

Abstract
Combination therapies have the potential to improve outcomes in melanoma patients but have not yet been clinically efficacious. Here, we used high-throughput flow cytometry-based screening to identify and characterize candidate therapies that might synergize with and augment T-cell immunotherapy efficacy. Two lead therapies, regorafenib and NU7441, were selected based on their ability to alter a variety of immunomodulatory proteins, including CD55, CD73, CD155, programmed death-ligand 1 (PD-L1), nerve growth factor receptor (NGFR), and HLA class I in a heterogeneous panel of melanomas. The therapies also upregulated several melanoma antigens, inhibited proliferation, and perturbed activation of oncogenic signaling pathways in melanomas. T cells treated with the therapies proliferated normally and exhibited a favorably altered phenotype, including increased CD25, CD28, inducible T-cell costimulator (ICOS), and reduced expression of co-inhibitory receptors. Cytokine production was also increased in treated T cells. When administered in mice, regorafenib suppressed melanoma progression in a CD8+ T cell–dependent manner when used alone and with various immunotherapies. Additionally, regorafenib altered the number, phenotype, and function of various T-cell subsets in the tumor microenvironment. These studies reveal that regorafenib and NU7441 influence the immunobiology of both tumor cells and T cells and enhance the efficacy of various immunotherapies.
Keywords: Combination therapy, immunotherapy, regorafenib, BRAF, melanoma
Introduction
New treatment options have improved prognoses for melanoma patients over the last half decade. These improvements stem from the development of two treatments: targeted therapies and immunotherapies. However, both classes pose unique challenges that limit their efficacy and application.
Targeted therapies for melanoma include BRAF and MEK inhibitors. Currently, targeted therapies are only available for the roughly 50% of melanoma patients who harbor BRAFV600 mutations. Of these patients, only 50% and 20% respond to BRAF or MEK inhibitors, respectively, and responses are typically short-lived (1–3). Indeed, acquired resistance develops rapidly in around half of patients treated with BRAF inhibitors with a median time of 6–8 months (1,2). BRAF and MEK inhibitor combinations improve response rates and durations, though only marginally (4,5). Moreover, targeted therapies are not available for patients without BRAF mutations. Thus, there is a need to identify new targeted therapies in order to expand application to a larger patient population and prolong response duration.
Immunotherapies, particularly antibodies blocking checkpoint receptors such as cytotoxic T-lymphocyte antigen-4 (CTLA-4) and programmed death-1 (PD-1), have revolutionized melanoma treatment. Durable and often complete tumor regressions in the setting of widespread metastatic disease have been observed. Yet, objective responses to PD-1 antibodies only occur in roughly one third of patients and complete regressions occur in approximately 5% of patients (6,7). Thus, strategies that increase response rates are necessary to improve the overall efficacy of these therapies.
Efforts have been made to combine targeted therapies and immunotherapies. Toxicities such as hepatotoxicity and gastrointestinal complications, however, have led to the termination of such clinical trials (8,9). Trials targeting PD-1 or its primary ligand, programmed death-ligand 1 (PD-L1), in combination with BRAF and/or MEK inhibitors appear more promising, but are still in progress (10). Other trials have shifted to establishing an optimal sequential application of the two treatments, which may not be as effective as simultaneous administration.
We sought to identify and characterize targeted therapies that would enhance immunotherapy efficacy. To achieve this goal, we performed flow cytometry-based high-throughput screening (HTS) on over 1,500 small molecule inhibitors, chemotherapies, medications, and natural compounds, examining their ability to alter cell surface molecules associated with immunosuppressive or immunostimulatory processes. Many of the screened compounds are already FDA-approved for various diseases. Using HTS, we identified two compounds, regorafenib (Reg) and NU7441 (NU), for further characterization.
Reg is a multikinase inhibitor which targets various tyrosine kinases, including VEGFR, c-KIT, PDGFR, and others (11). It is FDA-approved for pretreated colorectal carcinomas, gastrointestinal stromal tumors (GIST), and hepatocellular carcinomas. NU is a small molecule DNA-PK inhibitor (12). Reg and NU altered a variety of immunogenic and oncogenic properties of melanomas while favorably influencing T cells. In melanoma animal models, Reg decreased tumor growth alone and in combination with immunotherapies.
Materials and Methods
Cell culture
A375*, C32*, G-361*, MNT-1‡, SK-MEL-5*, SK-MEL-24*, SK-MEL-28*, and B16-F1* were maintained in DMEM with 10% FBS (12306C, Sigma Aldrich) and 1% antibiotics (15140122, Gibco). Malme-3M* were maintained in IMDM with 20% FBS and 1% antibiotics. All remaining cell lines (624-Mel‡, COLO-829†, LOX-IMVI†, RPMI-7951*, UACC-62†, UACC-257†, WM-115†, WM-266-4†, C8161‡, Mel-Juso†, MM415†, MM485†, SK-MEL-2*, SK-MEL-30†, SK-MEL-103†, SK-MEL-119†, SK-MEL-147†, SK-MEL-173†, and UCD-Mel-N†) were maintained in RPMI-1940 with 10% FBS and 1% antibiotics. Cell lines were obtained from ATCC (*) or generously provided by either Dr. Thomas Hornyak (†) (University of Maryland, Baltimore) or Dr. Suzanne Ostrand-Rosenberg (‡) (University of Maryland, Baltimore County) between 2009–2014. After thawing, cells were cultured for no longer than 6 weeks and tested for mycoplasma monthly. Cell lines were authenticated using quantitative PCR (qPCR) for melanoma transcripts (gp100, MART-1, TYRP1). Human PBMCs were purified by Ficoll gradient (17-1440-02, GE Healthcare) from buffy coats purchased from New York Blood Center. Purified PBMCs were cryopreserved, thawed, and cultured in AIM-V media (Gibco, 12055091) with 5% human serum (H4522, Sigma Aldrich), 1% non-essential amino acids (11140050, Gibco), and 1% antibiotics.
Flow cytometry antibodies
Antibodies from BioLegend for the following human molecules were used: PD-L1 (329718), CD155 (337610), HLA-A/B/C (311404), CD73 (344012), CD271 (345112), CD55 (311312), CD4 (317408), CD8 (300914), IFNγ (502509), TNFα (502912), and IL2 (500324). Antibodies from BioLegend for the following mouse molecules were used: CD45.2 (109828), CD8 (100725), CD4 (100406), Lag3 (125128), Tim3 (119718), PD-1 (135216), CD25 (102012), Foxp3 (126404), CD11b (101206), CD11c (117308), F4/80 (123124), Gr1 (108412), NK1.1 (108728), PD-L1 (124314), CD73 (127212), and CD155 (131508). Anti-H2-Db was from Abcam (ab112492). Anti-mouse IFNγ (12-7311-81, eBioscience), IL2 (560544, BD Biosciences), and TNFα (560658, BD Biosciences) were used for T-cell cytokine experiments. LEGENDScreen human and mouse cell screening kits (700001, 700005) from BioLegend were used for flow cytometry protein arrays.
Drugs
The SC200 drug library was generously provided by Dr. Paul Wilder (University of Maryland, Baltimore). The L1100 drug library was from SelleckChem. Regorafenib (BAY73-4506, S1178, SelleckChem) was dissolved in dimethyl sulfoxide (DMSO, 0231, Quality Biological) or 5% Kolliphor EL (C5135, Sigma Aldrich) for in vitro and in vivo studies, respectively. NU7441 (S2638, SelleckChem) and NU7026 (S2893, SelleckChem) were dissolved in DMSO or 10% DMSO for in vitro and in vivo studies, respectively.
High-throughput screening, drug modulation of surface molecules, and cytokine production
For HTS, C8161 cells were treated with the indicated compounds for 48 hours. Treated cells were analyzed by flow cytometry for expression of indicated molecules. Viable cells were gated using a fixable viability dye (423101, BioLegend) or using light scatter. For IFNγ experiments, cells were pretreated for 24 hours with 20 U/ml human recombinant IFNγ (14-8311-63, eBioscience), and IFNγ was maintained in the media throughout the experiment. For assessing drug effects on T-cell phenotype, PBMCs were stimulated with 20 ng/ml anti-CD3 (OKT3, 16-0037-85, eBioscience), 50 U/ml human recombinant IL2 (589106, BioLegend), and drug for five days and analyzed using CD4 and CD8 antibodies to distinguish between T-cell subsets. For T-cell cytokines, PBMCs were stimulated with anti-CD3 (100 ng/ml) for 72 hours. Some cells were restimulated with phorbol myristate acetate (PMA, 10 ng/ml) and ionomycin (0.5 μg/ml) for six hours. During the final 6 hours, cells were treated with Brefeldin A (GolgiPlug, 555029, BD Biosciences). A cell fixation and permeabilization kit (554714, BD Biosciences) was used.
Synergy analysis
Molecule expression was measured in cells treated with six concentrations of Reg, NU, or the combination and MFIs were compared to vehicle-treated cells to calculate fold change. Using the Chou-Talalay method, combination index (CI) values were calculated with CompuSyn software (ComboSyn, Inc). Synergy was defined as at least four of six concentrations yielding CI values below one, additive interactions as at least four CI values within 0.5–1.5, and antagonism as at least four of six CI values above two. Cell lines that met none of these classifications were defined as not determined. The fractionated product analysis method was used to calculate in vivo synergy. A ratio greater than one was considered synergistic, equal to one as additive, and less than one as antagonistic.
qPCR
RNA was collected from cells treated with Reg (2 μM) and/or NU (1 μM) for 48 hours using Qiagen RNeasy Mini kits (74104) following the manufacturer’s protocol. cDNA was prepared using a high-capacity reverse transcription kit (4368814, Applied Biosystems). qPCR was performed with iTaq Universal SYBR Green Supermix (1725121, Bio-Rad) and primers for gp100 (Fwd: CTGCCTCAATGTGTCTCTGGCT, Rev: CAAGGACCACAGCCATCAACAC), MART-1 (Fwd: GGACAGCAAAGTGTCTCTTCAAG, Rev: TCAGGTGTCTCGCTGGCTCTTA), TYRP1 (Fwd: TCTCAATGGCGAGTGGTCTGTG, Rev: CCTGTGGTTCAGGAAGACGTTG), and beta-actin (Fwd: CACCATTGGCAATGAGCGGTTC, Rev: AGGTCTTTGCGGATGTCCACGT). Relative fold changes were calculated using the ΔΔCt method normalizing to beta-actin.
Proliferation assays
Melanoma cell lines were treated with varying concentrations of Reg, NU, or vemurafenib for 48 hours. PBMCs were cultured with 20–50 ng/ml anti-CD3 and 50–100 U/ml human recombinant IL2 for five days. All cells were cultured with 3H-thymidine (0.1 μCi/ml, NET027E005MC, Perkin Elmer) for the final 16 hours of drug treatment to assess thymidine incorporation.
Immunoblot
Cells were treated with varying concentrations of Reg and NU for 24–48 hours, and lysed with RIPA buffer (R0278, Sigma Aldrich) containing protease and phosphatase inhibitors (78440, Thermo Scientific). Phospho-MEK1/2 (Clone 41G9, 9154S), MEK1/2 (Clone 47E6, 9126S), phospho-Akt (Clone D9E, 4060S), Akt (Clone 11E7, 4685S), β-Actin (Clone D6A8, 8457S), and GAPDH (Clone D16H11, 5174S) antibodies from Cell Signaling were used. gp100 (ab137078) was from Abcam. Polyclonal Tyrp1 antibody was generously provided by Dr. Thomas Hornyak (University of Maryland, Baltimore). Densitometry was performed using ImageJ.
Mice, tumor model, and ex vivo analyses
Studies were approved by the UMB Institutional Animal Care and Use Committee. C57BL/6J and pmel (B6.Cg-Thy1/Cy Tg(TcraTcrb)8Rest/J) mice were purchased from Jackson Laboratory. Animal experiments contained 5–7 animals per group for tumor growth and survival with 3–5 for ex vivo analyses. Six to eight week old C57BL/6J mice were injected with 2×105 B16-F1 melanoma cells subcutaneously on the right flank. Tumors were allowed to establish and reached approximately 50–100 mm2 before treatment. Animal weights and tumor sizes were monitored every 2–3 days. Tumor volumes were calculated using the following formula: volume = (height×width2)/2. For studies without immunotherapies, mice were treated with Reg (8 mg/kg) and/or NU7026 (6 mg/kg) on the following days post-tumor inoculation: 9–10, 12–15, 17–20, 22–25, 27–30, and 32–35. Drugs were delivered intraperitoneally (i.p) once per day. For studies using anti-CD40 and c-di-GMP, mice were treated with Reg (4–5 mg/kg) on days 8–9, 11–14, 16–19, 21–24, 26–29, 31–34, and 36–39. Some mice received 100 μg of anti-CD40 (clone FGK4.5, BE0016-2, BioXCell) i.p and 50 μg c-di-GMP (tlrl-nacdg, InvivoGen) peritumorally on days 10, 15, 20, and 25. For CD8+ T-cell depletion studies, mice were treated with 400 μg anti-CD8 (clone 53-6.7, BE0004-1, BioXCell) one day prior to tumor inoculation. Subsequently, mice were treated twice per week with 100 μg anti-CD8. For adoptive transfer experiments, mice were treated with Reg (5 mg/kg) on days 8–12 post-tumor inoculation, irradiated with 650 radians of γ-irradiation on day 12, and received intravenous delivery of 3 × 106 pmel T cells (from pmel splenocytes and activated six days prior with hgp10025–33) on day 13. Adoptive transfer included i.p injections of 200 μg anti–4-1BB (clone 3H3, BE0239, BioXCell) on days 14 and 19. For all ex vivo experiments, tumors were processed through 100 micron filters, cells were stained for flow cytometry, and fixed in 4% paraformaldehyde (19943, Affymetrix). Regulatory T cells were assessed using a Foxp3 / transcription factor staining buffer set (00-5523-00, eBioscience) following the manufacturer’s protocol. For ex vivo cytokines, intratumoral leukocytes were isolated by Ficoll gradient and activated with PMA (20 ng/ml) and ionomycin (1 μg/ml) for six hours.
Data and statistical analyses
For all flow cytometry based experiments, data was analyzed using FlowJo software (FlowJo, LLC). Viable populations were assessed using a fixable viability dye and/or using light scatter. Fold changes were determined using median fluorescent intensities (MFIs). Specifically, fold change = (experimental MFI – isotype MFI) / (vehicle MFI – isotype MFI). For all molecules, MFI values below 200 in vehicle-treated cells were considered not expressed. For ex vivo analyses normalized to tumor cell number, tumor cells were defined as CD45− cells. Mann-Whitney U tests were used to assess differences in responses between cells with different mutations. Kruskal-Wallis tests were used to evaluate overall differences in cell lines treated with different drugs. If P values for Kruskal-Wallis tests were less than 0.05, Mann-Whitney U tests were used to compare two drugs at a time. Paired t tests were used to examine differences in T-cell markers and cytokines between vehicle- and drug-treated T cells. One-way ANOVA with Tukey post-tests were used to determine differences in tumor growth and cell populations. Unpaired t tests were used to determine differences in T-cell levels and markers between ACT and ACT+Reg groups. Significant differences in survival were assessed using the log-rank test. P values below 0.05 were considered significant.
Results
High-throughput screening generates diverse candidate immunotherapies
To identify compounds that have immunomodulatory effects on melanomas, we examined the ability for compounds to alter PD-L1 and human leukocyte antigen class I (HLA-I) expression in C8161, a human melanoma cell line which lacks BRAFV600 or NRAS mutations. PD-L1 was selected because therapies directed at the PD-1-PD-L1 axis have generated clinical success. HLA-I is essential for target recognition by T cells and a central component of anti-tumor immunity. Two large drug libraries, SC200 and L1100, were evaluated. In the SC200 library, only 20 of the 1,120 FDA-approved drugs, chemicals, and natural compounds reduced PD-L1 by at least 10% (Supplementary Fig. S1). Of these, 17 were glucocorticoid or anti-inflammatory drugs that are unfit for immunotherapeutic application. Thus, compounds from the SC200 library were excluded from further studies. However, 38 compounds decreased PD-L1 by at least 20% and 44 compounds increased HLA-I by at least 50% in the 484-compound L1100 library, and 44 candidates were selected for further evaluation (Fig. 1A, representative examples in Fig. 1B–C).
Figure 1. Compounds from high-throughput screening altered PD-L1 and HLA-I.
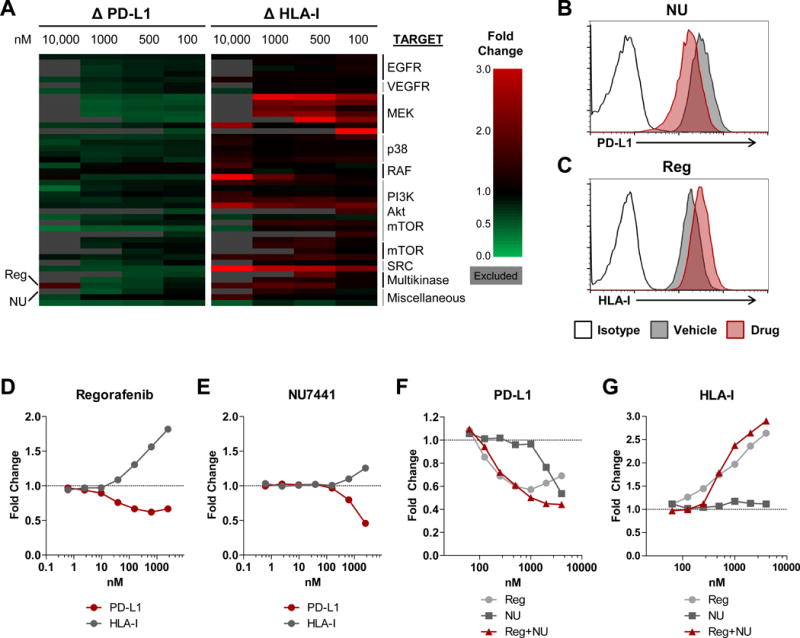
(A) Heat map depicting changes in PD-L1 and HLA-I expression from 44 selected compounds out of 484 compounds in the L1100 library and organized by target. PD-L1 and HLA-I levels were assessed by flow cytometry in C8161 cells treated with compounds at the four indicated concentrations and shown as fold change by comparing MFIs of PD-L1 and HLA-I to vehicle-treated cells. Values were excluded and shaded in gray when cell viability was 50% below vehicle-treated controls. Reg and NU are indicated on the left. (B–C) Representative histograms showing expression of PD-L1 (B) and HLA-I (C) in C8161 cells treated with vehicle, 1 μM NU (B), or 1 μM Reg (C). (D–E) Fold change in PD-L1 and HLA-I in C8161 treated with Reg (D) or NU (E) compared to vehicle-treated cells. (F–G) Fold change in PD-L1 (F) or HLA-I (G) in C8161 treated with Reg, NU, or the combination. HTS was performed once and selected candidates were verified. B–G are representative of three independent measurements.
The candidate inhibitors targeted proteins that act in the MAPK, PI3K/Akt/mTOR, and Src oncogenic signaling pathways, suggesting that these pathways may contribute to the regulation of both PD-L1 and HLA-I. To verify that the candidates were active in different melanomas, their effects on PD-L1 and HLA-I expression were tested in several BRAF- and NRAS-mutant melanomas (Supplementary Fig. S2–S3). In parallel, the effects of the candidate therapeutics on T-cell proliferation were examined (Supplementary Fig. S2–S3, right panels). EGFR inhibitors reduced PD-L1 in only a subset of cell lines and did not alter HLA-I. VEGFR inhibitors decreased PD-L1 by less than 25% and increased HLA-I. p38 inhibitors had no effect. MEK, PI3K, and Src inhibitors, which often reduced PD-L1 and increased HLA-I on melanomas, also potently inhibited T-cell proliferation. A selective BRAFV600 inhibitor, PLX-4720, did not alter PD-L1 or HLA-I. Therefore, despite positive results from HTS, these compounds were not studied further.
Two compounds, regorafenib and NU7441, altered PD-L1 and HLA-I and were chosen for additional evaluation. Regorafenib (Reg) is a multikinase inhibitor with anti-angiogenic properties due to its inhibition of VEGFR and TIE2. However, Reg also blocks the activity of RET, PDGFR, BRAF, and c-KIT, many of which are activated in melanomas (13–16). Reg decreased PD-L1 and induced HLA-I in C8161 (Fig. 1D). NU7441 (NU), a DNA-PK inhibitor that reduced PD-L1 and increased HLA-I expression in a dose-dependent manner was also selected (Fig. 1E). Reg and NU combination treatment further reduced PD-L1 and increased HLA-I at certain concentrations (Fig. 1F–G). Therefore, inhibition of commonly activated kinases and DNA-PK by Reg and NU, respectively, altered PD-L1 and HLA-I at a range of concentrations.
Regorafenib and NU7441 regulate immunologically active molecules
To characterize the effects that Reg and NU had on the “immunophenotype” of melanomas, a flow cytometry-based array was used to examine expression of 332 cell surface proteins on four melanoma cell lines treated with combination Reg (2 μM) and NU (1 μM) (Supplementary Fig. S4A). Sixty-four cell surface markers were expressed by at least three of four cell lines. Of these, several molecules involved in complement regulation, including CD46, CD55, and CD97 were downregulated by Reg and NU (Supplementary Fig. S4B). Other molecules involved in immunosuppressive processes, including CD73 (ecto-5′-nucleotidase, ecto-5′-NT), CD155 (poliovirus receptor, PVR), and CD271 (Nerve Growth Factor Receptor, NGFR) were also downregulated by drug treatment (Supplementary Fig. S4C).
The immunophenotypic effects of Reg (4 μM) and NU (2 μM) were evaluated in a panel of heterogeneous melanoma cell lines (Supplementary Table S1). Twenty-seven melanoma lines were examined including 16 harboring BRAFV600 mutations, 10 with NRAS mutations, and one BRAF and NRAS wild-type line. Reg and NU concentrations used correlated to physiologically achievable plasma concentrations (17–19). This approach allowed for analysis of molecule expression levels and changes induced by Reg and NU in melanomas with different mutations. The effects of a BRAFV600 inhibitor, vemurafenib (Vem, 1 μM), were also examined. BRAF inhibitors have been reported to increase melanoma antigen expression, decrease immunosuppressive cytokine secretion, and increase intratumoral T-cell infiltration in both preclinical and clinical studies (20–23). Therefore, the effects of Vem were compared to Reg and NU. A reduction was defined as a decrease in expression of at least 30% compared to vehicle-treated cells unless otherwise specified. Statistical analyses comparing responses in cell lines with different mutations and responses to different drugs are shown in Supplementary Tables S2–S8.
PD-L1 expression was measured in melanoma cell lines treated with Reg, NU, or Vem. The regulation of PD-L1 is not well characterized, and clinically, few melanomas express consistently high PD-L1 (24). All 27 cell lines expressed PD-L1, with 44.4% (12/27) expressing low PD-L1 (Supplementary Fig. S5A). Reg treatment reduced PD-L1 in 37% (10/27) of the cell lines (Fig. 2A, representative histograms in Supplementary Fig. S5B). NU reduced PD-L1 in 74.1% (20/27) of the lines. Combination Reg plus NU reduced PD-L1 in all but one of the melanomas (96.3%). Moreover, reductions in PD-L1 were augmented with combination treatment (Supplementary Fig. S5C). Responses to Reg and NU treatment were similar between BRAF- and NRAS-mutant cell lines. PD-L1 reductions were greater in NRAS-mutant compared to BRAF-mutant lines, though not significantly (P = 0.07). In line with previous studies, BRAFV600 inhibition with Vem did not consistently affect PD-L1 expression in melanomas (25). BRAF inhibitors can paradoxically activate MAPK signaling in BRAF wild-type cells (26). Accordingly, we observed an upregulation of PD-L1 in Vem-treated NRAS-mutant cell lines. Similar trends were observed for all of the examined molecules. Finally, Reg and NU synergistically or additively reduced PD-L1 in 59.3% (16/27) of cell lines, suggesting synergistic modulation of PD-L1 by the two therapies is common in melanomas (Fig. 2B; examples of synergistic, additive, and antagonistic effects in Supplementary Fig. S6).
Figure 2. Reg and NU altered immunomodulatory molecules PD-L1, CD155, HLA-I, CD73, NGFR, and CD55.
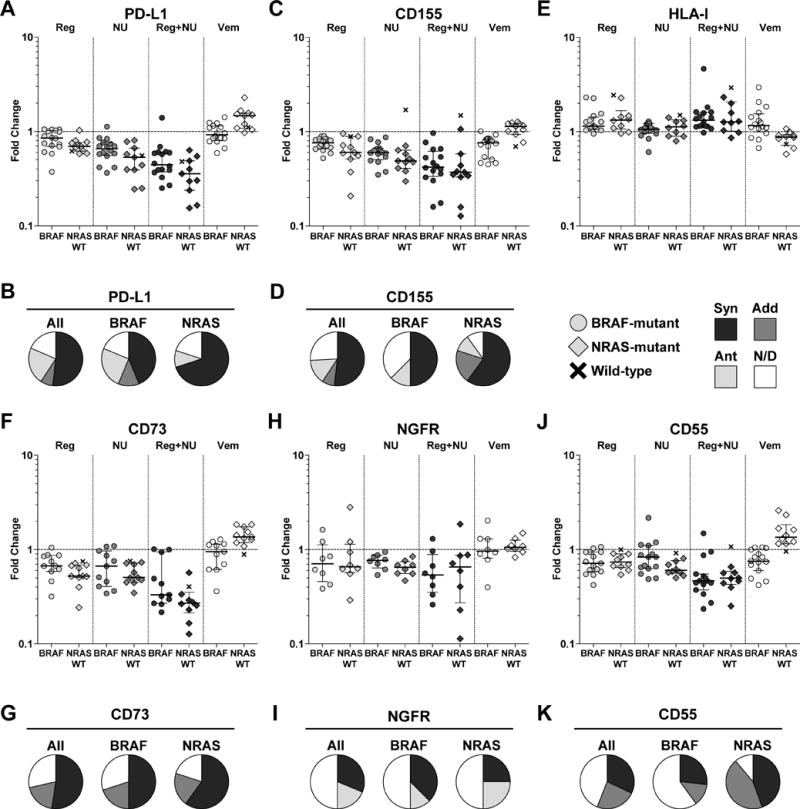
Human melanoma cell lines (n = 27) were treated with Reg (4 μM), NU (2 μM), Vem (1 μM), or a combination of Reg and NU for 48 hours and changes in the expression of PD-L1 (A), CD155 (C), HLA-I (E), CD73 (F), NGFR (H), and CD55 (J) were determined and shown as fold change compared to vehicle-treated cells. Each symbol represents the mean of a technical triplicate. Median and interquartile range are shown. BRAF-mutant cell lines are represented by circles, NRAS-mutant cell lines by diamonds, and BRAF/NRAS wild-type (WT) cell lines by ‘x’, and were separated into groups as indicated. Synergistic (Syn), additive (Add), and antagonistic (Ant) effects of combination Reg and NU treatment were determined for PD-L1 (B), CD155 (D), CD73 (G), NGFR (I), and CD55 (K) in cell lines treated with six concentrations of Reg, NU, and the combination. Cell lines were separated into BRAF- and NRAS-mutant groups as indicated. Synergy was determined using the Chou-Talalay method as described in the materials and methods. Combinatorial effects in some cell lines were unclear and were not determined (N/D). All results are representative of at least two independent measurements. Statistical analyses accompanying A, C, E, F, H, and J are shown in Supplementary Tables S2–S8.
CD155 is one of two major ligands for the checkpoint receptor TIGIT. Like PD-1, TIGIT has been associated with T-cell exhaustion – a phenomenon in which T cells progressively lose functional capacity and eventually undergo cell death (27). CD155 was expressed at high levels in all 27 melanoma lines (Supplementary Fig. S7A). Reg reduced CD155 in roughly half of the melanomas (48.1%, 13/27), whereas NU or combination treatment reduced expression in most melanomas (81.5%, 22/27 and 85.2%, 23/27, respectively) (Fig. 2C, Supplementary Fig. S7B). Despite high constitutive expression, combination treatment led to greater than 70% reductions in 14.8% (4/27) of the lines. Vem reduced CD155 in only 31.3% (5/16) of BRAF-mutant melanomas, but decreases of at least 10% occurred in 87.5% (14/16) of lines. Therefore, although factors regulating CD155 in melanoma are largely unknown, these data suggest that MAPK signaling may regulate CD155 to a limited degree. Like PD-L1, Reg and NU additively or synergistically reduced CD155 in a majority of melanomas (59.3%, 16/27), including similar proportions of BRAF- and NRAS-mutant lines (Fig. 2D).
HLA-I is crucial for target recognition by CD8+ T cells. HLA-I loss has been observed in a variety of malignancies, although complete loss appears rare in melanoma (28). Only one of 27 lines exhibited undetectable levels of HLA-I, and most remaining lines expressed medium or high levels (Supplementary Fig. S8A). Reg increased HLA-I expression by at least 50% in 18.5% (5/27) of melanomas, whereas NU did not increase HLA-I (Fig. 2E, Supplementary Fig. S8B). MAPK inhibition induces HLA-I expression (29). We also observed increased HLA-I expression following Vem treatment, but upregulation of at least 50% was uncommon, occurring in 25% (4/16) of BRAF-mutant lines. Taken together, these data suggest that Reg and NU reduce ligands of immunosuppressive checkpoint receptors while maintaining or increasing HLA-I, and collectively may enhance anti-tumor T-cell immunity.
PD-L1, CD155, and HLA-I alter TCR signaling or co-stimulation in T cells. Beyond these molecules, we also focused on others that modulate anti-melanoma immunity. CD73 catalyzes the conversion of extracellular AMP to adenosine. Adenosine suppresses immune cells, including various effector T cells, and enhances regulatory T-cell (Treg) function (30). CD73 is often overexpressed on tumors, including melanoma (30). CD73 was expressed at relatively low levels in a subset of melanoma cell lines, including 62.5% (10/16) of BRAF-mutant and all NRAS-mutant cell lines (Supplementary Fig. S9A). CD73 was downregulated in 66.7% (14/21) of lines treated with Reg or NU monotherapies (Fig. 2F, Supplementary Fig. S9B). Combination treatment was particularly effective where CD73 was reduced by greater than 50% in 81% (17/21) of lines. Vem downregulated CD73 in 40% (4/10) of the CD73-expressing BRAF-mutant lines. Finally, synergistic or additive combinatorial effects were observed in most lines (71.4%, 15/21) without evidence of antagonism (Fig. 2G).
NGFR is expressed on a variety of cell types and is often upregulated in cancer (31). In melanoma, NGFR expression has been associated with properties of cancer stem cells and immunosuppressive functions (32–35). NGFR expression was detected in 59.3% (16/27) of melanomas (Supplementary Fig. S10A). A higher proportion of NRAS-mutant cell lines expressed NGFR compared to BRAF-mutant lines (80% versus 50%). NGFR expression varied widely in melanomas. Although Reg or NU typically reduced NGFR, Reg also induced NGFR by more than 20% in 25% (4/16) of the cell lines (Fig. 2H, Supplementary Fig. S10B). NGFR was reduced in a synergistic manner by Reg and NU in only 31.3% (5/16) of cell lines (Fig. 2I). Thus, NGFR expression and regulation in melanoma is heterogeneous and complex, but NGFR can often be downregulated by Reg and NU treatment.
CD55, also known as decay accelerating factor (DAF), is expressed on most cells to prevent activation of the complement system and is frequently overexpressed on cancer cells (36). The functions of humoral immunity, cancer-specific antibodies, and the complement system remain unclear in cancer, but evidence for their roles in tumor immunology have increased (36–38). Detectable, but typically low CD55 expression was observed in 92.6% (25/27) of lines (Supplementary Fig. S11A). Reg and NU treatment consistently decreased CD55 expression and did so with equal effectiveness (Fig. 2J, Supplementary Fig. S11B). Vem also reduced CD55 expression in one-third (5/15) of the BRAF-mutant melanomas suggesting a role for the MAPK pathway in CD55 maintenance. Combination therapy resulted in synergistic or additive reduction of CD55 in over half of the melanomas examined (56%, 14/25) (Fig. 2K). Thus, Reg and NU can regulate a variety of molecules with varying immunomodulatory functions.
IFNγ induces PD-L1 and HLA-I expression. Indeed, PD-L1 expression in tumors is often observed at the interface of IFNγ-producing T cells (39). We confirmed that PD-L1 and HLA-I were upregulated by IFNγ treatment (Fig. 3A–B, Supplementary Fig. S5A and S8A). The magnitudes of PD-L1 reduction or HLA-I induction by Reg, NU, or combination treatment were often enhanced in the setting of IFNγ pretreatment. Thus, Reg and NU suppress PD-L1 and increase HLA-I in diverse melanomas and in the setting of IFNγ induction.
Figure 3. Reg and NU decreased PD-L1 and increased HLA-I in both unstimulated and IFNg-stimulated cells while increasing melanoma antigen expression.
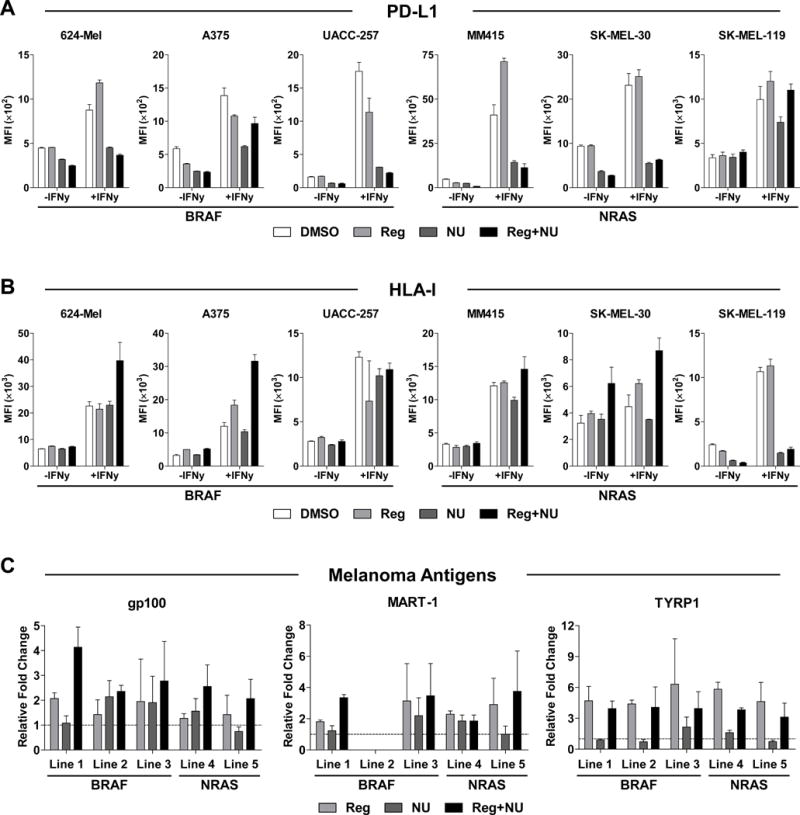
(A–B) Three BRAF-mutant and three NRAS-mutant melanoma cell lines were pretreated with or without 20 U/ml of IFNg for 24 hours and subsequently treated with 4 μM Reg and/or 2 μM NU for 48 hours. The treated cells were assessed for PD-L1 (A) and HLA-I (B) expression and shown as MFI. (C) Melanoma antigen transcript levels were assessed in five melanoma cell lines by qPCR. gp100 (left), MART-1 (middle), and TYRP1 (right) transcripts were measured in cells treated for 48 hours with Reg (2 μM), NU (1 μM) or the combination. Line 1 = 624-Mel, 2 = A375, 3 = Malme-3M, 4 = SK-MEL-2, 5 = SK-MEL-173. BRAF and NRAS mutations are indicated. MART-1 is not expressed in line 2 (A375). All results show mean ± SD from one of two independent measurements.
Regorafenib and NU7441 modulate antigens, proliferation, and signaling pathways
Studies with BRAF and MEK inhibitors have shown that MAPK pathway inhibition can upregulate various melanoma antigens (20,22,23). We examined three melanoma antigens – gp100, MART-1, and TYRP1 – in melanoma cell lines treated with Reg (2 μM) and/or NU (1 μM) by qPCR (Fig. 3C). All three melanoma antigens were upregulated by Reg and combination treatment. Increased protein levels were confirmed for gp100 and Tyrp1 (Supplementary Fig. S12). Antigens were upregulated in both BRAF- and NRAS-mutant cell lines.
We also measured melanoma proliferation after Reg, NU, or Vem treatment (Supplementary Fig. S13). Reg and NU suppressed melanoma proliferation with average 50% inhibitory concentrations around 3 μM. Vem reduced proliferation at lower concentrations in BRAF-mutant lines without affecting NRAS-mutant lines.
To understand the mechanisms underlying these findings, we examined activation of oncogenic signaling pathways in drug-treated melanoma cell lines (Supplementary Fig. S14). MEK1/2 phosphorylation was decreased in all cell lines and Akt phosphorylation was reduced in 63.3% (7/11) of melanomas assessed. Phosphorylation was decreased in both BRAF- and NRAS-mutant lines. Therefore, Reg and NU can induce antigen expression, inhibit proliferation, and reduce oncogenic signaling pathways in heterogeneous melanomas.
Effects of Regorafenib and NU7441 on T-cell proliferation, phenotype, and function
We sought to identify therapies that would enhance the efficacy of cancer immunotherapies. Consequently, the effects of Reg and NU on T-cell proliferation, phenotype, and function were assessed. Activation of T cells from peripheral blood mononuclear cells (PBMC) with CD3 stimulation in the presence of Reg and/or NU reduced T-cell proliferation by a maximum of 25% (Supplementary Fig. S15). In contrast, anti-proliferative effects were observed when T cells were treated with a variety of other targeted therapies (Supplementary Fig. S2–S3).
We also characterized the phenotypic impact Reg and NU had on T cells (Fig. 4A–C, Supplementary Fig. S16). Although CD44 was decreased, several other T-cell activation or co-stimulatory markers including CD25, CD28, and inducible T-cell costimulator (ICOS) were upregulated in CD4+ and CD8+ T cells (Fig. 4A–B). Additionally, co-inhibitory checkpoint receptors, including CD244 (2B4), PD-1, and Tim-3 were reduced on CD4+ and/or CD8+ T cells (Fig. 4C).
Figure 4. T cells treated with Reg and NU express increased activation markers, co-stimulatory molecules, cytokines, and decreased co-inhibitory receptors.
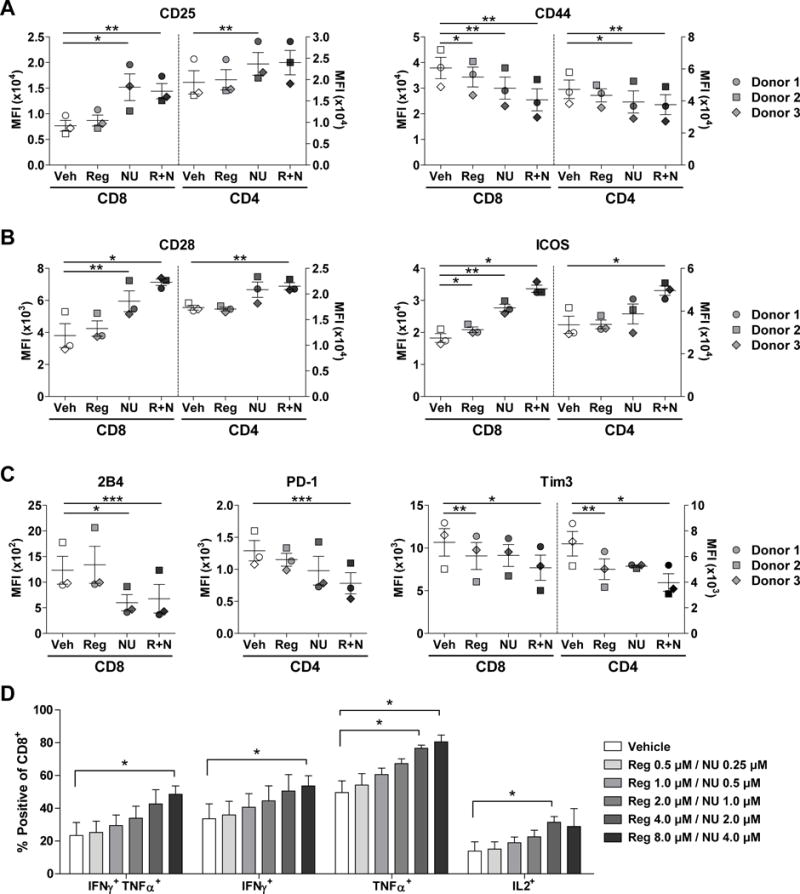
(A–C) Flow cytometry was used to assess expression levels of activation markers (CD25, CD44) (A), co-stimulatory markers (CD28, ICOS) (B), and co-inhibitory receptors (2B4, PD-1, Tim3) (C) in anti-CD3-stimulated PBMCs treated with Reg (2 μM) and/or NU (1 μM) and show either CD4+ or CD8+ populations. A–C show results from three PBMC donors represented by different symbols. ‘R+N’ = Reg plus NU. Mean ± SEM are shown for one of two independent measurements. (D) IFNg, TNFα, and IL2 production in CD8+ T cells from PBMCs stimulated with anti-CD3 and varying concentrations of Reg and NU for 72 hours. Cells were restimulated with PMA and ionomycin for the final six hours. Mean ± SEM are shown for three different independently assessed PBMC donors. Differences for A–D were assessed by paired, two-tailed t tests comparing drug-treated T cells to vehicle-treated T cells. *, P < 0.05, **, P < 0.01, ***, P < 0.001.
As a surrogate for T-cell function, production of IFNγ, TNFα, and IL2 was examined. Cytokine production was increased when T cells were treated with combination Reg and NU after CD3 stimulation with or without PMA and ionomycin reactivation (Fig. 4D, Supplementary Fig. S17–S18). Collectively, these studies suggest that Reg and NU may be suitable for enhancing T-cell cancer immunotherapies.
Regorafenib suppresses melanoma progression and alters the tumor microenvironment
In vitro studies cannot recapitulate the complex and diverse immunosuppressive mechanisms of the tumor microenvironment. Consequently, we examined the efficacy of Reg and NU in the B16 melanoma model with and without various immunotherapies. In vitro, B16 cells treated with Reg and/or NU expressed decreased PD-L1, without changes to CD155 or MHC-I (Supplementary Fig. S19). Mice with established B16 melanomas were treated with Reg, NU7026, or the combination. In vivo Reg doses were lower than those used in murine studies and in humans (11,18). NU7026 is a DNA-PK inhibitor related to NU7441 that was used for animal trials due to improved solubility. No evidence of toxicity was observed throughout animal experiments.
Daily Reg treatment delayed tumor progression and prolonged survival (Fig. 5A–B). NU7026 had no effect alone and did not enhance the efficacy of Reg. Responses in Reg- or combination-treated mice were associated with trends toward increased intratumoral CD45+ leukocytes and CD8+ T cells (Fig. 5C–D). CD8+ T cells were increased within the CD45+ population in animals treated with combination therapy (Fig. 5E). No changes in the levels of intratumoral dendritic cells, macrophages, myeloid-derived suppressor cells, or natural killer cells were observed (Supplementary Fig. S20). B16 tumor cells from treated mice expressed lower CD155 and trends toward increased MHC-I (Supplementary Fig. S21). PD-L1 was increased on B16 cells but decreased on leukocytes in treated mice. However, a variety of potentially immunosuppressive molecules, in vitro and/or in vivo, were altered on B16 (Supplemental Fig. S22). Specifically, semaphorin 4D, CEACAM1, and galectin-9 have immunosuppressive functions and were reduced in Reg-treated B16 cells (39–41). Thus, Reg alters B16 phenotype, suppresses tumor progression, and increases CD8+ T-cell accumulation in the tumor microenvironment.
Figure 5. Reg suppressed melanoma tumor progression and promoted intratumoral CD45+ leukocyte and CD8+ T-cell accumulation.
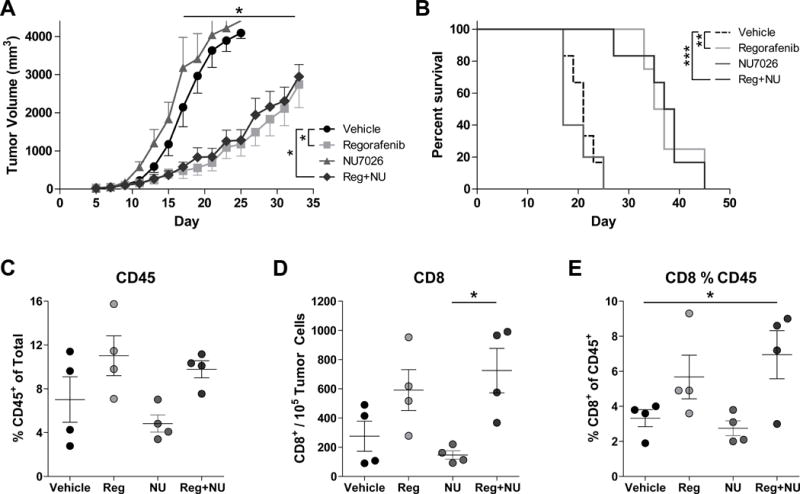
(A–B) B16-F1 tumor-bearing mice were treated with Reg (8 mg/kg) and/or NU7026 (6 mg/kg) and tumor volume (A) and survival (B) were monitored (n = 5–6/group). Differences for tumor volumes were determined on days 15–33 by one-way ANOVA with Tukey post-test and log-rank test for survival comparing vehicle to either Reg or Reg+NU groups. On days 15 and 16, intratumoral CD45+ leukocytes (C) and CD8+ T cells (D–E) were measured. CD8+ T cells were normalized to either tumor cell number (D) or as a percentage of the CD45+ population (E). Differences in C–E were assessed using one-way ANOVA with Tukey post-test. ‘NU’ = NU7026. All results are representative of two independent experiments and show mean ± SEM. *, P < 0.05, **, P < 0.01, ***, P < 0.001.
We next studied how low doses of Reg (4–5 mg/kg) affected the efficacy of two immunotherapeutic regimens. First, agonistic anti-CD40 was used in combination with c-di-GMP, a stimulator of interferon genes (STING) agonist. These therapies promote myeloid maturation and antigen presenting cell function and have shown promise in preclinical studies and clinical trials (42–44). We predicted that enhancing antigen presentation would further augment intratumoral CD8+ T-cell populations as seen with Reg alone. Anti-CD40 with STING agonist treatment resulted in tumor growth inhibition (Fig. 6A). Reg combined with anti-CD40 and STING agonist additively reduced tumor growth and prolonged animal survival (Fig. 6A–B, Supplementary Table S9). Combination therapy also increased intratumoral CD45+ leukocytes, CD4+ T cells, and CD8+ T cells, but did not increase CD4+ T cells within the CD45+ population (Fig. 6C–G). Thus, promoting antigen presentation in mice treated with Reg can reduce tumor progression.
Figure 6. Reg cooperated with anti-CD40 and STING agonist immunotherapies.
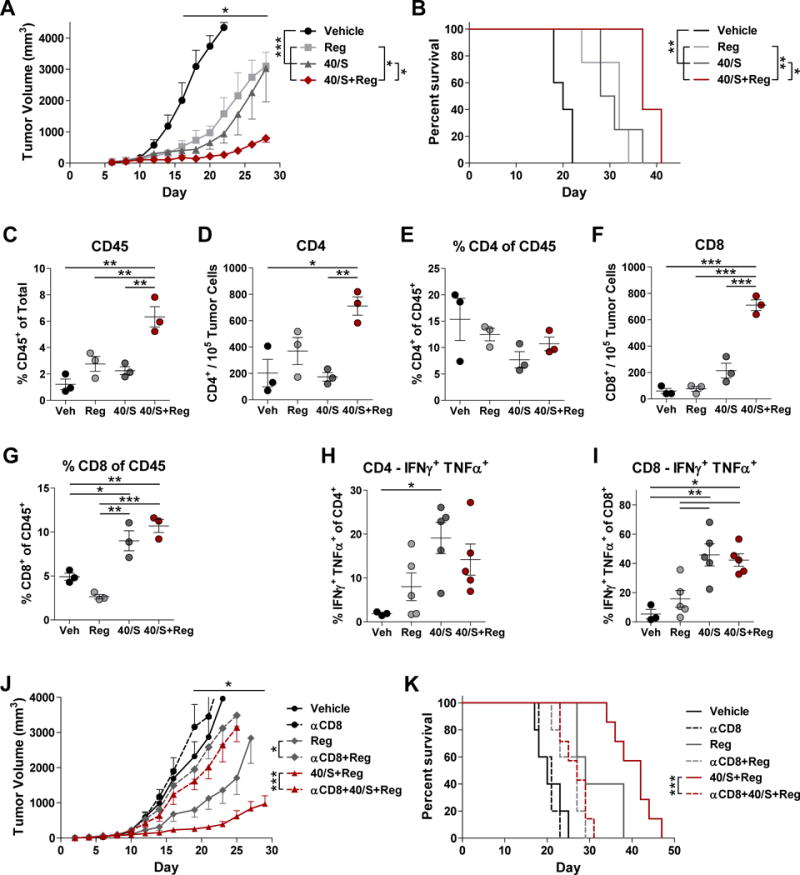
(A–B) B16-F1 tumor-bearing mice were treated with Reg (4 mg/kg) with or without a combination of anti-CD40 (40) and c-di-GMP (STING agonist, S) and tumor volume (A) and survival (B) were monitored (n = 5–6/group). Differences for tumor volumes were determined on days 16–28 by one-way ANOVA with Tukey post-test comparing vehicle to all groups, and comparing 40/S or Reg to 40/S+Reg. All survival comparisons except Reg vs. 40/S were significant according to log-rank tests. On day 21, intratumoral CD45+ leukocytes (C), CD4+ T cells (D–E) and CD8+ T cells (F–G) were measured. Intratumoral CD4+ (H) and CD8+ (I) T cells were stimulated with PMA and ionomycin and evaluated for cytokine production. Differences in C–I were assessed by one-way ANOVA. (J–K) B16-F1 tumor-bearing mice were treated with anti-CD8 to deplete CD8+ T cells, Reg (5 mg/kg), anti-CD40, and c-di-GMP and tumor volume (J) and survival (K) were monitored (n = 5–7/group). Reg and 40/S+Reg groups were significantly different compared to associated anti-CD8-treated mice on days 19–29 by one-way ANOVA with Tukey post-test. Log-rank tests were used to evaluate survival comparisons. A–G are representative of two independent experiments; H–K were performed once. All results show mean ± SEM. *, P < 0.05, **, P < 0.01, ***, P < 0.001.
In order to determine whether Reg efficacy was dependent on increases in CD8+ T cells, studies examining the function of intratumoral T cells were performed alongside CD8+ T-cell depletion experiments. IFNγ, TNFα, and IL2 production were increased in CD4+ and CD8+ T cells of treated mice (Fig. 6H–I, Supplementary Fig. S23). Additionally, CD8+ T-cell depletion resulted in near complete loss of efficacy of Reg alone and when administered with anti-CD40 and STING agonist (Fig. 6J–K). Thus, Reg treatment improves CD8+ T-cell function and these cells are necessary for therapeutic efficacy.
Finally, tumor-bearing mice were treated with Reg in combination with adoptive cell therapy (ACT) using tumor-specific T cells (Fig. 7). Pmel T cells, which recognize the gp10025–33 antigen expressed on B16 cells, were administered in mice treated with a five-day course of Reg. Animals receiving both ACT and Reg exhibited synergistically reduced tumor growth and prolonged survival compared to monotherapies (Fig. 7A–B, Supplementary Table S10). These responses were associated with decreased PD-L1 and CD73 expression on intratumoral leukocytes, and decreased CD155 on both leukocytes and tumor cells (Supplementary Fig. S24). No changes were detected in PD-L1 or MHC-I expression on tumor cells. Similarly to previous findings, the CD45+ population was increased in mice treated with ACT plus Reg compared to mice treated with ACT alone (Fig. 7C). Additionally, anti-tumor responses were associated with increased intratumoral CD4+ and CD8+ T cell and reduced Treg densities (Fig. 7D–H). Reductions in exhaustion marker (Lag-3, Tim3, and PD-1) co-expression on both CD4+ and CD8+ T cells were also observed (Fig. 7I–J).
Figure 7. Reg synergized with adoptive transfer.
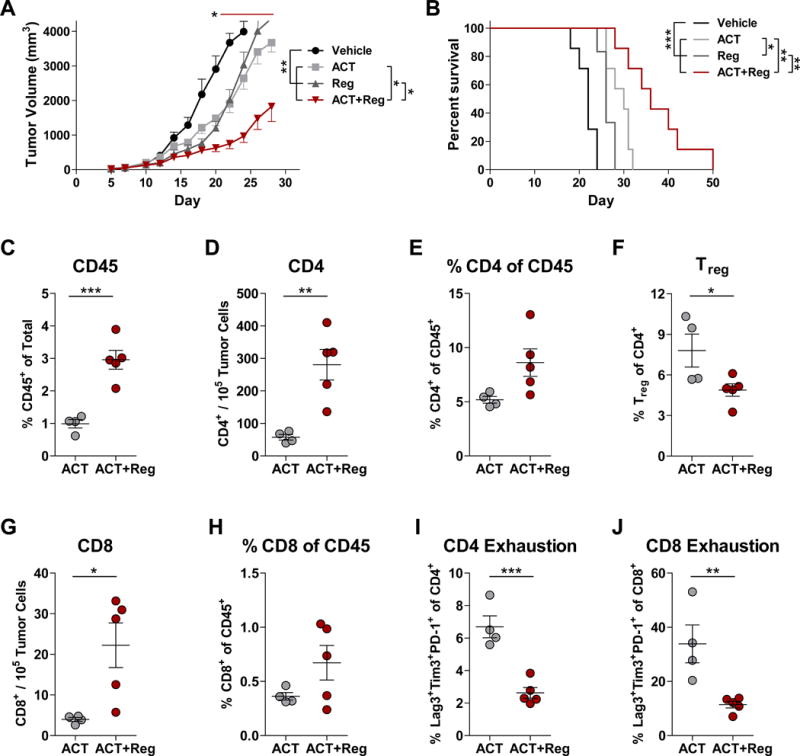
(A–B) B16-F1 tumor-bearing mice were treated with Reg (5 mg/kg) and adoptive transfer of pmel T cells and tumor volume (A) and survival (B) were monitored (n=6–7/group). Significant differences were determined by one-way ANOVA with Tukey post-test for vehicle vs. all groups (asterisk, day 20) and comparisons between ACT or Reg vs. ACT+Reg (red bar, days 20–28). All survival comparisons were significantly different by log-rank tests. On day 17, tumors from mice treated with ACT or ACT+Reg were evaluated for CD45+ leukocytes (C), CD4+ T cells (D–E), Treg (CD45+CD4+CD25HIFoxp3+) (F), and CD8+ T cells (G–H). Phenotypes associated with T-cell exhaustion, as defined by co-expression of Lag-3, Tim3, and PD-1 were also assessed (I–J). Significant differences in C-J were assessed using unpaired, two-tailed t tests. All results are representative of two independent experiments and show mean ± SEM. *, P < 0.05, **, P < 0.01, ***, P < 0.001.
Therefore, Reg cooperates with various immunotherapies to suppress melanoma progression and alter T cells in a manner associated with greater anti-tumor potential. These data serve as a proof-of-concept for using Reg as an immunotherapeutic targeted inhibitor along with additional established immunotherapies.
Discussion
Despite considerable progress, immunotherapeutics only benefit a small subset of melanoma patients. Attempts to address this by combining checkpoint blockade with targeted therapies have thus far proven unsuccessful. One major limitation is that trials have focused on BRAF inhibitors and checkpoint blockade. There remains potential for the identification and development of new combination therapies that rely on the ability for the immune system to eliminate melanoma. The findings herein demonstrate a translational approach to repurpose targeted therapies for use in combination with immunotherapies for melanoma.
Although our primary goal was to identify lead compounds and study their ability to alter a variety of molecules, our drug screening approach also allowed for the examination of PD-L1 regulation. PI3K, VEGFR, MEK, and Src inhibitors led to decreases in PD-L1 expression in a variety of cell lines, suggesting that many pathways may contribute to PD-L1 regulation.
The approach also delineated expression levels of several immunomodulatory molecules in a panel of melanomas. Expression of PD-L1, CD73, NGFR, and CD55 varied, and detectable expression of CD73 and NGFR was more frequently associated with NRAS mutations. These data contribute to gaining a more thorough understanding of the immunobiology of melanoma, and may become more valuable as therapies targeting these molecules are developed.
Animal models demonstrated that Reg is capable of suppressing melanoma growth alone and in combination with emerging immunotherapies. Although Reg reduced B16 PD-L1 expression in vitro, PD-L1 was increased in vivo. This may be a reflection of increased IFNγ derived from infiltrating lymphocytes. Despite this, Reg may still function by immunomodulating B16 tumors, as several potentially immunosuppressive molecules were decreased by Reg treatment. Low doses of Reg also enhanced the efficacy of immunotherapies. As a therapy already approved for the treatment of other cancers, future clinical trials using Reg could be accelerated. It remains difficult to predict whether immunotherapy combinations with Reg would be tolerable. Nevertheless, no evidence of toxicity was observed in animal trials, and Reg is well tolerated in humans (45,46). Moreover, dosing for combination with immunotherapies could be lower compared to standard Reg dosing. Finally, Reg has potential for further development in melanoma patients without BRAF mutations for which targeted therapies are unavailable. Thus, Reg is a candidate for further studies in melanoma with and without additional immunotherapies.
DNA-PK inhibitors failed to elicit changes in animal studies despite promising in vitro findings. One reason for these findings may be related to limited solubility and poor pharmacokinetic properties of DNA-PK inhibitors. NU7441 is poorly absorbed and rapidly metabolized in mice, and these factors have hampered the clinical application of DNA-PK inhibitors (19,47). Yet, targeting DNA-PK remains a promising approach to immunosensitize melanomas. Orally available DNA-PK inhibitors with improved pharmacokinetics are currently in development and clinical trials (48,49).
Combination therapies can improve treatment efficacy in melanoma. Our findings demonstrate that repurposing existing therapies, including regorafenib, in combination with T cell-based immunotherapies, is a viable strategy for controlling melanoma progression.
Supplementary Material
Acknowledgments
Additional Information: This research was supported by the University of Maryland Marlene and Stewart Greenebaum Comprehensive Cancer Center, Melanoma Research Foundation, National Cancer Institute R01CA140917 and T32CA154274, and VA Merit Award BX002142.
Footnotes
Conflicts of Interest: The authors declare no potential conflicts of interest.
References
- 1.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. The New England journal of medicine. 2011;364(26):2507–16. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380(9839):358–65. doi: 10.1016/S0140-6736(12)60868-X. [DOI] [PubMed] [Google Scholar]
- 3.Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. The New England journal of medicine. 2012;367(2):107–14. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 4.Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N Engl J Med. 2014;371(20):1877–88. doi: 10.1056/NEJMoa1406037. [DOI] [PubMed] [Google Scholar]
- 5.Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, et al. Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: a multicentre, double-blind, phase 3 randomised controlled trial. Lancet. 2015;386(9992):444–51. doi: 10.1016/S0140-6736(15)60898-4. [DOI] [PubMed] [Google Scholar]
- 6.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. The New England journal of medicine. 2015;372(4):320–30. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 7.Robert C, Ribas A, Wolchok JD, Hodi FS, Hamid O, Kefford R, et al. Anti-programmed-death-receptor-1 treatment with pembrolizumab in ipilimumab-refractory advanced melanoma: a randomised dose-comparison cohort of a phase 1 trial. Lancet. 2014;384(9948):1109–17. doi: 10.1016/S0140-6736(14)60958-2. [DOI] [PubMed] [Google Scholar]
- 8.Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. The New England journal of medicine. 2013;368(14):1365–6. doi: 10.1056/NEJMc1302338. [DOI] [PubMed] [Google Scholar]
- 9.Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. The Lancet Oncology. 2015;16(4):375–84. doi: 10.1016/S1470-2045(15)70076-8. [DOI] [PubMed] [Google Scholar]
- 10.Ribas A, Butler M, Lutzky J, Lawrence D, Robert C, Miller W, et al. Phase I study combining anti-PD-L1 (MEDI4736) with BRAF (dabrafenib) and/or MEK (trametinib) inhibitors in advanced melanoma. J Clin Oncol. 2015;33(suppl) abstr 3003. [Google Scholar]
- 11.Wilhelm SM, Dumas J, Adnane L, Lynch M, Carter CA, Schutz G, et al. Regorafenib (BAY 73-4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. International journal of cancer. 2011;129(1):245–55. doi: 10.1002/ijc.25864. [DOI] [PubMed] [Google Scholar]
- 12.Leahy JJ, Golding BT, Griffin RJ, Hardcastle IR, Richardson C, Rigoreau L, et al. Identification of a highly potent and selective DNA-dependent protein kinase (DNA-PK) inhibitor (NU7441) by screening of chromenone libraries. Bioorganic & medicinal chemistry letters. 2004;14(24):6083–7. doi: 10.1016/j.bmcl.2004.09.060. [DOI] [PubMed] [Google Scholar]
- 13.Narita N, Tanemura A, Murali R, Scolyer RA, Huang S, Arigami T, et al. Functional RET G691S polymorphism in cutaneous malignant melanoma. Oncogene. 2009;28(34):3058–68. doi: 10.1038/onc.2009.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dai J, Kong Y, Si L, Chi Z, Cui C, Sheng X, et al. Large-scale analysis of PDGFRA mutations in melanomas and evaluation of their sensitivity to tyrosine kinase inhibitors imatinib and crenolanib. Clin Cancer Res. 2013;19(24):6935–42. doi: 10.1158/1078-0432.CCR-13-1266. [DOI] [PubMed] [Google Scholar]
- 15.Omholt K, Grafstrom E, Kanter-Lewensohn L, Hansson J, Ragnarsson-Olding BK. KIT pathway alterations in mucosal melanomas of the vulva and other sites. Clin Cancer Res. 2011;17(12):3933–42. doi: 10.1158/1078-0432.CCR-10-2917. [DOI] [PubMed] [Google Scholar]
- 16.Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006;24(26):4340–6. doi: 10.1200/JCO.2006.06.2984. [DOI] [PubMed] [Google Scholar]
- 17.Zopf D, Fichtner I, Bhargava A, Steinke W, Thierauch KH, Diefenbach K, et al. Pharmacologic activity and pharmacokinetics of metabolites of regorafenib in preclinical models. Cancer Med. 2016;5(11):3176–85. doi: 10.1002/cam4.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strumberg D, Scheulen ME, Schultheis B, Richly H, Frost A, Buchert M, et al. Regorafenib (BAY 73-4506) in advanced colorectal cancer: a phase I study. Br J Cancer. 2012;106(11):1722–7. doi: 10.1038/bjc.2012.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao Y, Thomas HD, Batey MA, Cowell IG, Richardson CJ, Griffin RJ, et al. Preclinical evaluation of a potent novel DNA-dependent protein kinase inhibitor NU7441. Cancer research. 2006;66(10):5354–62. doi: 10.1158/0008-5472.CAN-05-4275. [DOI] [PubMed] [Google Scholar]
- 20.Kono M, Dunn IS, Durda PJ, Butera D, Rose LB, Haggerty TJ, et al. Role of the mitogen-activated protein kinase signaling pathway in the regulation of human melanocytic antigen expression. Molecular cancer research : MCR. 2006;4(10):779–92. doi: 10.1158/1541-7786.MCR-06-0077. [DOI] [PubMed] [Google Scholar]
- 21.Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. The Journal of experimental medicine. 2006;203(7):1651–6. doi: 10.1084/jem.20051848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CN, Sloss CM, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer research. 2010;70(13):5213–9. doi: 10.1158/0008-5472.CAN-10-0118. [DOI] [PubMed] [Google Scholar]
- 23.Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(5):1225–31. doi: 10.1158/1078-0432.CCR-12-1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Science translational medicine. 2012;4(127):127ra37. doi: 10.1126/scitranslmed.3003689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Atefi M, Avramis E, Lassen A, Wong DJ, Robert L, Foulad D, et al. Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20(13):3446–57. doi: 10.1158/1078-0432.CCR-13-2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464(7287):427–30. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wherry EJ. T cell exhaustion. Nature immunology. 2011;12(6):492–9. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- 28.Campoli M, Ferrone S. HLA antigen changes in malignant cells: epigenetic mechanisms and biologic significance. Oncogene. 2008;27(45):5869–85. doi: 10.1038/onc.2008.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sapkota B, Hill CE, Pollack BP. Vemurafenib enhances MHC induction in BRAFV600E homozygous melanoma cells. Oncoimmunology. 2013;2(1):e22890. doi: 10.4161/onci.22890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Antonioli L, Pacher P, Vizi ES, Hasko G. CD39 and CD73 in immunity and inflammation. Trends in molecular medicine. 2013;19(6):355–67. doi: 10.1016/j.molmed.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fanburg-Smith JC, Miettinen M. Low-affinity nerve growth factor receptor (p75) in dermatofibrosarcoma protuberans and other nonneural tumors: a study of 1,150 tumors and fetal and adult normal tissues. Human pathology. 2001;32(9):976–83. doi: 10.1053/hupa.2001.27602. [DOI] [PubMed] [Google Scholar]
- 32.Truzzi F, Marconi A, Lotti R, Dallaglio K, French LE, Hempstead BL, et al. Neurotrophins and their receptors stimulate melanoma cell proliferation and migration. The Journal of investigative dermatology. 2008;128(8):2031–40. doi: 10.1038/jid.2008.21. [DOI] [PubMed] [Google Scholar]
- 33.Boiko AD, Razorenova OV, van de Rijn M, Swetter SM, Johnson DL, Ly DP, et al. Human melanoma-initiating cells express neural crest nerve growth factor receptor CD271. Nature. 2010;466(7302):133–7. doi: 10.1038/nature09161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Redmer T, Welte Y, Behrens D, Fichtner I, Przybilla D, Wruck W, et al. The nerve growth factor receptor CD271 is crucial to maintain tumorigenicity and stem-like properties of melanoma cells. PloS one. 2014;9(5):e92596. doi: 10.1371/journal.pone.0092596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Furuta J, Inozume T, Harada K, Shimada S. CD271 on melanoma cell is an IFN-gamma-inducible immunosuppressive factor that mediates downregulation of melanoma antigens. J Invest Dermatol. 2014;134(5):1369–77. doi: 10.1038/jid.2013.490. [DOI] [PubMed] [Google Scholar]
- 36.Kolev M, Towner L, Donev R. Complement in cancer and cancer immunotherapy. Archivum immunologiae et therapiae experimentalis. 2011;59(6):407–19. doi: 10.1007/s00005-011-0146-x. [DOI] [PubMed] [Google Scholar]
- 37.Vollmers HP, Brandlein S. Natural antibodies and cancer. Journal of autoimmunity. 2007;29(4):295–302. doi: 10.1016/j.jaut.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 38.Affara NI, Ruffell B, Medler TR, Gunderson AJ, Johansson M, Bornstein S, et al. B cells regulate macrophage phenotype and response to chemotherapy in squamous carcinomas. Cancer cell. 2014;25(6):809–21. doi: 10.1016/j.ccr.2014.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nature reviews Cancer. 2012;12(4):252–64. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Evans EE, Jonason AS, Jr, Bussler H, Torno S, Veeraraghavan J, Reilly C, et al. Antibody Blockade of Semaphorin 4D Promotes Immune Infiltration into Tumor and Enhances Response to Other Immunomodulatory Therapies. Cancer immunology research. 2015;3(6):689–701. doi: 10.1158/2326-6066.CIR-14-0171. [DOI] [PubMed] [Google Scholar]
- 41.Gray-Owen SD, Blumberg RS. CEACAM1: contact-dependent control of immunity. Nature reviews Immunology. 2006;6(6):433–46. doi: 10.1038/nri1864. [DOI] [PubMed] [Google Scholar]
- 42.Hassan SB, Sorensen JF, Olsen BN, Pedersen AE. Anti-CD40-mediated cancer immunotherapy: an update of recent and ongoing clinical trials. Immunopharmacology and immunotoxicology. 2014;36(2):96–104. doi: 10.3109/08923973.2014.890626. [DOI] [PubMed] [Google Scholar]
- 43.Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015;11(7):1018–30. doi: 10.1016/j.celrep.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fu J, Kanne DB, Leong M, Glickman LH, McWhirter SM, Lemmens E, et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci Transl Med. 2015;7(283):283ra52. doi: 10.1126/scitranslmed.aaa4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grothey A, Van Cutsem E, Sobrero A, Siena S, Falcone A, Ychou M, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):303–12. doi: 10.1016/S0140-6736(12)61900-X. [DOI] [PubMed] [Google Scholar]
- 46.Demetri GD, Reichardt P, Kang YK, Blay JY, Rutkowski P, Gelderblom H, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295–302. doi: 10.1016/S0140-6736(12)61857-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davidson D, Amrein L, Panasci L, Aloyz R. Small Molecules, Inhibitors of DNA-PK, Targeting DNA Repair, and Beyond. Frontiers in pharmacology. 2013;4:5. doi: 10.3389/fphar.2013.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zenke F, Zimmermann A, Sirrenberg C, Dahmen H, Vassilev L, Pehl U, et al. M3814, a novel investigational DNA-PK inhibitor: enhancing the effect of fractionated radiotherapy leading to complete regression of tumors in mice. Cancer Res. 2016;76(14 Suppl) Abstract nr 1658. [Google Scholar]
- 49.Gavande NS, VanderVere-Carozza PS, Hinshaw HD, Jalal SI, Sears CR, Pawelczak KS, et al. DNA repair targeted therapy: The past or future of cancer treatment? Pharmacology & therapeutics. 2016;160:65–83. doi: 10.1016/j.pharmthera.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.