Drug resistant cancers like pancreatic ductal adenocarcinoma (PDAC) are difficult to treat, and nanoparticle drug delivery systems can overcome some of the limitations of conventional systemic chemotherapy. In this study, we demonstrate that FdUMP and dFdCMP, the bioactive, phosphorylated metabolites of the chemotherapy drugs 5-FU and gemcitabine, can be encapsulated into calcium phosphosilicate nanoparticles (CPSNPs). The non-phosphorylated drug analogs were not well encapsulated by CPSNPs, suggesting the phosphate modification is essential for effective encapsulation. In vitro proliferation assays, cell cycle analyses and/or thymidylate synthase inhibition assays verified that CPSNP-encapsulated phospho-drugs retained biological activity. Analysis of orthotopic tumors from mice treated systemically with tumor-targeted FdUMP-CPSNPs confirmed the in vivo up take of these particles by PDAC tumor cells and release of active drug cargos intracellularly. These findings demonstrate a novel methodology to efficiently encapsulate chemotherapeutic agents into the CPSNPs and to effectively deliver them to pancreatic tumor cells.
Keywords: FdUMP, 5-fluorouracil, gemcitabine, nanodelivery, dFdCMP
Graphical Abstract
By encapsulating chemotherapeutic agents into nanocarriers, such as calcium phosphosilicate nanoparticles (CPSNPs), metabolic inactivation of pro-drugs, for instance the inactivation of 5-FU by the liver enzyme dihydropyrimidine dehydrogenase (DPD), often can be avoided. Herein we demonstrate that the bioactive metabolite of 5-FU, FdUMP, can be effectively encapsulated into CPSNPs. Once taken up by tumor cells, the mPEG-FdUMP-CPSNPs dissolve in the late endosome to release the FdUMP cargo. In the presence of folate (CH2THF), FdUMP irreversibly binds to and inhibits thymidylate synthase (TS), resulting in a cellular depletion of dTMP, increased DNA damage, and ultimately cell death.
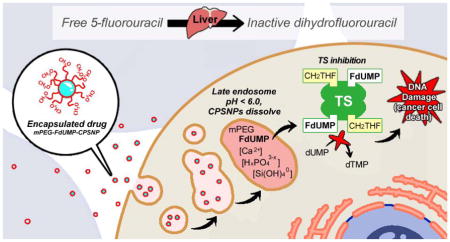
BACKGROUND
Applications of nanoparticle (NP) delivery systems to medical oncology include the use of NPs for encapsulated therapeutic drug delivery and for non-invasive tumor imaging. Due to the small size and versatile surface chemistry, NPs are ideal platforms to penetrate poorly vascularized and fibrotic tumors (1). Reports on NP-based delivery systems encompass a diversity of materials that include liposomes (2–4), dendrimers (5, 6), gold NPs (7), and quantum dots (8, 9). The design and composition of NPs reflect their abilities to load and protect agents in circulation and to provide a clearance mechanism for excess NPs (10).
Amorphous calcium phosphosilicate nanoparticles (CPSNPs) are composed of bioresorbable materials (10, 11) that have been used to deliver therapeutic (12–14) and imaging agents (13, 15–18) to tumor cells. The NP matrix provides protection of encapsulated bioactive agents (such as chemotherapeutic drugs) from metabolic breakdown and rapid clearance. Further, surface decoration of CPSNPs with polyethylene glycol (PEG) mitigates non-specific interactions that cause agglomeration and facilitates bioconjugation of tumor targeting agents such as peptides, antibodies or aptamers (14, 15, 17–20). After cellular uptake, the low pH of late-stage endosomes triggers CPSNP dissolution and the endosomes rupture from a change in osmotic pressure to release active agents into the cytosol (13, 18).
Treating pancreatic cancer with standard chemotherapeutic agents is challenging. Most pancreatic adenocarcinoma (PDAC) patients respond poorly to chemotherapeutic drugs, and even new drug combinations have demonstrated only a modest improvement in patient survival (21). This lack of efficacy has been attributed in part to inadequate drug delivery and metabolic drug inactivation (22). Two chemotherapeutics commonly used to treat PDAC, 5-fluorouracil (5-FU) and gemcitabine (dFdC), act by blocking key enzymes in nucleotide synthesis. 5-FU is metabolized to 5-fluorodeoxyuridine monophosphate (FdUMP) which, in the presence of 5,10-methylenetetrahydrofolate (CH2THF), irreversibly inhibits thymidylate synthase (TS) (23). TS inhibition results in nucleotide pool imbalances, impaired DNA synthesis, and a reduction in DNA repair (24). Similarly, the prodrug dFdC is phosphorylated intracellularly by deoxycytidine kinase to form gemcitabine monophosphate (dFdCMP), diphosphate (dFdCDP) and triphosphate (dFdCTP) (25). Gemcitabine has multiple modes of action; dFdCDP inhibits ribonucleotide reductase, which is responsible for producing the deoxynucleotides required for DNA synthesis and repair. This favors incorporation of dFdCTP into DNA, resulting in stalled DNA replication forks and apoptosis (26). While both 5-FU and dFdC are activated within tumor cells by conversion to phosphorylated metabolites, direct delivery of phosphorylated metabolites is limited by cell impermeability, lipid insolubility and precipitation.
Systemic administration of many chemotherapeutics is limited by the need for high dosing regimens due to metabolic inactivation and rapid clearance. For example, in vivo studies have shown that less than 20% of the 5-FU becomes activated to FdUMP while more than 80% of 5-FU is converted to the inactive 5-FU metabolite 5-fluorodihydrouracil by the liver enzyme dihydropyrimidine dehydrogenase (DPD) (27). Likewise, dFdC can be inactivated by cytidine deaminase and rapidly cleared from the body (28–30). Both dFdC and 5-FU are transported into tumor cells by nucleoside transporter systems, including human equilibrative nucleoside transporters (hENT), which have low affinity for FdUMP and dFdCMP (31). Tumor targeted, NP-based delivery of phosphorylated drug metabolites could avoid drug inactivation processes and enhance drug uptake by tumor cells. This study compares the encapsulation efficiency of both phosphorylated and non-phosphorylated forms of 5-FU and dFdC and outlines a novel approach to deliver the chemotherapeutics directly to pancreatic tumors using targeted CPSNPs.
METHODS
CPSNP Synthesis and Characterization
CPSNPs were prepared in reverse-micelles and isolated by HPLC as described by Barth et al. and Morgan et al. (13, 17, 18). The materials, synthetic procedure, and additional sample preparation methods are outlined in supplementary information. Citrate-functionalized (Cit) CPSNPs were preheated to 50°C and stirred at 550 rpm for PEGylation. To yield mPEG-CPSNPs, 1 ml of EDC (1 mg/ml) was first added drop-wise to 10 ml of Cit-CPSNPs. After 5 min, 1 ml of each Sulfo-NHS (15 mg/ml) and 2 kDa methoxy-PEG-amine (6 mg/ml) were transferred. The reaction proceeded for 15 hr. PEGylated CPSNPs were purified via filtration for 2–3 min at 5000 g (Amicon Ultra-15 30 kDa). Gastrin-16 peptide (G16, CSGGQQQQQQAYGWMDF, Genscript) was conjugated via 2 kDa maleimide-PEG-amine coupling (18). The peptide was added to the malPEG-CPSNPs after filtration and stored at 4°C overnight. The aptamer (AP, TriLink Biotechnologies) was conjugated via 2 kDa carboxy-PEG-amine coupling, first reported by Clawson et al. (19). A second EDC/NHS coupling reaction followed after filtration with an aliquot of 0.2 mM aptamer solution (5 ul per ml cPEG-CPSNPs).
Surface modification of CPSNPs was characterized by the Brookhaven Instruments zeta potential analyzer (ZetaPlus v. 3.23, Holtsville, NY). Particles were imaged on the FEI Tecnai G2 Spirit BioTWIN TEM operating at 120kV. The drug concentration in CPSNPs was quantified by LC-MS/MS (supplementary information). To calculate the payload, the total number of particles (Ntotal) was first determined on the Nanotrac Wave II DLS (Microtrac Flex 11.1.0.2, York, PA). Measurements were taken from an average of five runs, 10 s per run. Treating the NPs as spheres, the products of Ntotal and probability of particle sizes based on TEM distributions were used to determine the total NP volume in suspension. The number of drug molecules per nm3 of CPSNP were averaged with 95% confidence intervals.
Cell Proliferation Assay
PANC-1 (in Dulbecco’s modified Eagle medium/10% fetal bovine serum) and BxPC-3 (in RPMI medium/10% fetal bovine serum) cells (ATCC) were seeded onto 96-well plates at 5,000 cells/well. After 24 hr, treatment with vehicle (1x PBS), free drug, mPEG-CPSNPs or drug-loaded CPSNPs were initiated. Viable cell determinations were made after 72 hr using an alamarBlue® assay (Life Technologies). Relative proliferation for all treatment groups were normalized to vehicle controls.
Thymidylate Synthase Immunoblotting
PANC-1 and BxPC-3 cells were seeded onto 6-well dishes and incubated for 24 hr in one of the following treatment groups: no treatment, vehicle, free FdUMP, mPEG-FdUMP-CPSNPs, and mPEG-CPSNPs. Lysates were collected by aspirating the media, washing with 1x PBS, and adding RIPA buffer containing Complete Mini protease cocktail (Roche). Protein concentration was determined by microBCA assay (Thermo Scientific) and 20 μg of total protein was separated by gel electrophoresis. After transfer to HyBond ECL and blocking for 1 hr in 5% BSA, blots were probed overnight with anti-TS antibody (#9045 Cell Signaling Technology, 1:1000) or beta-actin antibody (#A2228; Sigma, 1:10,000). Membranes were washed, incubated with secondary antibody coupled to horseradish peroxidase (Amersham), and visualized using an enhanced chemiluminescent substrate (Pierce). Quantitation of scanned blots was done using Image-J software (NIH). After normalizing to β-actin, % active TS is the amount of uncomplexed TS divided by the amount of total TS (active TS and TS-FdUMP ternary complex).
Cell Cycle Analysis
PANC-1 cells were treated with vehicle, free FdUMP, mPEG-FdUMP-CPSNPs or an equal volume of mPEG-CPSNPs, or left untreated for 72 hr (32). Cells were fixed in 75% ethanol overnight and immediately prior to analysis treated with 1 μg/mL RNase A and 50 μg/mL propidium iodide. Cellular DNA content was determined using a FACSCalibur flow cytometer (BD Biosciences), and data were analyzed with Cellquest (Verity Software, Topsham, ME).
In vivo assessment of aptamer-targeted FdUMP-CPSNP up take by PDAC tumor cells
Animal protocols were approved by the Penn State College of Medicine IACUC committee. All procedures were done in accordance with institutional guidelines for the humane care of the animals. Orthotopic PANC-1 tumors were established in male, athymic (nu/nu) mice (Charles River). Pancreata were injected with 5×106 cells and treatment with CSPNPs was initiated at one week post-surgery. Treatment groups (n= 4–5 mice per group, with two experimental replicates) included mPEG-CPSNPs, mPEG-FdUMP-CPSNPS, gastrin-targeted G16-malPEG-FdUMP-CPSNPs, and aptamer-targeted AP-cPEG-FdUMP-CPSNPs. CPSNPs were administered at a FdUMP dose of 100 μg/kg, or an equal volume of mPEG-CPSNPs, twice weekly via tail vein injection. After six weeks of treatment, animals were sacrificed. Blood was removed by cardiac puncture, and complete blood cell counts and serum chemistries were done by the Department of Comparative Medicine Diagnostic Laboratory (PSU-Hershey). A portion of each tumor was flash frozen in liquid nitrogen and was powdered by grinding in a pre-chilled mortar and pestle. RIPA buffer containing Complete Mini protease cocktail (Roche) was added to each sample following grinding; the resuspended tissue was sonicated for 15 s and centrifuged to pellet debris. Protein concentrations in supernatants were determined by micro BCA protein assay (Thermo Scientific), and 20 μg of protein from each sample was separated by gel electrophoresis. Thymidylate synthase immunoblots were performed as described above. Another portion of each tumor was fixed and embedded in OCT, cryo-sectioned and stained for Ki-67 (Abcam #92742) according to the manufacturer’s instructions; nuclei were counterstained with Hoechst 33432. Images were acquired with a Leica AOBS SP8 laser scanning confocal microscope.
Statistical Analysis
Results were expressed as means ± standard error. Student t-tests were used to evaluate statistical significance with a p < 0.05 considered to be significant. To calculate EC50± 95% CI, nonlinear regression analysis was performed to generate the curve of best fit for the data according to a Sigmoidal regression using a 4-parameter logistic curve calibration [Y = Yo + (a/(1+((X/Xo)b))] in SigmaPlot 12 (Systat, Inc., San Jose, CA).
RESULTS
CPSNP Characterization
Preparation of CPSNPs (Figure 1) in reverse micelles yielded sub-100 nm particles (13–18, 33, 34); a size range that is optimal for NP penetration into fibrotic pancreatic tumors (18, 35). The lognormal mean of the size distribution for mPEG-FdUMP-CPSNPs was 8.4 nm (σz =0.2) and 10.5 nm (σz=0.4) for mPEG-dFdCMP-CPSNPs (Figures 2A and 2B). Aptamer-targeted AP-cPEG-FdUMP-CPSNPs in Figure 2C were about 8.0 nm (σz =0.3) and 17.2 nm (σz =0.1) for gastrin-targeted G16-malPEG-FdUMP-CPSNPs in Figure 2D. Because CPSNPs depend on citrate (Cit) and/or PEG to maintain dispersion (33), the zeta potential is a common metric to verify changes in surface terminal groups and colloidal stability (Figure 3). The mean values for Cit-CPSNPs, Cit-FdUMP-CPSNPs and Cit-dFdCMP-CPSNPs were −37±6 mV, −44±5 mV and −28±7 mV, respectively. After PEGylation, the uncharged methoxy group termination at the NP surface exhibits near-zero net charge, so mPEG-CPSNPs, mPEG-FdUMP-CPSNPs and mPEG-dFdCMP-CPSNPs displayed mean values of −1±5 mV, −4±6 mV and 1±9 mV, respectively. Bioconjugation of the aptamer to cPEG-FdUMP-CPSNPs resulted in a reduction of the zeta potential magnitude from −35±5 mV to −22±5 mV. An increased magnitude from −8±5 mV to −29±9 mV was observed for the conjugation of the gastrin to malPEG-FdUMP-CPSNPs.
Figure 1. Schematic of CPSNP synthesis.

Microemulsions A (calcium) and B (phosphate/silicate/X) are combined to form sub-particles that encapsulate bioactive agents (*high-angle angular dark field image of an osmium-stained CPSNP). Cit-X-CPSNPs are purified and PEGylated to obtain mPEG-X-CPSNPs, shown as an example. X = 5-FU, ATP:5-FU, FdUMP, FUdR, dFdC or dFdCMP.
Figure 2. Particle size distribution of CPSNPs.

TEM micrograph and lognormal particle distributions of (A) mPEG-FdUMP-CPSNPs, (B) mPEG-dFdCMP-CPSNPs, (C) AP-cPEG-FdUMP-CPSNPs, and (D) G16-malPEG-FdUMP-CPSNPs with enlarged insets.
Figure 3. PEGylation of CPSNPs.

A comparison of the mean zeta-potential values of Cit-CPSNPs, Cit-FdUMP-CPSNPS, and Cit-dFdCMP-CPSNPs, with mPEG-CPSNPs, mPEG-FdUMP-CPSNPs, mPEG-dFdCMP-CPSNPs, cPEG- and malPEG- FdUMP-CPSNPs after PEGylation. A reduction of the zeta potential magnitude from aptamer coupling to cPEG-FdUMP-CPSNPs and increased magnitude from gastrin-16 coupling to malPEG-FdUMP-CPSNPs are also shown. Error bars represent the mean±95% CI; n=10.
Encapsulation efficiency of chemotherapeutics by CPSNPs
We hypothesized that chemotherapeutic drugs such as 5-FU or dFdC would be less effectively encapsulated into CPSNPs than FdUMP and dFdCMP (Figure 4). To address this hypothesis, the encapsulation efficiencies (EE) for 5-FU, ATP:5-FU complex, FUdR (5-fluorodeoxyuridine), FdUMP, dFdC, and dFdCMP (Table 1) were experimentally determined. The EE is defined by the relationship,
Figure 4. Therapeutic nucleoside and nucleotide analogs.

Structures of 5-fluorouracil (5-FU), 5-fluorodeoxyuridine (FUdR), 5-fluorodeoxyuridine monophosphate (FdUMP), 2′,2′-difluorodeoxycytidine (dFdC, or gemcitabine), 2′,2′-difluorodeoxycytidine monophosphate (dFdCMP, or gemcitabine monophosphate), and the adenosine triphosphate:5-fluorouracil complex (ATP:5-FU).
TABLE 1.
Encapsulated concentrations, encapsulation efficiency (EE), and payload for 5-FU, ATP:5-FU, FUdR, FdUMP, dFdC or dFdCMP in citrate-functionalized (Cit) CPSNPs.
| Formulation | Drug Concentration (M)* | EE (mol%)* | Calculated Payload (molecules/nm3 CPSNP×10−1)† |
|---|---|---|---|
| Cit-5-FU-CPSNP | 4.1 (±1.8) ×10−7 | 0.11 (±0.04) | 0.10 (±0.07) |
| Cit-ATP:5-FU-CPSNP | 1.8 (±2.5) ×10−6 | 0.36 (±0.39) | 0.47 (±0.54) |
| Cit-FUdR-CPSNP | 2.8 (±1.2) ×10−8 | <0.01 | 0.07 (±0.03) |
| Cit-FdUMP-CPSNP | 3.0 (±1.4) ×10−4 | 41 (±16) | 32 (±16) |
| Cit-dFdC-CPSNP | 1.9 (±0.6) ×10−9 | <0.01 | 0.01 (±0.01) |
| Cit-dFdCMP-CPSNP | 2.3 (±0.4) ×10−5 | 22 (±6) | 3.6 (±1.6) |
Expressed as the mean ±95% CI of n=4–6 experimental replicates.
Particle number concentration by volume (particles/ml) is listed in Table S1 of supplementary information.
| (1) |
where mi is the initial moles of drug and mf is the moles present in the purified CPSNPs, quantified by LC-MS/MS. CPSNP encapsulation of 5-FU was only 4.1 (±1.8) ×10−7 M, or 0.11 (±0.04) mol% (Table 1), and extended reaction times failed to improve 5-FU encapsulation. FUdR, the deoxynucleoside analog of 5-FU, was incorporated in CPSNPs even less effectively than 5-FU, with an EE of <0.01 mol% or 2.8 (±1.2) ×10−8 M. Both double and triple microemulsion methods were implemented to create a core-shell CPSNP, which consisted of a calcium phosphosilicate core layered with both 5-FU and ATP. However, the weak hydrogen bonds between 5-FU and ATP were compromised by the high ionic strength within the micelles, and encapsulation efficiency of 5-FU was only marginally improved to 0.36 (±0.39) mol%, or 1.8 (±2.5) ×10−6 M of incorporated drug.
In contrast to the low encapsulation efficiencies of 5-FU and FUdR, FdUMP was reproducibly encapsulated into CPSNPs at 3.0 (±1.4) ×10−4 M and an EE of 41 (±16) mol%– several orders of magnitude higher than 5-FU or FUdR encapsulation. The increased EE for FdUMP versus FUdR suggests that encapsulation is enhanced by the formation of a bond between calcium from the NP and phosphate from the drug molecule. Similarly, dFdC was poorly encapsulated into CPSNPs, 1.9 (±0.6) ×10−9 M at an EE of <0.01 mol%, while dFdCMP was encapsulated with significantly higher efficiency, 2.3 (±0.4) ×10−5 M at an EE of 22 (±6) mol%.
A similar trend is observed in the payloads listed in Table 1. The payloads were estimated with measurements from TEM (for lognormal particle size distributions) and DLS (for total number of particles in suspension). For this purpose, TEM was the preferred method for true size analysis since DLS takes into account the NP interactions with surrounding molecules in a liquid medium to give the hydrodynamic radius. Determining the exact payload (drug molecules per particle) is generally a complex calculation for NPs due to the presence of various particle sizes in a distribution that influence drug-loading capacity. Thus, the payload was normalized to drug molecules per volume of CPSNP to allow comparison between drug formulations. As expected from the corresponding EEs and concentrations, Cit-FdUMP and Cit-dFdCMP-CPSNPs contain the highest number of drug molecules/nm3, 32 (±16) ×10−1 and 3.6 (±1.6) ×10−1, respectively. An improvement in payload for 5-FU with ATP-loaded CPSNPs is also evident as these samples averaged 0.47 (±0.54) ×10−1 5-FU molecules/nm3. The payloads for Cit-5FU, Cit-FUdR, and Cit-dFdC-CPSNPs fall within a lower narrow range of 0.10 (±0.7) ×10−1, 0.07 (±0.03) ×10−1, and 0.01 (±0.01) ×10−1 drug molecules/nm3, respectively. Thus, while the prodrugs 5-FU and dFdC were poorly encapsulated into CPSNPS, we have demonstrated efficient uptake of the bioactive monophosphate forms FdUMP and dFdCMP in the CPSNP system.
CPSNP-encapsulated phospho-drugs block PDAC cell proliferation in vitro
While others have reported that polymeric FdUMP has efficacy against other cancers (36–38), the efficacy of phosphorylated drug metabolites on the growth of pancreatic cancer cells has not been shown. In these studies, the anti-proliferative effects of 5-FU, FdUMP, dFdC and dFdCMP were compared to CPSNP-encapsulated FdUMP and CPSNP-encapsulated dFdCMP. The cultured human pancreatic cancer cell lines BxPC-3 and PANC-1 were chosen, since each demonstrates some resistance to these drugs (39). Against both cell lines, 5-FU and FdUMP had similar efficacy, with a low uM EC50 for both compounds and both cell lines (Figure 5). Similarly, dFdC and dFdCMP were both effective in blocking proliferation of BxPC-3 and PANC-1. The EC50 for free dFdCMP against both cell lines was in the nM range and was lower than for 5-FU or FdUMP.
Figure 5. Phospho-drug loaded-CPSNPs block pancreatic cancer cell proliferation.

In vitro growth of human PDAC cell lines BxPC-3 and PANC-1 is effectively blocked by mPEG-dFdCMPs, with EC50 values of 130 and 550 nM, respectively. BxPC-3 cells were more resistant to mPEG-FdUMP-CPSNPS than PANC-1 cells, which had an EC50 of 1.3 μM. Empty mPEG-CPSNPs (light hatched), free drug (black) or drug-containing CPSNPs (shaded hatched) are expressed as relative proliferation (percent of vehicle, white). Values are the mean of 3–4 independent experiments; ***p<0.001 and **p<0.01.
Comparing FdUMP to mPEG-FdUMP-CPSNPs, the response of BxPC-3 cells to encapsulated FdUMP plateaued between 2.5 uM and 50 nM, and higher doses failed to affect the proliferation of these cells (Figure 5). PANC-1 responded similarly to both free FdUMP and encapsulated FdUMP, with an EC50 for mPEG-FdUMP-CPSNPs of 1.3 μM. Both BxPC-3 and PANC-1 were more sensitive to mPEG-dFdCMP-CPSNPs than to mPEG-FdUMP-CPSNPs. The EC50 for mPEG-dFdCMP-CPSNPs against PANC-1 was 550 nM, and proliferation of BxPC-3 was decreased to a much greater degree by mPEG-dFdCMP-CPSNPs (EC50 130 nM) than by mPEG-FdUMP-CPSNPs. There was no significant difference between the efficacy of free dFdCMP and encapsulated dFdCMP for either cell line. Neither BxPC-3 nor PANC-1 proliferation was affected by mPEG-CPSNPs, consistent with previous work indicating that the particles themselves were non-toxic (11, 18). Thus, CPSNP encapsulation of FdUMP and dFdCMP did not affect the ability of these phospho-drugs to inhibit the growth of PDAC cell lines in vitro.
CPSNP-encapsulated FdUMP Inhibits Thymidylate Synthase (TS)
In the presence of folate, TS dimers and FdUMP create an irreversibly inactivated enzyme:drug complex (40, 41), and the inactive ternary complex (TS, FdUMP and THF) migrates more slowly than active TS on immunoblots (42). Immunoblot analysis of TS in untreated cells, cells treated with vehicle or cells treated with mPEG-CPSNPs did not show formation of a TS ternary complex (only the lower molecular weight, active form of TS was present in these cells; Figure 6, Lanes 1, 2, and 4). Treatment with free FdUMP shifted most of the active TS into the inactive ternary complex, as expected, although FdUMP was slightly less effective for PANC-1 cells (80% inhibition) than for BxPC-3 cells (90% inhibition; Figure 6, Lane 3). However, nearly all of the TS was in the catalytically inactive TS:FdUMP:THF ternary complex when BxPC-3 or PANC-1 cells were treated with mPEG-FdUMP-CPSNPs (89%–91% inhibition; Figure 6, Lane 5). Thus, when delivered in vitro, CPSNP-encapsulated FdUMP retained its capacity to bind to and inhibit its target enzyme, TS, in both BxPC-3 and PANC-1 cells.
Figure 6. CPSNP-encapsulated FdUMP inactivates TS.

Immunoblots of the FdUMP target enzyme thymidylate synthase (TS) from PANC-1 (upper panel) or BxPC-3 (lower panel) cells treated with (Lane 3) 250 μM free FdUMP or (Lane 5) 2 μM mPEG-FdUMP-CPSNPs. Both cell lines showed significant (>80%) conversion of TS to an inactive ternary complex (TS:FdUMP) with free drug and with mPEG-FdUMP-CPSNP treatment. Controls that received (Lane 1) no treatment, (Lane 2) vehicle or (Lane 4) mPEG-CPSNPs exhibited only active TS with no evidence of TS:FdUMP ternary complex formation.
Cell Cycle Arrest in FdUMP-CPSNP-Treated PANC-1 Cells
Since TS provides the sole de novo source of cellular thymidylate, which is necessary for DNA replication and repair, inhibition of TS depletes nucleotide pools and leads to DNA strand breaks and arrest of the cell cycle since cells are unable to synthesize new DNA or repair damaged DNA (43). To demonstrate that this cellular activity of FdUMP was retained after encapsulation, PANC-1 cells treated with various CPSNP formulations were analyzed for cell cycle progression (Figure 7). Untreated cells, vehicle-treated cells, and mPEG-CPSNP-treated cells all had equivalent percentages of cells in G0/G1, S, and G2/M-phase, indicating that the CPSNPs themselves had no effect on cell cycle progression. Compared to these controls, cells treated with free FdUMP demonstrated a partial S-phase arrest and a reduced number of cells in the G0/G1 phase. The percentage of free FdUMP-treated cells in the G0/G1 phase decreased from 54% to 20%, and the percentage of cells in the S phase of the cell cycle increased from 25% to 56% compared to untreated cells. Similarly, cells treated with mPEG-FdUMP-CPSNPs showed virtually no G0/G1 phase cells, and the percentage of cells in G2/M and in S phase was nearly equal, consistent with the known mechanism of action of 5-FU and FdUMP (44). Despite the fact that a significantly lower concentration of FdUMP was used, the arrest of cell division in cells treated with mPEG-FdUMP-CPSNPs was more pronounced than in free FdUMP-treated cells.
Figure 7. mPEG-FdUMP-CPSNPs block PANC-1 cell cycle progression.

The table indicates the percentage of cells in either G0/G1 or G2/M phase (red) or in S-phase (light hatched) after treatment. Control groups (no treatment, vehicle or mPEG-CPSNPs) have similar percentages of cells in all cell cycle phases. While cells treated with FdUMP demonstrated a reduced number of cells in G0/G1, only 0.1% of cells treated with mPEG-FdUMP-CPSNPs are in G0/G1. The percentage of cells in each cell cycle phase is expressed as the mean ±SEM of 3 independent experiments.
CCKBR-Targeted FdUMP-CPSNPS Deliver Active Drug to PDAC Tumor Cells in vivo
Recently, we described a DNA aptamer (AP) which binds to the cholecystokinin B receptor (CCKBR) with high affinity yet does not activate receptor signaling (19). NP uptake by pancreatic tumors in mice is enhanced with aptamer targeting, although the effect of aptamer targeting on uptake by tumor cells cultured in vitro is minimal (19). For these in vivo studies, either the selective CCKBR aptamer, or the endogenous CCKBR peptide ligand gastrin-16 (G16), were covalently attached to FdUMP-CPSNPS. The ability of these targeted CPSNPs to deliver active FdUMP to PDAC tumor cells in vivo was assessed by TS immunoblotting of tumor tissues. Active TS protein levels in tumors from mice treated with mPEG-CPSNPs and untargeted mPEG-FdUMP-CPSNPs were not significantly different from each other (Figure 8A), indicating that without tumor-specific targeting, CPSNPs are poorly taken up by PDAC tumors in vivo. Previous studies showed that gastrin-targeted fluorescent CPSNPS had enhanced PDAC tumor uptake in vivo (18). In this study, although the tumors from mice treated with G16-malPEG-FdUMP-CPSNPs appeared to have reduced TS protein levels compared to mPEG-FdUMP-CPSNPs, the differences between these two treatment groups did not reach statistical significance. Tumors from mice treated with AP-cPEG-FdUMP-CPSNPs had a significant reduction in active TS levels: a 60% reduction versus either mPEG-CPSNPS or mPEG-FdUMP-CPSNP treatments (*p<0.05; Figure 8A). This demonstrates that in mice treated with AP-targeted FdUMP-CPSNPs, FdUMP was internalized and released into PDAC tumor cells and that the biological activity of FdUMP against TS was retained. Consistent with the reduced TS levels, tumor staining for Ki-67, a nuclear protein which serves as an index of proliferation (45), also was significantly reduced in mice treated with AP-targeted FdUMP-CPSNPs (Figure 8B). These results are in keeping with other recent data showing that the CCKBR AP increased fluorescent CPSNP accumulation in PDAC tumors in vivo compared to both untargeted or G16-targeted particles (19). However, the studies herein extend our previous studies with AP-targeted fluorescent CPSNPs, which did not specifically address cellular internalization of CPSNP cargo (as the fluorescent CPSNPS could have been associated with CCKBR at the plasma membrane rather than inside of the tumor cells). The current study clearly demonstrates that AP targeted FdUMP-CPSNPs are internalized by PDAC tumor cells and release biologically active FdUMP into the cytoplasm.
Figure 8. CCKBR-targeted CPSNPs deliver active FdUMP to PDAC tumors in vivo without inducing systemic toxicity.

(A) Levels of active thymidylate synthase (unbound TS), as determined by immunoblotting, reflects that amount of the TS inhibitor FdUMP taken up by PANC-1 tumors in mice treated with various CPSNP formulations (n= 5 mice/treatment group). Tumors from mice treated with (1) empty mPEG-CPSNPs or (2) mPEG-FdUMP-CPSNPs had equivalent amounts of unbound, active TS, suggesting that untargeted particles were not efficiently taken up by tumor cells in vivo. Although the mean TS levels in tumors from (4) G16-malPEG-FdUMP-CPSNP-treated mice was decreased, only tumors in mice treated with (3) CCKBR AP-cPEG-FdUMP-CPSNPs had significantly reduced TS levels (*p<0.05) compared to empty or untargeted controls. (B) Ki-67 staining of PANC-1 tumor sections from mice treated with (1) mPEG-CPSNPs or (2) mPEG-FdUMP-CPSNPs was higher than in tumor sections from (3) AP-cPEG-FdUMP-CPSNP-treated mice; **=p<0.01, ns=not significant. (C) Complete blood cell counts, including white blood cells (WBC), red blood cells (RBC), platelets and neutrophils, were not significantly altered by FdUMP-loaded CPSNP treatments, either (2) without aptamer or (3) with aptamer targeting compared to (1) mPEG-CPSNPs. (D) Total serum albumin and blood urea nitrogen (BUN), as well as serum calcium and phosphate levels, were not different among treatment groups. (E) FdUMP-CPSNP treatment did not reduce mean body weight at necropsy or alter percent change in body weight during treatment. Bars represent the mean ±SEM of 2 independent experiments.
Previous work by our group has demonstrated that unloaded CPSNPs have no toxicity when administered systemically (46). To rule out non-specific effects of the drug-loaded CPSNPs on bone marrow or other organ systems, serum chemistries and complete blood cell counts were done for animals that had been treated with mPEG-CPSNPs, mPEG-FdUMP-CPSNPs, or AP-targeted FdUMP-CPSNPs. After six weeks of treatment, no significant differences in blood cell counts (WBCs, RBCs, platelets and neutrophils) or serum chemistries (albumin for hepatic function and BUN for renal function) were noted among the treatment groups (Figure 8C–D). Serum calcium and phosphate levels were unchanged by NP treatment (Figure 8D), suggesting there was no significant dissolution of the particles in circulation. Animals treated with drug-loaded CPSNPs also did not lose weight, either overall or during the course of treatment, compared to mPEG-CPSNP controls (Figure 8E 1 vs. 2, 3). This indicates there were no non-specific, whole animal toxicities that resulted from any of the CPSNP treatments.
DISCUSSION
Our results demonstrate that CPSNP encapsulation of bioactive chemotherapeutic drug metabolites, such as the 5-FU metabolite FdUMP or the gemcitabine metabolite dFdCMP, can be an effective means to deliver these drugs to pancreatic tumors. Due to metal-ligand complexes between phosphate group on the phospho-drug molecule and calcium in the CPSNPs, phosphorylated, bioactive drugs, which cannot be effectively administered systemically (47), can be encapsulated within CPSNPs without compromising cellular activity. This suggests a general strategy whereby other phosphorylated compounds can be effectively encapsulated into CPSNPs in addition to other drugs that have previously been challenging to incorporate into NPs.
These unique CPSNP-drug formulations could have a straightforward application to PDAC patient treatment. A recent meta-analysis suggested that PDAC patients may benefit from 5-FU/dFdC combination therapy (48), although significant toxicities, including neutropenia, thrombocytopenia and diarrhea, can occur (43). The toxicities are due mainly to the high systemic drug concentrations required to achieve a therapeutic response. A treatment strategy which combines delivery of NP encapsulated and targeted FdUMP-CPSNPs with dFdCMP-CPSNPs should increase the anti-tumor efficacy of this drug combination with less toxicity and reduced side effects. Other opportunities for improving treatment could include administering drug encapsulated-CPSNPs with a combination of agents that normalize tumor vasculature, such as low-dose Cilengitide and Verapamil – which have been shown to enhance gemcitabine efficacy (49).
Resistance to dFdC often occurs by down-regulating proteins that facilitate drug transport, such as hENT1 (39) or human concentrative nucleoside transporter 1 (hCNT1) (50), or reducing the expression of the dFdC activating enzyme deoxycytidine kinase (51). Patients with inherently low deoxycytidine kinase activity do not effectively convert dFdC to its metabolically active monophosphate form and thus do not respond well to dFdC treatment. A strategy to encapsulate dFdCMP into CCKBR-targeted CPSNPs should bypass these inherent or acquired PDAC resistance mechanisms (i.e. reduced dFdC uptake due to transporter dysregulation or lack of metabolic activation) and make this drug far more effective.
For those patients with the genetic DPD deficiency (i.e. those with the DPYD*2A SNP), encapsulation and delivery of FdUMP could give this chemotherapy treatment better efficacy without increasing the risk of toxicities (52, 53). Similarly, patients with low activity of cytidine deaminase, the enzyme that catabolizes dFdC to difluorodeoxyuridine, can develop severe adverse effects from dFdC treatment (54). Thus, replacing systemic dFdC with encapsulated dFdCMP should permit these PDAC patients to be more safely and effectively treated.
Beyond encapsulating effective concentrations of phospho-drugs within nanocarriers, this study demonstrated that CPSNPs deliver drugs such as FdUMP to tumor cells in a biologically active form both in vitro and in vivo. Evidence that CPSNP-encapsulated FdUMP retained activity included the formation of inactive thymidylate synthase ternary complexes and complete cell cycle arrest in mPEG-FdUMP-CPSNP-treated pancreatic cancer cells in vitro. When surface bioconjugated with a PDAC tumor-targeting agent, specifically the CCKBR-targeting aptamer (19), and administered via systemic circulation, AP-cPEG-FdUMP-CPSNPs were taken up by pancreatic tumors and delivered biologically active drug to tumor cells in vivo. The fact that active NP targeting with a CCKBR aptamer was most evident in vivo is consistent with other studies that have observed differing efficacy of targeted NPs using in vitro versus in vivo model systems (55).
Although the dose of FdUMP that was administered in these initial studies (100 μg/kg) was less than was needed to achieve a reduction in tumor mass, which requires 100 mg/kg for 5-FU, on average (56), significant aspects of this work include 1) the inferred stability of these particles in systemic circulation, 2) the ability of targeted-CPSNPs to be internalized by tumor cells and 3) the delivery of active drug to decrease the intracellular drug target, the enzyme TS, and tumor cell proliferation (Ki-67) without evidence of systemic toxicities. Having demonstrated CPSNP safety and efficacy in delivering active drug into PDAC tumor cells in vivo, future studies will examine dose-response curves with increased FdUMP (and dFdCMP) concentrations to further decrease tumor cell proliferation and achieve a therapeutic response.
This investigation also opens the possibility of encapsulating synthetically phosphorylated drug compounds to improve their delivery and efficacy. If CPSNPs can effectively encapsulate phosphorylated compounds that have systemic toxicities and/or poor cellular internalization, a similar approach can be applied to the encapsulation of many other phospho-drugs. During systemic administration, the plasma concentration of chemotherapeutic drugs can increase rapidly and then drop quickly as the drug is metabolized. By encapsulating chemotherapeutics into CPSNPs, which are stable in circulation, a more uniform systemic drug concentration can be achieved, thus reducing the chances of both under-dosing and over-dosing. Surface modification of drug-loaded CPSNPs with targeting aptamers that direct these particles to PDAC tumors adds specificity and should reduce off-target toxicities. This study demonstrates a promising new methodology for encapsulation and intracellular delivery of bioactive, phosphorylated chemotherapeutic agents to pancreatic cancer cells.
Supplementary Material
Acknowledgments
Statement of Funding:
This study was funded by the National Institutes of Health (NIH) grants R01CA167535 and R21CA170121 from the National Cancer Institute (NCI). The project was also supported in part under a grant with the Pennsylvania Department of Health using Tobacco CURE funds (SAP#4100072562 to GLM and JHA). The department specifically disclaims responsibility for any analyses, interpretations or conclusions. WSL was supported, in part, by the grants UL1 TR000127 and TL1 TR000125 from the National Center for Advancing Translational Sciences (NCATS). Penn State Research Foundation has licensed CPSNP technology to Keystone Nano, Inc. (PA, USA). JHA and MK are co-founders of Keystone Nano and are CSO and CMO, respectively.
The authors would like to thank Dr. Bernd Kabius for his assistance in HAADF TEM imaging (MCL), Dr. Jason Liao, Director of the Biostatistics Core of the Cancer Institute, for his help in designing these experiments and the Department of Comparative Medicine Diagnostic Laboratory for excellent technical assistance at Penn State.
ABBREVIATIONS
- CPSNP
calcium phosphosilicate nanoparticle
- mPEG
methoxy-polyethylene glycol
- cPEG
carboxy-polyethylene glycol
- malPEG
maleimide-polyethylene glycol
- Cit
citrate
- NT
no treatment
- TS
thymidylate synthase
- THF
5,10-methylene-tetrahydrofolate
- FdUMP
5-fluorodeoxyuridine monophosphate
- 5-FU
5-fluorouracil
- FUdR
5-fluorodeoxyuridine
- dFdC
gemcitabine or 2′,2′-difluorodeoxycytidine
- dFdCMP
gemcitabine monophosphate or 2′,2′-difluorodeoxycytidine monophosphate
- ATP
adenosine triphosphate
- 5-CU
5-chlorouracil
- SEM
standard error of the mean
- TEM
transmission electron microscopy
- CCKBR
cholecystokinin B receptor
Footnotes
Conflicts of Interest:
All other authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Spencer DS, Puranik AS, Peppas NA. Intelligent Nanoparticles for Advanced Drug Delivery in Cancer Treatment. Current opinion in chemical engineering. 2015 Feb;7:84–92. doi: 10.1016/j.coche.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kunstfeld R, Wickenhauser G, Michaelis U, Teifel M, Umek W, Naujoks K, et al. Paclitaxel encapsulated in cationic liposomes diminishes tumor angiogenesis and melanoma growth in a “humanized” SCID mouse model. The Journal of investigative dermatology. 2003 Mar;120(3):476–82. doi: 10.1046/j.1523-1747.2003.12057.x. [DOI] [PubMed] [Google Scholar]
- 3.Gregoriadis G. Drug entrapment in liposomes. FEBS letters. 1973 Nov 01;36(3):292–6. doi: 10.1016/0014-5793(73)80394-1. [DOI] [PubMed] [Google Scholar]
- 4.Juliano RL, Stamp D, McCullough N. Pharmacokinetics of liposome-encapsulated antitumor drugs and implications for therapy. Annals of the New York Academy of Sciences. 1978;308(1 Liposomes and):411–25. doi: 10.1111/j.1749-6632.1978.tb22038.x. [DOI] [PubMed] [Google Scholar]
- 5.Nigavekar SS, Sung LY, Llanes M, El-Jawahri A, Lawrence TS, Becker CW, et al. 3H dendrimer nanoparticle organ/tumor distribution. Pharmaceutical research. 2004 Mar;21(3):476–83. doi: 10.1023/B:PHAM.0000019302.26097.cc. [DOI] [PubMed] [Google Scholar]
- 6.Morgan MT, Carnahan MA, Finkelstein S, Prata CA, Degoricija L, Lee SJ, et al. Dendritic supramolecular assemblies for drug delivery. Chemical communications. 2005 Sep;14(34):4309–11. doi: 10.1039/b502411k. [DOI] [PubMed] [Google Scholar]
- 7.Loo C, Lin A, Hirsch L, Lee MH, Barton J, Halas N, et al. Nanoshell-enabled photonics-based imaging and therapy of cancer. Technology in cancer research & treatment. 2004 Feb;3(1):33–40. doi: 10.1177/153303460400300104. [DOI] [PubMed] [Google Scholar]
- 8.Smith AM, Duan H, Mohs AM, Nie S. Bioconjugated quantum dots for in vivo molecular and cellular imaging. Advanced drug delivery reviews. 2008 Aug 17;60(11):1226–40. doi: 10.1016/j.addr.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bagalkot V, Zhang L, Levy-Nissenbaum E, Jon S, Kantoff PW, Langer R, et al. Quantum dot-aptamer conjugates for synchronous cancer imaging, therapy, and sensing of drug delivery based on bi-fluorescence resonance energy transfer. Nano letters. 2007 Oct;7(10):3065–70. doi: 10.1021/nl071546n. [DOI] [PubMed] [Google Scholar]
- 10.Adair JH, Parette MP, Altinoglu EI, Kester M. Nanoparticulate alternatives for drug delivery. ACS Nano. 2010 Sep 28;4(9):4967–70. doi: 10.1021/nn102324e. [DOI] [PubMed] [Google Scholar]
- 11.Altinoglu EI, Adair JH. Calcium phosphate nanocomposite particles: a safer and more effective alternative to conventional chemotherapy? Future Oncol. 2009 Apr;5(3):279–81. doi: 10.2217/fon.09.4. [DOI] [PubMed] [Google Scholar]
- 12.Tabakovic A, Kester M, Adair JH. Calcium phosphate-based composite nanoparticles in bioimaging and therapeutic delivery applications. Wiley interdisciplinary reviews Nanomedicine and nanobiotechnology. 2012 Jan-Feb;4(1):96–112. doi: 10.1002/wnan.163. [DOI] [PubMed] [Google Scholar]
- 13.Morgan TT, Muddana HS, Altinoglu EI, Rouse SM, Tabakovic A, Tabouillot T, et al. Encapsulation of organic molecules in calcium phosphate nanocomposite particles for intracellular imaging and drug delivery. Nano Lett. 2008 Dec;8(12):4108–15. doi: 10.1021/nl8019888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barth BM, Shanmugavelandy SS, Kaiser JM, McGovern C, Altinoglu EI, Haakenson JK, et al. PhotoImmunoNanoTherapy reveals an anticancer role for sphingosine kinase 2 and dihydrosphingosine-1-phosphate. ACS nano. 2013 Mar 26;7(3):2132–44. doi: 10.1021/nn304862b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Altinoglu EI, Russin TJ, Kaiser JM, Barth BM, Eklund PC, Kester M, et al. Near-infrared emitting fluorophore-doped calcium phosphate nanoparticles for in vivo imaging of human breast cancer. ACS Nano. 2008 Oct 28;2(10):2075–84. doi: 10.1021/nn800448r. Epub 2009/02/12. eng. [DOI] [PubMed] [Google Scholar]
- 16.Muddana HS, Morgan TT, Adair JH, Butler PJ. Photophysics of Cy3-Encapsulated Calcium Phosphate Nanoparticles. Nano Lett. 2009;9(4):1559–66. doi: 10.1021/nl803658w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barth BM, EIA, Shanmugavelandy SS, Kaiser JM, Crespo-Gonzalez D, DiVittore NA, et al. Targeted indocyanine-green-loaded calcium phosphosilicate nanoparticles for in vivo photodynamic therapy of leukemia. ACS Nano. 2011 Jul 26;5(7):5325–37. doi: 10.1021/nn2005766. [DOI] [PubMed] [Google Scholar]
- 18.Barth BM, Sharma R, Altinoglu EI, Morgan TT, Shanmugavelandy SS, Kaiser JM, et al. Bioconjugation of calcium phosphosilicate composite nanoparticles for selective targeting of human breast and pancreatic cancers in vivo. ACS nano. 2010 Mar 23;4(3):1279–87. doi: 10.1021/nn901297q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clawson GA, Abraham T, Pan W, Tang X, Linton SS, McGovern CO, et al. A Cholecystokinin B Receptor-Specific DNA Aptamer for Targeting Pancreatic Ductal Adenocarcinoma. Nucleic Acid Ther. 2017 Feb;27(1):23–35. doi: 10.1089/nat.2016.0621. Epub 2016/10/19. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kester M, Heakal Y, Fox T, Sharma A, Robertson GP, Morgan TT, et al. Calcium phosphate nanocomposite particles for in vitro imaging and encapsulated chemotherapeutic drug delivery to cancer cells. Nano Lett. 2008 Dec;8(12):4116–21. doi: 10.1021/nl802098g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. The New England journal of medicine. 2011 May 12;364(19):1817–25. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 22.Neesse A, Michl P, Frese KK, Feig C, Cook N, Jacobetz MA, et al. Stromal biology and therapy in pancreatic cancer. Gut. 2011 Jun;60(6):861–8. doi: 10.1136/gut.2010.226092. [DOI] [PubMed] [Google Scholar]
- 23.Duschinsky R, Pleven E, Heidelberger C. The Synthesis of 5-Fluoropyrimidines. Journal of the American Chemical Society. 1957;79(16):4559–60. [Google Scholar]
- 24.Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nature reviews Cancer. 2003 May;3(5):330–8. doi: 10.1038/nrc1074. [DOI] [PubMed] [Google Scholar]
- 25.de Sousa Cavalcante L, Monteiro G. Gemcitabine: metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. European journal of pharmacology. 2014 Oct 15;741:8–16. doi: 10.1016/j.ejphar.2014.07.041. [DOI] [PubMed] [Google Scholar]
- 26.Ewald B, Sampath D, Plunkett W. Nucleoside analogs: molecular mechanisms signaling cell death. Oncogene. 2008 Oct 27;27(50):6522–37. doi: 10.1038/onc.2008.316. Epub 2008/10/29. eng. [DOI] [PubMed] [Google Scholar]
- 27.Diasio RB, Harris BE. Clinical pharmacology of 5-fluorouracil. Clinical pharmacokinetics. 1989 Apr;16(4):215–37. doi: 10.2165/00003088-198916040-00002. eng. [DOI] [PubMed] [Google Scholar]
- 28.Baker JA, Wickremsinhe ER, Li CH, Oluyedun OA, Dantzig AH, Hall SD, et al. Pharmacogenomics of gemcitabine metabolism: functional analysis of genetic variants in cytidine deaminase and deoxycytidine kinase. Drug metabolism and disposition: the biological fate of chemicals. 2013 Mar;41(3):541–5. doi: 10.1124/dmd.112.048769. [DOI] [PubMed] [Google Scholar]
- 29.Heinemann V, Xu YZ, Chubb S, Sen A, Hertel LW, Grindey GB, et al. Cellular elimination of 2′,2′-difluorodeoxycytidine 5′-triphosphate: a mechanism of self-potentiation. Cancer research. 1992 Feb 01;52(3):533–9. Epub 1992/02/01. eng. [PubMed] [Google Scholar]
- 30.Abbruzzese JL, Grunewald R, Weeks EA, Gravel D, Adams T, Nowak B, et al. A phase I clinical, plasma, and cellular pharmacology study of gemcitabine. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 1991 Mar;9(3):491–8. doi: 10.1200/JCO.1991.9.3.491. Epub 1991/03/01. eng. [DOI] [PubMed] [Google Scholar]
- 31.Wang WB, Yang Y, Zhao YP, Zhang TP, Liao Q, Shu H. Recent studies of 5-fluorouracil resistance in pancreatic cancer. World journal of gastroenterology: WJG. 2014 Nov 14;20(42):15682–90. doi: 10.3748/wjg.v20.i42.15682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rathos MJ, Joshi K, Khanwalkar H, Manohar SM, Joshi KS. Molecular evidence for increased antitumor activity of gemcitabine in combination with a cyclin-dependent kinase inhibitor, P276-00 in pancreatic cancers. Journal of translational medicine. 2012 Aug 08;10(1):161. doi: 10.1186/1479-5876-10-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morgan TT, Goff TM, Adair JH. The colloidal stability of fluorescent calcium phosphosilicate nanoparticles: the effects of evaporation and redispersion on particle size distribution. Nanoscale. 2011;3:2044–53. doi: 10.1039/c0nr00995d. [DOI] [PubMed] [Google Scholar]
- 34.Wang J, White WB, Adair JH. Dispersion of SiO2-based nanocomposites with high performance liquid chromatography. The journal of physical chemistry B. 2006 Mar 16;110(10):4679–85. doi: 10.1021/jp0547010. [DOI] [PubMed] [Google Scholar]
- 35.Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M, et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nature nanotechnology. 2011 Dec;6(12):815–23. doi: 10.1038/nnano.2011.166. [DOI] [PubMed] [Google Scholar]
- 36.Gmeiner WH, Boyacioglu O, Stuart CH, Jennings-Gee J, Balaji KC. The cytotoxic and pro-apoptotic activities of the novel fluoropyrimidine F10 towards prostate cancer cells are enhanced by Zn(2+) -chelation and inhibiting the serine protease Omi/HtrA2. The Prostate. 2015 Mar 1;75(4):360–9. doi: 10.1002/pros.22922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gmeiner WH, Jennings-Gee J, Stuart CH, Pardee TS. Thymineless death in F10-treated AML cells occurs via lipid raft depletion and Fas/FasL co-localization in the plasma membrane with activation of the extrinsic apoptotic pathway. Leukemia research. 2015 Feb;39(2):229–35. doi: 10.1016/j.leukres.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gmeiner WH, Lema-Tome C, Gibo D, Jennings-Gee J, Milligan C, Debinski W. Selective anti-tumor activity of the novel fluoropyrimidine polymer F10 towards G48a orthotopic GBM tumors. Journal of neuro-oncology. 2014 Feb;116(3):447–54. doi: 10.1007/s11060-013-1321-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farrell JJ, Elsaleh H, Garcia M, Lai R, Ammar A, Regine WF, et al. Human equilibrative nucleoside transporter 1 levels predict response to gemcitabine in patients with pancreatic cancer. Gastroenterology. 2009 Jan;136(1):187–95. doi: 10.1053/j.gastro.2008.09.067. Epub 2008/11/11. eng. [DOI] [PubMed] [Google Scholar]
- 40.van der Wilt CL, Backus HH, Smid K, Comijn L, Veerman G, Wouters D, et al. Modulation of both endogenous folates and thymidine enhance the therapeutic efficacy of thymidylate synthase inhibitors. Cancer Res. 2001 May 1;61(9):3675–81. [PubMed] [Google Scholar]
- 41.Lockshin A, Mondal K, Danenberg PV. Spectroscopic studies of ternary complexes of thymidylate synthetase, deoxyribonucleotides, and folate analogs. The Journal of biological chemistry. 1984 Sep 25;259(18):11346–52. [PubMed] [Google Scholar]
- 42.Patel K, Yerram SR, Azad NA, Kern SE. A thymidylate synthase ternary complex-specific antibody, FTS, permits functional monitoring of fluoropyrimidines dosing. Oncotarget. 2012 Jul;3(7):678–85. doi: 10.18632/oncotarget.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parker WB. Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chemical reviews. 2009 Jul;109(7):2880–93. doi: 10.1021/cr900028p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matuo R, Sousa FG, Escargueil AE, Grivicich I, Garcia-Santos D, Chies JA, et al. 5-Fluorouracil and its active metabolite FdUMP cause DNA damage in human SW620 colon adenocarcinoma cell line. Journal of applied toxicology: JAT. 2009 May;29(4):308–16. doi: 10.1002/jat.1411. [DOI] [PubMed] [Google Scholar]
- 45.Ammendola M, Sacco R, Marech I, Sammarco G, Zuccala V, Luposella M, et al. Microvascular density and endothelial area correlate with Ki-67 proliferative index in surgically-treated pancreatic ductal adenocarcinoma patients. Oncology letters. 2015 Aug;10(2):967–71. doi: 10.3892/ol.2015.3286. Epub 2015/12/02. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tacelosky DM, Creecy AE, Shanmugavelandy SS, Smith JP, Claxton DF, Adair JH, et al. Calcium phosphosilicate nanoparticles for imaging and photodynamic therapy of cancer. Discovery medicine. 2012 Apr;13(71):275–85. Epub 2012/05/01. eng. [PubMed] [Google Scholar]
- 47.Pradere U, Garnier-Amblard EC, Coats SJ, Amblard F, Schinazi RF. Synthesis of nucleoside phosphate and phosphonate prodrugs. Chemical reviews. 2014 Sep 24;114(18):9154–218. doi: 10.1021/cr5002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li Q, Yan H, Liu W, Zhen H, Yang Y, Cao B. Efficacy and safety of gemcitabine-fluorouracil combination therapy in the management of advanced pancreatic cancer: a meta-analysis of randomized controlled trials. PLoS One. 2014;9(8):e104346. doi: 10.1371/journal.pone.0104346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wong PP, Demircioglu F, Ghazaly E, Alrawashdeh W, Stratford MR, Scudamore CL, et al. Dual-action combination therapy enhances angiogenesis while reducing tumor growth and spread. Cancer Cell. 2015 Jan 12;27(1):123–37. doi: 10.1016/j.ccell.2014.10.015. [DOI] [PubMed] [Google Scholar]
- 50.Skrypek N, Duchene B, Hebbar M, Leteurtre E, van Seuningen I, Jonckheere N. The MUC4 mucin mediates gemcitabine resistance of human pancreatic cancer cells via the Concentrative Nucleoside Transporter family. Oncogene. 2013 Mar 28;32(13):1714–23. doi: 10.1038/onc.2012.179. Epub 2012/05/15. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ohhashi S, Ohuchida K, Mizumoto K, Fujita H, Egami T, Yu J, et al. Down-regulation of deoxycytidine kinase enhances acquired resistance to gemcitabine in pancreatic cancer. Anticancer research. 2008 Jul-Aug;28(4B):2205–12. Epub 2008/08/30. eng. [PubMed] [Google Scholar]
- 52.van Kuilenburg AB, Hausler P, Schalhorn A, Tanck MW, Proost JH, Terborg C, et al. Evaluation of 5-fluorouracil pharmacokinetics in cancer patients with a c.1905+1G>A mutation in DPYD by means of a Bayesian limited sampling strategy. Clin Pharmacokinet. 2012 Mar 1;51(3):163–74. doi: 10.1007/BF03257473. [DOI] [PubMed] [Google Scholar]
- 53.Kline CL, El-Deiry WS. Personalizing colon cancer therapeutics: targeting old and new mechanisms of action. Pharmaceuticals. 2013;6(8):988–1038. doi: 10.3390/ph6080988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ciccolini J, Dahan L, Andre N, Evrard A, Duluc M, Blesius A, et al. Cytidine deaminase residual activity in serum is a predictive marker of early severe toxicities in adults after gemcitabine-based chemotherapies. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2010 Jan 01;28(1):160–5. doi: 10.1200/JCO.2009.24.4491. Epub 2009/11/26. eng. [DOI] [PubMed] [Google Scholar]
- 55.CND, Tate JA, Kett WC, Batra J, Demidenko E, Lewis LD, et al. Tumor cell targeting by iron oxide nanoparticles is dominated by different factors in vitro versus in vivo. PloS one. 2015;10(2):e0115636. doi: 10.1371/journal.pone.0115636. Epub 2015/02/20. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shore S, Raraty MG, Ghaneh P, Neoptolemos JP. Review article: chemotherapy for pancreatic cancer. Alimentary pharmacology & therapeutics. 2003 Dec;18(11–12):1049–69. doi: 10.1111/j.1365-2036.2003.01781.x. Epub 2003/12/05. eng. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.