

ABSTRACT
Group B Streptococcus (GBS), a leading cause of neonatal sepsis and meningitis, asymptomatically colonizes up to 30% of women and can persistently colonize even after antibiotic treatment. Previous studies have shown that GBS resides inside macrophages, but the mechanism by which it survives remains unknown. Here, we examined the ability of 4 GBS strains to survive inside macrophages and then focused on 2 strains belonging to sequence type (ST)-17 and ST-12, to examine persistence in the presence of antibiotics. A multiple stress medium was also developed using several stressors found in the phagosome to assess the ability of 30 GBS strains to withstand phagosomal stress. The ST-17 strain was more readily phagocytosed and survived intracellularly longer than the ST-12 strain, but the ST-12 strain was tolerant to ampicillin unlike the ST-17 strain. Exposure to sub-inhibitory concentrations of ampicillin and erythromycin increased the level of phagocytosis of the ST-17 strain, but had no effect on the ST-12 strain. In addition, blocking acidification of the phagosome decreased the survival of the ST-17 strain indicating a pH-dependent survival mechanism for the ST-17 strain. Congruent with the macrophage experiments, the ST-17 strain had a higher survival rate in the multiple stress medium than the ST-12 strain, and overall, serotype III isolates survived significantly better than other serotypes. These results indicate that diverse GBS strains may use differing mechanisms to persist and that serotype III strains are better able to survive specific stressors inside the phagosome relative to other serotypes.
KEYWORDS: Group B Streptococcus, intracellular survival, macrophages, phagocytosis, Streptococcus agalactiae
Introduction
Commonly found as a commensal in the gastrointestinal and genitourinary tracts of up to 30% of adults, group B Streptococcus (GBS) is also a leading cause of neonatal sepsis and meningitis.1 GBS can be classified into 10 serotypes based on the capsular polysaccharide (CPS): Ia, Ib and II through IX. These ten antigenically distinct CPS types play a role in GBS virulence, with types Ia, Ib, II, III, and V most often causing disease.2,3 GBS isolates can be further characterized using a multilocus sequence typing (MLST) system that groups strains into phylogenetically distinct lineages, or sequence types (STs). Several studies have shown that serotype III strains belonging to ST-17 are more often associated with neonatal disease, indicating that ST-17 strains may be inherently more virulent than strains of other GBS lineages.4-8 Additionally, a previous study that examined the GBS colonization status of pregnant women before and after delivery showed that serotype III ST-19 and ST-17 strains were more likely to persistently colonize women after receiving intrapartum antibiotic prophylaxis (IAP), whereas ST-12 strains were more frequently lost.9 This ability of certain GBS STs to persist suggests an enhanced ability to evade the effect of antibiotics, either through enhanced antibiotic tolerance or protection via biofilm production or uptake into host cells. In addition, the capability of GBS to both colonize individuals as a commensal and cause disease in susceptible hosts indicates a heightened ability to evade host immune responses.
Several pathogens have developed mechanisms to survive inside macrophages, the innate immune cells designed to eliminate pathogens, thus evading the immune response of the host.10 Pathogens are taken up by macrophages and other phagocytes through a process called phagocytosis, which contains the pathogen in a vacuole inside the macrophage. The vacuole then goes through phagosome maturation and becomes highly acidic as antimicrobial peptides, reactive oxygen species (ROS), and reactive nitrogen species (RNS) are generated to kill the bacteria. A number of pathogens have developed a variety of ways to overcome these defense mechanisms, including disruption of cellular signaling to prevent or slow down phagosomal maturation, phagosomal escape by lysing the membrane to replicate in the more favorable cytosol, and the production of enzymes to protect against ROS and RNS.10 GBS was shown to survive inside macrophages within the fully mature phagosome,11 but little is known about the mechanism used for intracellular survival. Since many antibiotics poorly penetrate eukaryotic cells, the ability to survive inside macrophages may provide protection from antibiotics and allow the bacterium to recolonize the host following antibiotic cessation. Additionally, residing inside phagocytic cells has been suggested to facilitate the dissemination of bacterial cells to other sites of the body via the bloodstream or penetration of host tissue barriers.12,13
This study examines the ability of distinct lineages of GBS to tolerate antibiotics, survive inside macrophages, and express virulence genes important for survival in each condition. Survival in a synthetic multiple stress medium comprising common phagosomal stressors was also developed to compare strains of varying serotypes, genotypes, and sources. Identifying bacterial factors important for persistent colonization and dissemination could aid in the discovery of therapeutic targets aimed at eradicating the commensal GBS population, which is particularly critical for pregnant women with an increased risk of transmitting GBS to their newborns.
Results
GBS genotypes differ in level of phagocytosis by macrophages and intracellular survival
Since macrophages play an important role in controlling bacterial levels in the host, we examined phagocytic uptake and intracellular survival of 4 GBS strains: NEM316, GB00590, GB00112, and GB00653 (Table 1). Importantly, the GBS strains representing diverse lineages varied in their interactions with macrophages. NEM316 and GB112, which belong to ST-23 and ST-17, respectively, were phagocytosed the most followed by GB590 (ST-19) and GB653 (ST-12) (Fig. 1A). At 24 hr, the ST-23 strain had the greatest ability to survive inside the macrophages followed by the strains belonging to STs 17, 19, and 12, which had the lowest ability to survive (Fig. 1B). Together these data show that distinct lineages of GBS vary in their ability to be phagocytosed by macrophages and survive intracellularly for up to 24 hr.
Table 1.
GBS strains used in this study by sequence type (ST).
| ST | Strain | Serotype | Source |
|---|---|---|---|
| ST-1 | GB00020 | V | Colonizing-Persisted |
| GB00305 | Ia | Colonizing-Lost | |
| GB00620 | Ia | Colonizing-Persisted | |
| GB00037 | V | Invasive | |
| GB00310 | V | Invasive | |
| GB00686 | V | Invasive | |
| ST-12 | GB00285 | II | Colonizing-Lost |
| GB00555 | Ib | Colonizing-Persisted | |
| GB00653 | II | Colonizing-Lost | |
| GB00438 | Ib | Invasive | |
| GB00910 | II | Invasive | |
| GB01455 | II | Invasive | |
| ST-17 | GB00097 | III | Colonizing-Lost |
| GB00112 | III | Colonizing-Persisted | |
| GB00557 | III | Colonizing-Lost | |
| COH1 | III | Invasive | |
| GB00411 | III | Invasive | |
| GB00418 | III | Invasive | |
| ST-19 | GB00571 | III | Colonizing-Persisted |
| GB00590 | III | Colonizing | |
| GB00651 | Ib | Colonizing-Lost | |
| GB00036 | III | Invasive | |
| GB00079 | III | Invasive | |
| GB00377 | III | Invasive | |
| ST-23 | GB00002 | Ia | Colonizing-Persisted |
| GB00279 | II | Colonizing-Lost | |
| GB00644 | Ia | Colonizing-Persisted | |
| NEM 316 | III | Invasive | |
| GB00033 | Ia | Invasive | |
| GB00397 | III | Invasive |
Figure 1.
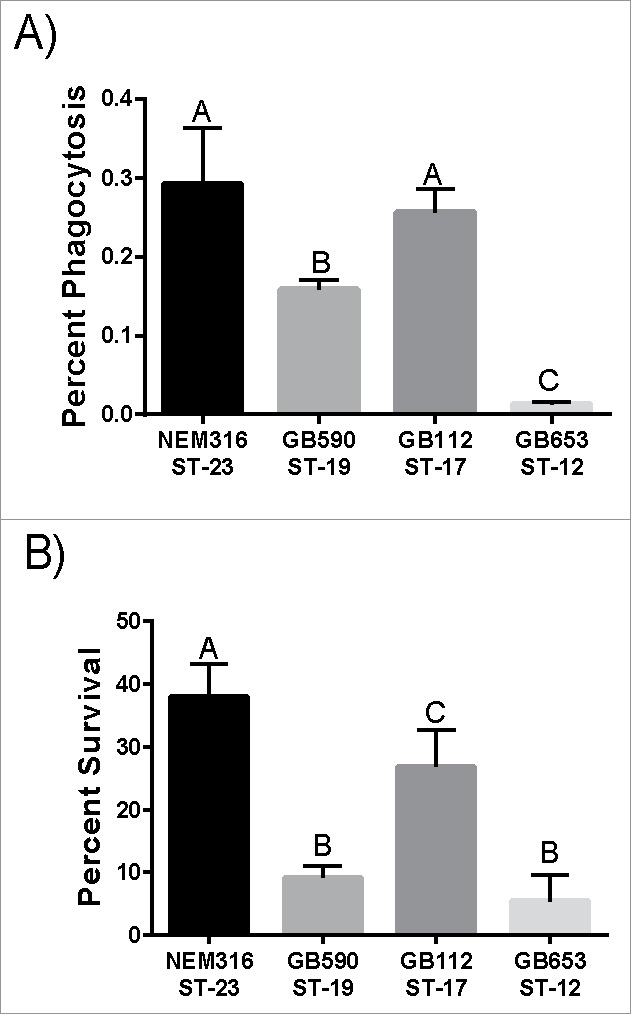
Phagocytosis and intracellular survival of diverse GBS strains in human macrophages. (A) Phagocytosis of GBS by macrophages after a 1 hr infection period. The number of phagocytosed bacteria was normalized to the total bacteria per well after the infection period to get percent phagocytosis. (B) Intracellular survival of GBS after 24 hr normalized to phagocytosis to account for differential uptake. Data represents the average of 3 separate experiments combined. Bars labeled with different letters are significantly different from each other.
Since ST-17 strains were previously found to persistently colonize pregnant women after receiving IAP and ST-12 strains were more commonly lost,9 we selected the ST-17 (GB00112) and ST-12 (GB00653) strains to determine whether persistence is due to antibiotic tolerance or protection from antibiotic effects by surviving inside host cells. Both strains were collected from women during a routine vaginal/rectal screen before or after childbirth9 and the administration of ampicillin for IAP. The ST-12 strain was recovered from a pregnant mother prior to delivery and IAP as the mother was GBS-negative at the postpartum visit. By contrast, the ST-17 strain was isolated from a mother at her postpartum visit and was found to be highly similar to the strain recovered prior to childbirth and IAP, indicating persistent colonization.9
We first compared the ability of these 2 strains to survive inside macrophages for up to 72 hr. Survival of the ST-17 strain was significantly higher than the ST-12 strain at 3 hr and 24 hr post infection. After 48 hr, the ST-17 strain was still detected inside the THP-1 cells, whereas the ST-12 strain was no longer detectable (Figs. 2A). Since GBS has been shown to induce apoptosis in macrophages,14,15 we wanted to confirm that the difference in survival between the 2 strains was not due to increased induction of apoptosis by the ST-12 strain. Using trypan blue staining followed by viable cell counting, we found that GBS infection significantly decreases macrophage viability over time compared to the uninfected control (Fig. 2B). It is important to note that overall viable cell counts per well did vary from approximately 3×105 at 1 hr to 5×105 at 3 hr. Variation was also observed across biological replicates and is likely indicative of variation in the recovery of cells from the well. Therefore, these results were confirmed using a lactate dehydrogenase (LDH) assay, which measures cell lysis with triton-X as a positive control (Fig. 2C). The LDH assay also showed reduced viability in the GBS treated macrophages versus the untreated; however, there was no difference in macrophage viability during infection between the 2 GBS strains for either method (Figs. 2B and C). These data indicate that the difference in the ability to survive intracellularly was not due to differential killing of the macrophages.
Figure 2.
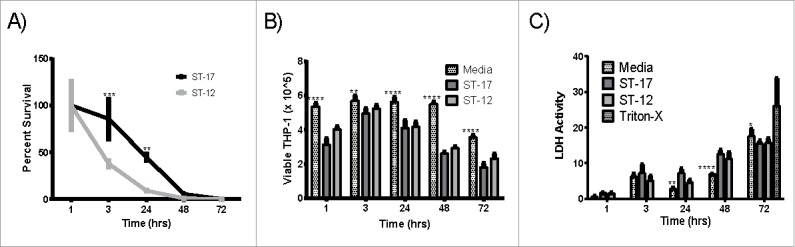
Intracellular survival of the ST-17 (GB00112) and ST-12 (GB00653) strains in PMA treated THP-1 macrophages. (A) Long-term intracellular survival of GBS. Data was normalized to the 1 h time point to calculate percent survival. (B and C) Assessment of macrophage viability during intracellular survival using trypan blue staining and viable cell counting (B) and measurement of LDH activity in milliunits/mL in the supernatant to detect cell lysis (C). Cells incubated with media alone were used as a negative control to assess spontaneous cell death. Cells were incubated with triton-X for 30 min before collecting supernatant for LDH assay as a positive control for complete cell death. Data shown are representative experiments of 3 biological replicates performed in triplicate. (**P < 0.01, ***P < 0.001, ****P < 0.0001).
Virulence gene expression varies temporally between the ST-17 and ST-12 strains during intracellular survival
Next, virulence gene expression during intracellular survival at 1 hr and 24 hr post infection was examined to quantify both short and long term survival as well as compare gene expression profiles between the 2 strains. The genes examined were selected based on previous studies showing their importance in either intracellular survival or GBS virulence (Table 2). Interestingly, 5 of the 8 genes (cylE, covS, scpB, lmb, and fbsA) were expressed significantly higher in the ST-12 strain compared to the ST-17 strain at 1 hr post infection (Fig. 3A). By 24 hr post infection, however, cylE, scpB, lmb, and fbsA were significantly higher in the ST-17 strain vs. the ST-12 strain (Fig. 3B). Since the ST-17 strain was able to survive beyond 24 hr unlike the ST-12 strain, it is likely that the prolonged upregulation of these genes in the ST-17 strain could contribute to resisting phagosomal stress for the long-term. Of note, sodA, which encodes superoxide dismutase, was downregulated during intracellular survival even though it has previously been shown to be important for survival inside macrophages.16
Table 2.
Virulence genes examined in this study.
| Gene | Product | Function/Characteristics | Reference |
|---|---|---|---|
| sodA | Mn-cofactored superoxide dismutase | Converts superoxide anions to molecular oxygen and hydrogen peroxide | 16 |
| ponA | Penicillin-binding protein 1a | Promotes resistance to antimicrobial peptides. | 52 |
| cylE | CylE protein | Predicted to function as a N-acyltransferase in the biosynthesis of the GBS pigment granadaene required for hemolytic/cytolytic activity of GBS. | 53 |
| covR | DNA-binding response regulator CovR | Two component regulatory system response regulator involved in virulence | 54 |
| covS | Sensor histidine kinase covS | Two component regulatory histidine kinase involved in virulence | 54 |
| scpB | C5a peptidase | Dual function: cleaves and inactivates complement; promotes binding to epithelial cells and fibronectin. | 28 |
| lmb | Laminin-binding protein | Promotes GBS colonization and translocation into bloodstream. | 29,52 |
| fbsA | Fibrinogen-binding protein A | Promotes adherence to epithelial cells | 30 |
Figure 3.
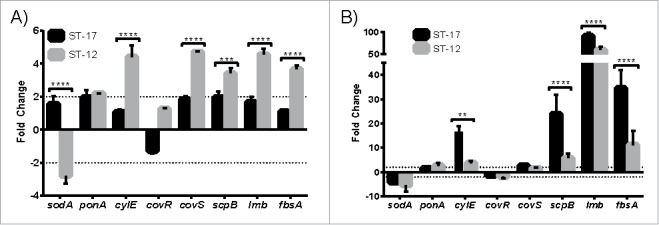
Virulence gene expression during intracellular survival. Differential expression of virulence genes (Table 2) relative to basal expression in culture medium was determined after 1 hr (A) and 24 hr (B) intracellular survival. Dashed lines mark 2-fold change in expression. Data shown are representative experiments of 3–4 biological replicates performed in triplicate. (**P < 0.01, ***P < 0.001, ****P < 0.0001).
The ST-12 strain is tolerant to ampicillin but exposure to sub-inhibitory ampicillin and erythromycin enhances phagocytosis of the ST-17 strain
We next determined the ability of these 2 strains to tolerate antibiotic exposure by developing an ampicillin tolerance assay in which GBS was exposed to 5µg/ml of ampicillin for 48 hr followed by 24 hr incubation with 1µg/ml, after which the culture was resuspended in fresh medium to detect tolerant bacteria. Surprisingly, the ST-12 strain was tolerant to ampicillin exposure while the ST-17 strain was effectively killed by ampicillin (Fig. 4A). It is important to note that the ST-12 strain was tolerant, not resistant, since now growth was detected during antibiotic incubation. Because the ST-17 strain is able to survive inside macrophages and cannot tolerate ampicillin exposure, we hypothesized that ampicillin exposure could induce the ST-17 strain to be more readily taken up by macrophages. Indeed, exposure to a sub-inhibitory concentration of ampicillin during macrophage infection significantly increased phagocytosis of the ST-17 strain, but had no effect on the ST-12 strain (Fig. 4B). Additionally, this ampicillin pretreatment had no effect on the ability of either strain to survive inside the macrophages over a 24 hr period (Fig. 4C). Since penicillin and erythromycin are also commonly used during IAP, the effect of exposure to sub-inhibitory concentrations of both drugs on phagocytosis and intracellular survival was also examined. Although penicillin exposure had no effect on phagocytosis or intracellular survival for either strain, erythromycin exposure significantly increased phagocytosis of the ST-17 strain.
Figure 4.
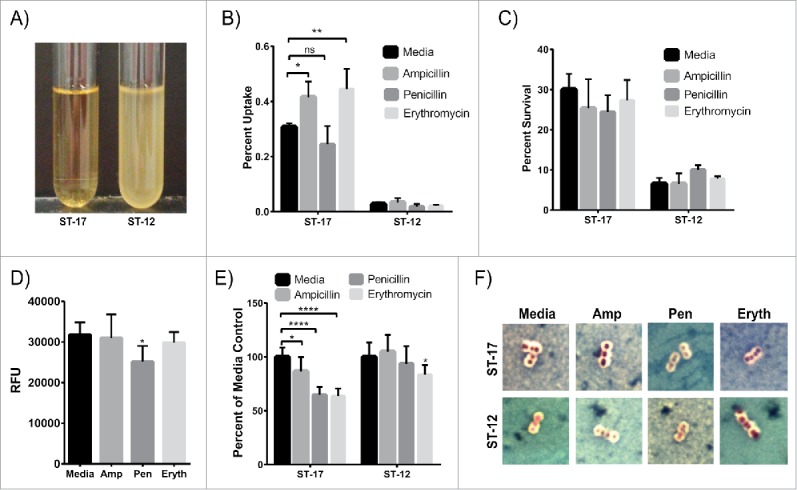
GBS exposure to antibiotics. (A) GBS cultures of the ST-17 strain (left) and ST-12 strain (right) were exposed to 5µg/ml ampicillin for 48 hr then 1µg/ml ampicillin for 24 hr. After ampicillin exposure, each culture was resuspended in fresh medium to detect any ampicillin tolerant bacteria. This figure shows representative results from 4 separate experiments (B and C) Phagocytosis (B) and 24 hr survival rate normalized to initial uptake (C) of GBS after cultures were exposed to a sub-inhibitory concentration of ampicillin (0.05µg/ml), penicillin (0.01µg/ml), or erythromycin (0.01µg/ml) during the macrophage infection period. (D) Phagocytosis of BioParticles after 1hr exposure to antibiotics as in B and C. B-D show all data combined from 3 separate experiments done in triplicate. (E) Relative capsule size of GBS after 1hr antibiotic exposure as described in B and C normalized to the media control. Data represents average relative capsule size from 3 separate experiments. (F) Representative images of capsule staining of GBS cultures used for relative capsule size shown in E. Capsule is represented by the clear zone around the cell. RFU: relative fluorescence units; Amp: Ampicillin, Pen: Penicillin; Eryth: Erythromycin (*P < 0.05, **P < 0.01, ****P < 0.0001).
To determine if this increase in phagocytosis was due to a change in the bacteria or a change in the macrophages, we assessed the ability of antibiotic-treated macrophages to phagocytose FITC-labeled BioParticles. Ampicillin and erythromycin treatment did not alter phagocytosis of the BioParticles, but penicillin treatment significantly reduced the ability of the macrophages to take up the BioParticles (Fig. 4D). This finding suggests that the enhanced uptake of the ST-17 strain after ampicillin and erythromycin exposure is due to a change in the GBS and not a change in the macrophages themselves. Moreover, since penicillin treatment reduced the phagocytic capacity of the macrophages, but phagocytosis of the ST-17 strain after penicillin exposure was the same as untreated, it is possible that penicillin impacts the bacterium resulting in enhanced uptake as well.
Since capsule is an important virulence factor in immune system evasion17 and a previous study showed a correlation between capsule alteration after antibiotic exposure and enhanced phagocytosis in E. coli,18 we hypothesized that changes in capsule could contribute to enhanced phagocytosis of the ST-17 strain. To test this, we examined the GBS cultures after exposure to antibiotics using a capsule stain (Figs. 4E and F). Interestingly, exposure to all 3 antibiotics significantly reduced capsule size relative to media treated for the ST-17 strain, with penicillin and erythromycin exposure showing the greatest reduction in capsule size. Ampicillin and penicillin treatment had no effect on capsule size, but erythromycin treatment caused a slight, but significant reduction in capsule size for the ST-12 strain.
The examined ST-17 and ST-12 strains differ in their ability to survive phagosomal stressors and alterations to phagosome acidification
To better understand how GBS withstands phagosomal stress, a multiple stress medium was developed using stressors commonly found in the phagosome. The following stressors were first tested individually to determine how the 2 strains survived each stress and then tested in combination to assess survival in multiple stress conditions: pH 4.5, H2O2, NO, lysozyme, and CuCl2. The ST-17 strain survived significantly better in acidic pH and CuCl2 compared to the ST-12 strain, but the ST-12 strain survived significantly better in H2O2 and NO than the ST-17 strain (Fig. 5A). Both strains were resistant to lysozyme at concentrations up to 10mg/ml. Congruent with the intracellular survival data, the ST-17 strain survived significantly better than the ST-12 strain in the combined multiple stress medium (Fig. 5A). Although the ST-12 strain was better able to survive some of these stresses individually compared to the ST-17 strain, the combination of all stresses contributed to enhanced survival for only the ST-17 strain. To further examine this, we measured survival of both strains in pH 4.5 and H2O2 stress combined (Fig. 5B). Interestingly, the survival rate in both pH 4.5 and H2O2 more closely resembled the survival rate in pH 4.5 alone. Moreover, the H2O2 was not breaking down more rapidly in the low pH (data not shown), ensuring that GBS was continuously encountering both stresses. Taken together, these data suggest that diverse strains of GBS use different mechanisms to survive phagosomal stress.
Figure 5.
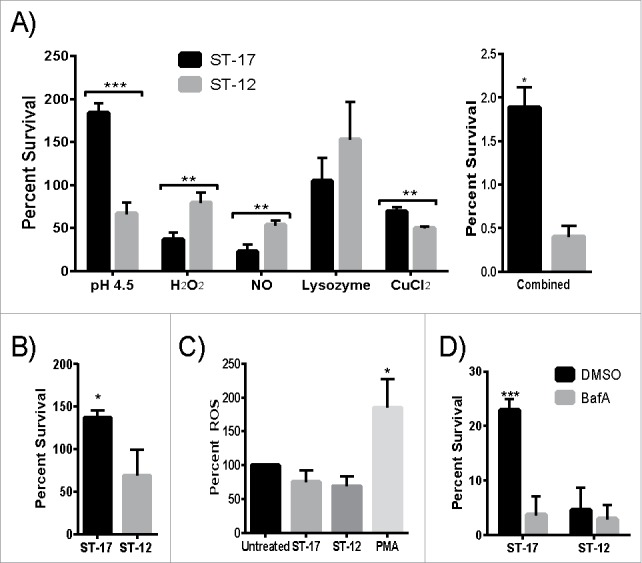
The GBS ST-17 strain shows an overall enhanced ability to survive a multiple stress environment and uses a pH dependent mechanism to survive the phagosome. (A) Survival of GBS after exposure to 5 stressors commonly found in the phagosome tested individually and combined in a single medium. Data is represented as percent of the untreated control. (B) Survival of GBS after exposure to both pH 4.5 and H2O2 combined. (C) Production of ROS by placental macrophages after exposure to medium alone (untreated), GBS, or PMA as a positive control. ROS production was measured every 30 min for 2 hr. The 2 hr time point is shown as representative data. A-C show all data combined from 3 separate experiments done in triplicate. (D) Representative data showing intracellular survival of GBS inside THP-1 macrophages pretreated with DMSO (control) or 100nM Bafilomycin A1 (BafA) to block acidification of the phagosome.(*P ≤ 0.5, **P < 0.01, ***P < 0.001).
Since the ST-12 strain survived H2O2 stress significantly better than the ST-17 strain and GBS was previously shown to prevent the induction of ROS production in the macrophage during infection,19 we hypothesized that the ST-17 strain would have an enhanced ability to inhibit ROS production. To test this, we examined the production of ROS in placental macrophages every 30 min for 2 hr after GBS infection. Placental macrophages were selected because these cells are likely to be involved in host defense against bacterial infections of the placenta and associated membranes during ascending chorioamnionitis. Both strains equally inhibited ROS production to levels significantly less than the PMA-treated positive control and slightly less than the uninfected control at all time points tested with the highest ROS production at 2 hr. (Fig. 5C). This ROS inhibition would not be due to overall cytotoxicity because we measured ROS activity up to 2 hr after infection and observed only a slight reduction in cell viability at 3 hr post infection (Figs. 2B and C). Since both strains were capable of infecting placental macrophages without initiating a strong oxidative burst compared to the positive control, this suggests ROS inhibition as a possible survival mechanism for GBS.
Since the ST-17 strain had higher CFUs/ml in the acid treated samples compared to the untreated control and the ST-12 strain was killed in acidic pH (Fig. 5A), we hypothesized that survival in acid is critical for the ST-17 strain to withstand phagosomal stress for a longer period of time relative to the ST-12 strain. To test this, we examined the 24 hr survival rate of each strain inside human macrophages treated with bafilomycin A1 (BafA), which inhibits acidification of the phagosome. Interestingly, inhibiting phagosome acidification significantly reduced the ability of the ST-17 strain to survive intracellularly, but had no effect on the ST-12 strain (Fig. 5D). Of note, the overall intracellular survival rate is less than that shown in Figs. 1B and 2A. This is due to the inherent variation of this assay and therefore, data shown is representative from at least 3 independent experiments. Although the exact survival rate varies between experiments, the overall trends and differences between the strains were similar. Moreover, the range in survival rates for the 2 strains never overlapped across replicates and the differences in survival were significant in each experiment.
Survival in phagosome-like conditions is dependent on CPS type
Since intracellular survival assays using macrophages can be time consuming and labor intensive, we used the multiple stress medium to rapidly assess the ability of a large number of strains to survive phagosome-like conditions. To do this, we selected 30 strains representing a range of CPS types, STs, and clinical types (Table 1). Clinical types included colonizing strains isolated from pregnant women that were either lost or persisted after IAP and invasive strains isolated from infected neonates. There was no significant difference in survival by clinical type or ST; however, CPS type III GBS strains survived significantly better than any other CPS type combined (Fig. 6A). Survival rates of 24.3%, 7.3%, 1.9%, and 0.4% were observed in the multiple stress medium for NEM316 (ST-23), GB00590 (ST-19), GB00112 (ST-17), and GB00653 (ST-12), respectively. Similar trends were observed using THP-1 cells with the exception of the ST-19 strain, which did not survive significantly better than the ST-17 strain after 24 hr.
Figure 6.
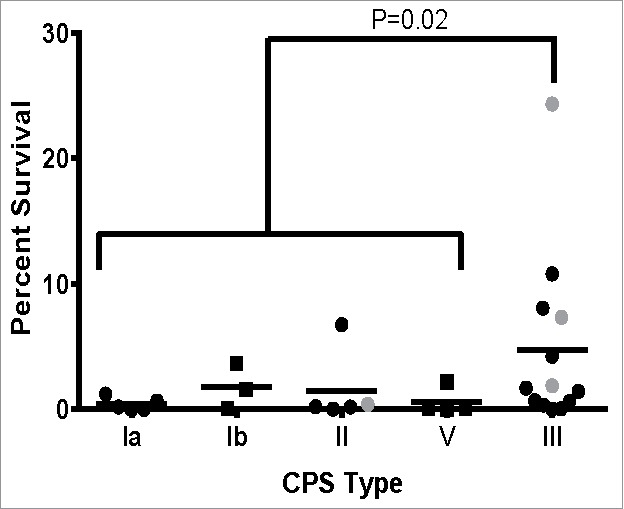
Survival of GBS clinical isolates in multiple stress medium. Thirty isolates representing a range of CPS types, STs, and clinical sources were assessed for their ability to survive a multiple stress medium. Data was represented as percent of the untreated control for each strain and stratified by CPS type. Data points in gray indicate the strains assessed in the macrophage survival assay in Figure 1. All data were combined from at least 3 independent experiments done in triplicate.
Since low pH survival was an important factor for the examined ST-17 strain (Fig. 5), we next determined if low pH survival played a role in the ability of other GBS strains to survive the multiple stress medium. To do this, we selected a subset of strains from each CPS type and ST for a total of 10 of the 30 strains tested in the multiple stress medium and examined their ability to withstand pH 4.5. These strains had a range of 0.01–6.75% survival in the multiple stress medium and a range of 70–190% survival in pH 4.5. Interestingly, we found no correlation between ability to survive multiple stress and low pH (R2=0.0202).
Discussion
In this study, we sought to better understand differences in the ability of diverse GBS strains to persist after antibiotic exposure by comparing a ST-17 strain that had persistently colonized a mother after IAP and a ST-12 strain that had been eradicated. We have shown that, in the case of the strains tested in this study, the ST-17 strain's ability to persist was more likely due to its ability to remain inside macrophages for an extended period of time rather than antibiotic tolerance. Although the ST-12 strain was tolerant to ampicillin, this strain was eradicated in the mother following IAP with ampicillin as it was not detectable at her postpartum visit. Since the strain was tolerant, but not resistant, it is possible that IAP limited the bacterial population enough for adequate clearance by the immune system. The differences in antibiotic tolerance and macrophage intracellular survival suggests varying mechanisms of persistence and warrants further investigation using a larger number of diverse strains to identify ST- or CPS type-specific mechanisms as well as other possible mechanisms not examined in this study.
Additionally, the ST-17 strain was more readily taken up by macrophages after ampicillin and erythromycin exposure. Similar to our findings, it has previously been reported that treatment with sub-inhibitory concentrations of antibiotics increased phagocytic uptake of Listeria monocytogenes,20 Staphylococcus aureus,21 and E. coli18 by macrophages, but this enhanced phagocytic activity was not due to a change in the macrophages themselves.20 In addition, ampicillin or erythromycin exposure did not alter phagocytosis of the BioParticles; therefore, the enhanced phagocytosis of the ST-17 strain is likely due to a change in the bacteria rather than a change in the macrophages. Since penicillin treatment reduced the phagocytic capacity of the macrophages, but phagocytosis of the ST-17 strain after penicillin exposure was the same as the untreated control, it is possible that penicillin exposure does result in enhanced phagocytic uptake.
Indeed, changes in the level of capsule production in response to antibiotics could increase the number of exposed surface proteins that are recognized by macrophages and result in increased phagocytosis. Similar findings of reduced encapsulation after antibiotic exposure were previously reported for Bacteroides fragilis after sub-inhibitory clindamycin.22 A link between encapsulation and enhanced phagocytic uptake after exposure to sub-inhibitory antibiotics in E. coli was also reported.18 Taken together, these data indicate a common response to sub-inhibitory concentrations of antibiotics across different bacterial species as well as antibiotics with different modes of action. However, ampicillin exposure resulted in a smaller reduction in capsule compared to penicillin and erythromycin. Since sub-inhibitory concentrations of antibiotics have also been shown to cause other morphological changes in bacteria,23 further investigation of the effect of antibiotics on GBS is warranted to have a full understanding of the molecular mechanisms behind the increased phagocytosis. In addition, the level of capsule production may vary across strains and could also influence differences in phagocytic uptake across strains.
Interestingly, antibiotic exposure had no effect on intracellular survival of either strain but enhanced phagocytic uptake, suggesting that antibiotics could increase the likelihood of persistent colonization, at least for the strain tested in this study. In a recent study, Lehar et. al. proposed a novel therapeutic using antibody-antibiotic conjugates to clear intracellular reservoirs of Staphylococcus aureus.24 Similar strategies could also be used to eliminate persistent maternal GBS colonization and to potentially reduce the risk of neonatal infection. A previous study reported that serotype Ib GBS treated with sub-inhibitory concentrations of β-lactam antibiotics, including penicillin, increased killing of GBS by phagocytes.25 However, our current study shows no difference in phagocytic killing of the 2 strains (serotypes II and III) examined here. The differing results suggest that the antibiotic effect on phagocytic killing of GBS could be dependent on serotype.
By examining gene expression during intracellular survival, we have shown that the ST-17 and ST-12 strains vary in their temporal response to macrophages. The finding that these genes are important for long term survival in macrophages is congruent with previous studies. Both strains, for example, had ∼2-fold upregulation of ponA, which encodes the penicillin-binding protein 1a that was linked to phagosomal stress resistance in a prior study by protecting GBS against cationic antimicrobial peptides.26 The cyl operon, which includes cylE, produces a pore-forming β-hemolysin/cytolysin (βH/C) and a carotenoid pigment that helps protect against reactive oxygen species. A previous study showed that a cylE deletion mutant does not produce either the βH/C or pigment and is more susceptible to both oxidative and macrophage killing.27 Another study has found that the CovR/S 2 component regulatory system is required for survival inside macrophages. This system regulates the expression of many virulence genes including cylE, scpB, and fbsA19 and could explain the upregulation of these genes in the ST-12 strain at 1hr since covS was also upregulated. However, the covR/S regulated genes are upregulated in the ST-17 strain during intracellular survival despite covR/S not being differentially expressed. This upregulation could be due to increased activity of CovR/S rather than upregulation of gene expression. Although scpB, lmb, and fbsA play important roles in GBS pathogenesis by aiding in attachment and invasion of host cells,28-30 their importance for survival inside macrophages has yet to be determined. Consequently, future studies should focus on constructing GBS mutants to better understand the role these genes play inside the macrophage.
Interestingly, sodA was found to be important for survival in oxidative stress and intracellular survival in mouse bone marrow-derived macrophages using a sodA-disrupted mutant.16 It is therefore likely that the downregulation of sodA, despite its importance in intracellular survival, results because the ROS burst occurs early on during phagosome maturation and the elevation in ROS is transient.31 Hence, by 1 and 24 hr, ROS concentration is reduced and expression of sodA would be turned off. Proper examination of sodA expression during intracellular survival would require sampling at earlier time points. Additionally, since GBS inhibits the ROS burst in macrophages, it is also possible that sodA upregulation is not necessary. Because mRNA expression does not always correlate with protein levels, future studies are necessary to examine protein levels for a more complete determination of their levels inside a macrophage for each of these strains.
In addition to enhanced intracellular survival, the ST-17 strain was phagocytosed significantly more than the ST-12 strain. Chattopadhyay et. al. demonstrated that GBS strains containing the pilus island 2b backbone protein, Spb1, had increased phagocytosis and intracellular survival compared to strains lacking Spb1.32 Since Spb1 is specific to ST-17 strains,33 this finding suggests that the enhanced phagocytosis and ability to survive intracellularly is ST-dependent. However, using our multiple stress medium, ST-17 strains did not survive significantly better than other STs. Since the mechanism of how Spb1 promotes intracellular survival is currently not known, it is possible that the factor Spb1 protects against was not included in our multiple stress medium.
Using the multiple stress medium we demonstrated that the strains with CPS type III examined in this study have an enhanced ability to survive multiple phagosomal stressors. Indeed, previous studies have reported that CPS type III GBS strains induce a lower maternal antibody response34 and are more virulent. Since newborn infants rely on the transfer of maternal antibodies for defense against infections early in life, the low antibody response to type III strains could explain the high rate of type III infections.35 This low antibody response could also be due to the enhanced ability of type III strains to survive inside macrophages, which are common antigen presenting cells that alert the immune system of invading pathogens.36 Future work, however, requires the examination of a larger set of strains representing varying genotypes and CPS types using both THP-1 and placental macrophages for a more complete understanding of intracellular survival in GBS.
Because the exact trends of survival in macrophages for the 4 strains tested in this system did not completely match survival in the synthetic multiple stress medium, it is important to note that this multiple stress medium does not fully represent the phagosomal environment. This synthetic medium is missing a number of factors present in the macrophage as well as the timing of the addition of each stressor as the phagosome matures. Nonetheless, this medium would work as an excellent first step in screening for novel factors important for surviving phagosomal stress, such as screening mutant strains, before proceeding to the more labor intensive assays with macrophages.
The ST-17 strain, which survived intracellularly longer than the ST-12 strain, had decreased survival when acidification of the phagosome was blocked with BafA treatment. BafA treatment possibly causes other changes in the cell in addition to inhibiting the vacuolar H+-ATPase to block phagosome acidification.37 Nonetheless, the increased intracellular survival in pH 4.5, the survival rate in both pH 4.5 and H2O2 most closely resembling low pH alone, and the decrease in intracellular survival with BafA treatment indicates a pH-dependent mechanism of intracellular survival for the ST-17 strain. This pH requirement is congruent with a previous study that found the CovR/S acid response regulator was needed for intracellular survival.19 When comparing the individual effect of different phagosomal stressors on each of the strains, the ST-17 strain survived better in low pH and CuCl2 but the ST-12 strain survived better under H2O2 and NO stress. Taken together, these results suggest that different GBS strains use different mechanisms to survive inside the phagosome.
Survival inside professional phagocytes can assist with the dissemination of a pathogen throughout the host to cause more severe disease. Pathogens can cross host cell barriers, such as the extraplacental membranes or the blood brain barrier, via phagocyte-facilitated invasion.38,39 Therefore, a better understanding of the mechanism by which GBS survives inside macrophages could aid in preventing GBS infection. Additionally, GBS has the ability to use intracellular survival as a mechanism to evade antibiotics and persistently colonize women, providing a possible explanation for the unchanged rate of late onset neonatal infections despite the implementation of IAP preventative measures.40
Materials and Methods
Bacterial culture
GBS strains (Table 1) were cultured in Todd-Hewitt broth (THB) or on agar (THA) at 37°C with 5% CO2. With the exception of NEM31641 and COH1,42 invasive strains were originally recovered from neonatal blood or cerebral spinal fluid in a prior study,43 and colonizing strains were recovered via vaginal/rectal swabs from women during and after pregnancy.44 The ST-17 strain (GB00112) was examined in our prior studies,45,46 and all strains were previously characterized by capsular genotyping and MLST as described.8,47
Cell culture
The human monocyte cell line, THP-1 (ATCC TIB-202), was maintained by incubating at 37°C with 5% CO2 in Roswell Park Memorial Institute 1640 (RPMI) growth medium (Gibco) containing 10% fetal bovine serum (FBS; Hyclone) and 2% penicillin/streptomycin (Gibco), referred to as full RPMI for the rest of the paper. For experiments, 106 cells were seeded into each well of a 24 well plate in the presence of 100 nM phorbol 12-myristate 13-acetate (PMA; Sigma) in RPMI 1640 with 2% FBS for 24 hr. The THP-1 cells were differentiated into adherent macrophages, which could be observed microscopically, after 24 hr.48
Intracellular survival assay
Bacterial strains were grown in THB to mid-log phase, washed once with PBS and resuspended in RPMI. PMA treated THP-1 cells were washed twice with PBS then infected with GBS strains at a multiplicity of infection of 10:1 for 1 hr, after which the wells were washed 3 times with PBS. Remaining extracellular bacteria were killed using RPMI containing 2% FBS, 100 µg/ml gentamicin (Gibco) and 5 µg/ml penicillin G (Sigma). After 1 hr, the number of intracellular bacteria was determined by washing each well with PBS to remove antibiotics and adding 0.1% triton X-100 (Sigma) to lyse the cells for 30 min. Lysates were diluted and plated onto THA and incubated at 37°C with 5% CO2 overnight to count colony forming units (CFUs). This was repeated at several time points to determine survival rate over time. The number of intracellular bacteria was normalized to the total number of bacteria in the well after the 1 hr infection period. The survival rate was calculated as follows: survival rate= (intracellular bacteria at time x / intracellular bacteria 1 hr after adding antibiotics) * 100. Macrophage viability was assessed throughout the assay using 2 methods: 1) Cells were removed from the wells using trypsin-EDTA (Gibco) and stained with trypan blue (Gibco); viable cells per well were counted using a hemocytometer; and 2) Supernatants were collected from each well and the amount of LDH, an indicator of cell death, was assessed using the colorimetric LDH activity assay kit (Sigma) following manufacturer's instructions. Since LDH reduces NAD to NADH, LDH activity was calculated by determining the amount of NADH generated by the sample supernatant by comparing the absorbance from that sample to a NADH standard curve. LDH activity is reported as milliunit/mL where one unit is the amount of enzyme needed to generate 1µmol per minute. To assess the effect of blocking acidification of the phagosome on intracellular survival, 100 nM BafA (Sigma) was added to the cells 1 hr prior to GBS infection and remained with the cells throughout the assay.
RNA isolation and RT-PCR
RNA preparation, cDNA synthesis, and RT-PCR were performed as previously described.45 RNA samples were collected from bacteria cultured in medium by adding samples to 2 volumes of RNAprotect Bacteria Reagent (Qiagen). RNA samples were collected from intracellular bacteria during the survival assay described above by washing the wells twice with PBS then adding 1ml RNAprotect Bacteria Reagent. RT-PCR analysis was performed using primers listed in Table 3 and the relative fold change of each gene was calculated using the 2−ΔΔCt method using gyrA as the internal control gene.49
Table 3.
Oligonucleotide primers used for qRT-PCR.
| Gene | Forward Primer (5′ to 3′) | Reverse Primer (5′ to 3′) |
|---|---|---|
| gyrA | CGGGACACGTACAGGCTACT | CGATACGAGAAGCTCCCACA |
| sodA | TGGGAATTGATGTCACCAGA | CCTGAACCAAAACGTCCTGT |
| ponA | AGGAAGTTTGGCTTGGGCTT | AGCGAGCAAAGCAAGTTGTG |
| cylE | TGGAAATTGCTAAGTTAGATAACG | AGCCCTCGTTAAGTTTGCCA |
| covR | TGCTCCACGATCAAGACCAG | ACGGTCGTGAAGGATTGGAC |
| covS | AGCTTCCTTGGACGCGAATC | TTAAGAACGCCTGTCGCTGT |
| scpB | GTAACTACGCTCAAGCTATC | CCCAAAGCTACTATCATTAC |
| lmb | GATCCCTTGCCCAAGCTTCT | TCCAATCAGGTGCAGGCATT |
| fbsA | GCGGTTTGAGACGCAATGAA | AAAAGTCACCCTAACCAACCT |
Ampicillin tolerance
Overnight GBS cultures were exposed to 5 μg/ml ampicillin in THB for 48 hr at 37°C. The same population of bacteria was then re-suspended in THB containing 1 μg/ml ampicillin for an additional 24 hr exposure at 37°C. In an attempt to recover any live cells, the culture was resuspended in fresh THB and grown for 24 hr at 37°C; full growth represented ampicillin tolerance.
Phagocytosis after antibiotic exposure
Sub-inhibitory concentrations of antibiotic were determined by exposing GBS to antibiotic in RPMI for 1 hr to mimic the infection period of the intracellular survival assay described above. The highest concentration without a significant reduction in cell viability compared to growth in medium alone was used for the assay. To assess the effect of exposure to a sub-inhibitory concentration of antibiotics on phagocytosis and intracellular survival of GBS, cultures were exposed to 0.05 µg/ml ampicillin, 0.01 µg/ml penicillin, or 0.01 µg/ml erythromycin where indicated during the 1 hr infection period of the intracellular survival assay; phagocytosis and intracellular survival were determined as described above. Phagocytic activity of the macrophages after antibiotic exposure was determined by measuring the uptake of FITC-labeled Staphylococcus aureus BioParticles (Molecular Probes). BioParticles were opsonized using the S. aureus BioParticles opsonizing reagent (Molecular Probes) following the manufacturer's instructions. BioParticles were added to PMA treated THP-1 cells at 50 particles per cell in 96 well plates in RPMI and incubated for 1 hr. Wells were then washed 3 times with PBS. Extracellular fluorescence was quenched using 0.25mg/ml trypan blue. Intracellular fluorescence was read using the Cytation™ 3 multi-mode microplate reader (Biotek Instruments, Inc.) at 480nm excitation and 520 nm emission.
Capsule staining
GBS capsules were stained using the Maneval method50 after 1 hr exposure to media alone or sub-inhibitory concentrations of penicillin, ampicillin, or erythromycin as described above. One drop of culture was mixed with one drop of 1% Congo red stain (Sigma). Once the smears were dry, they were counterstained with Maneval's stain (Carolina) for 2 min, drained and air dried. Antibiotic exposure and capsule staining were performed 3 separate times and representative images were taken from each. Capsule thickness was determined by measuring the diameter of the clear zone representing the capsule and subtracting the size of the cell to control for differential cell size. Three separate measurements were made for each cell examined and averaged to determine capsule thickness. Approximately 10–15 cells were analyzed for each treatment. Data was then normalized to the untreated media control and is reported as average percent of media control.
GBS survival in synthetic multiple stress medium
Survival of GBS in several phagosomal stressors was assessed as previously described.19 Briefly, stationary phase cultures were washed in PBS, then resuspended in 0.1M sodium phosphate buffer pH 7.5 alone or buffer with the indicated stressor at a final concentration of approximately 1×106 cells/ml. Cultures were incubated for 1 hr at 37°C then diluted and plated on THA to count viable bacteria. The number of viable bacteria in the treated samples was normalized to untreated samples to calculate percent survival. The ability of GBS to survive the following stressors at the indicated concentrations was tested individually: hydrogen peroxide (H2O2) at 5 mM, sodium nitrate (NaNO2) at 10 mM in sodium phosphate buffer at pH 4.5 (NaNO2 dissociates to yield nitric oxide (NO) at low pH), copper chloride (CuCl2) at 0.25 mM, lysozyme at 100 µg/ml, and acidic sodium phosphate buffer at pH 4.5. For the combined multiple stress medium, the concentrations of the stressors were 1.5 mM H2O2, 3 mM NaNO2, 100 µM CuCl2, and 100 µg/ml lysozyme in sodium phosphate buffer at pH 4.5. For the low pH and H2O2 survival, GBS cultures were exposed to 5mM H2O2 in sodium phosphate buffer at pH 4.5.
Isolation of placental macrophages
Placental macrophages were obtained from term, non-laboring placentas obtained at the time of Cesarean section as previously described.51 Samples were only obtained from healthy donors with no significant medical condition, aged 18–40 y. Briefly, the tissue was washed 3 times with PBS by centrifuging at 1,500 rpm for 10 min to remove circulating blood. The tissue was minced into small pieces and weighed to determine final grams collected. Tissue fragments were placed into 50 ml conical tubes with digestion solution containing 150 µg/ml deoxyribonuclease (Sigma), 1 mg/ml collagenase (Sigma) and 1 mg/ml hyaluronidase (Sigma) at 10 ml per gram of tissue. Cells were filtered through a 280 µm metal sieve, followed by 180 and 80 µm nylon screens (Millipore). Cells were centrifuged again and resuspended in 25% Percoll (Sigma) diluted in cold full RPMI and overlaid onto 50% Percoll, plus 2 ml of PBS on top of the density gradient. CD14+ macrophages were isolated by positive selection using the magnetic MACS® large cell separation column system according to the manufacturer's instructions (Miltenyi). Isolated CD14+ placental macrophages were rested overnight in full RPMI before experimentation.
Measurement of ROS production
The production of ROS following infection of placental macrophages by GBS was determined by seeding 2 × 105 human placental macrophages onto a 384-well cell culture-treated plate and rested overnight in full RPMI. The next day, placental macrophages were washed with PBS and labeled with 10 µM Carboxyl H2DCF-DA for 30 min following the manufacturer's instructions (Invitrogen). Placental macrophages were washed again with PBS before being inoculated with GBS at a MOI of 50:1 or PMA (300 nM) as a positive control. Cells were assessed for the generation of ROS every 30 min for 2 hr post infection, and ROS production was normalized to the percent of the untreated control.
Human subjects
These studies were approved by the Vanderbilt University Institutional Review Board (Protocol #131607). Tissue samples were provided by the Cooperative Human Tissue Network at Vanderbilt University, which is funded by the National Cancer Institute.
Statistical analysis
Data shown were either pooled from or representative of at least 3 independent experiments performed in triplicate. GraphPad Prism version 6 was used for statistical analysis. Significant differences between strains/treatments for 2 groups, multiple groups or 2 parameters comparisons was determined using Mann-Whitney U test, one-way ANOVA and 2-way ANOVA, respectively, along with Tukey's multiple comparison test where appropriate. Statistical significance was accepted at P ≤ 0.05.
Abbreviations
- BafA
Bafilomycin A
- CFU
colony forming units
- CPS
capsular polysaccharide
- FBS
fetal bovin serum
- GBS
Group B Streptococcus
- IAP
intrapartum antibiotic prophylaxis
- LDH
lactate dehydrogenase
- MLST
multilocus sequence typing
- PMA
phorbol 12-myristate 13-acetate
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- RPMI
Roswell Park Memorial Institute 1640
- ST
Sequence Type
- THA
Todd-Hewitt agar
- THB
Todd-Hewitt broth
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Robert Abramovitch for his assistance and guidance in the phagocytosis assays, Dr. H. Dele Davies for the bacterial isolates, and Dr. Poorna Viswanathan and Brian Snyder for their assistance with capsule staining.
Funding
This work was supported by the Global Alliance to Prevent Prematurity and Stillbirth (D.M.A., SDM.) in collaboration with the Bill and Melinda Gates Foundation (project N015615), the Burroughs Welcome Fund Investigators in the Pathogenesis of Infectious Disease award (D.M.A.), and the Thomas S. Whittam and Bertinna B. Wentworth graduate fellowship awards (M.L.K.).
References
- [1].Gibbs RS, Schrag S, Schuchat A. Perinatal infections due to group B streptococci. Obstet Gynecol 2004; 104:1062-76; PMID:15516403; http://dx.doi.org/ 10.1097/01.AOG.0000144128.03913.c2 [DOI] [PubMed] [Google Scholar]
- [2].Cieslewicz MJ, Chaffin D, Glusman G, Kasper D, Madan A, Rodrigues S, Fahey J, Wessels MR, Rubens CE. Structural and genetic diversity of group B Streptococcus capsular polysaccharides. Infect Immun 2005; 75:3096-103; http://dx.doi.org/ 10.1128/IAI.73.5.3096-3103.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Slotved H-C, Kong F, Lambertsen L, Sauer S, Gilbert GL. Serotype IX, a proposed new Streptococcus agalactiae serotype. J Clin Microbiol 2007; 45:2929-36; PMID:17634306; http://dx.doi.org/ 10.1128/JCM.00117-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Bohnsack JF, Whiting A, Gottschalk M, Dunn DM, Weiss R, Azimi PH, Philips JB, Weisman LE, Rhoads GG, Lin F-YC. Population structure of invasive and colonizing strains of Streptococcus agalactiae from neonates of six U.S. academic centers from 1995 to 1999. J Clin Microbiol 2008; 46:1285-91; PMID:18287314; http://dx.doi.org/ 10.1128/JCM.02105-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Lin FC, Whiting A, Adderson E, Takahashi S, Dunn DM, Weiss R, Azimi PH, Philips JB, Weisman LE, Regan J, et al.. Phylogenetic lineages of invasive and colonizing strains of serotype III group B streptococci from neonates: a multicenter prospective study. J Clin Microbiol 2006; 44:1257-61; PMID:16597848; http://dx.doi.org/ 10.1128/JCM.44.4.1257-1261.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Luan S, Granlund M, Sellin M, Lagergård T, Spratt BG, Norgren M. Multilocus sequence typing of swedish invasive group B Streptococcus isolates indicates a neonatally associated genetic lineage and capsule switching. J Clin Microbiol 2005; 43:3727-33; PMID:16081902; http://dx.doi.org/ 10.1128/JCM.43.8.3727-3733.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Manning SD, Springman C, Lehotzky E, Lewis M a, Whittam TS, Davies HD. Multilocus sequence types associated with neonatal group B streptococcal sepsis and meningitis in Canada. J Clin Microbiol 2009; 47:1143-8; PMID:19158264; http://dx.doi.org/ 10.1128/JCM.01424-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jones N, Bohnsack JF, Takahashi S, Karen A, Chan M, Kunst F, Glaser P, Rusniok C, Crook DWM, Rosalind M, et al.. Multilocus sequence typing system for group B Streptococcus. J Clin Microbiol 2003; 41:2530-6; PMID:12791877; http://dx.doi.org/ 10.1128/JCM.41.6.2530-2536.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Manning SD, Lewis MA, Springman C, Lehotzky E, Whittam TS, Davies HD. Genotypic diversity and serotype distribution of group B Streptococcus isolated from women before and after delivery. Clin Infect Dis 2008; 46:1829-37; PMID:18462173; http://dx.doi.org/ 10.1086/588296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Thi EP, Lambertz U, Reiner NE. Sleeping with the enemy: how intracellular pathogens cope with a macrophage lifestyle. PLoS Pathog 2012; 8:e1002551; PMID:22457616; http://dx.doi.org/ 10.1371/journal.ppat.1002551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Valentin-weigand P, Benkel P, Rohde M, Chhatwal GS. Entry and intracellular survival of group B streptococci in J774 macrophages. Infect Immun 1996; 64:2467-73; PMID:8698468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kaplan EL, Chhatwal GS, Rohde M. Reduced ability of penicillin to eradicate ingested group A streptococci from epithelial cells: clinical and pathogenetic implications. Clin Infect Dis 2006; 43:1398-406; PMID:17083011; http://dx.doi.org/ 10.1086/508773 [DOI] [PubMed] [Google Scholar]
- [13].Thwaites GE, Gant V. Are bloodstream leukocytes trojan horses for the metastasis of Staphylococcus aureus? Nat Rev Microbiol 2011; 9:215-22; PMID:21297670; http://dx.doi.org/ 10.1038/nrmicro2508 [DOI] [PubMed] [Google Scholar]
- [14].Fettucciari K, Rosati E, Scaringi L, Cornacchione P, Migliorati G, Sabatini R, Fetriconi I, Rossi R, Marconi P. Group B Streptococcus induces apoptosis in macrophages. J Immunol 2000; 165:3923-33; PMID:11034400; http://dx.doi.org/ 10.4049/jimmunol.165.7.3923 [DOI] [PubMed] [Google Scholar]
- [15].Ulett GC, Bohnsack JF, Armstrong J, Adderson EE. Beta-hemolysin-independent induction of apoptosis of macrophages infected with serotype III group B Streptococcus. J Infect Dis 2003; 188:1049-53; PMID:14513426; http://dx.doi.org/ 10.1086/378202 [DOI] [PubMed] [Google Scholar]
- [16].Poyart C, Pellegrini E, Gaillot O, Boumaila C, Baptista M, Trieu-cuot P. Contribution of Mn-cofactored superoxide dismutase (SodA) to the virulence of Streptococcus agalactiae. Infect Immun 2001; 68:5098-106; http://dx.doi.org/ 10.1128/IAI.69.8.5098-5106.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Henneke P, Berner R. Interaction of neonatal phagocytes with group B Streptococcus: recognition and response. Infect Immun 2006; 74:3085-95; PMID:16714536; http://dx.doi.org/ 10.1128/IAI.01551-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Raponi G, Keller N, Overbeek BP, Rozenberg-Arska M, Van Kessel KPM, Verhoef J. Enhanced phagocytosis of encapsulated Escherichia coli strains after exposure to sub-MICs of antibiotics is correlated to changes of the bacterial cell surface. Antimicrob Agents Chemother 1990; 34:332-6; PMID:2109580; http://dx.doi.org/ 10.1128/AAC.34.2.332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Cumley NJ, Smith LM, Anthony M, May RC. The CovS/CovR acid response regulator is required for intracellular survival of group B Streptococcus in macrophages. Infect Immun 2012; 80:1650-61; PMID:22331428; http://dx.doi.org/ 10.1128/IAI.05443-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Adam D, Schaffert W, Marget W. Enhanced in vitro phagocytosis of Listeria monocytogenes by human monocytes in the presence of ampicillin, tetracycline, and chloramphenicol. Infect Immun 1974; 9:811-4; PMID:4207515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Milatović D. Effect of subinhibitory antibiotic concentrations on the phagocytosis of Staphylococcus aureus. Eur J Clin Microbiol 1982; 1:97-101; PMID:6293812; http://dx.doi.org/ 10.1007/BF02014199 [DOI] [PubMed] [Google Scholar]
- [22].Gemmel CG, Peterson PK, Schmeling D, Mathews J, Quie PG. Antibiotic-induced modification of Bacteroides fragilis and its susceptibility to phagocytosis by human polymorphonuclear leukocytes. Eur J Clin Microbiol 1983; 2:327-34; PMID:6628373; http://dx.doi.org/ 10.1007/BF02019462 [DOI] [PubMed] [Google Scholar]
- [23].Washington JA. The effects and significance of subminimal inhibitory concentrations of antibiotics. Rev Infect Dis 1979; 1:781-6; PMID:396633; http://dx.doi.org/ 10.1093/clinids/1.5.781 [DOI] [PubMed] [Google Scholar]
- [24].Lehar SM, Pillow T, Xu M, Staben L, Kajihara KK, Vandlen R, DePalatis L, Raab H, Hazenbos WL, Hiroshi Morisaki J, et al.. Novel antibody–antibiotic conjugate eliminates intracellular S. aureus. Nature 2015; 527:323-8; PMID:26536114; http://dx.doi.org/ 10.1038/nature16057 [DOI] [PubMed] [Google Scholar]
- [25].Horne D, Tomasz A. Hypersusceptibility of penicillin-treated group B streptococci to bactericidal activity of human polymorphonuclear leukocytes. Antimicrob Agents Chemother 1981; 19:745-53; PMID:7027922; http://dx.doi.org/ 10.1128/AAC.19.5.745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hamilton A, Popham DL, Carl DJ, Lauth X, Nizet V, Jones AL. Penicillin-binding protein 1a promotes resistance of group B Streptococcus to antimicrobial peptides. Infect Immun 2006; 74:6179-87; PMID:17057092; http://dx.doi.org/ 10.1128/IAI.00895-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Liu GY, Doran KS, Lawrence T, Turkson N, Puliti M, Tissi L, Nizet V. Sword and shield: linked group B streptococcal beta-hemolysin/cytolysin and carotenoid pigment function to subvert host phagocyte defense. Proc Natl Acad Sci U S A 2004; 101:14491-6; PMID:15381763; http://dx.doi.org/ 10.1073/pnas.0406143101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Cheng Q, Stafslien D, Purushothaman SS, Cleary P. The group B streptococcal C5a peptidase is both a specific protease and an invasin. Infect Immun 2002; 70:2408-13; PMID:11953377; http://dx.doi.org/ 10.1128/IAI.70.5.2408-2413.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Spellerberg B, Rozdzinski E, Martin S, Weber-Heynemann J, Schnitzler N, Lütticken R, Podbielski A. Lmb, a protein with similarities to the LraI adhesin family, mediates attachment of Streptococcus agalactiae to human laminin. Infect Immun 1999; 67:871-8; PMID:9916102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Schubert A, Zakikhany K, Pietrocola G, Meinke A, Speziale P, Eikmanns BJ, Reinscheid DJ. The fibrinogen receptor FbsA promotes adherence of Streptococcus agalactiae to human epithelial cells. Infect Immun 2004; 72:6197-205; PMID:15501744; http://dx.doi.org/ 10.1128/IAI.72.11.6197-6205.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Forman HJ, Torres M. Reactive oxygen species and cell signaling. Am J Respir Crit Care Med 2002; 166:S4-8; PMID:12471082; http://dx.doi.org/ 10.1164/rccm.2206007 [DOI] [PubMed] [Google Scholar]
- [32].Chattopadhyay D, Carey AJ, Caliot E, Webb RI, Layton JR, Wang Y, Bohnsack JF, Adderson EE, Ulett GC. Phylogenetic lineage and pilus protein Spb1/SAN1518 affect opsonin-independent phagocytosis and intracellular survival of group B Streptococcus. Microbes Infect 2011; 13:369-82; PMID:21238599; http://dx.doi.org/ 10.1016/j.micinf.2010.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Tazi A, Bellais S, Tardieux I, Dramsi S, Trieu-Cuot P, Poyart C. Group B Streptococcus surface proteins as major determinants for meningeal tropism. Curr Opin Microbiol 2012; 15:44-9; PMID:22206860; http://dx.doi.org/ 10.1016/j.mib.2011.12.002 [DOI] [PubMed] [Google Scholar]
- [34].Davies HD, Adair C, McGeer A, Ma D, Robertson S, Mucenski M, Kowalsky L, Tyrell G, Baker CJ. Antibodies to capsular polysaccharides of group B Streptococcus in pregnant Canadian women: relationship to colonization status and infection in the neonate. J Infect Dis 2001; 184:285-91; PMID:11443553; http://dx.doi.org/ 10.1086/322029 [DOI] [PubMed] [Google Scholar]
- [35].Baker CJ, Kasper DL. Correlation of maternal antibody deficiency with susceptibility to neonatal group B Streptococcal infection. N Engl J Med 1976; 294:753-6; PMID:768760; http://dx.doi.org/ 10.1056/NEJM197604012941404 [DOI] [PubMed] [Google Scholar]
- [36].Flannagan RS, Jaumouillé V, Grinstein S. The cell biology of phagocytosis. Annu Rev Pathol 2012; 7:61-98; PMID:21910624; http://dx.doi.org/ 10.1146/annurev-pathol-011811-132445 [DOI] [PubMed] [Google Scholar]
- [37].Dmitriev RI, Zhdanov A V, Papkovsky DB. Uncoupling effect of bafilomycin A1 on HIF and cell bioenergetics. FASEB J 2011; 25:861.15 [Google Scholar]
- [38].Doran KS, Banerjee A, Disson O, Lecuit M. Concepts and mechanisms: crossing host barriers. Cold Spring Harb Perspect Med 2013; 3:1-20; http://dx.doi.org/ 10.1101/cshperspect.a010090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Robbins JR, Bakardjiev AI. Pathogens and the placental fortress. Curr Opin Microbiol 2012; 15:36-43; PMID:22169833; http://dx.doi.org/ 10.1016/j.mib.2011.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Brown HL, Ahmadzia HK, Heine RP. GBS screening: an update on guidelines and methods. Contemp Ob Gyn 2013; http://contemporaryobgyn.modernmedicine.com/contemporary-obgyn/content/tags/bd-geneohm/gbs-screening-update-guidelines-and-methods?page=full [Google Scholar]
- [41].Glaser P, Rusniok C, Buchrieser C, Chevalier F, Frangeul L, Msadek T, Zouine M, Couvé E, Lalioui L, Poyart C, et al.. Genome sequence of Streptococcus agalactiae, a pathogen causing invasive neonatal disease. Mol Microbiol 2002; 45:1499-513; PMID:12354221; http://dx.doi.org/ 10.1046/j.1365-2958.2002.03126.x [DOI] [PubMed] [Google Scholar]
- [42].Wilson CB, Weaver WM. Comparative susceptibility of group B streptococci and Staphylococcus aureus to killing by oxygen metabolites. J Infect Dis 1985; 152:323-9; PMID:2993435; http://dx.doi.org/ 10.1093/infdis/152.2.323 [DOI] [PubMed] [Google Scholar]
- [43].Davies HD, Raj S, Adair C, Robinson J, McGeer A. Population-based active surveillance for neonatal group B streptococcal infections in Alberta, Canada: implications for vaccine formulation. Pediatr Infect Dis J 2001; 20:879-84; PMID:11734768; http://dx.doi.org/ 10.1097/00006454-200109000-00011 [DOI] [PubMed] [Google Scholar]
- [44].Spaetgens R, DeBella K, Ma D, Robertson S, Mucenski M, Dele Davies H. Perinatal antibiotic usage and changes in colonization and resistance rates of group B Streptococcus and other pathogens. Obstet Gynecol 2002; 100:525-33; PMID:12220773 [DOI] [PubMed] [Google Scholar]
- [45].Korir ML, Knupp D, LeMerise K, Boldenow E, Loch-Caruso R, Aronoff DM, Manning SD. Association and virulence gene expression vary among serotype III group B Streptococcus isolates following exposure to decidual and lung epithelial cells. Infect Immun 2014; 82:4587-95; PMID:25135682; http://dx.doi.org/ 10.1128/IAI.02181-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Singh P, Springman AC, Davies HD, Manning SD. Whole-genome shotgun sequencing of a colonizing multilocus sequence type 17 Streptococcus agalactiae strain. J Bacteriol 2012; 194:6005-6005; PMID:23045509; http://dx.doi.org/ 10.1128/JB.01378-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Manning SD, Lacher DW, Davies HD, Foxman B, Whittam TS. DNA polymorphism and molecular subtyping of the capsular gene cluster of group B Streptococcus. J Clin Microbiol 2005; 43:6113-6; PMID:16333106; http://dx.doi.org/ 10.1128/JCM.43.12.6113-6116.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Schwende H, Fitzke E, Ambs P, Dieter P. Differences in the state of differentiation of THP-1 cells induced by phorbol ester and 1,25-dihydroxyvitamin D3. J Leukoc Biol 1996; 59:555-61; PMID:8613704 [PubMed] [Google Scholar]
- [49].Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative CT method. Nat Protoc 2008; 3:1101-8; PMID:18546601; http://dx.doi.org/ 10.1038/nprot.2008.73 [DOI] [PubMed] [Google Scholar]
- [50].Maneval WE. Staining bacteria and yeasts with acid dyes. Stain Technol 1941; 16:13-9; http://dx.doi.org/ 10.3109/10520294109106189 [DOI] [Google Scholar]
- [51].Soares EM, Mason KL, Rogers LM, Serezani CH, Faccioli LH, Aronoff DM. Leukotriene B4 enhances innate immune defense against the puerperal sepsis agent Streptococcus pyogenes. J Immunol 2013; 190:1614-22; PMID:23325886; http://dx.doi.org/ 10.4049/jimmunol.1202932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Rajagopal L. Understanding the regulation of group B streptococcal virulence factors. Future Microbiol 2009; 4:201-21; PMID:19257847; http://dx.doi.org/ 10.2217/17460913.4.2.201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Whidbey C, Harrell MI, Burnside K, Ngo L, Becraft AK, Iyer LM, Aravind L, Hitti J, Waldorf KMA, Rajagopal L. A hemolytic pigment of group B Streptococcus allows bacterial penetration of human placenta. J Exp Med 2013; 210:1265-81; PMID:23712433; http://dx.doi.org/ 10.1084/jem.20122753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Lamy M-C, Zouine M, Fert J, Vergassola M, Couve E, Pellegrini E, Glaser P, Kunst F, Msadek T, Trieu-Cuot P, et al.. CovS/CovR of group B Streptococcus: a two-component global regulatory system involved in virulence. Mol Microbiol 2004; 54:1250-68; PMID:15554966; http://dx.doi.org/ 10.1111/j.1365-2958.2004.04365.x [DOI] [PubMed] [Google Scholar]