

Abstract
Nonischemic dilated cardiomyopathy often has a genetic etiology. Because of the large number of genes and alleles attributed to dilated cardiomyopathy, comprehensive genetic testing encompasses ever-increasing gene panels. Genetic diagnosis can help predict prognosis, especially with regard to arrhythmia risk for certain subtypes. Moreover, cascade genetic testing in family members can identify those who are at-risk or with early stage disease, offering the opportunity for early intervention. This review will address diagnosis and management of dilated cardiomyopathy, including the role of genetic evaluation. We will also overview distinct genetic pathways linked to dilated cardiomyopathy and their pathogenetic mechanisms. Historically, cardiac morphology has been used to classify cardiomyopathy subtypes. Determining genetic variants is emerging as an additional adjunct to help further refine subtypes of dilated cardiomyopathy, especially where arrhythmia risk is increased, and ultimately contribute to clinical management.
Keywords: congestive heart failure, dilated cardiomyopathy, mutations, lamin A/C, titin, genetic testing, therapy, diagnostic method, arrhythmia
Subject Terms: Cardiomyopathy, Genetics
Prevalence and etiology of dilated cardiomyopathy
Cardiomyopathies are defined as myocardial disorders in which the heart is structurally and functionally abnormal. Morphologically defined subtypes include hypertrophic (HCM), dilated (DCM), arrhythmogenic (AC) and left ventricular noncompaction (LVNC) cardiomyopathies1, 2, and each of these subtypes can be genetically mediated (Figure 1). DCM is characterized by an enlarged and poorly contractile left ventricle (LV). DCM can be attributed to genetic and nongenetic causes including hypertension, valve disease, inflammatory/infectious causes and toxins3. Even these “nongenetic” forms of cardiomyopathy, can be influenced by an individual’s genetic profile and, furthermore, mixed etiologies may be present. In DCM, the degree of LV systolic dysfunction is variable, and LV systolic dysfunction is often progressive. DCM is a major risk factor for developing heart failure (HF) as the presence of reduced systolic function does not imply symptoms. Notably, DCM is often associated with an increased risk of severe arrhythmia indicating the pathological involvement of the cardiac conduction system.
Figure 1. Echocardiography demonstrates forms of cardiomyopathy.
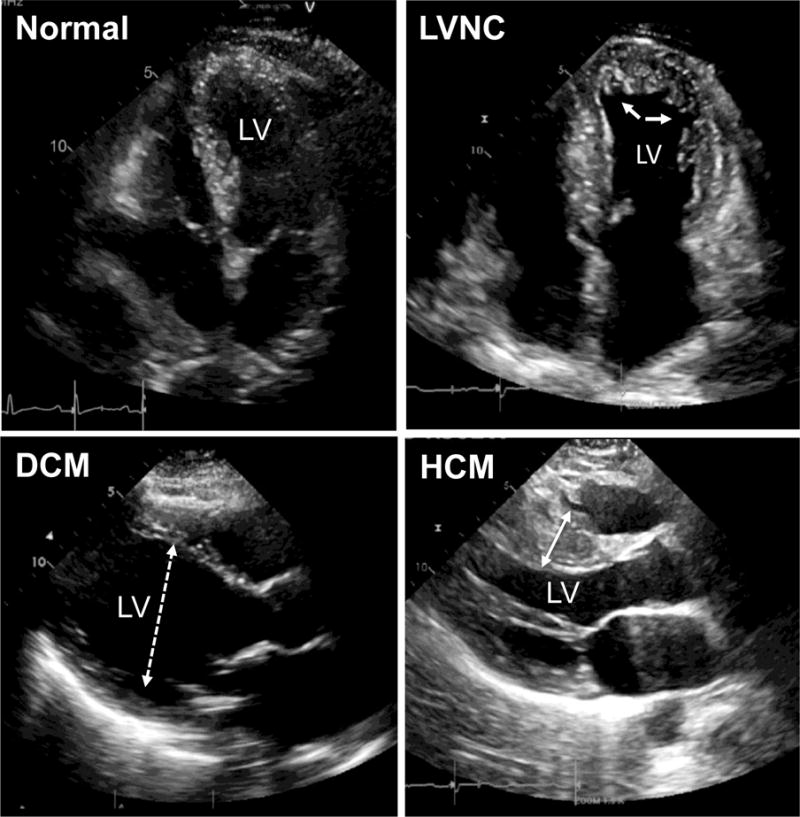
Left ventricular noncompaction cardiomyopathy (LVNC) is shown in the upper right (arrows indicate deep trabeculations in the left ventricle (LV). Dilated cardiomyopathy (DCM) is defined by enlarged LV diameters (dashed double sided arrow). Hypertrophic cardiomyopathy (HCM) is defined with a thickened LV, including the septum (marked with double sided arrow).
Randomized clinical heart failure trials typically report 30–40% of subjects with a nonischemic DCM compared to ischemic DCM3. Clinical trials are evaluating interventions to reduce CHF symptoms, these studies may focus on the later stages of disease. Similarly, a recent survey of hospitalized patients in the United States in which the mean age was 75 years (n=156,013) found that ischemic cardiomyopathy was more common than nonischemic (59% compared to 41%)4. Of nonischemic DCM, hypertension accounted for 48% and idiopathic was the next most common at 31%. In this study, individuals with nonischemic DCM were more likely to be female, nonwhite, and younger that those with ischemic cardiomyopathy.
The true prevalence of DCM, and of genetically mediated DCM, is not fully known. An early estimate of DCM prevalence derived from a study carried out from 1975 to 1984 in Olmstead County, MN, USA5. This epidemiological study relied on echocardiography, angiography or autopsy evaluation of DCM cases and resulted in a prevalence of 36.5/100,000 individuals, or 1 in 2,700 with a male to female ratio of 3.4 in a European-American population5. The prevalence of DCM varies likely reflecting geographic and ethnic differences, as well as the methodologies used6–10. Studies from England (8.3/100,000)11, Italy (7.0/100,000)12, and Japan (14/100,000)9, each report similar DCM prevalence. However, the prevalence of DCM is likely underestimated. The prior studies relied on older less sensitive imaging modalities13. More recently, Hershberger and colleagues used a different approach to estimate DCM prevalence, based on the known ratio of idiopathic DCM to HCM of approximately 2:1, prevalence estimates of heart failure and prevalence estimates of left ventricular dysfunction as a surrogate for DCM14. With this approach, a much higher prevalence of DCM is estimated, in the range of 1:250. Similarly, estimates of familial DCM prevalence varies: a meta-analysis of 23 studies found a prevalence estimate of 23% with a range of 2% to 65% indicating a significant heterogeneity in diagnostic criteria, and a frequency progressively increasing over time due to more systematic clinical screening15. In the clinical practice and in the current guidelines, the prevalence of familial DCM is assumed to be around 30 – 50%1, 3, 13, 16–18. In patients with familial DCM, approximately 40% has an identifiable genetic cause17. Also in sporadic DCM pathogenic genetic variants can be identified, although the frequency of genetic causes in this population is not well defined17.
Clinical diagnosis of DCM
DCM has been defined by the presence of: a) fractional shortening (FS) less than 25% (> 2SD) and/or ejection fraction less than 45% (> 2SD); and b) left ventricular end diastolic diameter (LVEDD) greater than 117% (>2SD of the predicted value of 112% corrected for age and body surface area, BSA), excluding any known cause of myocardial disease19. In the context of a familial DCM, these criteria are used to diagnose the proband in a family19 and the strategy for evaluation is shown in Figure 2. Familial DCM is defined by the presence of: a) two or more affected relatives with DCM meeting the above criteria; or b) a relative of a DCM patient with unexplained sudden death before the age of 35 years19. Family members may be classified as affected, unaffected, or unknown when subtle cardiac abnormalities are present but not sufficient for a definitive diagnosis. Less common forms of primary cardiomyopathies are peripartum, tachycardia-induced, stress-provoked Takotsubo cardiomyopathy and myocarditis, according to the 2006 AHA definition and classification2. Interestingly, myocarditis and peripartum cardiomyopathy can occur in a familial setting and are believed to have a genetic component20–22. Secondary forms of cardiomyopathies, in which cardiomyopathy arises from systemic disorders such as amyloidosis, hemochromatosis, and due to toxicity from agents like doxorubicin are also under genetic influence or may arise due to primary genetic mutations23, 24. Neuromuscular disease is also commonly associated with cardiomyopathy, and cardiomyopathy typically arises from the primary responsible genetic mutation exerting pathological effects directly in the myocardium (see below). With the increase in using genetic testing, especially testing in family members, it is now possible to use cardiac imaging modalities to ascertain early features of disease in gene mutation positive individual who do not fully manifest disease. Since many of these studies examine gene positive individuals at one point in time, a full view of DCM is progression has not been delineated. Definitive studies on DCM in progression in genetically at-risk individuals must span many years. Imaging studies have identified chamber size dimensions, strain abnormalities and contrast enhancement each as features of early DCM23.
Figure 2. Algorithm for the management of a patient with nonischemic DCM.
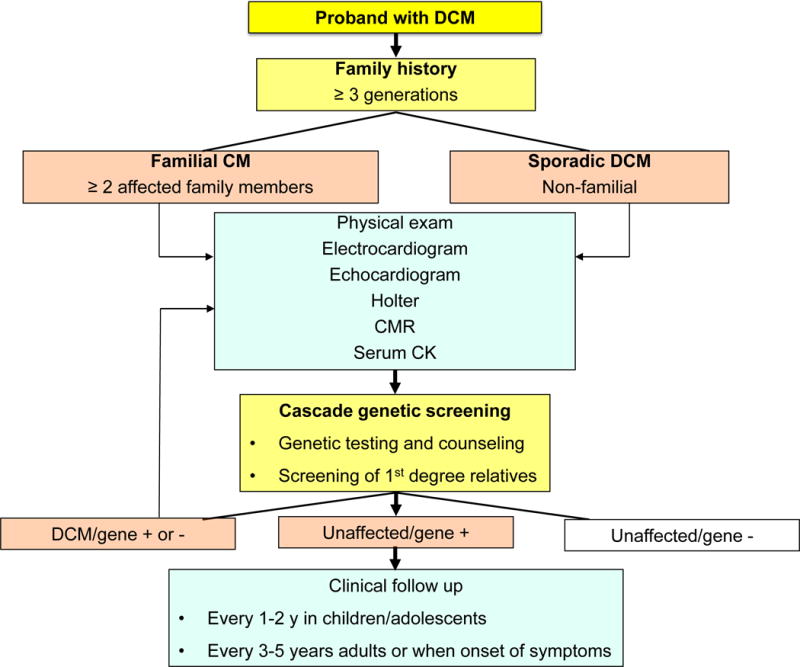
CMR: cardiac magnetic resonance, CK: creatine kinase. DCM patients should undergo an accurate family history examination. A comprehensive exam should include serum CK to evaluate skeletal muscle involvement. Genetic testing and genetic counseling should be offered to DCM patients, regardless of family history; 1st degree relatives should be examined (physical exam, ECG, echocardiogram). A positive genetic testing in the proband offers the possibility of a confirmatory genetic testing in relatives, which may guide follow up and need of further testing. Adapted from14, 19, 140, 164
Imaging - Echocardiography
To diagnose DCM, LV measurements can be determined using multiple imaging modalities. M-mode and 2-dimensional echocardiography are frequently used to determine LV internal dimensions in systole and diastole (LVIDd, LVIDs, respectively) (Figure 1). It was originally thought that LV dilation occurs in response to reduced function. However, in genetic DCM, where genetic markers make it feasible to monitor LV dimensions over many years, increased LV dimensions typically precede detectable reduction in function25–27. This state of LV enlargement is recognized as a prodome to DCM. Enlarged LV dimensions contrast with what is seen in hypertrophic cardiomyopathy (HCM), where the earliest findings in genetically mediated, sarcomere-positive HCM are reduced LV dimensions28. Strain and strain rate differences can be detected by echocardiography in first degree relatives of those with newly diagnosed nonischemic DCM, indicating that LV dimensions are not the earliest detectable differences in familial DCM29, 30.
Imaging - Cardiac Magnetic Resonance (CMR)
LV chamber dimensions and function, including strain measurements, are also accurately determined by CMR imaging. Contrast agents, mainly gadolinium, are used to evaluate fibrosis and therefore provide additional information on myocardial tissue quality. In DCM, the degree of fibrosis, marked by delayed gadolinium enhancement, is a predictor of all-cause mortality and need for future hospitalization31. Specifically, delayed enhancement is associated with increased risk for ventricular arrhythmias32–34. Delayed enhancement may also reflect features beyond fibrosis including edema and inflammatory infiltrate35.
DCM can also be associated with LV noncompaction seen using either echocardiography or CMR. A ratio of greater than 2.3:1 for the noncompacted to compacted layer of LV myocardium is considered abnormal, but notably can be detected in normal hearts36. Recent studies have suggested that hypertrabeculation is seen in a high fraction (36%) of adult DCM, although this was not associated with adverse outcomes compared to DCM without these noncompaction features37.
Endomyocardial Biopsy (EMB)
EMB has been used to confirm etiology in some forms of DCM, although with improved cardiac imaging, EMB is less frequently used. In some settings, for example, iron overload, amyloid, and other infiltrative processes myocardial biopsy may still be highly useful38. The complication rates with EMB range from 1–3%, and serious complications including perforation and tamponade occur at 0.5%38. EMB has been used to evaluate myocarditis and in the setting of unexplained HF39, 40. The nonuniform nature of infiltrative disease limits the sensitivity of myocardial biopsy, as the RV septum is targeted for sampling41. For the majority of genetic cardiomyopathies, genetic testing is favored over EMB. Of the genetic cardiomyopathies, ARVC may be evaluated by EMB, although more recent work suggests that alternative and more easily accessible cell types can be used to diagnose ARVC and avoid EMB42, 43.
Non-invasive arrhythmia monitoring
DCM is associated with an increased risk for cardiac arrhythmias and sudden cardiac death (SCD)44, and specific genetic DCM subtypes are especially prone to arrhythmias. CMR, especially the identification of delayed enhancement, can help risk stratify for sudden death34. Because of increased risk for SCD, there is need for arrhythmia surveillance in order to more appropriately deploy device management, including pacemakers and/or internal cardioverter defibrillators (ICDs). Symptomatic and even life-threatening bradycardia and tachycardia may occur in genetic DCM. Personal history of syncope or near syncope should be ascertained, and patient education to increase awareness of symptoms is needed. Holter monitoring, for its ease, remains a mainstay using 24–48 hour sampling. Other external event recorders are similarly transcutaneous and provide real-time, transmitted data as well as triggered monitoring. External loop recorders and now implantable loop recorders offer longer-term information. Those with familial DCM are likely to have more findings of ventricular ectopy and ventricular tachycardia (VT) on monitoring45. For primary prevention of SCD in DCM, risk stratification often relies on evaluating the specific genetic etiology (see below), family history, delayed enhancement and presence of VT on monitoring.
Clinical manifestations including neuromuscular findings
The range of clinical manifestations in DCM ranges from none to overt heart failure. With the increase in familial and genetic screening, it is now more common to identify the minimally to mildly affected stage of DCM in younger individuals. Using genetic markers, strain defects can be detected by echo or CMR. LGE may be present even when the heart appears still normal, suggesting that disease is ongoing. There is an emerging view that this represents an early stage of disease and one in which early institution of treatment should benefit. Although it is generally thought that arrhythmia risk scales with degree of LV dysfunction, for several subtypes of genetic cardiomyopathy, arrhythmias may be the earliest manifestation46–49. Specifically, in LMNA and SCN5A-mediated cardiomyopathies, arrhythmias including atrial fibrillation with slow ventricular response or ventricular arrhythmias may be the presenting finding. There is little in the clinical evaluation that makes it possible to distinguish one genetic subtype of DCM from another. This “phenocopying” is what has driven gene panel testing since with this approach multiple genes are screened at the same time. A DCM gene panel is shown in Figure 3, and a comprehensive list of DCM genes in Table 1.
Figure 3. DCM gene panels are used for genetic testing.
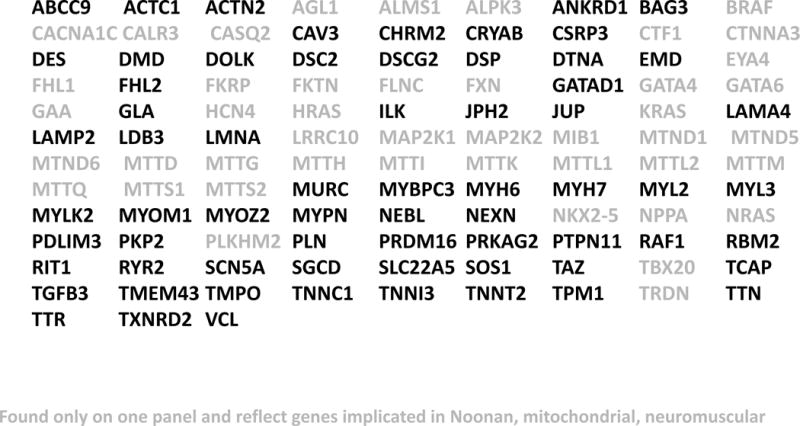
Shown is a list of 111 genes offered from multiple commercial testing laboratories for the evaluation of DCM. Those shown in black are commonly found on DCM panels from multiple sources, while those shown in gray are found on only some panels reflecting their role in syndromic cardiomyopathy such as Noonan syndrome, neuromuscular disease and/or mitochondrial myopathies.
Table 1.
Frequency and phenotype correlates of definitive and putative DCM genes
| Gene | Protein | Frequency and overlapping phenotypes |
|---|---|---|
| Sarcomere | Force generation/transmission | |
|
| ||
| MYH6 | Alpha-myosin heavy chain | HCM, CHD, Sick Sinus Syndrome |
| MYH7* | Beta-myosin heavy chain | 3–4% of DCM; HCM, LVNC |
| TPM1 | Alpha-tropomyosin | 1–2% of DCM; HCM, LVNC |
| ACTC1 | Alpha cardiac actin | HCM, LVNC |
| TNNT2* | Cardiac troponin T | 3% of DCM; HCM, LVNC |
| TNNC1 | Cardiac troponin C | HCM, LVNC |
| TNNI3 | Cardiac troponin I | HCM |
| MYBPC3 | Myosin-binding protein C | HCM, LVNC |
| TTN* | Titin | 12–25% of DCM; HCM, tibial muscle dystrophy |
| TNNI3K | Troponin I interacting kinase | Conduction defect, atrial fibrillation |
| MYL2 | myosin light chain2, regulatory | HCM |
| MYL3 | myosin light chain 3 | HCM |
| MYLK2 | Myosin Light Chain Kinase 2 | HCM; in panels, not reported as DCM gene |
| MYOM1 | myomesin 1 | Myofibrillar myopathy; in panels, not reported as DCM gene |
| MYOZ2 | myozenin 2 | HCM |
|
| ||
| Z-disk | Mechanosensing/mechanosignaling | |
|
| ||
| ACTN2 | Alpha-actinin 2 | LVNC |
| BAG3 | BCL2 Associated Athanogene 3 | Myofibrillar myopathy |
| CRYAB | Alpha-B-crystallin | Protein aggregation myopathy |
| TCAP | Titin-cap/telethonin | LGMD2G |
| MYPN | Myopalladin | HCM, RCM |
| CSRP3 | Muscle LIM protein | HCM |
| NEXN | Nexilin | HCM |
| FHL1 | Four-and-a-half LIM protein1 | EDMD, HCM |
| FHL2 | Four-and-a-half LIM protein 2 | HCM |
| ANKRD1 | Cardiac ankyrin repeat protein | Congenital heart disease |
| MURC | Muscle Related Coiled-Coil Protein | |
| LDB3 | Cypher/ZASP | LVNC |
| NEBL | nebulette | LVNC, HCM |
|
| ||
| Dystrophin complex | Sarcolemma, structural integrity | |
|
| ||
| DMD | Dystrophin | Duchenne/Becker muscular dystrophy |
| DTNA | Alpha-dystrobrevin | LVNC |
| SGCA | Alpha-sarcoglycan | LGMD2D |
| SGCB | Beta-sarcoglycan | LGMD2E |
| SGCD | Delta-sarcoglycan | LGMD2F |
| SGCG | Gamma-sarcoglycan | LGMD2C |
| CAV3 | Caveolin | HCM, LGMD1C, distal myopathy |
| ILK | Integrin-linked kinase | |
| FKTN | Fukutin | dystroglycanopathy, congenital muscular dystrophy |
| FKRP | Fukutin-related protein | dystroglycanopathy, LGMD |
|
| ||
| Cytoskeleton | Mechanotransduction/mechanosignaling/structural integrity | |
|
| ||
| DES | Desmin | <1% of DCM; Desminopathies, myofibrillar myopathy |
| VCL | Metavinculin | 1% of DCM |
| FLNC | Filamin C | 1% of DCM, AR-DCM; Myofibrillar myopathy, HCM, RCM |
| SYNM | Desmulin | |
| PDLIM3 | PDZ LIM domain protein 3 | |
| PLEC1 | Plectin-1 | LGMD2Q, epidermolysis bullosa |
|
| ||
| Desmosomes | Cell–cell adhesion/mechanotransmission/mechanosignaling | |
|
| ||
| DSC2 | Desmocollin 2 | ARVC, mild palmoplantar keratoderma |
| DSG2 | Desmoglein 2 | ARVC |
| DSP* | Desmoplakin | 2% of DCM; ARVC |
| PKP2 | Plakophilin 2 | ARVC |
| CTNNA3 | Catenin Alpha 3 | ARVC; in panels, not reported as DCM gene |
|
| ||
| Sarcoplasmic reticulum and cytoplasm | Ca homeostasis, contractility modulation, signaling | |
|
| ||
| PLN | Phospholamban | ARVC, HCM |
| RYR2 | Ryanodine receptor 2, Ca channel | ARVC, CPVT |
| CALR3 | Calreticulin-3 | ARVC, HCM; in panels, not reported as DCM gene |
| JPH2 | Junctophilin 2 | HCM |
| DOLK | Dolichol KINASE | Congenital disorder of glycosylation |
| MAP2K1 | Mitogen-Activated Protein Kinase Kinase 1 | Noonan Syndrome; in panels, not reported as DCM gene |
| MAP2K2 | Mitogen-Activated Protein Kinase Kinase 2 | HCM, Noonan Syndrome; in panels, not reported as DCM gene |
| NRAS | Neuroblastoma RAS Viral Oncogene Homolog | HCM, Noonan Syndrome |
| PRKAG2 | Protein Kinase AMP-Activated Non-Catalytic Subunit Gamma 2 | HCM, WPW |
| PTPN11 | Protein Tyrosine Phosphatase, Non-Receptor Type 11 | HCM, Noonan and Leopard Syndromes |
| RAF1 | Proto-Oncogene | HCM, Noonan and Leopard Syndromes |
| RIT1 | Ras Like Protein | Noonan Syndrome; in panels, not reported as DCM gene |
| SOS1 | SOS Ras/Rac Guanine Nucleotide Exchange Factor 1 | HCM, Noonan Syndrome; in panels, not reported as DCM gene |
| TRDN | Triadin | CVPT; in panels, not reported as DCM gene |
|
| ||
| Nuclear envelope | Nuclear structural integrity, mechanotransduction, mechanosignaling | |
|
| ||
| LMNA* | Lamin A/C | 4–8% of DCM; multiple phenotypes, LGMD1B, EDMD, progeria |
| EMD | Emerin | EDMD |
| LAP2/TMPO | Lamin-associated polypeptide 2 | |
| SYNE1/2 | Nesprin 1/2 | EDMD, ataxia |
|
| ||
| Nucleus | Transcription cofactors, gene expression | |
|
| ||
| EYA4 | Eyes absent 4 | Deafness |
| FOXD4 | Forkhead box D4 | |
| HOPX | Homeobox only protein | |
| NFKB1 | NF-kappa B1 | |
| PRDM16 | PR domain containing 16 | LVNC |
| TBX20 | T-box 20 | Atrial septal defect |
| ZBTB17 | Zinc finger and BTB domain containing protein 17 | |
| RBM20* | RNA-binding protein 20 | 2% of DCM; RNA-binding protein of spliceosome of TTN and other proteins |
| GATA4 | GATA binding protein 4 | Atrial, ventricular septal defects, Fallot |
| GATA5 | GATA binding protein 6 | Atrial, ventricular septal defects, Fallot |
| GATAD1 | GATA zinc finger domain-protein 1 | |
| NKX2–5 | Cardiac specific homeobox 1 | Atrial, ventricular septal defects, Fallot, hypoplastic left heart |
| ALSM1 | Centrosome And Basal Body Associated Protein | Alstrom Syndrome (phenocopy) |
| ALPK3 | Alpha kinase 3 | Pediatric cardiomyopathy |
| LRRC10 | Leucine Rich Repeat Containing 10 | |
| NPPA | Natriuretic paptide A | Atrial fibrillation |
| PLEKHM2 | Pleckstrin Homology Domain | LVNC |
| TGFB3 | Transforming Growth Factor Beta 3 | ARVC; in panels, not reported as DCM gene |
| TMEM43 | Transmembrane Protein 43 | ARVC, EDMD; in panels, not reported as DCM gene |
|
| ||
| Ion channels | ||
|
| ||
| SCN5A* | Type V Voltage-gated cardiac Na channel | 2–3% of DCM; LQT, Brugada, atrial fibrillation, conduction defects |
| ABCC9 | Sulfonylurea receptor 2A, component of ATP-sensitive potassium channel | Atrial fibrillation, osteochondrodysplasia |
| KCNQ1 | Potassium channel | Atrial fibrillation, LQT1, Short QT1, Jervell and Lange-Nielsen syndrome |
| CACNA1C | Calcium Voltage-Gated Channel Subunit Alpha1 C | Brugada Syndrome, Timothy Sindrome |
| HCN4 | Hyperpolarization Activated Cyclic Nucleotide Gated Potassium Channel 4 | Brugada, Sick Sinus Syndrome |
|
| ||
| Mitochondria | Supply and/or regulation of energy metabolism | |
|
| ||
| CPT2 | Carnitine palmitoyltransferase 2 | carnitine deficiency, myopathy, lethal neonatal |
| FRDA/FXN | Frataxin | Fredreich ataxia |
| DNAJC19 | HSP40 homolog, C19 | 3-methylglutaconic aciduria, type V |
| SDHA | Succinate dehydrogenase | Leigh syndrome |
| SOD2 | Superoxide dismutase | |
| TAZ/G4.5 | Tafazzin | LVNC, Barth syndrome, endocardial fibroelastosis 2 |
| CTF1 | Cardiotrophin 1 cytokine | |
| mtDNA | Mitochondrially Encoded TRNA genes | Typically syndromic, mitochondial myopathy; in panels, not always reported as DCM genes |
| TXNRD2 | Thioredoxin Reductase 2 | |
|
| ||
| Extracellular matrix | Cell adhesion and mechanosignaling | |
|
| ||
| LAMA2 | Laminin 2, merosin | Congenital muscular dystrophy |
| LAMA4 | Laminin 4 | |
| Lysosome | ||
| LAMP2 | Lysosome-associated membrane protein 2 | Danon Disease |
| AGL | Amylo-Alpha-1, 6-Glucosidase, 4-Alpha-Glucanotransferase | Glycogen storage disease |
| BRAF | B-Raf Proto-Oncogene, Serine/Threonine Kinase | Cardiofaciocutaneous, Leopard, Noonan Syndromes |
| GAA | Glucosidase Alpha, Acid | Glycogen storage disease |
| GLA | Alpha galactosidase | Fabry Disease |
|
| ||
| Other | ||
|
| ||
| PSEN1 | Presenillin 1, Gamma secretase intramembrane protease complex | Alzheimer disease |
| PSEN2 | Presenillin 2, Gamma secretase intramembrane protease complex | Alzheimer disease |
| CHRM2 | Cholinergic Receptor Muscarinic 2 | |
| HFE | Hemochromatosis | Phenocopy |
| HRAS | HRas Proto-Oncogene, GTPase | HCM, Costello Syndrome; in panels, not reported as DCM gene |
| KRAS | KRAS Proto-Oncogene, GTPase | HCM, Costello Syndrome; in panels, not reported as DCM gene |
| MIB1 | Mitogen-Activated Protein Kinase Kinase 2 | LVNC; in panels, not reported as DCM gene |
| SLC22A5 | Cation/Carnitine Transporter | Skeletal myopathy |
| TTR | Transthyretin | Amyloidosis (phenocopy) |
Legend: frequent (asterisk), definitive (bold) and putative DCM genes,14, 23, 58, 164, 165 OMIM (www.omim.org, accessed 01/19/2017); GeneCards (www.genecards.org, accessed 01/19/2017). AR-DCM: arrhythmogenic DCM; ARVC: arrhythmogenic right ventricular cardiomyopathy; CPVT: cathecolaminergic polymorphic ventricular tachycardia; DCM: dilated cardiomyopathy, EDMD: Emery-Dreyfuss muscular dystrophy, HCM: hypertrophic cardiomyopathy, LGMD: limb-girdle muscular dystrophy, LVNC: left ventricular noncompaction, RCM: restrictive cardiomyopathy.
Neuromuscular disease may accompany cardiomyopathy, and in some forms of neuromuscular disease, the presenting feature may be irregular heart rhythms. LMNA mutations can present with or without muscle disease, and the muscle disease ranges from limb girdle muscular dystrophy to Emery Dreifuss Muscular Dystrophy, which is typically associated with contractures of the elbows and Achilles tendons50. LMNA mutations are inherited in an autosomal manner, seen as multiple affected family members in each generation. Both X-linked and autosomal neuromuscular diseases can also associate with cardiomyopathy, and this includes Duchenne Muscular Dystrophy as well as the autosomal recessive forms of sarcoglycanopathies51. In these disorders, skeletal muscle disease usually appears in childhood with a typical dilated cardiomyopathy arising in the teenage years or early twenties. DCM in neuromuscular disease is highly amenable to treatment and responds well to guideline directed medical therapy. Both forms of myotonic muscular dystrophy, type 1 and type 2, can also be associated with DCM52–55. Atrial and ventricular arrhythmias are common in these tri- and tetra-nucleotide repeat expansion disorders, and should be aggressively managed. Myotonic dystrophy type 2 usually present in older individuals and in this case, genetic testing panels usually do not include these genes and thus the diagnosis can be easily missed especially if neuromuscular symptoms are not so pronounced.
DCM Genetics
The majority of genetic DCM is inherited in an autosomal dominant pattern with variable expressivity and penetrance (Figure 4), although specific forms of autosomal recessive, X-linked recessive and mitochondrial inheritance each occur14, 19, 56. De novo mutations also contribute to genetic cardiomyopathy and are defined when neither biological parent carries the offspring’s mutation. De novo mutations have been described in many different genes, and the presence of a de novo variant can be used to define the pathogenic status of genetic variants since the frequency of de novo variation in each genome is exceedingly rare. Thus, a novel mutation introducing a protein altering change in a cardiomyopathy gene is typically considered pathogenic.
Figure 4.
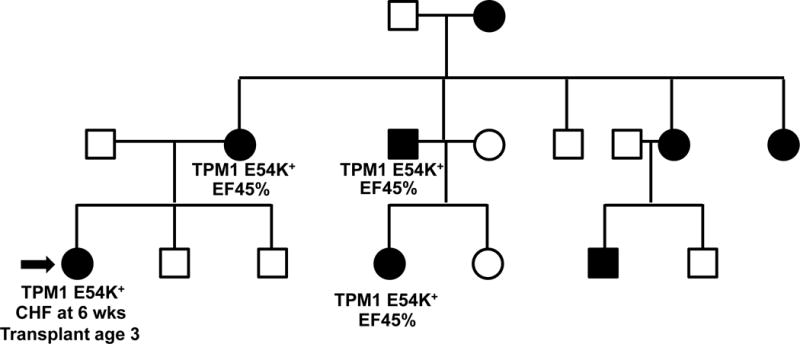
A typical DCM pedigree is shown highlighting variable expressivity. Most DCM is inherited in an autosomal dominant pattern. Affected individuals with DCM are shown in black. A gene panel revealed the previously reported pathogenic TPM1 E54K variant. The proband (arrow) presented in early life requiring heart transplant during early childhood. Other members of the family are in their 3rd to 6th decade with LVEF 45%, demonstrating variable expressivity of the primary mutation. Environmental and additional genetic factors may contribute to variable expressivity.
Interpreting whether genetic variants are pathogenic is increasing complex, owing to the vast amount of rare variation in each human genome. The emerging consensus around interpretation of genetic variation and its effect on phenotype relies on a classification system ranging from pathogenic, likely pathogenic, variant of uncertain significance (VUS), likely benign and benign57. The availability of large control cohorts provides invaluable information of the frequency of variants, and the largest available data set is currently ExAC (Exome Aggregation Consortium) (http://exac.broadinstitute.org), which collected exome sequencing data of over 60,000 individuals from a series of studies including the 1000 Genomes Project and the Exome Sequencing Project (http://evs.gs.washington.edu/EVS). The current trend is to consider putative pathogenic variants as those that are either unique to the DCM patient or family58, or extremely rare, (MAF <1 × 10−4)59. The recently adopted, more stringent criteria for genetic testing have prompted the reclassification of variants and indicate the needs of a continuous reanalysis of data58. The use of whole exome/genome sequencing in clinical laboratories warrants strong criteria to discriminate common variants. At present, genetic testing typically relies on self-reported ethnicity testing, and it is important to match ethnicity between the proband and testing databases. However, at this point, this integration of common and rare variation is not routinely being used in cardiomyopathy genetic testing, potentially contributing to false positive interpretation60.
DCM is genetically heterogeneous, and DCM genes encode proteins of broad cellular functions. Mutations in genes encoding cytoskeletal, sarcomeric, mitochondrial, desmosomal, nuclear membrane and RNA binding proteins have all been linked to DCM. Thus, the pathological mechanisms that lead to DCM are very diverse. The genes below are listed in order of frequency for their contribution to genetic DCM with focus on the most commonly implicated genes and their mechanism of action if known.
TTN
The discovery of the role of TTN truncating variants in DCM has been major advance61. The TTN gene encodes the giant protein titin, which is the largest known protein expressed in the heart. Titin functions as a spring, providing passive force and regulating sarcomere contraction and signaling62. Titin is a large ~35,000 aa protein that spans half the length of the sarcomere from Z-disc to M-band, and is referred to as a “third” filament with the thin and thick filaments that comprise the sarcomere. Proposed as a molecular rule for the sarcomere, titin has domains that can accommodate passive stiffness63. Titin’s I band region includes the PEVK (proline-glutamate-valine-lysine) repetitive region, which is thought to directly regulate passive tension. The I band region of the TTN gene is encoded by 220 of TTN’s 360 exons. The large size, repetitive nature, and extensive alternative splicing of TTN makes it challenging for genetic analysis. The PEVK region is just carboxyl to the N2A and N2B regions that interact with the FHL (four and half LIM protein), identified as a modifier for HCM64. Notably, TTN is differentially spliced throughout heart development and adaptively to distinct physiological states including HF65. The larger N2A form is associated with a more compliant ventricle (Figure 5). In contrast, the smaller N2B form lacks more of the repetitive units and is associated with stiffer heart. Deep RNA sequencing of TTN from failed hearts suggests highly variable exon usage in this region consistent with even subtler defects in cardiac elasticity that may be variable across regions of the LV66.
Figure 5.
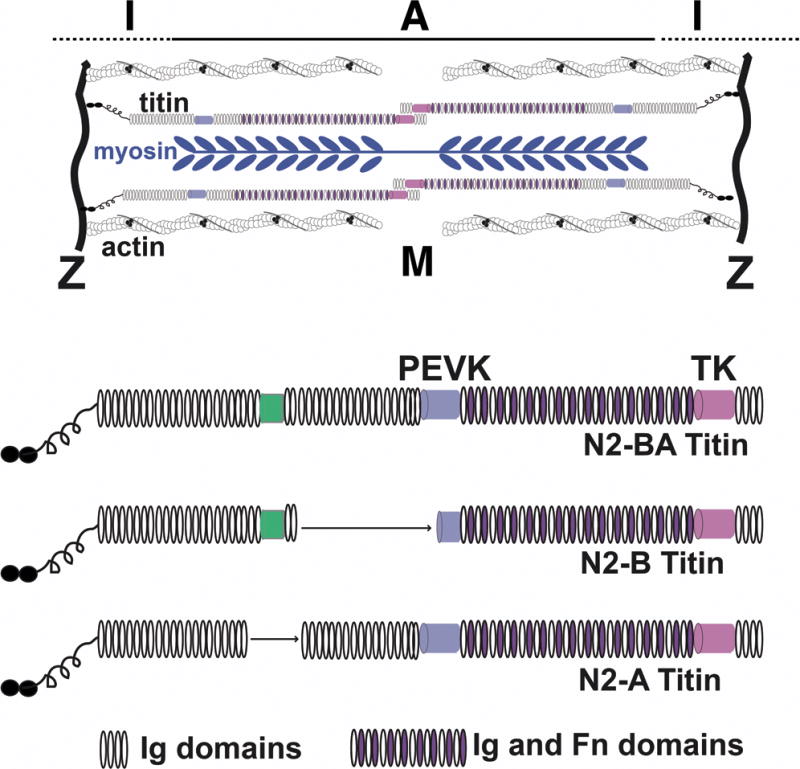
Shown in the top is a schematic of the sarcomere with the position of the thick myosin-containing filaments and the thin actin-containing filaments. Titin is considered a third filament of the sarcomere since its spans from Z disk to M band. The lower schematics show the major splice forms of titin (N2-BA, N2-B, N2-A). The green box represents a unique sequence domain. The PEVK region is named for the repetitive amino acid sequences (proline, glutamine, valine, lysine).
Using a TTN specific array designed to capture all TTN exons, it was shown that truncating variants of TTN contribute to 20–25% of nonischemic DCM61. Prior to this, only a few missense TTN variants had been described linked to DCM67. Induced pluripotent stem cells (iPSC) differentiated into cardiomyocytes in culture demonstrate a paucity of sarcomeres, suggesting that force may be impaired directly through sarcomere loss in TTN truncations68. In these studies and others it has been shown that TTN truncations are observed at a low frequency in the general population, ranging from 1–3%61, 66, 69. There is a tendency for TTN truncations in DCM to distribute to the A band, rather than the I band66, and TTN truncations can also be associated with mild DCM70. A recent study showed that truncating variants in the general population are linked to eccentric cardiac remodeling, suggesting that TTN truncations may be “at-risk” alleles71.
Peripartum cardiomyopathy is a heterogeneous syndrome of mixed etiology. Yet a subset of peripartum cardiomyopathy is attributable to TTN truncating variants72, 73. Peripartum cardiomyopathy can be associated with recovered LV function after pregnancy. Moreover, the observation that TTN truncating variants can be associated with recovery of function in DCM after LV assist device (LVAD) placement also suggests a dynamic state of TTN truncating variants74. Additional genes with mutations beyond TTN have also been described in peripartum cardiomyopathy21. Overall, the presence of TTN truncating variants in the general population argues for caution in interpreting these variants and again underscores the importance of familial segregation analysis. At this point, until more is known, the presence of a TTN truncation variant should trigger at least intermittent cardiac imaging and management aimed at reducing other stressors to the heart.
TTN has a high prevalence of missense variants, both rare and common75. TTN missense variants have been reported in ARVC and other forms of cardiomyopathy67, 75–78. Additionally, TTN missense variants have also been reported in skeletal myopathy, including the common tibial myopathy79, 80. The enormous number of TTN missense variants makes these variants exceedingly complex to interpret in the context of broad genetic testing on individuals with DCM.
Zebrafish have been used successfully to model myopathies due to TTN mutations, demonstrating both cardiac and skeletal muscle defects81. A mouse models with an in frame deletion in the PEVK region of TTN develops diastolic dysfunction, consistent with the complex role of titin for both systolic and diastolic dysfunction82. In both rats and mice with heterozygous TTN truncation mutations, additional stressors like transaortic constriction are used to promote the development of DCM71, 83.
LMNA
LMNA missense and truncating mutations account for 5–8% of genetic DCM84, 85. Like TTN, LMNA mutations are inherited in an autosomal dominant manner. The single LMNA gene encodes lamins A and C, and differential splicing at the 3′ end results in two proteins that are identical across their first 566 amino acids; mutations in LMNA lead to a constellation of diseases from premature aging, to myopathies and DCM86. Mutations that alter processing of lamin A lead to accumulation of prelamin A (sometimes called progerin) and these have been associated with the premature aging syndrome Hutchinson Gilford Progeria87. LMNA mutations linked to autosomal dominant DCM are both missense and frameshifting in nature, and these mutations may occur anywhere along the length of the coding region. DCM-associated mutations are not specifically associated with Prelamin A accumulation; thus, the basic mechanisms underlying the premature aging syndrome versus DCM appear to be distinct. The mechanisms responsible for autosomal dominant DCM LMNA mutations may be a mix of multiple defects including dominant-negative function as well as haploinsufficiency88, 89. Lamins A and C are implicated in many different cellular processes from regulating gene expression, mechanosensing, DNA replication, and nuclear to cytoplasmic transport.
Loss of LMNA leads to a defect in mechanosignaling90, 91. Mechanosignaling defects were observed in cell with a homozygous deletion in the mouse Lmna gene. Male mice heterozygously deleted for Lmna exhibit cardiomyopathy features in later life, suggesting that mice can be used to model laminopathy92. In LMNA associated adult-onset DCM, the mTOR pathway can be activated and, in animal models, inhibition mTOR by temsirolimus or rapamycin was able to rescue the DCM phenotype93, 94. Mitogen activated protein kinase (MAPK) signaling is increased in these models, leading to clinical trials testing compounds aimed at reducing this signaling95. Recently, early phase, encouraging results were reported from a Phase 2 registration trial on A797 (Array Biopharma), an oral, selective p38 mitogen-activated protein kinase inhibitor in 12 patients with LMNA–associated DCM96.
LMNA mutations associate frequently with a signature of dysrhythmias that includes sinus node dysfunction, atrial fibrillation, AV node dysfunction, VT and ventricular fibrillation and SCD46, 47. Notably, cardiac conduction system disease may precede the development of LV dilation and dysfunction, and the presence of early conduction system disease may suggest and LMNA mutation. Aspects of the arrhythmia and LV dysfunction phenotypes are not fully replicated in the mouse models; namely atrial fibrillation and ventricular arrhythmias are not frequently seen and a homozygous mutation is often needed to generate DCM. Heterozygous truncating LMNA mutations have a higher arrhythmia risk than missense variants47, and a prolonged PR interval indicates cardiac conduction system disease in laminopathy97. The susceptibility of the cardiac conduction system to LMNA mutations is not well understood.
PLN
The PLN gene encodes phospholamban, a 52 aa residue transmembrane protein that, when unphosphorylated, inhibits sarcoplasmic reticulum Ca2+-ATPase (SERCA). Several dominant mutations in PLN have been associated with DCM, including the R14del mutation that appears to be a founder mutation in the Netherlands and Germany. Thus, in some populations the percentage of DCM due to PLN mutations is quite high. The phenotype with PLN mutations is variable. Early onset DCM with lethal ventricular arrhythmias was described98, 99. Similarly, individuals from the Netherlands with the R14del founder mutation have a severe phenotype100. However, other reports suggest a milder phenotype101, 102. Identifying the same primary mutation(s) with a range of phenotype dependent on genetic background supports that other factors, including genetic factors, may modify the outcome of DCM due to PLN mutations.
iPSCs with the R14del PLN mutation were generated and found to display features of cellular cardiomyopathy including aberrant Ca2+ handling after caffeine and a higher percentage of irregular Ca2+ transients, and these features were reversed after gene editing to correct the primary mutation103. These same cells were used to engineer three dimensional human heart tissues and in this setting, more clear cut cardiomyopathic features were seen including reduced developed force that was improved after genetic correction104. In iPSC-derived cardiomyocytes and in hearts from PLN-mutation carriers, aggregates of phospholamban were seen in a perinuclear and cytoplasmic pattern suggesting that aggregated phospholamban contributes to the pathology possibly through aberrant autophagy105.
RBM20
RNA binding motif 20 is an RNA binding protein expressed highly in both atria and ventricle. Dominant mutations in the RBM20 gene were first described in DCM, where they contribute to 1–5% of DCM106, 107. RBM20 is 1227 aa in length and contains a ribonucleic acid recognition motif (RRM) domain between residues 525 to 600 aa. A second conserved domain is found between 650 and 725, and the mutations originally described in nonischemic DCM fall within or near these domains. More recently, mutations in a third conserved region were identified in a glutamate rich region108. As an RNA binding protein, RBM20 is implicated in tissue specific splicing relevant to development and adaptation to disease states. In the heart, RBM20 regulates cardiac splicing including the splicing of TTN109–111. Thus, the downstream molecular consequences of RBM20 mutations may share similarities to those occurring from TTN truncating variants.
iPSC-derived cardiomyocytes with the RBM20 R636S mutation develop a gene expression and splicing profile consistent with cardiomyopathy affecting not only TTN but also the CAMK2D and CACNA1C genes112. Sarcomeres within these RBM20 mutant lines were thinner, similar to what was described for TTN mutant iPSC-cardiomyocytes68. Recently, RBM20 was implicated in the production of circular RNAs from the TTN locus, and mice deleted for RBM20 failed to produce these Ttn-derived circular RNAs113. Although the function of circular RNAs is not known, the authors described that a subset of Ttn-derived circular RNAs were misregulated in DCM.
SCN5A
SCN5A encodes the major sodium channel expressed in the heart and heterozygous dominant mutations in SCN5A are also found in primary arrhythmia syndromes including the Long QT and Brugada syndromes. Missense mutations in SCN5A have also been described in familial DCM, and these mutations carry a higher risk for arrhythmias48, 49. There is considerable genetic heterogeneity in the SCN5A gene in the general population, making it challenging to interpret rare variation in the SCN5A gene59. Genotype-phenotype association studies may guide genotype-based therapies. For example, the SCN5A R222Q falls within the S4 voltage sensor and is thought to enhance excitability. Treatment with lidocaine was observed to suppress the bigeminy associated with cardiomyopathy114.
iPSCs have been used to model SCN5A mutations associated with primary human arrhythmia syndrome115–117. Transgenic directed inducible expression of the Scn5A F1759A in mice leads to atrial fibrillation and persistent sodium currents in atria and ventricles118. Along with atrial fibrillation, these mice have progressive reduced LVEF consistent with a model for DCM. Thus, the distinct roles of SCN5A in the myocardium and the conduction system lead to a combination of arrhythmia and myocyte dysfunction.
Cytoskeletal genes
Genes encoding cardiac cytoskeletal proteins have been implicated in DCM (Figure 6). For example, mutations in dystrophin link to X-linked DCM and cardiomyopathy in Duchenne Muscular Dystrophy119–121. Along with dystrophin mutations, mutations in the sarcoglycan genes produce cardiomyopathy, usually associated with muscular dystrophy51, 122. In these disorders, the gene products play a normal and essential role in managing sarcolemmal stability. Thus, in the absence of these genes, the sarcolemma becomes unstable leading to cardiomyocytes loss and heart dysfunction. A number of emerging therapies for restoring dystrophin expression are being tested or have been approved recently123, 124. Antisense oligonucleotides are being delivered to produce internally truncated dystrophin proteins, and stop codon suppression compounds promote read through of premature stop codons. The degree to which these drugs access the human heart is not well known and ongoing studies will be in a position to assess this in humans.
Figure 6.
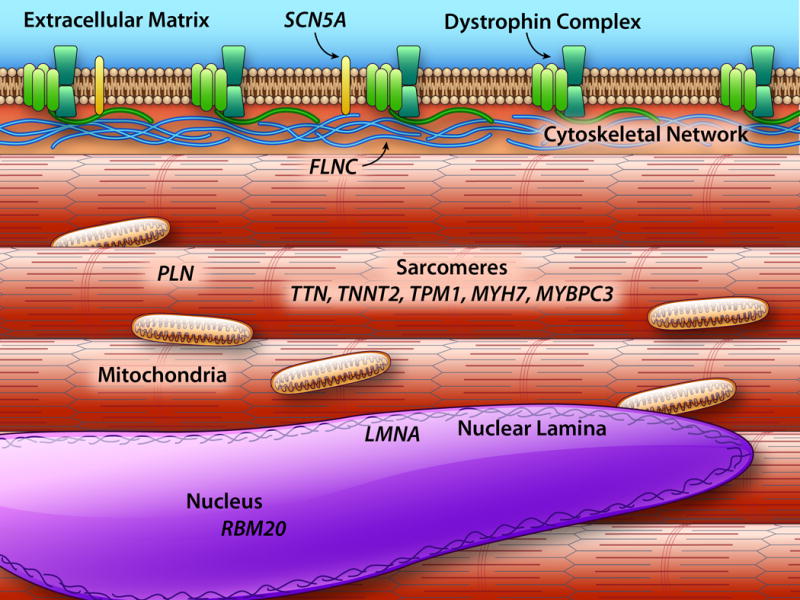
Shown are major components within the cardiomyocyte with emphasis on compartments that contribute to genetically mediated DCM. The extracellular matrix is shown in gray. The dystrophin complex that includes the sarcoglycans (green) is mutated in forms of DCM with neuromuscular disease. The sarcomeres (pink) include components that are mutated in both HCM and DCM. Z band (dark red) is a mechanosensing hub that serves to transmit force from the sarcomeres. Mutations in both mitochondrially encoded (purple) and nuclear encoded mitochondria proteins lead to cardiomyopathy. The nuclear lamina include lamins A and C, and the gene LMNA is commonly mutated in DCM.
More recently FLNC mutations, in the gene encoding filamin C, have been described in DCM125–127. Filamin C interacts with the dystrophin complex, and deletion of Flnc from the mouse leads to skeletal myopathy128, 129. In humans, truncating mutations lead to cardiomyopathy that is associated with a high rate of ventricular arrhythmias and SCD suggesting that filamin C has a role in the cardiac conduction system in addition to the cardiomyocyte.
Mitochondrial mutations
Both nuclear encoded and mitochondrially encoded mitochondrial genes lead to cardiomyopathy130–133. Mutations in the mitochondrial genomes may be difficult to identify and interpret given the role of heteroplasmy and the fact that most genetic testing relies on peripheral blood DNA, which may or may not match what occurs in the heart. Nuclear encoded mitochondrial genes follow either autosomal dominant or recessive inheritance, while mitochondrially encoded genes shows maternal inheritance.
Additional genetic mutations in DCM
The proteins encoding the sarcomere, the unit of contraction, are also implicated in DCM. Recent data from clinical genetic testing indicates that MYH7, TNNT2 and TPM1 are the most frequent mutated sarcomere genes in DCM, ranging from approximately 2 to 4%, while MYBPC3 mutations are rare58. Recently, truncations of the gene encoding obscurin have been found in both LVNC and DCM phenotypes134. DCM may be best considered as a cardiomyopathy of mixed origin, familial in approximately 30–50% of patients, consistent with its being a genetic disease. Non-familial or idiopathic DCM may still have a genetic origin, albeit a complex one. Since all DCM mutations have variable expressivity, this may support a model of oligogenic contribution along with environmental or other pathogenic stimuli. As it currently, stands not all DCM genes have been discovered. Thus, the etiology in nonfamilial cases could be as yet undiscovered genes, low penetrance, de novo mutations, “missing heritability” due to multiple genes with weaker effect, copy number variations, enhancer region mutations, and intronic variants, or may be the result of the interaction between modifier genes and the environment13, 135.
Digenic/oligogenic etiology
Occasionally, digenic variants have been reported; while it is possible that some of these variants may in fact be VUS or benign, in other cases the digenic condition have been found to segregate in informative kindred and associate with a more severe phenotype. This is the case of a large family cosegregating a LMNA mutation, where relatives with an additional TTN truncation showed worse outcome and distinct pathological changes136. With the growth of gene panels for cardiomyopathy genetic testing, it is not uncommon to identify more than one potential pathogenic variant. Segregation analysis may be helpful to clarify “primary” versus other variants. Compound mutations MYBPC3 lead to early onset, sometime neonatal cardiomyopathy137–139. Thus, in families with multiple affected members with DCM, a broad gene panel on a younger, affected proband may provide a more comprehensive view of potential variation. Segregation testing for variants of interest can then help clarify those variants with greatest effect.
Genetic testing
Family screening
Genetic evaluation of DCM should begin with extensive and accurate evaluation of the patient’s family history, involving at least three generations and including history of cardiomyopathy as well as history of sudden unexpected death at young age (<35)3, 19, 140. This information will guide genetic testing, provide good care to family members, and aid in the interpretation of the results and help identifying relatives at risk of disease.
Clinical cascade screening of relatives is recommended per AHA and ESC guidelines3, 141. First-degree relatives of affected family members should be clinically evaluated (Figure 2). First line of screening usually relies on ECG and echocardiography to evaluate ventricular size and function. Clinical history should evaluate signs and symptoms for arrhythmias and any history of neuromuscular disease. Other cost effective tools to consider in family member screening include ascertaining arrhythmia history and Holter monitoring45. A diagnosis of familial DCM is made where two or more family members are affected by DCM19.
Clinical Genetic testing
According to the 2016 AHA Scientific Statement on DCM3, genetic testing is recommended (with moderate level of consensus) in patients with familial (Level of Evidence A) and non-familial ‘idiopathic’ cardiomyopathy in conjunction with genetic counseling (Level of Evidence B), while there is strong level of consensus in recommending mutation-specific genetic testing for family members after the identification of a DCM-causative mutation in the proband (Level of Evidence B). Reflecting lack of full consensus in the field, the 2016 ESC Position Statement141 is slightly different and recommends genetic testing in all familial DCM, or non-familial with clinical clues (such as atrio-ventricular block or CK elevation). Interestingly, the ESC Position Statement recommends that genetic testing should be oriented by clinical diagnostic clues and restricted to genes known to cause DCM, while considering large panels of genes only when the family structure is large enough to permit segregation analysis.
Multiple commercial and academic vendors provide genetic testing gene panels under certification of the Clinical Laboratory Improvement Amendment (CLIA). A typical DCM gene panel includes approximately 40–50 genes (Figure 3)58, 142. The probability of positive genetic testing in familial DCM is overall in the range of 40% with the current next-generation sequencing panels, and seems not different compared to sporadic cases13, 58. A pancardiomyopathy panel, as opposed to a DCM panel, is chosen when the phenotype is unclear and a more comprehensive screening is preferred. The level of evidence to support testing for some genes has been questioned, and this is largely based on a high level of genetic variation in those genes in the population at large143. Larger panels may yield greater difficulty in interpreting the results because of variant of unknown significance (VUS). Indeed, in a survey of 766 patients screened in a clinical laboratory, with the introduction of next generation sequencing technology and larger panels, while the rate of positive testing increased from 10% to 40%, the number of VUS increased 10 fold13, 58, 144.
Genetic Counseling
Genetic counselors, especially those with experience in cardiovascular testing, provide both pre- and post-test counseling. With the increasing complexity of cardiomyopathy genetic testing, referral to specialized cardiovascular genetic clinics should be considered140. Pre-test genetic counseling should involve discussion of potential genetic results, (pathogenic mutations, variants of uncertain significance and benign genetic variants). This counseling should also discuss the impact on insurability, reproduction and lifestyle. Post-test counseling focuses on variant interpretation/possible reinterpretation, reproductive risks to offspring and family testing.
If a genetic mutation is identified, genetic cascade screening can be conducted with family members. Cascade genetic testing evaluates the specific family mutation, rather than a gene panel. Site specific testing is of low cost and rapid turnaround so that this can be a cost-effective strategy to eliminate the need to serially follow gene mutation-negative family members. For gene mutation-positive family members, current guidelines suggest an annual clinical follow up with ECG and echocardiography3, 140, 141.
In the absence of an informative genetic testing, asymptomatic first degree relatives should be examined every 3 to 5 years140. This strategy may allow prompt therapeutic measures in carriers showing sign of myocardial dysfunction. Even in the absence of a positive genetic testing, longitudinal studies have shown the benefit of family screening and monitoring. In approximately 10% of cases, mild myocardial dysfunction may progress into overt DCM within 5 years27, 145. Furthermore, clinical family screening in DCM helps to identify affected relatives at earlier stages of disease, and this associates with improved survival as compared to sporadic DCM146.
Management of DCM
Established medical therapies
Management of DCM is focused on 1) LV dimension and function, 2) arrhythmia surveillance and treatment, and 3) reducing congestive symptoms if present. Symptomatic DCM and heart failure with reduced LVEF (HFrEF) is managed following current AHA/ACC and ESC guidelines. Guideline directed therapy includes angiotensin-converting enzyme inhibitors (ACEi) or angiotensin receptor blockers (ARB), in association with beta-blockers, aldosterone antagonists, and in selected cases, vasodilators147–149. Medications should be titrated to the dose used in clinical trials unless limited by side effects150. DCM patients on optimal therapy with complete left bundle branch block (LBBB) may benefit from cardiac resynchronization147, 148, 151, 152. The improvement of survival with ICDs in patients with LVEF less than 35% is also well established147, 148. Patients with refractory heart failure may require advanced therapies including left ventricular assist devices or cardiac transplant147, 148.
Two newer medications for those not responding to optimal medical therapy include the angiotensin receptor-neprylisin inhibitor (ARNI, valsartan/sacubitril) and the sinoatrial modulator, ivabradine3, 148, 151. Updated guidelines recommend switching from ACEi/ARB to ARNI in class II-III patients who are not responding to optimal medical therapy (ESC guidelines148) or even in those responding to optimal therapy, considering the evidence of superior benefit of ARNI over ACEi/ARBS in terms of mortality and morbidity (AHA/ACC/HFSA guidelines153). Ivabradine instead can be added to optimal medical therapy to reduce morbidity in patients with sinus rhythm and a heart rate over 70 bmp3, 148. The treatment of patients with a LVEF between 40% and 50%, defined in the ESC guidelines HF with midrange ejection fraction (HFmrEF) and in the ACC/AHA/HFSA guidelines HF with improved ejection fraction remain less clear151.
Arrhythmia management
Arrhythmia management in genetic DCM patients follows the general recommendations for prevention of SCD and ICD implantation based on the reduced LVEF (<35%). However, there are notable exceptions. First, a subset of patients with DCM present early in the disease course with life-threatening ventricular arrhythmias (2%)154 or with frequent ventricular arrhythmias (30%), which are unrelated to the severity of left ventricular dysfunction45. These patients mirror the arrhythmogenic presentation of ARVC and are described as arrhythmogenic DCM (AR-DCM)45. AR-DCM patients who present with syncope, NSVT and frequent premature ventricular contractions, show a higher incidence of life-threatening arrhythmic events (SCD, sustained ventricular tachycardia, cardiac arrest) compared with the other DCM patients, while they show no difference in outcome of heart failure. The coexistence of a family history of SCD and the AR-DCM phenotype predicts a high risk of SCD events (Figure 7). These recent data suggest that ventricular arrhythmias should be systematically and carefully evaluated with monitoring, and that family history of ventricular arrhythmias predicts a poor prognosis and increased risk of SCD.
Figure 7. Sudden cardiac death and life-threatening ventricular arrhythmias in DCM.
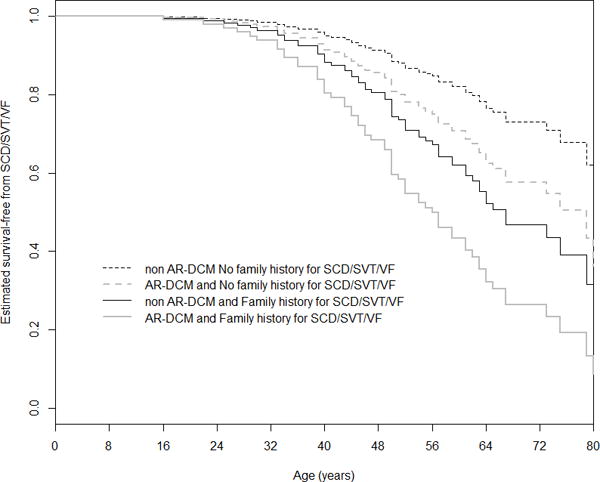
Cox-estimated event-free survival stratified by 2 risk factors, family history of SCD or ventricular arrhythmias (SVT or VF) and AR-DCM diagnosis, in a cohort of 285 DCM patients. The AR-DCM phenotype (p=0.02) and family history of SCD or ventricular arrhythmias (SCD/SVT/VF) (p=0.038) showed an additive prognostic effect on mortality for arrhythmic events. AR-DCM: Arrhythmogenic Dilated Cardiomyopathy; SCD/SVT/VF: sudden cardiac death, sustained ventricular tachycardia and ventricular fibrillation. From Spezzacatene et al.,45 with permission.
AR-DCM can be genetically determined, and a clear association between risk of SCD and gene has been established with the LMNA gene. Indeed, the 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of SCD155, recommend an ICD in patients with DCM and a confirmed disease-causing LMNA mutation and clinical risk factors (NSVT during ambulatory electrocardiogram monitoring, LVEF 45%, male sex and truncating mutations (class IIa, level B). Likewise, the HRS/ACC/AHA Expert Consensus Statement on the use of ICD therapy inpatients highlighted the increased risk for SCD in LMNA carriers156. Risk factors for SCD identified in two large LMNA carrier cohorts in Europe47 and in the U.S.157 include NSVT during ambulatory electrocardiogram monitoring47, LVEF < 4547 to 50%157, male sex and truncating mutations47, 157. Kumar et al. reported life-threatening ventricular arrhythmia rates of 3% to 7%/year, which is higher or comparable to other known groups of high-risk patients including those with LVEF < 25%, ARVC, HCM and high-risk ischemic cardiomyopathy.
DCM also carries an increased stroke risk, although less is known about the specific risk in genetic cardiomyopathy. As such, anticoagulation should be considered to reduce the risk of stroke in DCM with atrial fibrillation in particular in the presence of additional risk factor for cardioembolic events such as history of hypertension, diabetes mellitus, previous stroke or transient ischemic attack, or ≥75 years of age147.
Management of genotype-positive/ phenotype-negative patients
Genetic testing identifies genotype-positive/ phenotype-negative family members, and this information is useful for prevention strategies, lifestyle recommendations including participation in competitive sports, and possible arrhythmia management. The current guidelines recommend observation in asymptomatic at-risk relatives with yearly clinical assessment140. There is less consensus concerning the medical management, timing and the type of intervention in these patients. Current guidelines recommend control of risk factors in this stage, such as hypertension147. ESC and AHA suggest restriction from competitive sports in DCM genotype-positive/ phenotype-negative, although evidence is lacking to support these recommendations158. When initial signs of ventricular dysfunction present during follow-up, earlier institution of medical therapy can begin, although the exact timing of this is not known140.
Cardiac Regeneration for DCM?
DCM is usually associated with cardiomyocytes loss, and the human heart has limited regenerative capacity. Strategies for regeneration and repair include the application of a cell suspension, growth factors, miRNAs and the implantation of an engineered tissue159–161. Various studies and clinical trials have tested cardiac progenitor cells (CPCs), bone marrow-derived stem cells, and pluripotent stem cells162. Frustratingly, these clinical interventions demonstrated safety but often failed to prove functional improvement. The mechanism underlying the potential effects of bone marrow derived stem cells (BMSCs) is unclear; the injected cells do not appear to remain in the cardiac tissue, but may release paracrine factors and recruit CPCs159.
Stem cells are now being used to provide cellular models of DCM. The absence of human cardiomyocyte cell lines has been a problem for advancing research in human cardiomyopathy. iPSCs can be more readily generated from human patients with DCM, and these cells can be differentiated into cardiomyocytes for study. A major limitation of iPSC derived cardiomyocytes is their relative immaturity and variability from culture to culture, but nonetheless these cells can be used to study cellular properties reflective of DCM features. As yet, these stem cells are not yet sufficiently mature for treating cardiomyopathy159, although they are yielding an important platform for understanding mechanisms and testing therapies163.
Conclusions and future directions
Better understanding of the DCM phenome and recent improvements in sequencing technology to define DCM genome will eventually improve the diagnosis, prevention and therapy of this disease. Next generation sequencing technology provides a cost-effective and accurate diagnostic method to yield biomarkers that indicate disease risk, especially within families. With this progress, criteria for pathogenic mutations are evolving and becoming more and more stringent, and may require re-evaluation of the molecular diagnosis over time. Several questions still remain in DCM management that prompt future investigations, such as the interpretation of genetic testing, the correct treatment of pre-clinical asymptomatic DCM gene carriers, and the development of gene- and mechanism-specific therapies.
Acknowledgments
NIH HL128075 to EMB; NIH UL1 TR001082, R01 HL69071, R01 116906 and in part by a Trans-Atlantic Network of Excellence grant from the Leducq Foundation (14-CVD 03) to LM.
Nonstandard Abbreviations
- AC
arrhythmogenic cardiomyopathy
- ACEi
angiotensin converting enzyme inhibitor
- ARB
angiotensin receptor blocker
- ARNI
angiotensin receptor-neprylisin inhibitor
- BMSC
bone marrow derived stem cell
- CLIA
Clinical Laboratory Improvement Amendment
- CMR
cardiac magnetic resonance
- CPC
cardiac progenitor cell
- DCM
dilated cardiomyopathy
- EMB
endomyocardial biopsy
- ExAC
exome aggregation consortium
- FHL
four and half LIM
- FS
fractional shortening
- HCM
hypertrophic cardiomyopathy
- HFrEF
heart failure with reduced ejection fraction
- HFmrEF
heart failure with midrange ejection fraction
- ICD
implantable cardioverter-defibrillator
- iPSC
induced pluripotent stem cell
- LVAD
left ventricular assist device
- LVEDD
left ventricular end diastolic dimension
- LVIDd
left ventricular internal dimension in diastole
- LVIDs
left ventricular internal dimension in systole
- LVNC
left ventricular noncompaction
- LV
left ventricle
- MAF
minor allele frequency
- NSVT
nonsustained ventricular tachycardia
- PEVK
proline-glutamate-valine-lysine
- RRM
ribonucleic acid recognition motif
- SCD
sudden cardiac death
- VUS
variant of uncertain significance
- VT
ventricular tachycardia
Footnotes
Disclosures: EMM is a consultant to Invitae; LM is consultant to Array Biopharma.
References
- 1.Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, Kuhl U, Maisch B, McKenna WJ, Monserrat L, Pankuweit S, Rapezzi C, Seferovic P, Tavazzi L, Keren A. Classification of the cardiomyopathies: A position statement from the european society of cardiology working group on myocardial and pericardial diseases. European heart journal. 2008;29:270–276. doi: 10.1093/eurheartj/ehm342. [DOI] [PubMed] [Google Scholar]
- 2.Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB. Contemporary definitions and classification of the cardiomyopathies: An american heart association scientific statement from the council on clinical cardiology, heart failure and transplantation committee; quality of care and outcomes research and functional genomics and translational biology interdisciplinary working groups; and council on epidemiology and prevention. Circulation. 2006;113:1807–1816. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- 3.Bozkurt B, Colvin M, Cook J, Cooper LT, Deswal A, Fonarow GC, Francis GS, Lenihan D, Lewis EF, McNamara DM, Pahl E, Ramachandran VS, Ramasubbu K, Rasmusson K, Towbin JA, Yancy C, American Heart Association Committee on Heart F, Transplantation of the Council on Clinical C, Council on Cardiovascular Disease in the Y, Council on C, Stroke N, Council on E, Prevention, Council on Quality of C, Outcomes R Current diagnostic and treatment strategies for specific dilated cardiomyopathies: A scientific statement from the american heart association. Circulation. 2016 doi: 10.1161/CIR.0000000000000455. [DOI] [PubMed] [Google Scholar]
- 4.Shore S, Grau-Sepulveda MV, Bhatt DL, Heidenreich PA, Eapen ZJ, Hernandez AF, Yancy CW, Fonarow GC. Characteristics, treatments, and outcomes of hospitalized heart failure patients stratified by etiologies of cardiomyopathy. JACC Heart Fail. 2015;3:906–916. doi: 10.1016/j.jchf.2015.06.012. [DOI] [PubMed] [Google Scholar]
- 5.Codd MB, Sugrue DD, Gersh BJ, Melton LJ., 3rd Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population-based study in olmsted county, minnesota, 1975–1984. Circulation. 1989;80:564–572. doi: 10.1161/01.cir.80.3.564. [DOI] [PubMed] [Google Scholar]
- 6.Torp A. Incidence of congestive cardiomyopathy. Postgrad Med J. 1978;54:435–439. doi: 10.1136/pgmj.54.633.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gillum RF. Idiopathic cardiomyopathy in the united states, 1970–1982. Am Heart J. 1986;111:752–755. doi: 10.1016/0002-8703(86)90111-0. [DOI] [PubMed] [Google Scholar]
- 8.Manolio TA, Baughman KL, Rodeheffer R, Pearson TA, Bristow JD, Michels VV, Abelmann WH, Harlan WR. Prevalence and etiology of idiopathic dilated cardiomyopathy (summary of a national heart, lung, and blood institute workshop. Am J Cardiol. 1992;69:1458–1466. doi: 10.1016/0002-9149(92)90901-a. [DOI] [PubMed] [Google Scholar]
- 9.Miura K, Nakagawa H, Morikawa Y, Sasayama S, Matsumori A, Hasegawa K, Ohno Y, Tamakoshi A, Kawamura T, Inaba Y. Epidemiology of idiopathic cardiomyopathy in japan: Results from a nationwide survey. Heart. 2002;87:126–130. doi: 10.1136/heart.87.2.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amoah AG, Kallen C. Aetiology of heart failure as seen from a national cardiac referral centre in africa. Cardiology. 2000;93:11–18. doi: 10.1159/000006996. [DOI] [PubMed] [Google Scholar]
- 11.Williams DG, Olsen EG. Prevalence of overt dilated cardiomyopathy in two regions of england. Br Heart J. 1985;54:153–155. doi: 10.1136/hrt.54.2.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rakar S, Sinagra G, Di Lenarda A, Poletti A, Bussani R, Silvestri F, Camerini F. Epidemiology of dilated cardiomyopathy. A prospective post-mortem study of 5252 necropsies. The heart muscle disease study group. European heart journal. 1997;18:117–123. doi: 10.1093/oxfordjournals.eurheartj.a015092. [DOI] [PubMed] [Google Scholar]
- 13.Sweet M, Taylor MR, Mestroni L. Diagnosis, prevalence, and screening of familial dilated cardiomyopathy. Expert Opin Orphan Drugs. 2015;3:869–876. doi: 10.1517/21678707.2015.1057498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10:531–547. doi: 10.1038/nrcardio.2013.105. [DOI] [PubMed] [Google Scholar]
- 15.Petretta M, Pirozzi F, Sasso L, Paglia A, Bonaduce D. Review and metaanalysis of the frequency of familial dilated cardiomyopathy. Am J Cardiol. 2011;108:1171–1176. doi: 10.1016/j.amjcard.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 16.Grunig E, Tasman JA, Kucherer H, Franz W, Kubler W, Katus HA. Frequency and phenotypes of familial dilated cardiomyopathy. Journal of the American College of Cardiology. 1998;31:186–194. doi: 10.1016/s0735-1097(97)00434-8. [DOI] [PubMed] [Google Scholar]
- 17.Ganesh SK, Arnett DK, Assimes TL, Basson CT, Chakravarti A, Ellinor PT, Engler MB, Goldmuntz E, Herrington DM, Hershberger RE, Hong Y, Johnson JA, Kittner SJ, McDermott DA, Meschia JF, Mestroni L, O’Donnell CJ, Psaty BM, Vasan RS, Ruel M, Shen WK, Terzic A, Waldman SA, American Heart Association Council on Functional G, Translational B, American Heart Association Council on E, Prevention, American Heart Association Council on Basic Cardiovascular S, American Heart Association Council on Cardiovascular Disease in the Y, American Heart Association Council on C, Stroke N, American Heart Association Stroke C Genetics and genomics for the prevention and treatment of cardiovascular disease: Update: A scientific statement from the american heart association. Circulation. 2013;128:2813–2851. doi: 10.1161/01.cir.0000437913.98912.1d. [DOI] [PubMed] [Google Scholar]
- 18.Mestroni L, Rocco C, Gregori D, Sinagra G, Di Lenarda A, Miocic S, Vatta M, Pinamonti B, Muntoni F, Caforio AL, McKenna WJ, Falaschi A, Giacca M, Camerini Familial dilated cardiomyopathy: Evidence for genetic and phenotypic heterogeneity. Heart muscle disease study group. Journal of the American College of Cardiology. 1999;34:181–190. doi: 10.1016/s0735-1097(99)00172-2. [DOI] [PubMed] [Google Scholar]
- 19.Mestroni L, Maisch B, McKenna WJ, Schwartz K, Charron P, Rocco C, Tesson F, Richter A, Wilke A, Komajda M. Guidelines for the study of familial dilated cardiomyopathies. Collaborative research group of the european human and capital mobility project on familial dilated cardiomyopathy. European heart journal. 1999;20:93–102. doi: 10.1053/euhj.1998.1145. [DOI] [PubMed] [Google Scholar]
- 20.Lopez-Ayala JM, Pastor-Quirante F, Gonzalez-Carrillo J, Lopez-Cuenca D, Sanchez-Munoz JJ, Oliva-Sandoval MJ, Gimeno JR. Genetics of myocarditis in arrhythmogenic right ventricular dysplasia. Heart Rhythm. 2015;12:766–773. doi: 10.1016/j.hrthm.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 21.Morales A, Painter T, Li R, Siegfried JD, Li D, Norton N, Hershberger RE. Rare variant mutations in pregnancy-associated or peripartum cardiomyopathy. Circulation. 2010;121:2176–2182. doi: 10.1161/CIRCULATIONAHA.109.931220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Spaendonck-Zwarts KY, van Tintelen JP, van Veldhuisen DJ, van der Werf R, Jongbloed JD, Paulus WJ, Dooijes D, van den Berg MP. Peripartum cardiomyopathy as a part of familial dilated cardiomyopathy. Circulation. 2010;121:2169–2175. doi: 10.1161/CIRCULATIONAHA.109.929646. [DOI] [PubMed] [Google Scholar]
- 23.Japp AG, Gulati A, Cook SA, Cowie MR, Prasad SK. The diagnosis and evaluation of dilated cardiomyopathy. Journal of the American College of Cardiology. 2016;67:2996–3010. doi: 10.1016/j.jacc.2016.03.590. [DOI] [PubMed] [Google Scholar]
- 24.Burridge PW, Li YF, Matsa E, Wu H, Ong SG, Sharma A, Holmstrom A, Chang AC, Coronado MJ, Ebert AD, Knowles JW, Telli ML, Witteles RM, Blau HM, Bernstein D, Altman RB, Wu JC. Human induced pluripotent stem cell-derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat Med. 2016;22:547–556. doi: 10.1038/nm.4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fatkin D. Guidelines for the diagnosis and management of familial dilated cardiomyopathy. Heart Lung Circ. 2011;20:691–693. doi: 10.1016/j.hlc.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 26.Crispell KA, Hanson EL, Coates K, Toy W, Hershberger RE. Periodic rescreening is indicated for family members at risk of developing familial dilated cardiomyopathy. Journal of the American College of Cardiology. 2002;39:1503–1507. doi: 10.1016/s0735-1097(02)01788-6. [DOI] [PubMed] [Google Scholar]
- 27.Mahon NG, Murphy RT, MacRae CA, Caforio AL, Elliott PM, McKenna WJ. Echocardiographic evaluation in asymptomatic relatives of patients with dilated cardiomyopathy reveals preclinical disease. Ann Intern Med. 2005;143:108–115. doi: 10.7326/0003-4819-143-2-200507190-00009. [DOI] [PubMed] [Google Scholar]
- 28.Captur G, Lopes LR, Mohun TJ, Patel V, Li C, Bassett P, Finocchiaro G, Ferreira VM, Esteban MT, Muthurangu V, Sherrid MV, Day SM, Canter CE, McKenna WJ, Seidman CE, Bluemke DA, Elliott PM, Ho CY, Moon JC. Prediction of sarcomere mutations in subclinical hypertrophic cardiomyopathy. Circ Cardiovasc Imaging. 2014;7:863–871. doi: 10.1161/CIRCIMAGING.114.002411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sefa Okten M, Tuluce K, Yakar Tuluce S, Kilic S, Soner Kemal H, Sayin A, Vuran O, Yagmur B, Mutlu I, Simsek E, Soydas Cinar C, Gurgun C. Screening first-degree relatives of patients with idiopathic dilated cardiomyopathy. Herz. 2016 doi: 10.1007/s00059-016-4498-1. [DOI] [PubMed] [Google Scholar]
- 30.Cordero-Reyes AM, Youker K, Estep JD, Torre-Amione G, Nagueh SF. Molecular and cellular correlates of cardiac function in end-stage dcm: A study using speckle tracking echocardiography. JACC Cardiovasc Imaging. 2014;7:441–452. doi: 10.1016/j.jcmg.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 31.Assomull RG, Prasad SK, Lyne J, Smith G, Burman ED, Khan M, Sheppard MN, Poole-Wilson PA, Pennell DJ. Cardiovascular magnetic resonance, fibrosis, and prognosis in dilated cardiomyopathy. Journal of the American College of Cardiology. 2006;48:1977–1985. doi: 10.1016/j.jacc.2006.07.049. [DOI] [PubMed] [Google Scholar]
- 32.Gao P, Yee R, Gula L, Krahn AD, Skanes A, Leong-Sit P, Klein GJ, Stirrat J, Fine N, Pallaveshi L, Wisenberg G, Thompson TR, Prato F, Drangova M, White JA. Prediction of arrhythmic events in ischemic and dilated cardiomyopathy patients referred for implantable cardiac defibrillator: Evaluation of multiple scar quantification measures for late gadolinium enhancement magnetic resonance imaging. Circ Cardiovasc Imaging. 2012;5:448–456. doi: 10.1161/CIRCIMAGING.111.971549. [DOI] [PubMed] [Google Scholar]
- 33.Perazzolo Marra M, De Lazzari M, Zorzi A, Migliore F, Zilio F, Calore C, Vettor G, Tona F, Tarantini G, Cacciavillani L, Corbetti F, Giorgi B, Miotto D, Thiene G, Basso C, Iliceto S, Corrado D. Impact of the presence and amount of myocardial fibrosis by cardiac magnetic resonance on arrhythmic outcome and sudden cardiac death in nonischemic dilated cardiomyopathy. Heart Rhythm. 2014;11:856–863. doi: 10.1016/j.hrthm.2014.01.014. [DOI] [PubMed] [Google Scholar]
- 34.Gulati A, Jabbour A, Ismail TF, Guha K, Khwaja J, Raza S, Morarji K, Brown TD, Ismail NA, Dweck MR, Di Pietro E, Roughton M, Wage R, Daryani Y, O’Hanlon R, Sheppard MN, Alpendurada F, Lyon AR, Cook SA, Cowie MR, Assomull RG, Pennell DJ, Prasad SK. Association of fibrosis with mortality and sudden cardiac death in patients with nonischemic dilated cardiomyopathy. Jama. 2013;309:896–908. doi: 10.1001/jama.2013.1363. [DOI] [PubMed] [Google Scholar]
- 35.Ordovas KG, Higgins CB. Delayed contrast enhancement on mr images of myocardium: Past, present, future. Radiology. 2011;261:358–374. doi: 10.1148/radiol.11091882. [DOI] [PubMed] [Google Scholar]
- 36.Dawson DK, McLernon DJ, Raj VJ, Maceira AM, Prasad S, Frenneaux MP, Pennell DJ, Kilner PJ. Cardiovascular magnetic resonance determinants of left ventricular noncompaction. Am J Cardiol. 2014;114:456–462. doi: 10.1016/j.amjcard.2014.05.017. [DOI] [PubMed] [Google Scholar]
- 37.Amzulescu MS, Rousseau MF, Ahn SA, Boileau L, de Meester de Ravenstein C, Vancraeynest D, Pasquet A, Vanoverschelde JL, Pouleur AC, Gerber BL. Prognostic impact of hypertrabeculation and noncompaction phenotype in dilated cardiomyopathy: A cmr study. JACC Cardiovasc Imaging. 2015;8:934–946. doi: 10.1016/j.jcmg.2015.04.015. [DOI] [PubMed] [Google Scholar]
- 38.Gil KE, Pawlak A, Gil RJ, Frontczak-Baniewicz M, Bil J. The role of invasive diagnostics and its impact on the treatment of dilated cardiomyopathy: A systematic review. Adv Med Sci. 2016;61:331–343. doi: 10.1016/j.advms.2016.07.001. [DOI] [PubMed] [Google Scholar]
- 39.Caforio AL, Pankuweit S, Arbustini E, Basso C, Gimeno-Blanes J, Felix SB, Fu M, Helio T, Heymans S, Jahns R, Klingel K, Linhart A, Maisch B, McKenna W, Mogensen J, Pinto YM, Ristic A, Schultheiss HP, Seggewiss H, Tavazzi L, Thiene G, Yilmaz A, Charron P, Elliott PM. Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: A position statement of the european society of cardiology working group on myocardial and pericardial diseases. European heart journal. 2013;34:2636–2648. 2648a–2648d. doi: 10.1093/eurheartj/eht210. [DOI] [PubMed] [Google Scholar]
- 40.Cooper LT, Baughman KL, Feldman AM, Frustaci A, Jessup M, Kuhl U, Levine GN, Narula J, Starling RC, Towbin J, Virmani R. The role of endomyocardial biopsy in the management of cardiovascular disease: A scientific statement from the american heart association, the american college of cardiology, and the european society of cardiology. Circulation. 2007;116:2216–2233. doi: 10.1161/CIRCULATIONAHA.107.186093. [DOI] [PubMed] [Google Scholar]
- 41.Cooper LT, Baughman KL, Feldman AM, Frustaci A, Jessup M, Kuhl U, Levine GN, Narula J, Starling RC, Towbin J, Virmani R. The role of endomyocardial biopsy in the management of cardiovascular disease: A scientific statement from the american heart association, the american college of cardiology, and the european society of cardiology endorsed by the heart failure society of america and the heart failure association of the european society of cardiology. European heart journal. 2007;28:3076–3093. doi: 10.1093/eurheartj/ehm456. [DOI] [PubMed] [Google Scholar]
- 42.Asimaki A, Tandri H, Huang H, Halushka MK, Gautam S, Basso C, Thiene G, Tsatsopoulou A, Protonotarios N, McKenna WJ, Calkins H, Saffitz JE. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2009;360:1075–1084. doi: 10.1056/NEJMoa0808138. [DOI] [PubMed] [Google Scholar]
- 43.Asimaki A, Protonotarios A, James CA, Chelko SP, Tichnell C, Murray B, Tsatsopoulou A, Anastasakis A, te Riele A, Kleber AG, Judge DP, Calkins H, Saffitz JE. Characterizing the molecular pathology of arrhythmogenic cardiomyopathy in patient buccal mucosa cells. Circ Arrhythm Electrophysiol. 2016;9:e003688. doi: 10.1161/CIRCEP.115.003688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Felker GM, Thompson RE, Hare JM, Hruban RH, Clemetson DE, Howard DL, Baughman KL, Kasper EK. Underlying causes and long-term survival in patients with initially unexplained cardiomyopathy. N Engl J Med. 2000;342:1077–1084. doi: 10.1056/NEJM200004133421502. [DOI] [PubMed] [Google Scholar]
- 45.Spezzacatene A, Sinagra G, Merlo M, Barbati G, Graw SL, Brun F, Slavov D, Di Lenarda A, Salcedo EE, Towbin JA, Saffitz JE, Marcus FI, Zareba W, Taylor MR, Mestroni L, Familial Cardiomyopathy R Arrhythmogenic phenotype in dilated cardiomyopathy: Natural history and predictors of life-threatening arrhythmias. J Am Heart Assoc. 2015;4:e002149. doi: 10.1161/JAHA.115.002149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Berlo JH, de Voogt WG, van der Kooi AJ, van Tintelen JP, Bonne G, Yaou RB, Duboc D, Rossenbacker T, Heidbuchel H, de Visser M, Crijns HJ, Pinto YM. Meta-analysis of clinical characteristics of 299 carriers of lmna gene mutations: Do lamin a/c mutations portend a high risk of sudden death? J Mol Med (Berl) 2005;83:79–83. doi: 10.1007/s00109-004-0589-1. [DOI] [PubMed] [Google Scholar]
- 47.van Rijsingen IA, Arbustini E, Elliott PM, Mogensen J, Hermans-van Ast JF, van der Kooi AJ, van Tintelen JP, van den Berg MP, Pilotto A, Pasotti M, Jenkins S, Rowland C, Aslam U, Wilde AA, Perrot A, Pankuweit S, Zwinderman AH, Charron P, Pinto YM. Risk factors for malignant ventricular arrhythmias in lamin a/c mutation carriers a european cohort study. Journal of the American College of Cardiology. 2012;59:493–500. doi: 10.1016/j.jacc.2011.08.078. [DOI] [PubMed] [Google Scholar]
- 48.McNair WP, Sinagra G, Taylor MR, Di Lenarda A, Ferguson DA, Salcedo EE, Slavov D, Zhu X, Caldwell JH, Mestroni L. Scn5a mutations associate with arrhythmic dilated cardiomyopathy and commonly localize to the voltage-sensing mechanism. Journal of the American College of Cardiology. 2011;57:2160–2168. doi: 10.1016/j.jacc.2010.09.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mann SA, Castro ML, Ohanian M, Guo G, Zodgekar P, Sheu A, Stockhammer K, Thompson T, Playford D, Subbiah R, Kuchar D, Aggarwal A, Vandenberg JI, Fatkin D. R222q scn5a mutation is associated with reversible ventricular ectopy and dilated cardiomyopathy. Journal of the American College of Cardiology. 2012;60:1566–1573. doi: 10.1016/j.jacc.2012.05.050. [DOI] [PubMed] [Google Scholar]
- 50.Brown CA, Lanning RW, McKinney KQ, Salvino AR, Cherniske E, Crowe CA, Darras BT, Gominak S, Greenberg CR, Grosmann C, Heydemann P, Mendell JR, Pober BR, Sasaki T, Shapiro F, Simpson DA, Suchowersky O, Spence JE. Novel and recurrent mutations in lamin a/c in patients with emery-dreifuss muscular dystrophy. Am J Med Genet. 2001;102:359–367. doi: 10.1002/ajmg.1463. [DOI] [PubMed] [Google Scholar]
- 51.McNally EM. New approaches in the therapy of cardiomyopathy in muscular dystrophy. Annu Rev Med. 2007;58:75–88. doi: 10.1146/annurev.med.58.011706.144703. [DOI] [PubMed] [Google Scholar]
- 52.Tanawuttiwat T, Wagner KR, Tomaselli G, Nazarian S. Left ventricular dysfunction and conduction disturbances in patients with myotonic muscular dystrophy type i and ii. JAMA Cardiol. 2016 doi: 10.1001/jamacardio.2016.4145. [DOI] [PubMed] [Google Scholar]
- 53.Sommerville RB, Vincenti MG, Winborn K, Casey A, Stitziel NO, Connolly AM, Mann DL. Diagnosis and management of adult hereditary cardio-neuromuscular disorders: A model for the multidisciplinary care of complex genetic disorders. Trends Cardiovasc Med. 2017;27:51–58. doi: 10.1016/j.tcm.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 54.McNally EM, Sparano D. Mechanisms and management of the heart in myotonic dystrophy. Heart. 2011;97:1094–1100. doi: 10.1136/hrt.2010.214197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wahbi K, Babuty D, Probst V, Wissocque L, Labombarda F, Porcher R, Becane HM, Lazarus A, Behin A, Laforet P, Stojkovic T, Clementy N, Dussauge AP, Gourraud JB, Pereon Y, Lacour A, Chapon F, Milliez P, Klug D, Eymard B, Duboc D. Incidence and predictors of sudden death, major conduction defects and sustained ventricular tachyarrhythmias in 1388 patients with myotonic dystrophy type 1. European heart journal. 2016 doi: 10.1093/eurheartj/ehw569. [DOI] [PubMed] [Google Scholar]
- 56.McNally EM, Golbus JR, Puckelwartz MJ. Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin Invest. 2013;123:19–26. doi: 10.1172/JCI62862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McNally EM, Barefield DY, Puckelwartz MJ. The genetic landscape of cardiomyopathy and its role in heart failure. Cell Metab. 2015;21:174–182. doi: 10.1016/j.cmet.2015.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pugh TJ, Kelly MA, Gowrisankar S, Hynes E, Seidman MA, Baxter SM, Bowser M, Harrison B, Aaron D, Mahanta LM, Lakdawala NK, McDermott G, White ET, Rehm HL, Lebo M, Funke BH. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med. 2014;16:601–608. doi: 10.1038/gim.2013.204. [DOI] [PubMed] [Google Scholar]
- 59.Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, Mazzarotto F, Blair E, Seller A, Taylor JC, Minikel EV, Exome Aggregation C. MacArthur DG, Farrall M, Cook SA, Watkins H. Reassessment of mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60, 706 reference samples. Genet Med. 2016 doi: 10.1038/gim.2016.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li Q, Gruner C, Chan RH, Care M, Siminovitch K, Williams L, Woo A, Rakowski H. Genotype-positive status in patients with hypertrophic cardiomyopathy is associated with higher rates of heart failure events. Circ Cardiovasc Genet. 2014;7:416–422. doi: 10.1161/CIRCGENETICS.113.000331. [DOI] [PubMed] [Google Scholar]
- 61.Herman DS, Lam L, Taylor MR, Wang L, Teekakirikul P, Christodoulou D, Conner L, DePalma SR, McDonough B, Sparks E, Teodorescu DL, Cirino AL, Banner NR, Pennell DJ, Graw S, Merlo M, Di Lenarda A, Sinagra G, Bos JM, Ackerman MJ, Mitchell RN, Murry CE, Lakdawala NK, Ho CY, Barton PJ, Cook SA, Mestroni L, Seidman JG, Seidman CE. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–628. doi: 10.1056/NEJMoa1110186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.LeWinter MM, Granzier HL. Cardiac titin and heart disease. J Cardiovasc Pharmacol. 2014;63:207–212. doi: 10.1097/FJC.0000000000000007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Anderson BR, Granzier HL. Titin-based tension in the cardiac sarcomere: Molecular origin and physiological adaptations. Prog Biophys Mol Biol. 2012;110:204–217. doi: 10.1016/j.pbiomolbio.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Christodoulou DC, Wakimoto H, Onoue K, Eminaga S, Gorham JM, DePalma SR, Herman DS, Teekakirikul P, Conner DA, McKean DM, Domenighetti AA, Aboukhalil A, Chang S, Srivastava G, McDonough B, De Jager PL, Chen J, Bulyk ML, Muehlschlegel JD, Seidman CE, Seidman JG. 5’rna-seq identifies fhl1 as a genetic modifier in cardiomyopathy. J Clin Invest. 2014;124:1364–1370. doi: 10.1172/JCI70108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nagueh SF, Shah G, Wu Y, Torre-Amione G, King NM, Lahmers S, Witt CC, Becker K, Labeit S, Granzier HL. Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation. 2004;110:155–162. doi: 10.1161/01.CIR.0000135591.37759.AF. [DOI] [PubMed] [Google Scholar]
- 66.Roberts AM, Ware JS, Herman DS, Schafer S, Baksi J, Bick AG, Buchan RJ, Walsh R, John S, Wilkinson S, Mazzarotto F, Felkin LE, Gong S, MacArthur JA, Cunningham F, Flannick J, Gabriel SB, Altshuler DM, Macdonald PS, Heinig M, Keogh AM, Hayward CS, Banner NR, Pennell DJ, O’Regan DP, San TR, de Marvao A, Dawes TJ, Gulati A, Birks EJ, Yacoub MH, Radke M, Gotthardt M, Wilson JG, O’Donnell CJ, Prasad SK, Barton PJ, Fatkin D, Hubner N, Seidman JG, Seidman CE, Cook SA. Integrated allelic, transcriptional, and phenomic dissection of the cardiac effects of titin truncations in health and disease. Sci Transl Med. 2015;7:270ra276. doi: 10.1126/scitranslmed.3010134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gerull B, Gramlich M, Atherton J, McNabb M, Trombitas K, Sasse-Klaassen S, Seidman JG, Seidman C, Granzier H, Labeit S, Frenneaux M, Thierfelder L. Mutations of ttn, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat Genet. 2002;30:201–204. doi: 10.1038/ng815. [DOI] [PubMed] [Google Scholar]
- 68.Hinson JT, Chopra A, Nafissi N, Polacheck WJ, Benson CC, Swist S, Gorham J, Yang L, Schafer S, Sheng CC, Haghighi A, Homsy J, Hubner N, Church G, Cook SA, Linke WA, Chen CS, Seidman JG, Seidman CE. Heart disease. Titin mutations in ips cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science. 2015;349:982–986. doi: 10.1126/science.aaa5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Golbus JR, Puckelwartz MJ, Fahrenbach JP, Dellefave-Castillo LM, Wolfgeher D, McNally EM. Population-based variation in cardiomyopathy genes. Circ Cardiovasc Genet. 2012;5:391–399. doi: 10.1161/CIRCGENETICS.112.962928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jansweijer JA, Nieuwhof K, Russo F, Hoorntje ET, Jongbloed JD, Lekanne Deprez RH, Postma AV, Bronk M, van Rijsingen IA, de Haij S, Biagini E, van Haelst PL, van Wijngaarden J, van den Berg MP, Wilde AA, Mannens MM, de Boer RA, van Spaendonck-Zwarts KY, van Tintelen JP, Pinto YM. Truncating titin mutations are associated with a mild and treatable form of dilated cardiomyopathy. Eur J Heart Fail. 2016 doi: 10.1002/ejhf.673. [DOI] [PubMed] [Google Scholar]
- 71.Schafer S, de Marvao A, Adami E, Fiedler LR, Ng B, Khin E, Rackham OJ, van Heesch S, Pua CJ, Kui M, Walsh R, Tayal U, Prasad SK, Dawes TJ, Ko NS, Sim D, Chan LL, Chin CW, Mazzarotto F, Barton PJ, Kreuchwig F, de Kleijn DP, Totman T, Biffi C, Tee N, Rueckert D, Schneider V, Faber A, Regitz-Zagrosek V, Seidman JG, Seidman CE, Linke WA, Kovalik JP, O’Regan D, Ware JS, Hubner N, Cook SA. Titin-truncating variants affect heart function in disease cohorts and the general population. Nat Genet. 2016 doi: 10.1038/ng.3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.van Spaendonck-Zwarts KY, Posafalvi A, van den Berg MP, Hilfiker-Kleiner D, Bollen IA, Sliwa K, Alders M, Almomani R, van Langen IM, van der Meer P, Sinke RJ, van der Velden J, Van Veldhuisen DJ, van Tintelen JP, Jongbloed JD. Titin gene mutations are common in families with both peripartum cardiomyopathy and dilated cardiomyopathy. European heart journal. 2014;35:2165–2173. doi: 10.1093/eurheartj/ehu050. [DOI] [PubMed] [Google Scholar]
- 73.Ware JS, Li J, Mazaika E, Yasso CM, DeSouza T, Cappola TP, Tsai EJ, Hilfiker-Kleiner D, Kamiya CA, Mazzarotto F, Cook SA, Halder I, Prasad SK, Pisarcik J, Hanley-Yanez K, Alharethi R, Damp J, Hsich E, Elkayam U, Sheppard R, Kealey A, Alexis J, Ramani G, Safirstein J, Boehmer J, Pauly DF, Wittstein IS, Thohan V, Zucker MJ, Liu P, Gorcsan J, 3rd, McNamara DM, Seidman CE, Seidman JG, Arany Z. Shared genetic predisposition in peripartum and dilated cardiomyopathies. N Engl J Med. 2016;374:233–241. doi: 10.1056/NEJMoa1505517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Felkin LE, Walsh R, Ware JS, Yacoub MH, Birks EJ, Barton PJ, Cook SA. Recovery of cardiac function in cardiomyopathy caused by titin truncation. JAMA Cardiol. 2016;1:234–235. doi: 10.1001/jamacardio.2016.0208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Norton N, Li D, Rampersaud E, Morales A, Martin ER, Zuchner S, Guo S, Gonzalez M, Hedges DJ, Robertson PD, Krumm N, Nickerson DA, Hershberger RE. Exome sequencing and genome-wide linkage analysis in 17 families illustrate the complex contribution of ttn truncating variants to dilated cardiomyopathy. Circ Cardiovasc Genet. 2013;6:144–153. doi: 10.1161/CIRCGENETICS.111.000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gigli M, Begay RL, Morea G, Graw SL, Sinagra G, Taylor MR, Granzier H, Mestroni L. A review of the giant protein titin in clinical molecular diagnostics of cardiomyopathies. Front Cardiovasc Med. 2016;3:21. doi: 10.3389/fcvm.2016.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Akinrinade O, Ollila L, Vattulainen S, Tallila J, Gentile M, Salmenpera P, Koillinen H, Kaartinen M, Nieminen MS, Myllykangas S, Alastalo TP, Koskenvuo JW, Helio T. Genetics and genotype-phenotype correlations in finnish patients with dilated cardiomyopathy. European heart journal. 2015;36:2327–2337. doi: 10.1093/eurheartj/ehv253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Begay RL, Graw S, Sinagra G, Merlo M, Slavov D, Gowan K, Jones KL, Barbati G, Spezzacatene A, Brun F, Di Lenarda A, Smith JE, Granzier HL, Mestroni L, Taylor M. Role of titin missense variants in dilated cardiomyopathy. J Am Heart Assoc. 2015;4 doi: 10.1161/JAHA.115.002645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ceyhan-Birsoy O, Agrawal PB, Hidalgo C, Schmitz-Abe K, DeChene ET, Swanson LC, Soemedi R, Vasli N, Iannaccone ST, Shieh PB, Shur N, Dennison JM, Lawlor MW, Laporte J, Markianos K, Fairbrother WG, Granzier H, Beggs AH. Recessive truncating titin gene, ttn, mutations presenting as centronuclear myopathy. Neurology. 2013;81:1205–1214. doi: 10.1212/WNL.0b013e3182a6ca62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hackman P, Vihola A, Haravuori H, Marchand S, Sarparanta J, De Seze J, Labeit S, Witt C, Peltonen L, Richard I, Udd B. Tibial muscular dystrophy is a titinopathy caused by mutations in ttn, the gene encoding the giant skeletal-muscle protein titin. Am J Hum Genet. 2002;71:492–500. doi: 10.1086/342380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zou J, Tran D, Baalbaki M, Tang LF, Poon A, Pelonero A, Titus EW, Yuan C, Shi C, Patchava S, Halper E, Garg J, Movsesyan I, Yin C, Wu R, Wilsbacher LD, Liu J, Hager RL, Coughlin SR, Jinek M, Pullinger CR, Kane JP, Hart DO, Kwok PY, Deo RC. An internal promoter underlies the difference in disease severity between n- and c-terminal truncation mutations of titin in zebrafish. Elife. 2015;4:e09406. doi: 10.7554/eLife.09406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Granzier HL, Radke MH, Peng J, Westermann D, Nelson OL, Rost K, King NM, Yu Q, Tschope C, McNabb M, Larson DF, Labeit S, Gotthardt M. Truncation of titin’s elastic pevk region leads to cardiomyopathy with diastolic dysfunction. Circ Res. 2009;105:557–564. doi: 10.1161/CIRCRESAHA.109.200964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhou Q, Kesteven S, Wu J, Aidery P, Gawaz M, Gramlich M, Feneley MP, Harvey RP. Pressure overload by transverse aortic constriction induces maladaptive hypertrophy in a titin-truncated mouse model. Biomed Res Int. 2015;2015:163564. doi: 10.1155/2015/163564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.van Tintelen JP, Hofstra RM, Katerberg H, Rossenbacker T, Wiesfeld AC, du Marchie Sarvaas GJ, Wilde AA, van Langen IM, Nannenberg EA, van der Kooi AJ, Kraak M, van Gelder IC, van Veldhuisen DJ, Vos Y, van den Berg MP. High yield of lmna mutations in patients with dilated cardiomyopathy and/or conduction disease referred to cardiogenetics outpatient clinics. Am Heart J. 2007;154:1130–1139. doi: 10.1016/j.ahj.2007.07.038. [DOI] [PubMed] [Google Scholar]
- 85.Parks SB, Kushner JD, Nauman D, Burgess D, Ludwigsen S, Peterson A, Li D, Jakobs P, Litt M, Porter CB, Rahko PS, Hershberger RE. Lamin a/c mutation analysis in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Am Heart J. 2008;156:161–169. doi: 10.1016/j.ahj.2008.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lu JT, Muchir A, Nagy PL, Worman HJ. Lmna cardiomyopathy: Cell biology and genetics meet clinical medicine. Dis Model Mech. 2011;4:562–568. doi: 10.1242/dmm.006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Young SG, Meta M, Yang SH, Fong LG. Prelamin a farnesylation and progeroid syndromes. J Biol Chem. 2006;281:39741–39745. doi: 10.1074/jbc.R600033200. [DOI] [PubMed] [Google Scholar]
- 88.Narula N, Favalli V, Tarantino P, Grasso M, Pilotto A, Bellazzi R, Serio A, Gambarin FI, Charron P, Meder B, Pinto Y, Elliott PM, Mogensen J, Bolognesi M, Bollati M, Arbustini E. Quantitative expression of the mutated lamin a/c gene in patients with cardiolaminopathy. Journal of the American College of Cardiology. 2012;60:1916–1920. doi: 10.1016/j.jacc.2012.05.059. [DOI] [PubMed] [Google Scholar]
- 89.Dittmer TA, Sahni N, Kubben N, Hill DE, Vidal M, Burgess RC, Roukos V, Misteli T. Systematic identification of pathological lamin a interactors. Mol Biol Cell. 2014;25:1493–1510. doi: 10.1091/mbc.E14-02-0733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nikolova V, Leimena C, McMahon AC, Tan JC, Chandar S, Jogia D, Kesteven SH, Michalicek J, Otway R, Verheyen F, Rainer S, Stewart CL, Martin D, Feneley MP, Fatkin D. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin a/c-deficient mice. J Clin Invest. 2004;113:357–369. doi: 10.1172/JCI19448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, Stewart CL, Lee RT. Lamin a/c deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest. 2004;113:370–378. doi: 10.1172/JCI19670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chandar S, Yeo LS, Leimena C, Tan JC, Xiao XH, Nikolova-Krstevski V, Yasuoka Y, Gardiner-Garden M, Wu J, Kesteven S, Karlsdotter L, Natarajan S, Carlton A, Rainer S, Feneley MP, Fatkin D. Effects of mechanical stress and carvedilol in lamin a/c-deficient dilated cardiomyopathy. Circ Res. 2010;106:573–582. doi: 10.1161/CIRCRESAHA.109.204388. [DOI] [PubMed] [Google Scholar]
- 93.Ramos FJ, Chen SC, Garelick MG, Dai DF, Liao CY, Schreiber KH, MacKay VL, An EH, Strong R, Ladiges WC, Rabinovitch PS, Kaeberlein M, Kennedy BK. Rapamycin reverses elevated mtorc1 signaling in lamin a/c-deficient mice, rescues cardiac and skeletal muscle function, and extends survival. Sci Transl Med. 2012;4:144ra103. doi: 10.1126/scitranslmed.3003802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Choi JC, Muchir A, Wu W, Iwata S, Homma S, Morrow JP, Worman HJ. Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin a/c gene mutation. Sci Transl Med. 2012;4:144ra102. doi: 10.1126/scitranslmed.3003875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wu W, Muchir A, Shan J, Bonne G, Worman HJ. Mitogen-activated protein kinase inhibitors improve heart function and prevent fibrosis in cardiomyopathy caused by mutation in lamin a/c gene. Circulation. 2011;123:53–61. doi: 10.1161/CIRCULATIONAHA.110.970673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.MacRae C, Taylor MRG, Mestroni L, Moses JR, Ashley EA, Wheeler MT, Lakdawala NK, Hershberger RE, Ptaszynski M, Sandor V, Saunders ME, Oliver C, Lee PA, Judge DP. Phase 2 study of a797, an oral, selective p38 mitogen-activated protein kinase inhibitor, in patients with lamin a/c-related dilated cardiomyopathy. European heart journal. 2016;37:1011. [Google Scholar]
- 97.Hasselberg NE, Edvardsen T, Petri H, Berge KE, Leren TP, Bundgaard H, Haugaa KH. Risk prediction of ventricular arrhythmias and myocardial function in lamin a/c mutation positive subjects. Europace. 2014;16:563–571. doi: 10.1093/europace/eut291. [DOI] [PubMed] [Google Scholar]
- 98.Haghighi K, Kolokathis F, Gramolini AO, Waggoner JR, Pater L, Lynch RA, Fan GC, Tsiapras D, Parekh RR, Dorn GW, 2nd, MacLennan DH, Kremastinos DT, Kranias EG. A mutation in the human phospholamban gene, deleting arginine 14, results in lethal, hereditary cardiomyopathy. Proc Natl Acad Sci U S A. 2006;103:1388–1393. doi: 10.1073/pnas.0510519103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liu GS, Morales A, Vafiadaki E, Lam CK, Cai WF, Haghighi K, Adly G, Hershberger RE, Kranias EG. A novel human r25c-phospholamban mutation is associated with super-inhibition of calcium cycling and ventricular arrhythmia. Cardiovasc Res. 2015;107:164–174. doi: 10.1093/cvr/cvv127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.van der Zwaag PA, van Rijsingen IA, Asimaki A, Jongbloed JD, van Veldhuisen DJ, Wiesfeld AC, Cox MG, van Lochem LT, de Boer RA, Hofstra RM, Christiaans I, van Spaendonck-Zwarts KY, Lekanne dit Deprez RH, Judge DP, Calkins H, Suurmeijer AJ, Hauer RN, Saffitz JE, Wilde AA, van den Berg MP, van Tintelen JP. Phospholamban r14del mutation in patients diagnosed with dilated cardiomyopathy or arrhythmogenic right ventricular cardiomyopathy: Evidence supporting the concept of arrhythmogenic cardiomyopathy. Eur J Heart Fail. 2012;14:1199–1207. doi: 10.1093/eurjhf/hfs119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.DeWitt MM, MacLeod HM, Soliven B, McNally EM. Phospholamban r14 deletion results in late-onset, mild, hereditary dilated cardiomyopathy. Journal of the American College of Cardiology. 2006;48:1396–1398. doi: 10.1016/j.jacc.2006.07.016. [DOI] [PubMed] [Google Scholar]
- 102.Truszkowska GT, Bilinska ZT, Kosinska J, Sleszycka J, Rydzanicz M, Sobieszczanska-Malek M, Franaszczyk M, Bilinska M, Stawinski P, Michalak E, Malek LA, Chmielewski P, Foss-Nieradko B, Machnicki MM, Stoklosa T, Poninska J, Szumowski L, Grzybowski J, Piwonski J, Drygas W, Zielinski T, Ploski R. A study in polish patients with cardiomyopathy emphasizes pathogenicity of phospholamban (pln) mutations at amino acid position 9 and low penetrance of heterozygous null pln mutations. BMC Med Genet. 2015;16:21. doi: 10.1186/s12881-015-0167-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Karakikes I, Stillitano F, Nonnenmacher M, Tzimas C, Sanoudou D, Termglinchan V, Kong CW, Rushing S, Hansen J, Ceholski D, Kolokathis F, Kremastinos D, Katoulis A, Ren L, Cohen N, Gho JM, Tsiapras D, Vink A, Wu JC, Asselbergs FW, Li RA, Hulot JS, Kranias EG, Hajjar RJ. Correction of human phospholamban r14del mutation associated with cardiomyopathy using targeted nucleases and combination therapy. Nat Commun. 2015;6:6955. doi: 10.1038/ncomms7955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Stillitano F, Turnbull IC, Karakikes I, Nonnenmacher M, Backeris P, Hulot JS, Kranias EG, Hajjar RJ, Costa KD. Genomic correction of familial cardiomyopathy in human engineered cardiac tissues. European heart journal. 2016;37:3282–3284. doi: 10.1093/eurheartj/ehw307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Te Rijdt WP, van Tintelen JP, Vink A, van der Wal AC, de Boer RA, van den Berg MP, Suurmeijer AJ. Phospholamban p.Arg14del cardiomyopathy is characterized by phospholamban aggregates, aggresomes, and autophagic degradation. Histopathology. 2016;69:542–550. doi: 10.1111/his.12963. [DOI] [PubMed] [Google Scholar]
- 106.Brauch KM, Karst ML, Herron KJ, de Andrade M, Pellikka PA, Rodeheffer RJ, Michels VV, Olson TM. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. Journal of the American College of Cardiology. 2009;54:930–941. doi: 10.1016/j.jacc.2009.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Li D, Morales A, Gonzalez-Quintana J, Norton N, Siegfried JD, Hofmeyer M, Hershberger RE. Identification of novel mutations in rbm20 in patients with dilated cardiomyopathy. Clin Transl Sci. 2010;3:90–97. doi: 10.1111/j.1752-8062.2010.00198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Beqqali A, Bollen IA, Rasmussen TB, van den Hoogenhof MM, van Deutekom HW, Schafer S, Haas J, Meder B, Sorensen KE, van Oort RJ, Mogensen J, Hubner N, Creemers EE, van der Velden J, Pinto YM. A mutation in the glutamate-rich region of rna-binding motif protein 20 causes dilated cardiomyopathy through missplicing of titin and impaired frank-starling mechanism. Cardiovasc Res. 2016;112:452–463. doi: 10.1093/cvr/cvw192. [DOI] [PubMed] [Google Scholar]
- 109.Guo W, Schafer S, Greaser ML, Radke MH, Liss M, Govindarajan T, Maatz H, Schulz H, Li S, Parrish AM, Dauksaite V, Vakeel P, Klaassen S, Gerull B, Thierfelder L, Regitz-Zagrosek V, Hacker TA, Saupe KW, Dec GW, Ellinor PT, MacRae CA, Spallek B, Fischer R, Perrot A, Ozcelik C, Saar K, Hubner N, Gotthardt M. Rbm20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat Med. 2012;18:766–773. doi: 10.1038/nm.2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li S, Guo W, Dewey CN, Greaser ML. Rbm20 regulates titin alternative splicing as a splicing repressor. Nucleic Acids Res. 2013;41:2659–2672. doi: 10.1093/nar/gks1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Maatz H, Jens M, Liss M, Schafer S, Heinig M, Kirchner M, Adami E, Rintisch C, Dauksaite V, Radke MH, Selbach M, Barton PJ, Cook SA, Rajewsky N, Gotthardt M, Landthaler M, Hubner N. Rna-binding protein rbm20 represses splicing to orchestrate cardiac pre-mrna processing. J Clin Invest. 2014;124:3419–3430. doi: 10.1172/JCI74523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wyles SP, Li X, Hrstka SC, Reyes S, Oommen S, Beraldi R, Edwards J, Terzic A, Olson TM, Nelson TJ. Modeling structural and functional deficiencies of rbm20 familial dilated cardiomyopathy using human induced pluripotent stem cells. Hum Mol Genet. 2016;25:254–265. doi: 10.1093/hmg/ddv468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Khan MA, Reckman YJ, Aufiero S, van den Hoogenhof MM, van der Made I, Beqqali A, Koolbergen DR, Rasmussen TB, van der Velden J, Creemers EE, Pinto YM. Rbm20 regulates circular rna production from the titin gene. Circ Res. 2016;119:996–1003. doi: 10.1161/CIRCRESAHA.116.309568. [DOI] [PubMed] [Google Scholar]
- 114.Nair K, Pekhletski R, Harris L, Care M, Morel C, Farid T, Backx PH, Szabo E, Nanthakumar K. Escape capture bigeminy: Phenotypic marker of cardiac sodium channel voltage sensor mutation r222q. Heart Rhythm. 2012;9:1681–1688.e1681. doi: 10.1016/j.hrthm.2012.06.029. [DOI] [PubMed] [Google Scholar]
- 115.Davis RP, Casini S, van den Berg CW, Hoekstra M, Remme CA, Dambrot C, Salvatori D, Oostwaard DW, Wilde AA, Bezzina CR, Verkerk AO, Freund C, Mummery CL. Cardiomyocytes derived from pluripotent stem cells recapitulate electrophysiological characteristics of an overlap syndrome of cardiac sodium channel disease. Circulation. 2012;125:3079–3091. doi: 10.1161/CIRCULATIONAHA.111.066092. [DOI] [PubMed] [Google Scholar]
- 116.Liang P, Sallam K, Wu H, Li Y, Itzhaki I, Garg P, Zhang Y, Vermglinchan V, Lan F, Gu M, Gong T, Zhuge Y, He C, Ebert AD, Sanchez-Freire V, Churko J, Hu S, Sharma A, Lam CK, Scheinman MM, Bers DM, Wu JC. Patient-specific and genome-edited induced pluripotent stem cell-derived cardiomyocytes elucidate single-cell phenotype of brugada syndrome. Journal of the American College of Cardiology. 2016;68:2086–2096. doi: 10.1016/j.jacc.2016.07.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Malan D, Friedrichs S, Fleischmann BK, Sasse P. Cardiomyocytes obtained from induced pluripotent stem cells with long-qt syndrome 3 recapitulate typical disease-specific features in vitro. Circ Res. 2011;109:841–847. doi: 10.1161/CIRCRESAHA.111.243139. [DOI] [PubMed] [Google Scholar]
- 118.Wan E, Abrams J, Weinberg RL, Katchman AN, Bayne J, Zakharov SI, Yang L, Morrow JP, Garan H, Marx SO. Aberrant sodium influx causes cardiomyopathy and atrial fibrillation in mice. J Clin Invest. 2016;126:112–122. doi: 10.1172/JCI84669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Feng J, Yan J, Buzin CH, Towbin JA, Sommer SS. Mutations in the dystrophin gene are associated with sporadic dilated cardiomyopathy. Mol Genet Metab. 2002;77:119–126. doi: 10.1016/s1096-7192(02)00153-1. [DOI] [PubMed] [Google Scholar]
- 120.Jefferies JL, Eidem BW, Belmont JW, Craigen WJ, Ware SM, Fernbach SD, Neish SR, Smith EO, Towbin JA. Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation. 2005;112:2799–2804. doi: 10.1161/CIRCULATIONAHA.104.528281. [DOI] [PubMed] [Google Scholar]
- 121.Ortiz-Lopez R, Li H, Su J, Goytia V, Towbin JA. Evidence for a dystrophin missense mutation as a cause of x-linked dilated cardiomyopathy. Circulation. 1997;95:2434–2440. doi: 10.1161/01.cir.95.10.2434. [DOI] [PubMed] [Google Scholar]
- 122.Tsubata S, Bowles KR, Vatta M, Zintz C, Titus J, Muhonen L, Bowles NE, Towbin JA. Mutations in the human delta-sarcoglycan gene in familial and sporadic dilated cardiomyopathy. J Clin Invest. 2000;106:655–662. doi: 10.1172/JCI9224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Haas M, Vlcek V, Balabanov P, Salmonson T, Bakchine S, Markey G, Weise M, Schlosser-Weber G, Brohmann H, Yerro CP, Mendizabal MR, Stoyanova-Beninska V, Hillege HL. European medicines agency review of ataluren for the treatment of ambulant patients aged 5 years and older with duchenne muscular dystrophy resulting from a nonsense mutation in the dystrophin gene. Neuromuscul Disord. 2015;25:5–13. doi: 10.1016/j.nmd.2014.11.011. [DOI] [PubMed] [Google Scholar]
- 124.Niks EH, Aartsma-Rus A. Exon skipping: A first in class strategy for duchenne muscular dystrophy. Expert Opin Biol Ther. 2016:1–12. doi: 10.1080/14712598.2017.1271872. [DOI] [PubMed] [Google Scholar]
- 125.Begay RL, Tharp CA, Martin A, Graw SL, Sinagra G, Miani D, Sweet ME, Slavov DB, Stafford N, Zeller MJ, Alnefaie R, Rowland TJ, Brun F, Jones KL, Gowan KLM, Garrity DM, Taylor MRG. Flnc gene splice mutations cause dilated cardiomyopathy. JACC Basic to Translational Science. 2016;1:344–359. doi: 10.1016/j.jacbts.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ortiz-Genga MF, Cuenca S, Dal Ferro M, Zorio E, Salgado-Aranda R, Climent V, Padron-Barthe L, Duro-Aguado I, Jimenez-Jaimez J, Hidalgo-Olivares VM, Garcia-Campo E, Lanzillo C, Suarez-Mier MP, Yonath H, Marcos-Alonso S, Ochoa JP, Santome JL, Garcia-Giustiniani D, Rodriguez-Garrido JL, Dominguez F, Merlo M, Palomino J, Pena ML, Trujillo JP, Martin-Vila A, Stolfo D, Molina P, Lara-Pezzi E, Calvo-Iglesias FE, Nof E, Calo L, Barriales-Villa R, Gimeno-Blanes JR, Arad M, Garcia-Pavia P, Monserrat L. Truncating flnc mutations are associated with high-risk dilated and arrhythmogenic cardiomyopathies. Journal of the American College of Cardiology. 2016;68:2440–2451. doi: 10.1016/j.jacc.2016.09.927. [DOI] [PubMed] [Google Scholar]
- 127.Golbus JR, Puckelwartz MJ, Dellefave-Castillo L, Fahrenbach JP, Nelakuditi V, Pesce LL, Pytel P, McNally EM. Targeted analysis of whole genome sequence data to diagnose genetic cardiomyopathy. Circ Cardiovasc Genet. 2014;7:751–759. doi: 10.1161/CIRCGENETICS.113.000578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Thompson TG, Chan YM, Hack AA, Brosius M, Rajala M, Lidov HG, McNally EM, Watkins S, Kunkel LM. Filamin 2 (fln2): A muscle-specific sarcoglycan interacting protein. J Cell Biol. 2000;148:115–126. doi: 10.1083/jcb.148.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dalkilic I, Schienda J, Thompson TG, Kunkel LM. Loss of filaminc (flnc) results in severe defects in myogenesis and myotube structure. Mol Cell Biol. 2006;26:6522–6534. doi: 10.1128/MCB.00243-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Arbustini E, Diegoli M, Fasani R, Grasso M, Morbini P, Banchieri N, Bellini O, Dal Bello B, Pilotto A, Magrini G, Campana C, Fortina P, Gavazzi A, Narula J, Vigano M. Mitochondrial DNA mutations and mitochondrial abnormalities in dilated cardiomyopathy. Am J Pathol. 1998;153:1501–1510. doi: 10.1016/S0002-9440(10)65738-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ashrafian H, Docherty L, Leo V, Towlson C, Neilan M, Steeples V, Lygate CA, Hough T, Townsend S, Williams D, Wells S, Norris D, Glyn-Jones S, Land J, Barbaric I, Lalanne Z, Denny P, Szumska D, Bhattacharya S, Griffin JL, Hargreaves I, Fernandez-Fuentes N, Cheeseman M, Watkins H, Dear TN. A mutation in the mitochondrial fission gene dnm1l leads to cardiomyopathy. PLoS Genet. 2010;6:e1001000. doi: 10.1371/journal.pgen.1001000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Grasso M, Diegoli M, Brega A, Campana C, Tavazzi L, Arbustini E. The mitochondrial DNA mutation t12297c affects a highly conserved nucleotide of trna(leu(cun)) and is associated with dilated cardiomyopathy. Eur J Hum Genet. 2001;9:311–315. doi: 10.1038/sj.ejhg.5200622. [DOI] [PubMed] [Google Scholar]
- 133.Sibbing D, Pfeufer A, Perisic T, Mannes AM, Fritz-Wolf K, Unwin S, Sinner MF, Gieger C, Gloeckner CJ, Wichmann HE, Kremmer E, Schafer Z, Walch A, Hinterseer M, Nabauer M, Kaab S, Kastrati A, Schomig A, Meitinger T, Bornkamm GW, Conrad M, von Beckerath N. Mutations in the mitochondrial thioredoxin reductase gene txnrd2 cause dilated cardiomyopathy. European heart journal. 2011;32:1121–1133. doi: 10.1093/eurheartj/ehq507. [DOI] [PubMed] [Google Scholar]
- 134.Rowland TJ, Graw SL, Sweet ME, Gigli M, Taylor MR, Mestroni L. Obscurin variants in patients with left ventricular noncompaction. Journal of the American College of Cardiology. 2016;68:2237–2238. doi: 10.1016/j.jacc.2016.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Marian AJ. The case of “missing causal genes” and the practice of medicine: A sherlock holmes approach of deductive reasoning. Circ Res. 2016;119:21–24. doi: 10.1161/CIRCRESAHA.116.308830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Roncarati R, Viviani Anselmi C, Krawitz P, Lattanzi G, von Kodolitsch Y, Perrot A, di Pasquale E, Papa L, Portararo P, Columbaro M, Forni A, Faggian G, Condorelli G, Robinson PN. Doubly heterozygous lmna and ttn mutations revealed by exome sequencing in a severe form of dilated cardiomyopathy. Eur J Hum Genet. 2013;21:1105–1111. doi: 10.1038/ejhg.2013.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wessels MW, Herkert JC, Frohn-Mulder IM, Dalinghaus M, van den Wijngaard A, de Krijger RR, Michels M, de Coo IF, Hoedemaekers YM, Dooijes D. Compound heterozygous or homozygous truncating mybpc3 mutations cause lethal cardiomyopathy with features of noncompaction and septal defects. Eur J Hum Genet. 2015;23:922–928. doi: 10.1038/ejhg.2014.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Zahka K, Kalidas K, Simpson MA, Cross H, Keller BB, Galambos C, Gurtz K, Patton MA, Crosby AH. Homozygous mutation of mybpc3 associated with severe infantile hypertrophic cardiomyopathy at high frequency among the amish. Heart. 2008;94:1326–1330. doi: 10.1136/hrt.2007.127241. [DOI] [PubMed] [Google Scholar]
- 139.Dellefave LM, Pytel P, Mewborn S, Mora B, Guris DL, Fedson S, Waggoner D, Moskowitz I, McNally EM. Sarcomere mutations in cardiomyopathy with left ventricular hypertrabeculation. Circ Cardiovasc Genet. 2009;2:442–449. doi: 10.1161/CIRCGENETICS.109.861955. [DOI] [PubMed] [Google Scholar]
- 140.Hershberger RE, Lindenfeld J, Mestroni L, Seidman CE, Taylor MR, Towbin JA, Heart Failure Society of A Genetic evaluation of cardiomyopathy–a heart failure society of america practice guideline. J Card Fail. 2009;15:83–97. doi: 10.1016/j.cardfail.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 141.Pinto YM, Elliott PM, Arbustini E, Adler Y, Anastasakis A, Bohm M, Duboc D, Gimeno J, de Groote P, Imazio M, Heymans S, Klingel K, Komajda M, Limongelli G, Linhart A, Mogensen J, Moon J, Pieper PG, Seferovic PM, Schueler S, Zamorano JL, Caforio AL, Charron P. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: A position statement of the esc working group on myocardial and pericardial diseases. European heart journal. 2016;37:1850–1858. doi: 10.1093/eurheartj/ehv727. [DOI] [PubMed] [Google Scholar]
- 142.van Spaendonck-Zwarts KY, van Rijsingen IA, van den Berg MP, Lekanne Deprez RH, Post JG, van Mil AM, Asselbergs FW, Christiaans I, van Langen IM, Wilde AA, de Boer RA, Jongbloed JD, Pinto YM, van Tintelen JP. Genetic analysis in 418 index patients with idiopathic dilated cardiomyopathy: Overview of 10 years’ experience. Eur J Heart Fail. 2013;15:628–636. doi: 10.1093/eurjhf/hft013. [DOI] [PubMed] [Google Scholar]
- 143.Garcia J, Tahiliani J, Johnson NM, Aguilar S, Beltran D, Daly A, Decker E, Haverfield E, Herrera B, Murillo L, Nykamp K, Topper S. Clinical genetic testing for the cardiomyopathies and arrhythmias: A systematic framework for establishing clinical validity and addressing genotypic and phenotypic heterogeneity. Front Cardiovasc Med. 2016;3:20. doi: 10.3389/fcvm.2016.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Harakalova M, Kummeling G, Sammani A, Linschoten M, Baas AF, van der Smagt J, Doevendans PA, van Tintelen JP, Dooijes D, Mokry M, Asselbergs FW. A systematic analysis of genetic dilated cardiomyopathy reveals numerous ubiquitously expressed and muscle-specific genes. Eur J Heart Fail. 2015;17:484–493. doi: 10.1002/ejhf.255. [DOI] [PubMed] [Google Scholar]
- 145.Fatkin D, Yeoh T, Hayward CS, Benson V, Sheu A, Richmond Z, Feneley MP, Keogh AM, Macdonald PS. Evaluation of left ventricular enlargement as a marker of early disease in familial dilated cardiomyopathy. Circ Cardiovasc Genet. 2011;4:342–348. doi: 10.1161/CIRCGENETICS.110.958918. [DOI] [PubMed] [Google Scholar]
- 146.Moretti M, Merlo M, Barbati G, Di Lenarda A, Brun F, Pinamonti B, Gregori D, Mestroni L, Sinagra G. Prognostic impact of familial screening in dilated cardiomyopathy. Eur J Heart Fail. 2010;12:922–927. doi: 10.1093/eurjhf/hfq093. [DOI] [PubMed] [Google Scholar]
- 147.Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Jr, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJ, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WH, Tsai EJ, Wilkoff BL, American College of Cardiology F, American Heart Association Task Force on Practice G 2013 accf/aha guideline for the management of heart failure: A report of the american college of cardiology foundation/american heart association task force on practice guidelines. Journal of the American College of Cardiology. 2013;62:e147–239. doi: 10.1016/j.jacc.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 148.Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, Falk V, Gonzalez-Juanatey JR, Harjola VP, Jankowska EA, Jessup M, Linde C, Nihoyannopoulos P, Parissis JT, Pieske B, Riley JP, Rosano GM, Ruilope LM, Ruschitzka F, Rutten FH, van der Meer P, Authors/Task Force M 2016 esc guidelines for the diagnosis and treatment of acute and chronic heart failure: The task force for the diagnosis and treatment of acute and chronic heart failure of the european society of cardiology (esc)developed with the special contribution of the heart failure association (hfa) of the esc. European heart journal. 2016;37:2129–2200. doi: 10.1093/eurheartj/ehw128. [DOI] [PubMed] [Google Scholar]
- 149.Zannad F, Gattis Stough W, Rossignol P, Bauersachs J, McMurray JJ, Swedberg K, Struthers AD, Voors AA, Ruilope LM, Bakris GL, O’Connor CM, Gheorghiade M, Mentz RJ, Cohen-Solal A, Maggioni AP, Beygui F, Filippatos GS, Massy ZA, Pathak A, Pina IL, Sabbah HN, Sica DA, Tavazzi L, Pitt B. Mineralocorticoid receptor antagonists for heart failure with reduced ejection fraction: Integrating evidence into clinical practice. European heart journal. 2012;33:2782–2795. doi: 10.1093/eurheartj/ehs257. [DOI] [PubMed] [Google Scholar]
- 150.Mestroni L, Brun F, Spezzacatene A, Sinagra G, Taylor MR. Genetic causes of dilated cardiomyopathy. Prog Pediatr Cardiol. 2014;37:13–18. doi: 10.1016/j.ppedcard.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Jessup M, Marwick TH, Ponikowski P, Voors AA, Yancy CW. 2016 esc and acc/aha/hfsa heart failure guideline update - what is new and why is it important? Nat Rev Cardiol. 2016;13:623–628. doi: 10.1038/nrcardio.2016.134. [DOI] [PubMed] [Google Scholar]
- 152.Ruschitzka F, Abraham WT, Singh JP, Bax JJ, Borer JS, Brugada J, Dickstein K, Ford I, Gorcsan J, 3rd, Gras D, Krum H, Sogaard P, Holzmeister J, Echo CRTSG Cardiac-resynchronization therapy in heart failure with a narrow qrs complex. N Engl J Med. 2013;369:1395–1405. doi: 10.1056/NEJMoa1306687. [DOI] [PubMed] [Google Scholar]
- 153.Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Jr, Colvin MM, Drazner MH, Filippatos G, Fonarow GC, Givertz MM, Hollenberg SM, Lindenfeld J, Masoudi FA, McBride PE, Peterson PN, Stevenson LW, Westlake C. 2016 acc/aha/hfsa focused update on new pharmacological therapy for heart failure: An update of the 2013 accf/aha guideline for the management of heart failure: A report of the american college of cardiology/american heart association task force on clinical practice guidelines and the heart failure society of america. Journal of the American College of Cardiology. 2016;68:1476–1488. doi: 10.1016/j.jacc.2016.05.011. [DOI] [PubMed] [Google Scholar]
- 154.Losurdo P, Stolfo D, Merlo M, Barbati G, Gobbo M, Gigli M, Ramani R, Pinamonti B, Zecchin M, Finocchiaro G, Mestroni L, Sinagra G. Early arrhythmic events in idiopathic dilated cardiomyopathy. JACC Clin EP. 2016;2:535–543. doi: 10.1016/j.jacep.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Priori SG, Blomstrom-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, Elliott PM, Fitzsimons D, Hatala R, Hindricks G, Kirchhof P, Kjeldsen K, Kuck KH, Hernandez-Madrid A, Nikolaou N, Norekval TM, Spaulding C, Van Veldhuisen DJ. 2015 esc guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The task force for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the european society of cardiology (esc). Endorsed by: Association for european paediatric and congenital cardiology (aepc) European heart journal. 2015;36:2793–2867. doi: 10.1093/eurheartj/ehv316. [DOI] [PubMed] [Google Scholar]
- 156.Kusumoto FM, Calkins H, Boehmer J, Buxton AE, Chung MK, Gold MR, Hohnloser SH, Indik J, Lee R, Mehra MR, Menon V, Page RL, Shen WK, Slotwiner DJ, Stevenson LW, Varosy PD, Welikovitch L. Hrs/acc/aha expert consensus statement on the use of implantable cardioverter-defibrillator therapy in patients who are not included or not well represented in clinical trials. Circulation. 2014;130:94–125. doi: 10.1161/CIR.0000000000000056. [DOI] [PubMed] [Google Scholar]
- 157.Kumar S, Baldinger SH, Gandjbakhch E, Maury P, Sellal JM, Androulakis AF, Waintraub X, Charron P, Rollin A, Richard P, Stevenson WG, Macintyre CJ, Ho CY, Thompson T, Vohra JK, Kalman JM, Zeppenfeld K, Sacher F, Tedrow UB, Lakdawala NK. Long-term arrhythmic and nonarrhythmic outcomes of lamin a/c mutation carriers. Journal of the American College of Cardiology. 2016;68:2299–2307. doi: 10.1016/j.jacc.2016.08.058. [DOI] [PubMed] [Google Scholar]
- 158.Saberi S, Day SM. Exercise prescription for the athlete with cardiomyopathy. Cardiol Clin. 2016;34:591–601. doi: 10.1016/j.ccl.2016.06.008. [DOI] [PubMed] [Google Scholar]
- 159.Rosen MR, Myerburg RJ, Francis DP, Cole GD, Marban E. Translating stem cell research to cardiac disease therapies: Pitfalls and prospects for improvement. Journal of the American College of Cardiology. 2014;64:922–937. doi: 10.1016/j.jacc.2014.06.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Eulalio A, Mano M, Dal Ferro M, Zentilin L, Sinagra G, Zacchigna S, Giacca M. Functional screening identifies mirnas inducing cardiac regeneration. Nature. 2012;492:376–381. doi: 10.1038/nature11739. [DOI] [PubMed] [Google Scholar]
- 161.Mann DL, Lee RJ, Coats AJ, Neagoe G, Dragomir D, Pusineri E, Piredda M, Bettari L, Kirwan BA, Dowling R, Volterrani M, Solomon SD, Sabbah HN, Hinson A, Anker SD. One-year follow-up results from augment-hf: A multicentre randomized controlled clinical trial of the efficacy of left ventricular augmentation with algisyl in the treatment of heart failure. Eur J Heart Fail. 2016;18:314–325. doi: 10.1002/ejhf.449. [DOI] [PubMed] [Google Scholar]
- 162.Feric NT, Radisic M. Strategies and challenges to myocardial replacement therapy. Stem Cells Transl Med. 2016;5:410–416. doi: 10.5966/sctm.2015-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sun N, Yazawa M, Liu J, Han L, Sanchez-Freire V, Abilez OJ, Navarrete EG, Hu S, Wang L, Lee A, Pavlovic A, Lin S, Chen R, Hajjar RJ, Snyder MP, Dolmetsch RE, Butte MJ, Ashley EA, Longaker MT, Robbins RC, Wu JC. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci Transl Med. 2012;4:130ra147. doi: 10.1126/scitranslmed.3003552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Burke MA, Cook SA, Seidman JG, Seidman CE. Clinical and mechanistic insights into the genetics of cardiomyopathy. Journal of the American College of Cardiology. 2016;68:2871–2886. doi: 10.1016/j.jacc.2016.08.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Marian AJ, van Rooij E, Roberts R. Genetics and genomics of single-gene cardiovascular diseases: Common hereditary cardiomyopathies as prototypes of single-gene disorders. Journal of the American College of Cardiology. 2016;68:2831–2849. doi: 10.1016/j.jacc.2016.09.968. [DOI] [PMC free article] [PubMed] [Google Scholar]