

Supplemental Digital Content is available in the text.
Keywords: antipsychotic, cariprazine, post-hoc analysis, safety and tolerability, schizophrenia
Abstract
Cariprazine, a potent dopamine D3 and D2 receptor partial agonist antipsychotic with preferential binding to D3 receptors, is Food and Drug Administration approved for treating schizophrenia and manic or mixed episodes of bipolar I disorder. A post-hoc safety/tolerability analysis of data from the four acute trials in the cariprazine schizophrenia clinical development program (NCT00404573; NCT00694707; NCT01104766; NCT01104779) was carried out using the overall safety population (all patients who received ≥1 dose of study drug) and modal daily dose subgroups (1.5–3, 4.5–6, and 9–12 mg/day). These exploratory findings were summarized using descriptive statistics. Cariprazine was generally well tolerated. The incidence of treatment-emergent adverse events versus placebo was similar for cariprazine 1.5–3 mg/day and higher for cariprazine 4.5–6 and 9–12 mg/day; a dose–response relationship was observed for akathisia, extrapyramidal symptoms, and diastolic blood pressure. The mean changes in metabolic parameters were generally similar in cariprazine-treated and placebo-treated patients. There was no prolactin level increase or QTc value greater than 500 ms; small increases in mean body weight (∼1to2 kg) versus placebo were observed. Within the Food and Drug Administration-approved dose range (1.5−6 mg/day), cariprazine was generally safe and well tolerated in patients with schizophrenia.
Introduction
Schizophrenia is a severe neuropsychiatric disorder with high rates of medical morbidity and excess mortality (Goff et al., 2005). Although the introduction of first-generation typical antipsychotic medications was a significant advancement in the treatment of schizophrenia, serious acute and chronic safety and tolerability concerns, including extrapyramidal symptoms (EPS), tardive dyskinesia, and hyperprolactinemia, limited their use (Tandon, 2002). There is no consensus on what constitutes atypicality in antipsychotic medications. Clozapine was the first atypical antipsychotic approved for use in the USA and all antipsychotics introduced since have been categorized as atypical (Farah, 2005). Atypical antipsychotics have efficacy that is at least equal to typical agents; some may offer advantages in improving negative and cognitive symptoms of schizophrenia, and they induce fewer EPS than haloperidol, a potent first-generation agent (Leucht et al., 2009). Atypical antipsychotics have different pharmacological profiles, which affect the likelihood that they will cause certain treatment-induced side effects (Farah, 2005; Miyamoto et al., 2005) including metabolic effects, weight gain, akathisia, and sedation (Stanniland and Taylor, 2000; Newcomer, 2007; Henderson and Doraiswamy, 2008; Kumar and Sachdev, 2010; Cha and Mcintyre, 2012). Individuals with schizophrenia who experience an acute and sustained response with antipsychotic medication should continue treatment to maintain symptom relief and reduce relapse risk (Kreyenbuhl et al., 2010). As such, safe and tolerable antipsychotic treatment is a medical necessity.
Cariprazine, a potent dopamine D3 and D2 receptor partial agonist with preferential binding to D3 receptors, is an Food and Drug Administration (FDA)-approved treatment for schizophrenia (1.5–6 mg/day) and manic or mixed episodes associated with bipolar I disorder (3–6 mg/day). Cariprazine shows high and balanced occupancy of D3 and D2 receptors at doses effective for the treatment of psychosis (Kiss et al., 2010; Gyertyan et al., 2011; Girgis et al., 2016), which may provide benefits in treating cognitive impairment, negative symptoms, and mood symptoms associated with schizophrenia (Schwartz et al., 2000; Laszy et al., 2005; Gyertyan et al., 2008; Kiss et al., 2008). The efficacy and safety of cariprazine in schizophrenia has been supported in three positive randomized, placebo-controlled, phase II/III clinical studies (Durgam et al., 2014; Durgam et al., 2015; Kane et al., 2015). In this investigation, pooled data from these studies and one additional exploratory study (Durgam et al., 2016) were analyzed to characterize the safety and tolerability of cariprazine in patients with schizophrenia. This exploratory reanalysis of the original database of safety information from the phase II/III controlled studies in schizophrenia was based on the overall pooled safety population and cariprazine modal daily doses (most frequent dose taken by a patient during double-blind treatment); it was carried out to better characterize potential dose–response relationships for various safety parameters.
Participants and methods
The constituent study protocols were approved by the institutional review board (US sites) or the ethics committee (non-US sites); ICH-E6 Good Clinical Practice guidelines were followed and written informed consent was obtained from all participants.
Study design and patient population
The detailed methods of the included studies have been published previously; studies were similarly designed and included a 6-week double-blind treatment period (Supplementary Table 1, Supplemental digital content 1, http://links.lww.com/ICP/A28). In RGH-MD-03 (NCT00404573) (Durgam et al., 2016), patients were randomized to placebo or cariprazine 1.5–4.5 or 6–12 mg/day. In RGH-MD-16 (NCT00694707) (Durgam et al., 2014), patients were randomized to placebo, cariprazine 1.5, 3, or 4.5 mg/day, or risperidone 4 mg/day. In RGH-MD-04 (Durgam et al., 2015) (NCT01104766), patients were randomized to placebo, cariprazine 3 or 6 mg/day, or aripiprazole 10 mg/day. Risperidone and aripiprazole were included in these studies for assay sensitivity. In RGH-MD-05 (NCT01104779) (Kane et al., 2015), patients were randomized to placebo or cariprazine 3–6 or 6–9 mg/day.
The patients included (18–60 years, inclusive [18–65 years for RGH-MD-03]) had a Diagnostic and Statistical Manual of Mental Disorders, 4th ed., text revision (DSM-IV-TR) (American Psychiatric Association, 2000) diagnosis of schizophrenia for 1 year or more and a current exacerbation less than 2 weeks in duration (<4 weeks for RGH-MD-03). Patients were required to fulfill the clinical criteria for inclusion: Clinical Global Impressions-Severity (Guy, 1976b) baseline score of at least 4 (moderately ill or worse), Positive and Negative Syndrome Scale (Kay et al., 1987) total score of at least 80 and less than or equal to 120, and a score of at least 4 (moderate or higher) on at least two specified Positive and Negative Syndrome Scale items (delusions, hallucinatory behavior, suspiciousness/persecution, and conceptual disorganization).
Exclusion criteria were typical of clinical studies in schizophrenia and included the presence of various other DSM-IV-TR diagnoses (e.g. schizoaffective disorder, bipolar disorders), treatment-resistant schizophrenia, substance abuse, and active suicidal intent or past attempt within 2 years. Concurrent medical conditions that could interfere with the conduct of the study, confound the interpretation of results, or endanger the patient’s well-being were exclusionary. Drugs with psychotropic activity were not allowed, except for lorazepam for agitation, hostility, and restlessness; eszopiclone, zolpidem, chloral hydrate, or zaleplon for insomnia; and diphenhydramine, benztropine, or propranolol for EPS.
Safety assessments
Safety was assessed by adverse event (AE) recordings (all study weeks), clinical laboratory tests (screening, weeks 2, 4, 6), vital sign parameters (screening, baseline, all study weeks), weight change (baseline, all study weeks), ECGs (screening, weeks 2, 4, 6), and EPS scales (baseline, all study weeks). An AE was defined as any unfavorable and unintended sign, symptom, or disease temporally associated with the use of an investigational product, whether or not it was considered related to the use of the product. The mean changes in EPS and treatment-emergent EPS were evaluated using the Abnormal Involuntary Movement Scale (Guy, 1976a), the Barnes Akathisia Rating Scale (BARS) (Barnes, 1989), and the Simpson–Angus Scale (SAS) (Simpson and Angus, 1970). In some of the studies, ophthalmologic parameters (RGH-MD-03, RGH-MD-04, RGH-MD-05 at screening, week 6) and suicidality (RGH-MD-04, RGH-MD-05 at screening, baseline, all study weeks) were also evaluated; suicidal ideation and behavior were assessed using the Columbia-Suicide Severity Rating Scale (C-SSRS) (Posner et al., 2011). The schedule of some safety assessments differed slightly among studies (Supplementary Table 2, Supplemental digital content 2, http://links.lww.com/ICP/A29).
Statistical evaluation
Safety was evaluated in the overall safety population (all patients who received ≥1 dose of study drug) and in cariprazine subgroups stratified by modal daily dose. Modal daily dose was used to assess dose response as some of the studies included in the analyses used a flexible-dose design, which makes dose response difficult to evaluate. Three modal daily dose groups were analyzed: 1.5–3, 4.5–6, and 9–12 mg/day. The safety baseline was the last nonmissing value before the first dose of the study drug; end of treatment was the last nonmissing value during double-blind treatment. Because of the exploratory nature of these analyses, all safety parameters were summarized using descriptive statistics; between-group inferential statistical analyses were not carried out. A treatment-emergent adverse event (TEAE) was an AE that started during double-blind treatment or safety follow-up and was not present before the first dose of double-blind study drug or an AE that was present before the first dose of double-blind study drug, but increased in severity during the double-blind treatment phase or the safety follow-up phase. Treatment-emergent parkinsonism was defined as an SAS score less than or equal to 3 at baseline and greater than 3 postbaseline; treatment-emergent akathisia was defined as a BARS score less than or equal to 2 at baseline and greater than 2 postbaseline.
Results
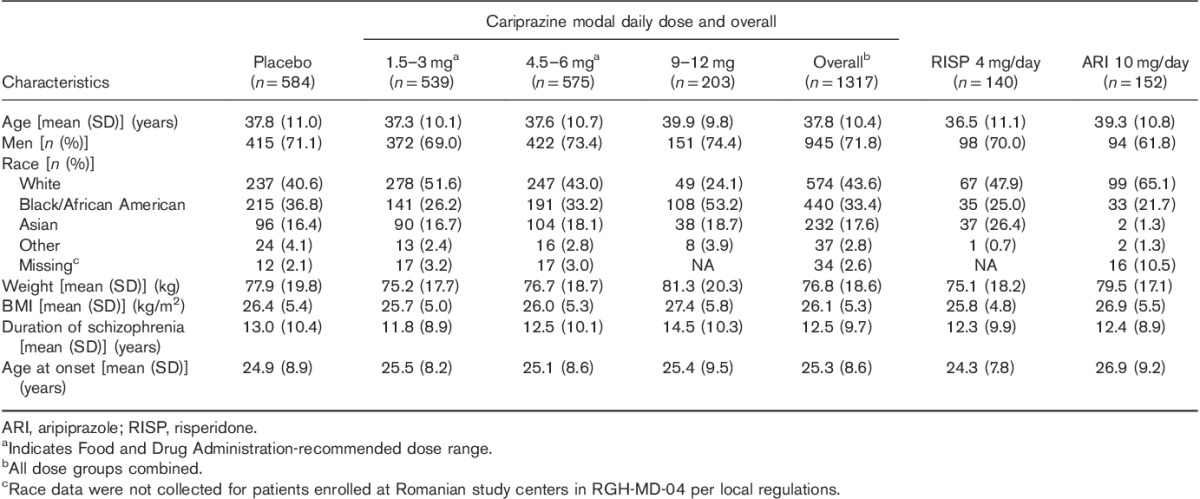
The pooled safety population included 2193 patients. Completion rates were slightly higher in cariprazine-treated patients than in placebo-treated patients, with the exception of the cariprazine 1.5–3 mg/day group, which had higher rates of withdrawn consent (Table 1). Discontinuations because of AEs and insufficient therapeutic response were greater for the placebo group than for any of the cariprazine-dose groups; withdrawal of consent was greater in the cariprazine-dose groups than in the placebo group. Baseline demographics and patient characteristics are presented in Table 2.
Table 1.
Patient disposition (safety population)
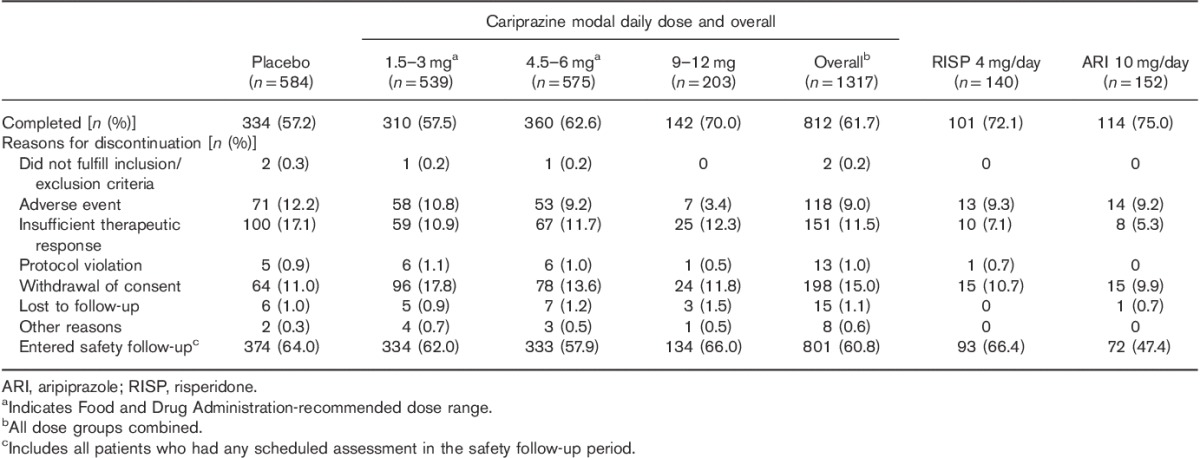
Table 2.
Patient demographic characteristics and schizophrenia history (safety population)
Safety and tolerability findings
Extent of exposure
The mean treatment duration was similar for placebo-treated and cariprazine-treated patients (32 and 33 days, respectively); the mean treatment duration in the cariprazine 1.5–3, 4.5–6, and 9–12 mg/day dose groups was 31, 33, and 36 days, respectively. The overall cariprazine exposure was 117.9 patient-years.
Adverse events
TEAEs were reported in slightly more cariprazine-treated than placebo-treated patients, and showed a dose–response relationship, with the highest rates of TEAEs observed in the 9–12 mg/day group (Table 3). Discontinuation because of AEs was higher in the placebo group (12.2%) than in the cariprazine-dose groups (1.5–3 mg/day=10.8%; 4.5–6 mg/day=9.2%; 9–12 mg/day=3.4%); the only AE leading to discontinuation in 2% of patients or greater in any treatment group was exacerbated schizophrenia. There was no observed cariprazine dose response for discontinuations because of AEs.
Table 3.
Treatment-emergent adverse events during double-blind treatment (safety population)
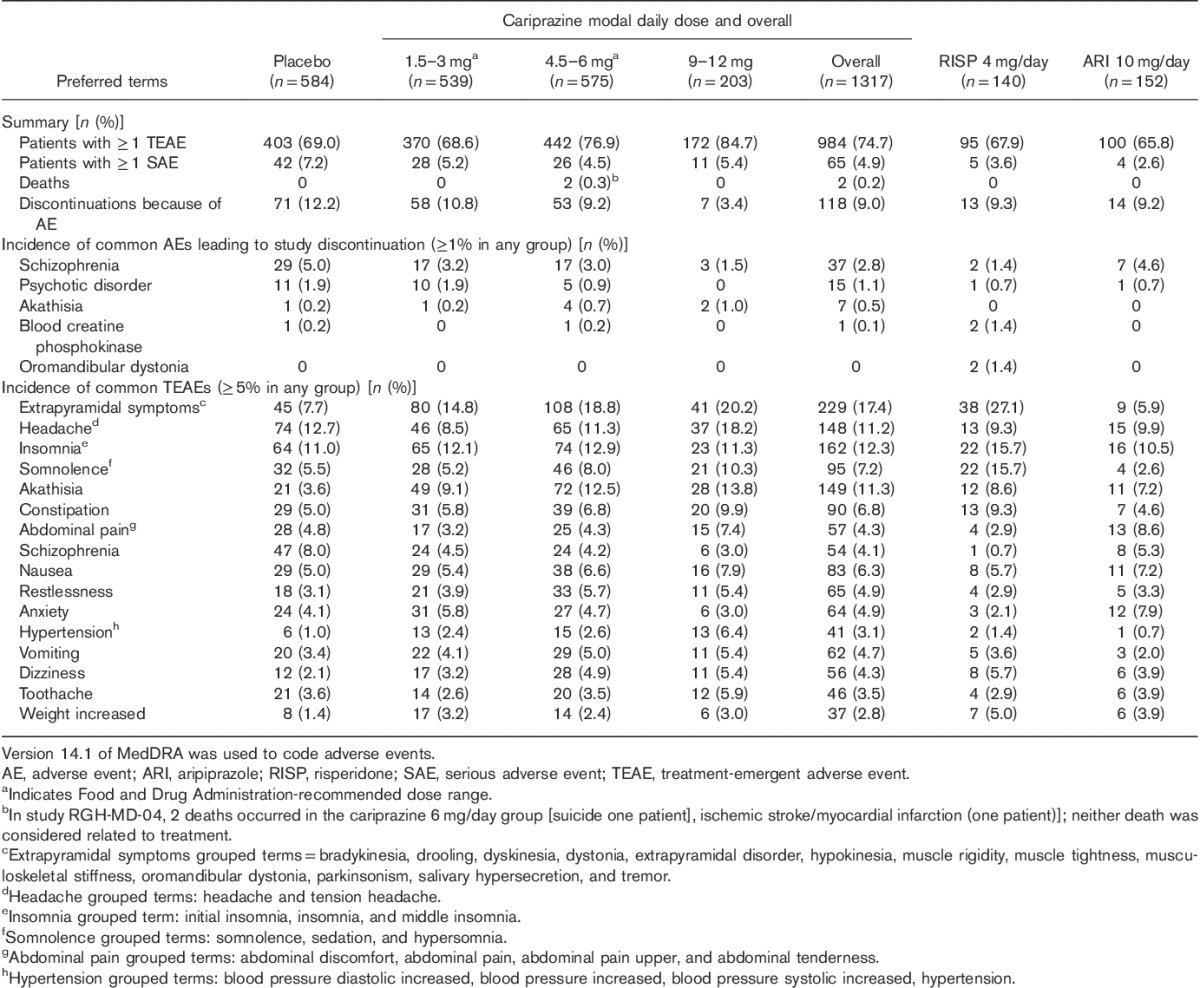
Individual TEAEs occurring at an incidence of 5% or greater in any group are presented in Table 3; many of the TEAEs showed a dose–response relationship. AEs that occurred in the overall cariprazine group at an incidence of at least 5% and twice the rate of placebo were EPS (grouped terms) and akathisia; the risk of these TEAEs was lower at doses less than or equal to 6 mg/day. A dose response was observed for the grouped term for somnolence, with similar rates observed for cariprazine 1.5–3 mg/day (5.2%) and placebo (5.5%) and higher rates for cariprazine 4.5–6 mg/day (8.0%) and 9–12 mg/day (10.3%). The majority of TEAEs were considered mild or moderate in severity; some severe TEAEs were reported in all treatment groups (placebo=8.7%; cariprazine: 1.5–3 mg/day=6.1%, 4.5–6 mg/day=5.4%, 9–12 mg/day=3.9%; risperidone=8.6%; aripiprazole=3.9%). Most TEAEs were considered to be related to treatment in all groups. A complete listing of patients in the pooled population with a TEAE is presented in Supplementary Table 3 (Supplemental digital content 3, http://links.lww.com/ICP/A30); the occurrence of a TEAE may or may not be related to the study drug.
Serious adverse events (SAEs) occurred in 4.9% of cariprazine-treated and 7.2% of placebo-treated patients, with no dose response observed for the incidence of SAEs. Schizophrenia exacerbation and worsening of psychotic disorder were the most commonly reported SAEs (placebo=2.9 and 1.5%; cariprazine=1.4 and 0.8%, respectively).
Metabolic, laboratory, ECG, and additional safety parameters
A summary of safety parameters is presented in Table 4.
Table 4.
Safety parameters: change from baseline to the end of the double-blind treatment (safety population)
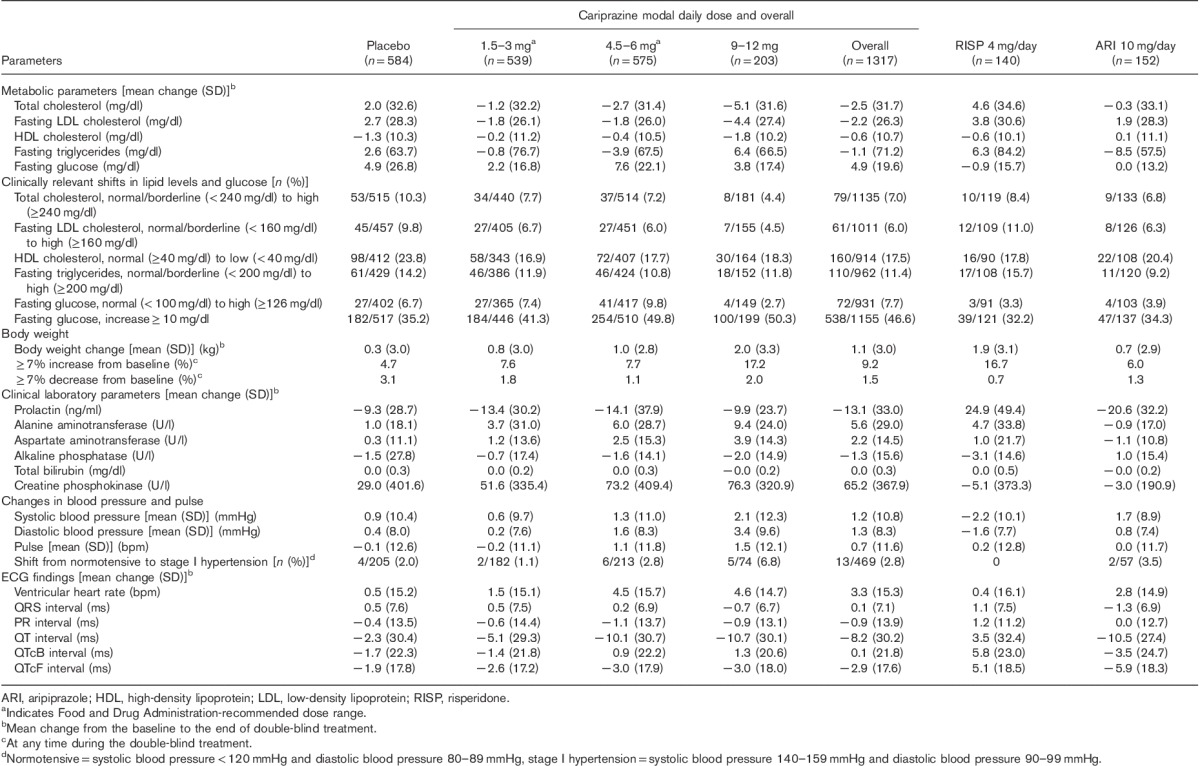
Metabolic parameters
Overall, cariprazine-treated patients showed a decrease in mean total cholesterol, fasting low-density lipoprotein cholesterol, and triglyceride levels, whereas placebo-treated patients showed increases from baseline in these parameters. The decrease in high-density lipoprotein cholesterol was similar for cariprazine and placebo, as was the increase in the mean fasting blood glucose. Clinically relevant shifts in lipid and glucose parameters were similar for cariprazine and placebo. The percentage of patients with a fasting glucose increase 10 mg/dl or more was higher in the cariprazine group than in the placebo group; no clear dose response was observed.
Greater mean increases in weight occurred in the cariprazine-treatment groups versus the placebo group. The overall cariprazine weight increase was 1.1 kg for cariprazine-treated patients compared with 0.3 kg for placebo-treated patients; weight increase of 7% or more occurred in 9.2% of cariprazine-treated patients and 4.7% of placebo-treated patients. Both mean weight change and shifts in weight of 7% or more were larger in the 9–12 mg/day dose group. Within the recommended cariprazine-dose range (1.5–6 mg/day), weight gain was 0.8–1.0 kg for cariprazine; the proportion of patients with weight increase of 7% or more was 7.6 and 7.7% for the 1.5–3 and 4.5–6 mg/day groups, respectively.
Clinical laboratory values
Small increases occurred in alanine aminotransferase and aspartate aminotransferase levels with cariprazine relative to placebo; there were no mean increases in bilirubin levels with cariprazine or placebo, and alkaline phosphatase levels decreased slightly in both groups. No cariprazine patient fulfilled Hy’s Law criteria [alanine aminotransferase or aspartate aminotransferase≥3×upper limit of normal (ULN) with concurrent total bilirubin≥2×ULN and alkaline phosphatase<2×ULN]. The mean prolactin levels decreased in all treatment groups, except for risperidone; decreases were greater for cariprazine-treated women (−26.0) than for cariprazine-treated men (−8.1).
There was a modest dose-related increase in the mean creatine phosphokinase (CPK) level with cariprazine. However, large fluctuations in CPK levels occurred over time as evidenced by large SDs and a relatively high percentage of patients entered the studies with elevated CPK levels. Treatment-emergent CPK levels more than 1000 U/l were similar for placebo (3.5%), overall cariprazine (4.7%), and risperidone (4.5%) and lower for aripiprazole (2.0%); ∼60% of events resolved during the study for patients in all treatment groups. Elevations greater than 1.5×ULN occurred in patients in all treatment groups (placebo=14.2%, cariprazine=22.7%, risperidone=23.0%, aripiprazole=18.9%). Most elevations greater than 1.5×ULN did not resolve during treatment, but data should be interpreted with caution as laboratory tests were not scheduled after the end of treatment. CPK elevations were not associated with altered renal function or renal failure.
Cardiac measures
Overall, no clinically significant changes in pulse or blood pressure were observed in cariprazine-treated patients, with the exception of patients in the 9–12 mg/day dose group, where the mean increase in diastolic blood pressure was 3.4 mmHg and five (6.8%) patients shifted from baseline normotensive values to stage I hypertension. No mean increases were observed in the overall cariprazine group versus the placebo group on ECG parameters; no cariprazine patient had a QTc interval greater than 500 ms (Bazett or Fridericia formula).
Orthostatic hypotension
The incidence of orthostatic hypotension (reduction of ≥20 mmHg in systolic blood pressure or ≥10 mmHg in diastolic blood pressure while changing from a supine position to standing) was similar for placebo (12.3%) and cariprazine (13.4%). No syncopal episodes were reported in the controlled schizophrenia studies.
Extrapyramidal symptoms
A dose–response relationship was observed for the incidence of akathisia and EPS AEs (excluding akathisia and restlessness). Most events of akathisia and EPS were considered mild or moderate in severity; AEs of akathisia that were considered severe occurred in one (4.8%) of 21 placebo-treated patients and four (2.7%) of 149 cariprazine-treated patients (1.5–3 and 9–12 mg/day=one patient each; 4.5–6 mg=two patients). Severe EPS-related AEs (excluding akathisia and restlessness) were reported in one (2.2%) of 45 placebo-treated patients and three (1.3%) of 229 cariprazine-treated patients (4.5–6 mg/day=two patients; 9–12 mg/day=one patient). Rates of discontinuation for EPS TEAEs, including akathisia and restlessness, were less than or equal to 2% among cariprazine-treated patients and no akathisia, restlessness, or EPS SAEs were reported. EPS was assessed by rating scales and TEAEs (Table 5); a greater percentage of cariprazine-treated than placebo-treated patients had treatment-emergent parkinsonism (SAS total score ≤3 at baseline and >3 postbaseline) and akathisia (BARS total score ≤2 at baseline and >2 postbaseline).
Table 5.
Extrapyramidal symptoms (safety population)
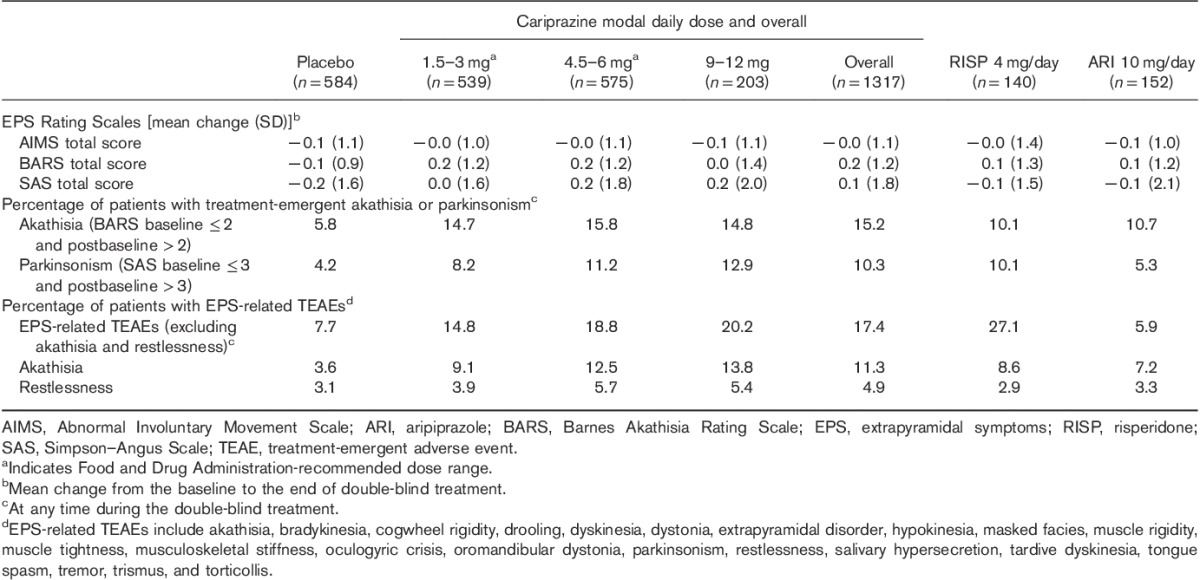
During double-blind treatment, the mean duration of akathisia was 24 days for cariprazine-treated and 29 days for placebo-treated patients; the peak onset of akathisia generally occurred within 2–3 weeks of initiation of cariprazine treatment. A greater percentage of cariprazine-treated patients (21.9%) than placebo-treated patients (8.7%) received EPS medication. In accordance with the constituent study protocols, investigators used beta-blocking agents (e.g. propranolol) and antiparkinson medications (e.g. benztropine and trihexyphenidyl) to manage symptoms of akathisia and EPS. Among active-treatment groups, the use of any EPS medication was the highest for risperidone (39/140, 27.9%) and the lowest for aripiprazole (18/152, 11.8%); EPS medications were used by 21.9% (288/1317) of cariprazine-treated patients. A lower rate of medication use for EPS-related AEs (excluding akathisia and restlessness) was observed in patients with mild AEs (96/158; 60.8%) than in patients with moderate (58/68; 85.3%) or severe (3/3; 100%) AEs. Antiparkinson drugs were used to control EPS in 25.0% (35/140) of risperidone-treated patients, 17.3% (228/1317) of cariprazine-treated patients, and 7.9% (12/152) of aripiprazole-treated patients.
Suicidal behavior
Per C-SSRS assessment, suicidal ideation was reported in 4.7% of placebo-treated and 3.6% of cariprazine-treated patients, with the majority of instances in the least severe category. No suicidal behavior was recorded by the C-SSRS in the placebo-treatment or the cariprazine-treatment groups, including patients who had akathisia or restlessness. The incidence of suicidality TEAEs for cariprazine (0.4%) was low and similar to placebo (0.2%); suicidal ideation was reported as a TEAE in one placebo-treated patient and four cariprazine-treated patients. There were two suicidality SAEs in the cariprazine group, including one completed suicide that was not considered to be treatment related.
Ophthalmologic parameters
Change from baseline in ophthalmologic parameters is presented in Supplementary Table 4 (Supplemental digital content 4, http://links.lww.com/ICP/A31). The most commonly reported ocular TEAE was blurred vision (placebo=0.3%; cariprazine=1.5%; risperidone=2.1%, aripiprazole=1.3%); all other ocular AEs were reported in less than 1% of patients in any group. No AEs of cataract or lens opacity were reported.
Discussion
In this pooled analysis of safety data from 4 short-term randomized, controlled studies in adult patients with acute exacerbation of schizophrenia, cariprazine at doses up to 12 mg/day was generally safe and well tolerated; patients receiving doses above 6 mg/day showed higher likelihood for AEs, EPS, akathisia, and blood pressure changes. The most frequent TEAEs in cariprazine overall (reported in ≥5% of cariprazine-treated patients and at least twice the rate of placebo) were EPS reported by grouped terms and akathisia. A dose–response relationship was observed for akathisia and EPS in cariprazine-treated patients. No cariprazine dose response was observed for SAEs and discontinuations because of AEs. The most commonly reported SAEs and discontinuations because of AEs were associated with worsening of schizophrenia or psychotic symptoms.
Of the distinct TEAEs associated with atypical antipsychotics, those that increase the risk for cardiometabolic disease are among the most important (Hennekens, 2007). Cardiovascular disease is the second leading cause of premature death for patients with schizophrenia, surpassed only by suicide (Goff et al., 2005). Compared with the general population, the prevalence of several modifiable risk factors associated with cardiovascular disease, including obesity, smoking, diabetes, and hypertension, is high in patients with schizophrenia (Newcomer, 2007). The propensity for medication-induced metabolic and weight TEAEs in this at-risk population varies among atypical antipsychotics (Newcomer, 2007; Riordan et al., 2011). For example, the incidence of clinically significant weight gain (≥7%) associated with atypical antipsychotics has been reported to range from 8.1% for aripiprazole (Abilify, 2014) to 22.2% for olanzapine (Zyprexa, 2011).
As such, the cardiometabolic profile of cariprazine is important to patients with schizophrenia. In our analyses, the mean changes and clinically relevant shifts in lipid parameters and fasting glucose were generally similar for placebo and cariprazine, suggesting that short-term cariprazine exposure had minimal effects on these parameters; glucose increase of more than 10 dl was greater in cariprazine than in the other treatment groups. No QTc values greater than 500 ms were observed; overall, there were no clinically significant changes in pulse or blood pressure in cariprazine-treated patients, however dose-related increases in blood pressure were observed. The mean body weight increase was greater in all active-treatment groups than in the placebo group. For cariprazine, the mean increase in body weight was lower in the lower-dose groups than in the 9–12 mg group. Weight increase for cariprazine in the approved dose range was similar to aripiprazole, and in the 9–12 mg/day group, it was similar to risperidone. In the approved dose range, weight gain of 7% or more occurred in ∼8% of cariprazine patients, which was less than that in the cariprazine 9–12 mg (17.2%) and risperidone (16.7%) groups, and similar to the aripiprazole (6.0%) group. Collectively, these values suggest that cariprazine in the approved dose range is on the lower end of the atypical antipsychotic weight gain hierarchy.
Despite improved safety profiles for atypical antipsychotics versus first-generation agents, EPS and akathisia are consistently noted in the literature (Caroff et al., 2002; Kane et al., 2009; Kumar and Sachdev, 2009; Kane et al., 2010; Poyurovsky, 2010). Analyses from the CATIE study found no significant differences in the incidence of EPS for some second-generation antipsychotics (olanzapine, quetiapine, risperidone, ziprasidone) relative to each other or to perphenazine, a mid-potency first-generation agent (Miller et al., 2008). In the current analyses, akathisia was among the most frequent TEAEs for cariprazine and both active-comparator groups. A dose–response relationship was observed for cariprazine, with a lower risk for akathisia and EPS (grouped terms) in the approved dose range. The incidence of akathisia TEAEs in the lowest cariprazine-dose group (9.1%) was slightly higher than aripiprazole (7.2%) and risperidone (8.6%), and EPS across the cariprazine-dose groups (14.8–20.2%) was lower than risperidone (27.1%). At doses up to 6 mg/day, the rates of akathisia with cariprazine were similar to some antipsychotic agents marketed in the USA (e.g. lurasidone=6–22%, asenapine=4–11%) (Latuda, 2013; Saphris, 2014) and EPS was lower than others (risperidone and olanzapine=20–30%) (Zyprexa, 2011; Risperdal, 2014).
In clinical practice, akathisia is a routinely managed treatment-induced symptom (Lehman et al., 2004). For cariprazine, most EPS-related TEAEs were considered mild or moderate and resulted in relatively few discontinuations. Medication use to manage EPS generally correlated with symptom severity. In short-term aripiprazole schizophrenia trials, the rates of akathisia that were considered severe (<10%) or led to study discontinuation (0.3%) (Kane et al., 2010) were consistent with this cariprazine analysis. Collectively, these results suggest that akathisia and EPS were manageable for most cariprazine-treated patients.
Another noteworthy safety consideration for cariprazine-treated patients was the absence of increase in the prolactin level, which is important since hyperprolactinemia increases the risk of sexual function side effects (Henderson and Doraiswamy, 2008). In addition, although some atypical antipsychotics produce undesirable sedation and somnolence (Kane and Sharif, 2008), these effects for overall cariprazine were similar to placebo, less than risperidone, and slightly greater than (sedation) or equal to (somnolence) aripiprazole. The rates of sedation were lower in the cariprazine approved dose range than in the 9–12 mg/day group. A modest dose-related increase in the mean CPK level, which did not result in altered renal function, was observed in cariprazine-treated patients; large SDs suggest considerable fluctuations in CPK level over time, although cariprazine may cause transient increases that normalize with continued treatment. Cariprazine was not associated with an increased risk of suicidality, an essential finding considering that individuals with schizophrenia compared with the general population have a 12-fold higher risk of dying from suicide (Saha et al., 2007). Ophthalmologic testing indicated no evidence of retinal toxicity or clinically significant lenticular changes.
These analyses were subject to the limitations inherent in post-hoc analyses. Safety parameters were analyzed by descriptive statistics; no inferential statistical analyses were carried out. A tolerability bias in favor of patients in the higher modal daily dose groups may exist as patients with tolerability issues at lower doses may have been more likely to discontinue the study and less likely to increase their dosage. Short treatment duration and the inclusion of active comparators in only two studies are additional limitations.
Conclusion
In this pooled analysis of four short-term clinical trials, cariprazine at doses of 1.5–12 mg/day was generally safe and well tolerated. Patients in the modal dose group above 6 mg/day showed a higher likelihood for AEs, EPS, akathisia, weight increases, and blood pressure changes, as well as higher rates of CPK and transaminase elevations. This observation, together with the beneficial effects observed on efficacy, resulted in the FDA approving a recommended dose range for schizophrenia of 1.5–6 mg/day. Within the FDA-approved dose range, cariprazine was a generally safe and well-tolerated treatment option for patients with schizophrenia.
Supplementary Material
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website (www.intclinpsychopharm.com).
Acknowledgements
Writing assistance and editorial support for the preparation of this manuscript were provided by Carol Brown, MS, of Prescott Medical Communications Group, a contractor of Allergan.
This study was funded by Forest Research Institute Inc., an Allergan affiliate (Jersey City, New Jersey), and Gedeon Richter Plc. (Budapest, Hungary). Forest Research Institute Inc., an Allergan affiliate, and Gedeon Richter Plc. were involved in the study design, collection (through contracted clinical investigator sites), analysis, and interpretation of data, and the decision to present these results.
Conflicts of interest
Willie Earley, Suresh Durgam, and Kaifeng Lu are employees of Allergan. István Laszlovszky is an employee of Gedeon Richter Plc. Marc Debelle is a former employee of Gedeon Richter Plc. John M. Kane serves or has served as a consultant for Alkermes Plc.; Bristol-Myers Squibb Company; Eli Lilly and Company; Forest Laboratories Inc.; FORUM Pharmaceuticals; Genentech Inc., a Member of the Roche Group; H. Lundbeck A/S; Hoffmann-La Roche Inc.; Intra-Cellular Therapies Inc.; Janssen Pharmaceuticals Inc.; Johnson & Johnson Services Inc.; Otsuka Pharmaceutical Co. Ltd; Pierre Fabre; Reviva Pharmaceuticals Inc.; and Teva. He also serves or has served on speaker bureaus for Bristol-Myers Squibb Company; Genentech Inc., a member of the Roche Group; Janssen Pharmaceuticals Inc.; and Otsuka Pharmaceutical Co. Ltd. He owns or has owned shares in LB Pharmaceuticals; MedAvante Inc.; and Vanguard Research Group.
References
- Abilify (2014). Prescribing information; Princeton, NJ. Rockville, MD: Otsuka America Pharmaceuticals Inc. [Google Scholar]
- American Psychiatric Association (2000). Diagnostic and Statistical Manual of Mental Disorders, fourth edition, text revision. Washington, DC: American Psychiatric Association. [Google Scholar]
- Barnes TR. (1989). A rating scale for drug-induced akathisia. Br J Psychiatry 154:672–676. [DOI] [PubMed] [Google Scholar]
- Caroff SN, Mann SC, Campbell EC, Sullivan KA. (2002). Movement disorders associated with atypical antipsychotic drugs. J Clin Psychiatry 63 (Suppl 4): 12–19. [PubMed] [Google Scholar]
- Cha DS, Mcintyre RS. (2012). Treatment-emergent adverse events associated with atypical antipsychotics. Expert Opin Pharmacother 13:1587–1598. [DOI] [PubMed] [Google Scholar]
- Durgam S, Starace A, Li D, Migliore R, Ruth A, Nemeth G, et al. (2014). An evaluation of the safety and efficacy of cariprazine in patients with acute exacerbation of schizophrenia: a phase II, randomized clinical trial. Schizophr Res 152:450–457. [DOI] [PubMed] [Google Scholar]
- Durgam S, Cutler AJ, Lu K, Migliore R, Ruth A, Laszlovszky I, et al. (2015). Cariprazine in acute exacerbation of schizophrenia: a fixed-dose, phase 3, randomized, double-blind, placebo- and active-controlled trial. J Clin Psychiatry 76:e1574–e1582. [DOI] [PubMed] [Google Scholar]
- Durgam S, Litman RE, Papadakis K, Li D, Nemeth G, Laszlovszky I. (2016). Cariprazine in the treatment of schizophrenia: a proof-of-concept trial. Int Clin Psychopharmacol 31:61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farah A. (2005). Atypicality of atypical antipsychotics. Prim Care Companion J Clin Psychiatry 7:268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girgis RR, Slifstein M, D'Souza D, Lee Y, Periclou A, Ghahramani P, et al. (2016). Preferential binding to dopamine D3 over D2 receptors by cariprazine in patients with schizophrenia using PET with the D3/D2 receptor ligand [11C]-(+)-PHNO. Psychopharmacology (Berl) 233:3503–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff DC, Cather C, Evins AE, Henderson DC, Freudenreich O, Copeland PM, et al. (2005). Medical morbidity and mortality in schizophrenia: guidelines for psychiatrists. J Clin Psychiatry 66:183–194. [DOI] [PubMed] [Google Scholar]
- Guy W. (1976a). The Abnormal Movement Scale, ECDEU assessment manual for psychopharmacology. Rockville, MD: National Institutes of Mental Health; 218–222. [Google Scholar]
- Guy W.Guy W. (1976b). Clinical global impressions. ECDEU assessment manual for psychopharmacology: publication ADM. Rockville, MD: National Institute of Mental Health, Psychopharmacology Research Branch; 76–338. [Google Scholar]
- Gyertyan I, Saghy K, Laszy J, Elekes O, Kedves R, Gemesi LI, et al. (2008). Subnanomolar dopamine D3 receptor antagonism coupled to moderate D2 affinity results in favourable antipsychotic-like activity in rodent models: II. behavioural characterisation of RG-15. Naunyn Schmiedebergs Arch Pharmacol 378:529–539. [DOI] [PubMed] [Google Scholar]
- Gyertyan I, Kiss B, Saghy K, Laszy J, Szabo G, Szabados T, et al. (2011). Cariprazine (RGH-188), a potent D3/D2 dopamine receptor partial agonist, binds to dopamine D3 receptors in vivo and shows antipsychotic-like and procognitive effects in rodents. Neurochem Int 59:925–935. [DOI] [PubMed] [Google Scholar]
- Henderson DC, Doraiswamy PM. (2008). Prolactin-related and metabolic adverse effects of atypical antipsychotic agents. J Clin Psychiatry 69 (Suppl 1): 32–44. [PubMed] [Google Scholar]
- Hennekens CH. (2007). Increasing global burden of cardiovascular disease in general populations and patients with schizophrenia. J Clin Psychiatry 68 (Suppl 4):4–7. [PubMed] [Google Scholar]
- Kane JM, Sharif ZA. (2008). Atypical antipsychotics: sedation versus efficacy. J Clin Psychiatry 69 (Suppl 1):18–31. [PubMed] [Google Scholar]
- Kane JM, Fleischhacker WW, Hansen L, Perlis R, Pikalov A, 3rd, Assuncao-Talbott S. (2009). Akathisia: an updated review focusing on second-generation antipsychotics. J Clin Psychiatry 70:627–643. [DOI] [PubMed] [Google Scholar]
- Kane JM, Barnes TR, Correll CU, Sachs G, Buckley P, Eudicone J, et al. (2010). Evaluation of akathisia in patients with schizophrenia, schizoaffective disorder, or bipolar I disorder: a post hoc analysis of pooled data from short- and long-term aripiprazole trials. J Psychopharmacol 24:1019–1029. [DOI] [PubMed] [Google Scholar]
- Kane JM, Zukin S, Wang Y, Lu K, Ruth A, Nagy K, et al. (2015). Efficacy and safety of cariprazine in acute exacerbation of schizophrenia: results from an international, phase III clinical trial. J Clin Psychopharmacol 35:367–373. [DOI] [PubMed] [Google Scholar]
- Kay SR, Fiszbein A, Opler LA. (1987). The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr Bull 13:261–276. [DOI] [PubMed] [Google Scholar]
- Kiss B, Laszlovszky I, Horvath A, Nemethy Z, Schmidt E, Bugovics G, et al. (2008). Subnanomolar dopamine D3 receptor antagonism coupled to moderate D2 affinity results in favourable antipsychotic-like activity in rodent models: I. neurochemical characterisation of RG-15. Naunyn Schmiedebergs Arch Pharmacol 378:515–528. [DOI] [PubMed] [Google Scholar]
- Kiss B, Horvath A, Nemethy Z, Schmidt E, Laszlovszky I, Bugovics G, et al. (2010). Cariprazine (RGH-188), a dopamine D(3) receptor-preferring, D(3)/D(2) dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther 333:328–340. [DOI] [PubMed] [Google Scholar]
- Kreyenbuhl J, Buchanan RW, Dickerson FB, Dixon LB. (2010). The Schizophrenia Patient Outcomes Research Team (PORT): updated treatment recommendations 2009. Schizophr Bull 36:94–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Sachdev PS. (2009). Akathisia and second-generation antipsychotic drugs. Curr Opin Psychiatry 22:293–299. [DOI] [PubMed] [Google Scholar]
- Laszy J, Laszlovszky I, Gyertyan I. (2005). Dopamine D3 receptor antagonists improve the learning performance in memory-impaired rats. Psychopharmacology (Berl) 179:567–575. [DOI] [PubMed] [Google Scholar]
- Latuda (2013). Prescribing information. Marlborough, MA: Sunovion Pharmaceuticals Inc. [Google Scholar]
- Lehman AF, Lieberman JA, Dixon LB, Mcglashan TH, Miller AL, Perkins DO, et al. (2004). Practice guideline for the treatment of patients with schizophrenia, second edition. Am J Psychiatry 161:1–56. [PubMed] [Google Scholar]
- Leucht S, Corves C, Arbter D, Engel RR, Li C, Davis JM. (2009). Second-generation versus first-generation antipsychotic drugs for schizophrenia: a meta-analysis. Lancet 373:31–41. [DOI] [PubMed] [Google Scholar]
- Miller DD, Caroff SN, Davis SM, Rosenheck RA, Mcevoy JP, Saltz BL, et al. (2008). Extrapyramidal side-effects of antipsychotics in a randomised trial. Br J Psychiatry 193:279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto S, Duncan GE, Marx CE, Lieberman JA. (2005). Treatments for schizophrenia: a critical review of pharmacology and mechanisms of action of antipsychotic drugs. Mol Psychiatry 10:79–104. [DOI] [PubMed] [Google Scholar]
- Newcomer JW. (2007). Antipsychotic medications: metabolic and cardiovascular risk. J Clin Psychiatry 68 (Suppl 4):8–13. [PubMed] [Google Scholar]
- Posner K, Brown GK, Stanley B, Brent DA, Yershova KV, Oquendo MA, et al. (2011). The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry 168:1266–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poyurovsky M. (2010). Acute antipsychotic-induced akathisia revisited. Br J Psychiatry 196:89–91. [DOI] [PubMed] [Google Scholar]
- Riordan HJ, Antonini P, Murphy MF. (2011). Atypical antipsychotics and metabolic syndrome in patients with schizophrenia: risk factors, monitoring, and healthcare implications. Am Health Drug Benefits 4:292–302. [PMC free article] [PubMed] [Google Scholar]
- Risperdal (2014). Prescribing information. Titusville, NJ: Janssen LP. [Google Scholar]
- Saha S, Chant D, Mcgrath J. (2007). A systematic review of mortality in schizophrenia: is the differential mortality gap worsening over time? Arch Gen Psychiatry 64:1123–1131. [DOI] [PubMed] [Google Scholar]
- Saphris (2014). Prescribing information. St Louis, MO: Forest Pharmaceuticals Inc. [Google Scholar]
- Schwartz JC, Diaz J, Pilon C, Sokoloff P. (2000). Possible implications of the dopamine D(3) receptor in schizophrenia and in antipsychotic drug actions. Brain Res Brain Res Rev 31:277–287. [DOI] [PubMed] [Google Scholar]
- Simpson GM, Angus JW. (1970). A rating scale for extrapyramidal side effects. Acta Psychiatr Scand Suppl 212:11–19. [DOI] [PubMed] [Google Scholar]
- Stanniland C, Taylor D. (2000). Tolerability of atypical antipsychotics. Drug Saf 22:195–214. [DOI] [PubMed] [Google Scholar]
- Tandon R. (2002). Safety and tolerability: how do newer generation "atypical" antipsychotics compare? Psychiatr Q 73:297–311. [DOI] [PubMed] [Google Scholar]
- Zyprexa (2011). Prescribing information. Indianapolis, IN: Eli Lily and Company. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website (www.intclinpsychopharm.com).