

ABSTRACT
Staphylococcus aureus is a leading infectious cause of life-threatening disease in humans, yet there is currently no vaccine to combat this bacterium. The pathogenesis of S. aureus is mediated by a diverse array of protein toxins including a large family of secreted pyrogenic superantigens. Neutralization of superantigens, including SEB and TSST-1, has proven to be protective in several animal models of toxic shock and sepsis. We demonstrate, for the first time, that a far more prevalent staphylococcal superantigen, SEK, can also induce lethal shock in mice. Additionally, we describe monoclonal antibodies (mAbs) that inhibit SEK-induced mitogenicity as well as protect against SEK-induced lethality, and enhance survival from S. aureus septicemia in murine models. MAb-4G3 (IgG2b), mAb-5G2 (IgG1), and mAb-9H2 (IgG1), all inhibit SEK-induced proliferation and cytokine production of human immune cells. We then demonstrate that passive immunization with a combination of mAb-4G3 and mAb-5G4, 2 mAbs that do not compete for epitope(s) on SEK, significantly enhance survival in a murine model of SEK-induced toxic shock (p = 0.006). In the setting of sepsis, passive immunization with this combination of mAbs also significantly enhances survival in mice after challenge with CA-MRSA strain USA300 (p = 0.03). Furthermore, septic mice that received mAb treatment in conjunction with vancomycin exhibit less morbidity than mice treated with vancomycin alone. Taken together, these findings suggest that the contribution of SEK to S. aureus pathogenesis may be greater than previously appreciated, and that adjunctive therapy with passive immunotherapy against SEs may be beneficial.
KEYWORDS: Staphylococcal Enterotoxin B, Staphylococcal Enterotoxin K, toxic shock syndrome toxin 1
Introduction
Staphylococcus aureus is the leading cause of bacterial infections in the United States, contributing to more deaths than AIDS, tuberculosis, and viral hepatitis combined.1 There is currently no vaccine available for the prevention or treatment of staphylococcal disease. The recent failures of staphylococcal vaccines in phase-II and phase-III clinical trials, all targeting prominent surface antigens, has increased interest in the development of immunotherapies against the secreted toxins of S. aureus.2 However, the exceptionally large number of toxins secreted by S. aureus coupled with the diversity of toxin repertoires between strains suggests that a vaccine targeting multiple common antigens may be more likely to succeed than previous vaccine attempts directed against single antigens.
The family of staphylococcal enterotoxins (SEs) is comprised of over 20 secreted protein toxins with demonstrable superantigenic activity. Their mechanism of action is mediated by their direct binding of TCR and MHC-II, resulting in an overwhelming immune response, a breakdown of the circulatory system, and a potentially fatal condition known as toxic shock syndrome (TSS). Almost all isolates of S. aureus encode at least 1 SE, and some harbor up to a dozen.3,4 The prominent members of this toxin family, including SEB and TSST-1, can individually cause TSS, and their associations with other immune mediated diseases are being investigated.5,6 Prior studies have shown that the toxigenic effects of SEB and TSST-1 can be mitigated in animal models by the administration of antibodies or synthetic molecules that prevent their engagement of immune cells.7-10 However, seb and tsst-1 genes are present in less than 10% of clinical isolates.4
Advancements in sequencing technology over the last 15 years have enabled the discovery of additional staphylococcal enterotoxins that have yet to be fully described in regards to prevalence or biological significance.11 One such enterotoxin, SEK, has demonstrable superantigenic properties against human immune cells in-vitro, and is lethal in a rabbit model of TSS.12 Moreover, recent epidemiological studies reveal that SEK is among the most prevalent superantigens in clinical isolates, particularly in methicillin resistant S. aureus (MRSA), and is thus far the only SE to be associated with CA-MRSA.4,13-17 We have previously shown that SEK was secreted by all examined S. aureus strains that encode the toxin (n = 36), and, in a murine soft-tissue infection model, we demonstrated that clinical isolates cause comparable accumulation of SEK within abscesses regardless of the amount of SEK they secrete in-vitro.18
Here, we utilize our previously described SEK-binding mAbs to further explore the role of SEK in immune activation and disease. We show that mAbs to SEK efficiently inhibit SEK-induced proliferation of human PBMCs in a dose-dependent manner. Moreover, we demonstrate that passive immunization with mAbs can protect mice from SEK-induced lethal shock, as well as from sepsis induced by a community-acquired MRSA USA300 strain.
Materials and methods
Purification of SEK
Recombinant SEK was cloned using the conserved USA300 sequence of SEK as previously described.18 However, the previously described protocol for the purification of SEK was modified to include ion-exchange chromatography with 20nM Tris buffer (pH7.5) as the final step of toxin purification.18
Monoclonal antibodies
SEK-specific mAbs 4G3 (IgG2b), 5G2 (IgG1), and 9H2 (IgG1) were previously described.18 These mAbs were selected for further study based on their estimated binding affinities. Supernatants from hybridoma cultures were sterilized by filtration using a 0.2mm pore syringe filter (Thermo Scientific). mAb concentrations were determined by ELISA using commercial antibodies as standards.
S.aureus strains
Two previously described clinical isolates of S. aureus were used in these studies: B-155 and W-132.4,18 Strain B-155 is a blood-borne USA300 isolate that encodes only one enterotoxin (sek), and W-132 is an MRSA wound isolate that encodes 2 enterotoxins (seb and sek). Each strain was streaked on brain heart infusion (BHI) agar and a single colony was inoculated into 25mL of BHI medium for growth in a shaking incubator for 16 hours at 37°C. The bacteria were then centrifuged at 2.9 × g at 4°C, washed with PBS, and suspended in 10mL of sterile PBS with 5% glycerol. One mL aliquots were stored at −80°C for no more than 2 weeks prior to their use in animals. The number of Colony Forming Units (CFU) of each frozen stock used was verified by serial dilution and plating on agar dishes.
Characterization of monoclonal antibodies
Relative binding affinities (KD) were determined by ELISA, using concentration gradients of mAb and SEK. These ELISAs were performed by coating 96-well plates with SEK, followed by unlabelled mAb (4G3 or 5G2 or 9H2). Wells were then treated with AP-conjugated anti-mouse IgG1 or IgG2b, and developed with PnPP tablets. Prism software was then used to fit a non-linear regression line to the resulting saturation curves, from which the KD values were calculated.
The 50% inhibitory concentration (IC50) for each mAb was determined using the PBMC proliferation-inhibition data from Fig. 3-1 (b-d). The data was transformed into log form and then fitted to a 4 parameter logistic function using the non-linear least squares Gauss-Newton algorithm. The analyses were then performed with R software.
Figure 3.
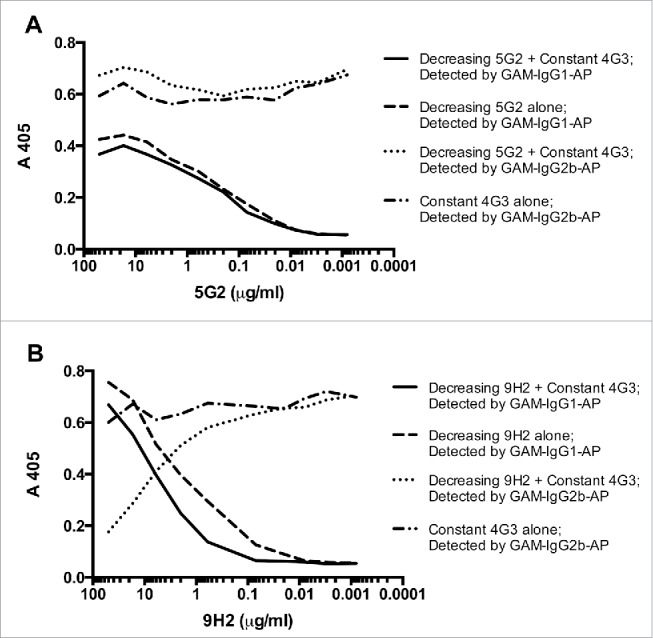
SEK binding competition between mAb combinations. A 96-well ELISA plate was coated with 25ng of SEK per well, blocked with 1% BSA, and then simultaneously treated with (A) the combination of mAb-4G3 (IgG2b) and mAb-5G2 (IgG1) or (B) the combination of mAb-9H2 (IgG1). MAb-4G3 and mAb-5G2 do no exhibit competition for epitope(s) on SEK. mAb-4G3 and mAb-9H2 exhibit competition for epitope(s) on SEK. All experiments were performed in duplicate. GAM = Goat anti-mouse, AP = Alkaline phosphatase.
A modified competition assay was performed to determine if 2 mAbs could bind simultaneously (non-competitively) to SEK. Briefly, a 96-well plate was coated with 25ng of SEK per well, blocked with 1% BSA in PBS, and then simultaneously treated with a concentration gradient of mAb-4G3 (IgG2b) and either mAb-5G2 (IgG1) or mAb-9H2 (IgG1).
Human PBMC proliferation assays
Human peripheral blood mononuclear cells (PBMCs) were obtained from whole blood of one healthy adult volunteer. PBMCs were seeded in 96-well plates at 1 × 105 cells per well in a final volume of 100μL per well, using RPMI + Glutamine media with 5% FBS. Serial dilutions of monoclonal antibodies were then added to the wells containing cells, followed immediately by the addition of SEK or control media. Cells were incubated in 10% CO2 for 96 hours at 37°C. All treatments were performed in triplicate. Cell proliferation was measured using the Via-Light HS cell proliferation kit (Lonza) according to the manufacturer's instructions.
Human T cell cytokine induction assays
Human T cells were isolated from whole blood of one healthy adult volunteer using RosettaSep CD4+ T cell enrichment mixture (Stemcell Tech), and cytokine production was measured by ELISA for human IL-2 and IFN-γ. Purified T cells were mixed 1:1 with donor matched PBMCs, and seeded in 96-well plates at 5 × 104 cells per well, using RPMI + Glutamine media with 5% FBS. After co-incubation of cells with SEK and mAbs in 10% CO2 for 8 hours at 37°C, the supernatants were removed, and cytokines were measured by ELISA for human IL-2 and IFN-γ (7). Experiments were performed in triplicate.
Mouse experiments
SEK intoxication model
All experiments involved 6-8 weeks old female BALB/c mice (NCI). Animals were injected intraperitoneally with 100μL of D-galactosamine (25mg) in PBS, followed within 10 minutes by intraperitoneal injection with 100μL of mAb (500μg or 1mg; single mAb or combination mAb) or PBS control, and then 100μL of SEK (50μg or 100μg) in PBS. All deaths occurred within 24 hours after administration of SEK.
Sepsis model
All experiments involved 6-8 weeks old female BALB/c mice (NCI). Animals were injected intravenously with either 100μL of mAb (1mg, single mAb or combination of mAbs) or PBS control, followed within 1 hour by intravenous injection with 100μL of bacteria. Strain B-155 was administered at 5.1 × 106 CFUs, and strain W-132 was administered at 1.1 × 108 and 1.2 × 108 CFUs, where indicated. Also, where indicated, a second injection of single or combined mAb solutions or PBS control was administered intravenously at 5 days post-infection. Vancomycin (Hospira) was injected intraperitoneally at a dose of 15mg/kg once daily for 3 days, starting 24 hours before challenge with S. aureus. Outcome parameters (survival, temperature, and weight) were measured daily.
Ethics statement
All animal experiments were conducted under the approval of the Animal Institute Committee (AIC), in accordance with the rules and regulations outlined by the Albert Einstein College of Medicine (Einstein) AIC. All blood from human volunteers was collected according to the Einstein Institutional Review Board (IRB) approved protocol for this study.
Statistical analysis
All statistical analyses were performed with either GraphPad Prism 6 software (GraphPad Software, Inc.) or R software (http://www.r-project.org/). SEK-induced lethality data was analyzed by Fisher's Exact Test. Inhibition of immune cell proliferation was analyzed by Student's t-tests (2-tailed, unpaired). Survival data in septicemia was analyzed by Log-rank (Mantel-Cox) test.
Results
MAbs neutralize SEK-induced immune cell proliferation and cytokine secretion
To assess the capacity of mAbs to neutralize the superantigenic effects of SEK, we performed proliferation assays with human PBMCs and cytokine production assays with human T cells. SEK induces proliferation of human PBMCs in a dose-dependent manner (Fig. 1a). Concurrent treatment with mAb-4G3, mAb-5G2, or mAb-9H2 at concentrations greater than 1μg/ml significantly inhibits this proliferative effect (Fig. 1, b-d). Additionally, these mAbs exhibit a differential capacity to inhibit SEK-induced proliferation, with mAb-4G3 inhibiting up to 50% of PBMC proliferation (IC50) at a lower concentration than mAb-5G2 or mAb-9H2 (Table 1). Concurrent treatment with these mAbs at 1ug/mL also inhibits SEK-induced production of pro-inflammatory cytokines. T cells treated with mAb-4G3 or mAb-5G2 produce significantly less IL-2 and IFN-γ than cells treated with an equivalent amount of isotype control antibody, and treatment with mAb-9H2 alone significantly inhibits secretion of IL-2 when compared with negative control treated T cells (Fig. 2). Similar trends of mAb-mediated inhibition of SEK were documented with immune cells from other adult donors and murine splenocytes (data not shown).
Figure 1.

MAbs inhibit SEK-induced proliferation of human immune cells. PBMCs from human whole blood were plated at 1 × 105 cells per well and incubated in the presence of SEK for 96 hours at 37°C and 10% CO2. (A) SEK induces maximal proliferation of immune cells (up to 6-fold greater than controls) at 10ng/ml. Immune cell proliferation after treatment with 10ng/ml of SEK is inhibited in a dose-dependent manner by concurrent incubation with 1, 10, and 100ug/ml of (B) mAb-4G3, (C) mAb-5G2, and (D) mAb-9H2. Relative luminescence units (RLU); Treatment with media alone (RPMI); Murine antibody isotype control (IC); Student's t-test: * = p < 0.05; ** = p < 0.01.
Table 1.
Characterization of SEK monoclonal antibodies.
| mAb | Isotype | KD (M) | IC50 (M) |
|---|---|---|---|
| 4G3 | IgG2b | 9.4 × 10−10 | 1.04 × 10−7 |
| 5G2 | IgG1 | 4.3 × 10−9 | 2.58 × 10−7 |
| 9H2 | IgG1 | 2.4 × 10−9 | 1.49 × 10−6 |
Note. Relative binding affinities to SEK (KD) were determined by ELISA. The 50% inhibitory concentration (IC50) were determined by analysis of the PBMC proliferation data from Fig. 3-1 (b-d). M = molar concentration.
Figure 2.

MAbs inhibit SEK-induced cytokine secretion by human immune cells. T cells from human whole blood were plated at 5 × 104 cells per well and incubated in the presence of SEK and 1ug/ml of mAb(s) for 8 hours at 37°C and 10% CO2. Supernatants were subsequently collected and pro-inflammatory cytokines were measured by ELISA for human (A) IL-2 or (B) INF-γ. Treatment with media alone (RPMI); Murine antibody isotype control (IC). Student's t-test: * = p < 0.05; ** = p < 0.01; *** < 0.001.
MAb combination protects mice from SEK-induced lethal shock
To produce SEK-induced lethal shock, BALB/c mice were treated with 25mg of D-galactosamine followed by intraperitoneal injection of SEK, analogous to previously published models of SEB induced lethal shock.7 Treatment with 50ug of SEK resulted in 90% mortality within 24 hours after challenge (Table 2). At the same time, we tested the protective efficacy of SEK-specific mAbs. Monotherapy with 500ug of either mAb-4G3 or mAb-5G2 failed to protect mice from SEK-induced lethality. However, mice that were passively immunized with a 1:1 combination (1mg total) of mAb-4G3 and mAb-5G2 prior to challenge with SEK exhibited significantly greater survival (80%) than mice that received toxin and were sham treated with PBS (10%) (p = 0.006). Our ELISA data indicates that these 2 mAbs recognize distinct non-overlapping epitopes, and can bind simultaneously to SEK (Fig. 3a). In a subsequent experiment, passive immunization with 1mg of mAb-4G3, 1mg of mAb-9H2, or a 1:1 combination of mAb-4G3 and mAb-9H2 (1mg total) failed to protect mice from SEK-induced lethality (Table 3), including when the mAbs were injected together intravenously to maximize their systemic dispersal. The mAbs in this latter combination exhibit competition for epitopes on SEK (Fig. 3b) suggesting that binding to different epitopes on SEK may facilitate the protective effect.
Table 2.
The combination of non-competing mAbs protects mice from SEK-induced lethal shock.
| Treatment | Total | Survived | Died | p-value |
|---|---|---|---|---|
| SEK + PBS | 10 | 1 | 9 | |
| SEK + mAb-4G3 | 10 | 1 | 9 | ns |
| SEK + mAb-5G2 | 10 | 2 | 8 | ns |
| SEK + mAb-4G3 + mAb-5G2 | 10 | 8 | 2 | 0.006 |
Note. Six to eight week-old female BALB/c mice were treated with a series of 3 intraperitoneal injections: first with 25mg of D-galactosamine, followed within 10 minutes by either antibody treatment or an equivalent volume of PBS, and then immediately followed by 50ug of SEK. All deaths occurred within 24 hours after SEK administration, and all surviving mice remained without symptoms for up to 30 days of monitoring. Data analyzed by Fisher's Exact Test (GraphPad Prism 6 software). NS = no significant difference.
Table 3.
Competitive mAb combination does not protect from SEK lethal shock.
| Treatment | Total | Survived | Died | p-value |
|---|---|---|---|---|
| SEK + PBS | 5 | 0 | 5 | |
| SEK + mAb-4G3 | 10 | 2 | 8 | NS |
| SEK + mAb-9H2 | 10 | 1 | 9 | NS |
| SEK + mAb-4G3 + mAb-9H2 | 10 | 3 | 7 | NS |
| SEK + mAb-4G3 + mAb-9H2 (intravenous) | 5 | 0 | 5 | NS |
Note. Six to eight week-old female BALB/c mice were treated with a series of 3 intraperitoneal injections, first with 25mg of D-galactosamine, followed 10 minutes later by either antibody treatment or an equivalent volume of PBS, and then immediately followed by 100ug of SEK. All deaths occurred within 24 hours after SEK administration, and all surviving mice remained without symptoms for up to 30 days of monitoring. Data analyzed by Fisher's Exact Test (GraphPad Prism 6 software). NS = no significant difference.
mAb combination protects mice from sepsis by USA300 S. aureus
To assess the capacity of mAbs to protect mice from sepsis by S. aureus, we used a previously characterized USA300 blood-borne isolate, B-155, that encodes only one enterotoxin, SEK.4 Groups of mice (n = 8) were challenged intravenously with 5 × 106 CFUs of S. aureus 1 hour after intravenous administration of mAb-4G3, mAb-5G2, combined mAbs-4G3/5G2, or PBS control (Fig. 4a). Passive immunization with a 1:1 combination of mAb-4G3 and mAb-5G2 (1mg total) resulted in significantly enhanced survival (75%) in comparison to mice that received PBS control (13%) (p = 0.0251). Monotherapy with either mAb-4G3 or mAb-5G2 (1mg) also resulted in enhanced survival (63% and 50%, respectively) relative to PBS treatment (13%), although these differences failed to be statistically significant (p = 0.10 and 0.14, respectively). There was no difference in survival between mice that received vancomycin alone or vancomycin and combined mAb-4G3/5G2 (90%). However, mice that were passively immunized with combined mAb-4G3/5G2 as an adjunctive therapy to vancomycin exhibited significantly less weight-loss than mice that received vancomycin alone (Fig. 4b).
Figure 4.
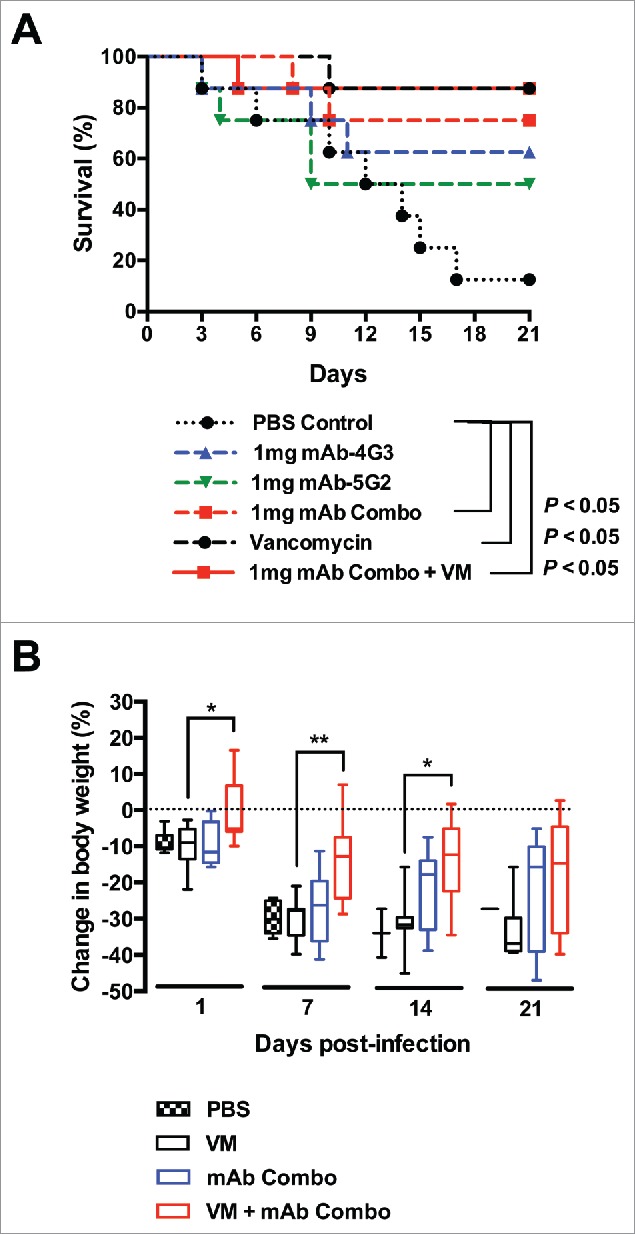
MAb combination is protective against sepsis by S. aureus USA300. (A) 6-8 week old mice female BALB/c mice were intravenously administered 1mg of mAb-4G3 or mAb-5G2 or both (1:1: ratio) or an equivalent volume of PBS control, followed within 1 hr by intravenous administration of 5.1 × 106 CFUs of USA300 strain, B-155. Where indicated, vancomycin was administered once daily for 3 days starting 24 hrs before infection. Survival curves were analyzed by Log-rank (Mantel-Cox) test (GraphPad Prism 6 software). (B) Body weight was monitored daily during the course of 21-day experiment. Change in body weight is relative to weight of same group of mice on Day 0, prior to bacterial challenge. Mann-Whitney test (GraphPad Prism 6 software): * P ≤ 0.05, ** P ≤ 0.01.
Next, we assessed the capacity of mAbs to protect mice from sepsis by an MRSA wound isolate, W-132, that encodes 2 enterotoxins, SEK and SEB. Groups of mice (n = 10) were challenged intravenously with 1 × 108 CFUs of S. aureus 1 hour after intravenous administration of mAb-4G3, mAb-5G2, combined mAbs-4G3/5G2, or PBS control (Fig. 5a). Passive immunization with any of these mAb treatments failed to protect mice from sepsis by this strain. A repetition of this experiment, this time, with a second administration of mAb treatments at 5 days post-infection, resulted in a minor, but statistically significant, enhancement of survival (Fig. 5b). In addition, an experiment was performed combining mAb-4G3 with a previously published anti-SEB mAb (20B1), and although an additive protective effect was observed, the trend did not achieve statistical significance7 (Fig. S1).
Figure 5.
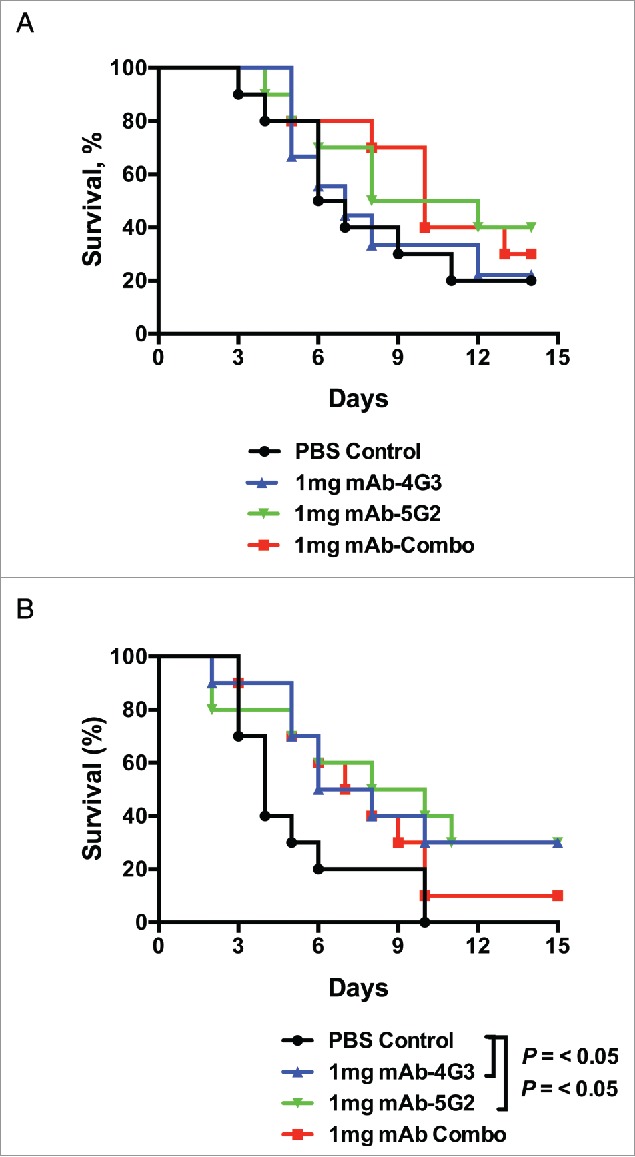
mAb booster is mildly protective from sepsis with SEB+ s. aureus. (A) Six to 8 week-old female BALB/c mice were intravenously administered 1mg of mAb-4G3 (IgG2b) or mAb-5G2 (IgG1) or both (1:1 ratio) or an equivalent volume of PBS control, followed within 1 hr by intravenous administration of 1.1 × 108 CFUs of MRSA strain W-132. (B) Similarly to panel A, mice were intravenously administered 1.2 × 108 CFUs of W-132, and received an additional intravenous administration of antibody or PBS control at 5 days post-infection. Survival curves were analyzed by Log-rank (Mantel-Cox) test (GraphPad Prism 6 software).
Discussion
SEK and other recently discovered staphylococcal enterotoxins are commonly categorized as “enterotoxin-like” due to their relatively unknown contribution to S. aureus pathogenesis.11 Here, we present data supporting the classification of SEK as a major staphylococcal enterotoxin, with superantigenic properties comparable to those exhibited by the classical enterotoxins. Published studies have now shown that SEK is secreted in high concentrations, and can induce mitogenic effects in T cells, toxic shock in rabbits, and most recently, emesis in primates.12,18,19 Our data further corroborate these findings by demonstrating mAb-mediated inhibition of SEK mitogenicity, and protection from SEK induced lethal shock. Moreover, passive immunization with these SEK-specific mAbs protects mice from morbidity and mortality resulting from sepsis by USA300 S. aureus.
The mAbs described in this study inhibit SEK-induced proliferation of PBMCs to varying degrees, with 100 μg/ml treatments of mAb-4G3, mAb-5G2, and mAb-9H2 inhibiting up to 97%, 60%, and 54% of proliferation, respectively. Combinations of these mAbs did not produce a clear enhancement of the inhibitory effect (data not shown). It is noteworthy that in previous studies with SEB, certain mAb combinations also did not show greater in-vitro neutralization than individual mAbs despite consistently demonstrating a protective synergy in in-vivo experiments.7 Micromolar concentrations for all 3 individual mAbs were required to achieve 50% inhibition. These micromolar IC50 values are substantially higher than the nanomolar IC50 values of our previously published neutralizing mAbs against SEB.20 The reason these relatively moderate neutralization capacities may be sufficient to protect mice from SEK remains unclear. Several potential explanations must be explored to reconcile these seemingly disparate findings. First, the IC50 of combination therapy with mAb-4G3 and mAb-5G2 may be greater than the IC50 of the individual mAbs. Second, the IC50 values of our mAbs may be artificially low. The proliferation assay we employed measures ATP as a marker of cell viability, and does not distinguish between the different cellular sources of ATP present within a mixture of PBMCs. Therefore, flow cytometry-based measurement of CD4+ T cells, or application of commercially available reagents for enrichment of CD4+ cells in PBMCs, may more accurately measure the capacity of these mAbs to inhibit T cell proliferation. Third, if these moderate IC50 values are accurate, suggesting that the protective mAb treatment of 4G3/5G2 is not efficiently inhibiting T cell proliferation, then the in-vivo protection provided by these mAbs may be mediated at least partly by effects other than T cell proliferation. Staphylococcal superantigens have been demonstrated to directly stimulate various cell types, including macrophages, B cells, fibroblasts, and mast cells.21-24 However, the effects of SEK on these other cell types have not been described.
Passive immunization with mAb-4G3 and mAb-5G2 in combination, but not individually, protects mice from SEK induced lethal shock. The mechanism by which this mAb mixture synergizes to protect from lethality will have to be resolved by in-depth structural analysis because the epitopes are likely not linear but conformational epitopes. One plausible explanation is that this particular combination of mAbs may be binding to suboptimal epitopes on SEK, but sterically inhibiting neighboring critical epitope(s) from binding the V-β chain of T cells. Previous studies have shown that protective mAb combinations against SEB can mediate protection by 2 possible mechanisms.25 First, simultaneous binding by 2 mAbs may promote greater clearance of toxin by Fc-mediated phagocytosis than binding by either mAb alone. Second, protective mAb combinations may induce allosteric conformational changes in the toxin that perturb the formation of stable MHC-SE-TCR complexes. Fc-mediated phagocytosis assays and structural biology studies may be pursued to address these hypotheses.
Additionally, the lack of protection exhibited by monotherapy with these mAbs may be a consequence of insufficient binding affinity. Of the 3 mAbs, only mAb-4G3 exhibits sub-nanomolar binding affinity, a degree of affinity normally associated with therapeutic mAbs and secondary immune response antibodies.26,27 Increasing the binding affinities of these mAbs by affinity maturation may result in protective monotherapies. As previous studies from our laboratory have shown, these epitopes are commonly complex conformational epitopes that are best identified by structural studies because simple mutational studies have given incorrect results in the past.7,25 Structural information will then allow site directed mutation, which could greatly improve affinity. Increasing the binding affinity of these protective mAbs would also reduce the necessary dose for therapeutic benefit. Previous studies also suggest that obtaining isotype switch variants of these mAbs, without affecting their sensitivities or specificities, may greatly enhance their protective ability.20
Passive immunization with combined mAb-4G3/5G2 also provided significant protection from sepsis by a blood-borne USA300 strain that produces SEK (p = 0.03). Moreover, mice that received combined mAb-4G3/5G2 as an adjunctive therapy to vancomycin experienced significantly less weight loss than mice that received vancomycin alone. These findings suggest that immunotherapy against SEK may be protective against other SEK+ strains, which represent the majority of clinical isolates in our previous census of S. aureus isolates from New York hospitals.4 However, in the setting of sepsis by an MRSA strain that produces both SEK and SEB, these mAbs were only mildly protective, and, only after a booster injection of mAbs at 5 days post-infection. These two strains, W-132 and B-155, possess heterologous toxin profiles that differ by more than the production of SEB. Further studies are needed to determine the relative contribution to pathogenesis by SEK versus SEB, and the amount of each toxin secreted in the setting of sepsis. In addition, previous studies indicate that at least 6 variants of SEK exist, among which, one is conserved and unique to the USA300 clone.18 Thus, the SEK variants expressed by these 2 strains may differ in toxicity, or binding affinity to the mAbs we've generated. The relatively lower degree of protection provided by these mAbs against an MRSA strain that produces both SEK and SEB also highlights a potentially important shortcoming of immunotherapeutic strategies that target a single toxin. Given the multitude of enterotoxins normally expressed by S. aureus, neutralizing a single SE through passive immunization is unlikely to affect the overall phenotype, especially when the infection strain encode multiple potent SEs. We postulate that targeting multiple common and important SEs, including SEK, may represent a more successful immunotherapeutic strategy.
In conclusion, our findings are encouraging because they demonstrate for the first time that immunotherapy targeting SEK, a prevalent but relatively uncharacterized enterotoxin, can be protective in the setting of life-threatening sepsis due to S. aureus. Moreover, our findings lend increasing plausibility to the prospect of successfully treating severe staphylococcal infections by toxin-targeted immunotherapy.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the NIH National Institute of Allergy and Infectious 348 Diseases, grant U54-AI057158, and the NIH MSTP training grant, T3GM07288.
References
- [1].van Hal SJ, Jensen SO, Vaska VL, Espedido BA, Paterson DL, Gosbell IB. Predictors of mortality in Staphylococcus aureus Bacteremia. Clin Microbiol Rev 2012; 25:362-86; PMID:22491776; http://dx.doi.org/ 10.1128/CMR.05022-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Fowler VG Jr., Proctor RA. Where does a Staphylococcus aureus vaccine stand? Clin Microbiol Infect 2014; 20 (Suppl 5):66-75; PMID:24476315; http://dx.doi.org/ 10.1111/1469-0691.12570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Becker K, Friedrich AW, Lubritz G, Weilert M, Peters G, Von Eiff C. Prevalence of genes encoding pyrogenic toxin superantigens and exfoliative toxins among strains of Staphylococcus aureus isolated from blood and nasal specimens. J Clin Microbiol 2003; 41:1434-9; PMID:12682126; http://dx.doi.org/ 10.1128/JCM.41.4.1434-1439.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Varshney AK, Mediavilla JR, Robiou N, Guh A, Wang X, Gialanella P, Levi MH, Kreiswirth BN, Fries BC. Diverse enterotoxin gene profiles among clonal complexes of Staphylococcus aureus isolates from the Bronx, New York. App Envir Microbiol 2009; 75:6839-49; http://dx.doi.org/ 10.1128/AEM.00272-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Xu SX, McCormick JK. Staphylococcal superantigens in colonization and disease. Front Cell Infect Microbiol 2012; 2:52; PMID:22919643; http://dx.doi.org/ 10.3389/fcimb.2012.00052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Li J, Yang J, Lu YW, Wu S, Wang MR, Zhu JM. Possible Role of Staphylococcal Enterotoxin B in the Pathogenesis of Autoimmune Diseases. Viral Immunol 2015; 7:354-359 [DOI] [PubMed] [Google Scholar]
- [7].Varshney AK, Wang X, Cook E, Dutta K, Scharff MD, Goger MJ, Fries BC. Generation, characterization, and epitope mapping of neutralizing and protective monoclonal antibodies against staphylococcal enterotoxin B-induced lethal shock. J Biol Chem 2011; 286:9737-47; PMID:21233204; http://dx.doi.org/ 10.1074/jbc.M110.212407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kum WW, Chow AW. Inhibition of staphylococcal enterotoxin A-induced superantigenic and lethal activities by a monoclonal antibody to toxic shock syndrome toxin-1. J Infect Dis 2001; 183:1739-48; PMID:11372026; http://dx.doi.org/ 10.1086/320732 [DOI] [PubMed] [Google Scholar]
- [9].Bonventre PF, Thompson MR, Adinolfi LE, Gillis ZA, Parsonnet J. Neutralization of toxic shock syndrome toxin-1 by monoclonal antibodies in vitro and in vivo. Infect Immun 1988; 56:135-41; PMID:3257201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Buonpane RA, Churchill HR, Moza B, Sundberg EJ, Peterson ML, Schlievert PM, Kranz DM. Neutralization of staphylococcal enterotoxin B by soluble, high-affinity receptor antagonists. Nat Med 2007; 13:725-9; PMID:17515896; http://dx.doi.org/ 10.1038/nm1584 [DOI] [PubMed] [Google Scholar]
- [11].Lina G, Bohach GA, Nair SP, Hiramatsu K, Jouvin-Marche E, Mariuzza R, International Nomenclature Committee for Staphylococcal S . Standard nomenclature for the superantigens expressed by Staphylococcus. J Infect Dis 2004; 189:2334-6; PMID:15181583; http://dx.doi.org/ 10.1086/420852 [DOI] [PubMed] [Google Scholar]
- [12].Orwin PM, Leung DY, Donahue HL, Novick RP, Schlievert PM. Biochemical and biological properties of Staphylococcal enterotoxin K. Infect Immun 2001; 69:360-6; PMID:11119525; http://dx.doi.org/ 10.1128/IAI.69.1.360-366.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Djahmi N, Messad N, Nedjai S, Moussaoui A, Mazouz D, Richard JL, Sotto A, Lavigne JP. Molecular epidemiology of Staphylococcus aureus strains isolated from inpatients with infected diabetic foot ulcers in an Algerian University Hospital. Clin Microbiol Infect 2013; 19:E398-404; PMID:23521557; http://dx.doi.org/ 10.1111/1469-0691.12199 [DOI] [PubMed] [Google Scholar]
- [14].Machuca MA, Sosa LM, Gonzalez CI. Molecular typing and virulence characteristic of methicillin-resistant Staphylococcus aureus isolates from pediatric patients in Bucaramanga, Colombia. PloS one 2013; 8:e73434; PMID:24058415; http://dx.doi.org/ 10.1371/journal.pone.0073434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Indrawattana N, Sungkhachat O, Sookrung N, Chongsa-nguan M, Tungtrongchitr A, Voravuthikunchai SP, Kong-ngoen T, Kurazono H, Chaicumpa W. Staphylococcus aureus clinical isolates: antibiotic susceptibility, molecular characteristics, and ability to form biofilm. BioMed Res Int 2013; 2013:314654; PMID:24069597; http://dx.doi.org/ 10.1155/2013/314654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shukla SK, Karow ME, Brady JM, Stemper ME, Kislow J, Moore N, Wroblewski K, Chyou PH, Warshauer DM, Reed KD, et al.. Virulence genes and genotypic associations in nasal carriage, community-associated methicillin-susceptible and methicillin-resistant USA400 Staphylococcus aureus isolates. J Clin Microbiol 2010; 48:3582-92; PMID:20668125; http://dx.doi.org/ 10.1128/JCM.00657-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wu D, Li X, Yang Y, Zheng Y, Wang C, Deng L, Liu L, Li C, Shang Y, Zhao C, et al.. Superantigen gene profiles and presence of exfoliative toxin genes in community-acquired meticillin-resistant Staphylococcus aureus isolated from Chinese children. J Med Microbiol 2011; 60:35-45; PMID:20829395; http://dx.doi.org/ 10.1099/jmm.0.023465-0 [DOI] [PubMed] [Google Scholar]
- [18].Aguilar JL, Varshney AK, Wang X, Stanford L, Scharff M, Fries BC. Detection and measurement of staphylococcal enterotoxin-like K (SEl-K) secretion by Staphylococcus aureus clinical isolates. J Clin Microbiol 2014; 52:2536-43; PMID:24808237; http://dx.doi.org/ 10.1128/JCM.00387-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Omoe K, Hu DL, Ono HK, Shimizu S, Takahashi-Omoe H, Nakane A, Uchiyama T, Shinagawa K, Imanishi K. Emetic potentials of newly identified staphylococcal enterotoxin-like toxins. Infect Immun 2013; 81:3627-31; PMID:23876808; http://dx.doi.org/ 10.1128/IAI.00550-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Varshney AK, Wang X, Aguilar JL, Scharff MD, Fries BC. Isotype switching increases efficacy of antibody protection against staphylococcal enterotoxin B-induced lethal shock and Staphylococcus aureus sepsis in mice. mBio 2014; 5:e01007-14; PMID:24917594; http://dx.doi.org/ 10.1128/mBio.01007-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Khan AA, Martin S, Saha B. SEB-induced signaling in macrophages leads to biphasic TNF-alpha. J Leukocyte Biol 2008; 83:1363-9; PMID:18364436; http://dx.doi.org/ 10.1189/jlb.1007686 [DOI] [PubMed] [Google Scholar]
- [22].Domiati-Saad R, Lipsky PE. Staphylococcal enterotoxin A induces survival of VH3-expressing human B cells by binding to the VH region with low affinity. J Immunol 1998; 161:1257-66; PMID:9686586 [PubMed] [Google Scholar]
- [23].Perez-Novo CA, Waeytens A, Claeys C, Cauwenberge PV, Bachert C. Staphylococcus aureus enterotoxin B regulates prostaglandin E2 synthesis, growth, and migration in nasal tissue fibroblasts. J Infect Dis 2008; 197:1036-43; PMID:18419541; http://dx.doi.org/ 10.1086/528989 [DOI] [PubMed] [Google Scholar]
- [24].Ackermann L, Pelkonen J, Harvima IT. Staphylococcal enterotoxin B inhibits the production of interleukin-4 in a human mast-cell line HMC-1. Immunology 1998; 94:247-52; PMID:9741348; http://dx.doi.org/ 10.1046/j.1365-2567.1998.00508.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dutta K, Varshney AK, Franklin MC, Goger M, Wang X, Fries BC. Mechanisms mediating enhanced neutralization efficacy of staphylococcal enterotoxin B by combinations of monoclonal antibodies. J Biol Chem 2015; 290:6715-30; PMID:25572397; http://dx.doi.org/ 10.1074/jbc.M114.630715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ponsel D, Neugebauer J, Ladetzki-Baehs K, Tissot K. High affinity, developability and functional size: the holy grail of combinatorial antibody library generation. Molecules 2011; 16:3675-700; PMID:21540796; http://dx.doi.org/ 10.3390/molecules16053675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Vaughan TJ, Williams AJ, Pritchard K, Osbourn JK, Pope AR, Earnshaw JC, McCafferty J, Hodits RA, Wilton J, Johnson KS. Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nat Biotechnol 1996; 14:309-14; PMID:9630891; http://dx.doi.org/ 10.1038/nbt0396-309 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.