

Abstract
Idiosyncratic liver toxicity represents an important problem in drug research and pharmacotherapy. Reactive drug metabolites that modify proteins are thought to be a principal factor in drug-induced liver injury. Here we describe a quantitative chemical proteomic method to identify the targets of reactive drug metabolites in vivo. Treating mice with clickable analogues of four representative hepatotoxic drugs, we demonstrate extensive covalent binding that is confined primarily to liver. Each drug exhibited a distinct target profile that, in certain cases, showed strong enrichment for specific metabolic pathways (e.g., lipid/sterol pathways for troglitazone). Site-specific proteomics revealed that acetaminophen reacts with high stoichiometry with several conserved, functional (seleno)cysteine residues throughout the liver proteome. Our findings thus provide an advanced experimental framework to characterize the proteomic reactivity of drug metabolites in vivo, revealing target profiles that may help to explain mechanisms and identify risk factors for drug-induced liver injury.
Graphical Abstract
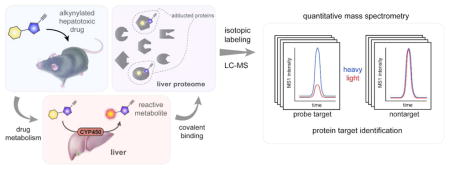
Adverse drug reactions are a common and often dangerous complication that can lead to unpredictable life-threatening injury and drug withdrawal at advanced stages of development1, 2. Some toxicological effects reflect parent drug action at primary targets or off-targets, leading to predictable and monitorable outcomes. A distinct type of high-risk adverse drug effect occurs primarily in the liver, where enzymatic metabolism can convert drugs to chemically reactive metabolites that covalently modify proteins. The chemical stress produced by reactive drug metabolites is considered to be a primary cause of drug-induced liver injury (DILI)3.
DILI is the most common adverse drug reaction and the cause of over half of all cases of acute liver failure in the United States4. Hepatotoxicity can manifest as a reproducible and dose-dependent phenomenon (as with acetaminophen) or can arise idiosyncratically in small numbers of individuals after a drug is used in large populations5. The appearance of idiosyncratic DILI during late clinical testing or post-marketing can lead to the unexpected withdrawal of drugs after significant investment and thus constitutes an impediment to drug development. Despite these serious issues, our understanding of the mechanisms of DILI remains limited6, and there is consequently a need for new methodologies to identify and characterize drugs at risk for reactive metabolism in vivo.
Strong correlative evidence supports that reactive metabolites play an underlying causative role in many cases of DILI7, 8. The reactive metabolites are thought to promote toxicity through the covalent modification and functional perturbation of liver proteins. Covalent protein adduction by reactive metabolites may also promote diverse immunogenic responses by generating drug-derived antigens7, 9. Concerns over the functional perturbation of liver proteins and aberrant promotion of autoimmune responses has motivated the drug discovery industry to adopt a strategy where chemical functionalities with the potential to undergo reactive metabolism are designated as “structural alerts” and avoided in lead optimization, as well as the implementation of in vitro screens for reactive metabolites and covalent drug-protein adducts10. Strict avoidance of drugs with reactive metabolites, however, can place an undue burden on drug research programs and halt the development of useful new medicines. A more nuanced approach that considers parameters such as drug dose, alternative routes of metabolism and clearance, and the risk-benefit related to specific therapeutic indications can guide decisions on advancing drugs with reactive metabolites6. Such judgments would also benefit from a more complete understanding of the proteins targeted by reactive metabolites in vivo and how the adduction of these proteins mechanistically relates to the endpoint of liver toxicity
The identification of protein targets of reactive metabolites commonly involves the use of radiolabeled drugs, which are administered to animal or cell models and drug-adducted proteins then detected by one- or two-dimensional SDS-PAGE and autoradiography. Proteins co-migrating with radioactive signals are then excised from the gel and identified by mass spectrometry (MS) methods11, 12. While this general approach has been employed to identify proteins modified by several drugs that produce DILI13, 14, it suffers from limitations that include poor sensitivity and resolution, resulting in a bias toward detecting high-abundance proteins and ambiguity in assigning the identified proteins as targets of reactive metabolites. These deficiencies could be addressed by incorporating latent affinity handles into drugs of interest to facilitate the enrichment of reactive metabolite-modified proteins over background proteins. We, and others, have adopted this type of chemical proteomic strategy to globally map the protein targets of electrophilic drugs15, environmental chemicals16, and natural products17. In these studies, drugs are modified with simple alkyne or azide groups so that reactive proteins can then be conjugated to reporter tags (fluorophore or biotin) using the copper-catalyzed azide-alkyne cycloaddition (CuAAC) reaction18 or subsequent proteomic profiling by gel- or mass spectrometry (MS)-based methods19. Incorporating isotopic labels into the MS-based proteomic workflow further enables quantification of electrophilic drug-protein reactions with good precision and often site (residue)-specific resolution20.
Here, we have applied quantitative chemical proteomics to globally map the protein targets of several hepatotoxic drugs in mice. Alkyne analogues of each drug were synthesized and found to react with distinct sets of liver proteins in vivo, indicating that reactive drug metabolites retain a substantial recognition component that influences their protein interactions. Some drugs showed a strong preference for targeting specific biochemical pathways, and, in the case of acetaminophen, we provide evidence for high-occupancy modification of functional (seleno)cysteine residues in diverse liver proteins. Our findings, taken together, thus highlight the value of in-depth mapping of protein targets of reactive drug metabolites in vivo to identify new risk factors and possible mechanistic underpinnings for DILI.
Results
Clickable probes for in vivo profiling of reactive metabolites
As case studies for examining the in vivo proteomic reactivity of hepatotoxic compounds, we selected three drugs known to cause idiosyncratic DILI – troglitazone (1), clozapine (2), tienilic acid (3) – as well as the acute hepatotoxin acetaminophen (4) Figure 1A). Each of these drugs undergoes metabolic activation, mostly mediated by cytochrome P450 (CYP) enzymes, to produce reactive metabolites that covalently bind to proteins21–24. We designed and synthesized alkynylated analogues of each drug where the installed alkynes were positioned to avoid interference with established pathways for metabolic activation (Figure 1A and Figure S1A). Metabolite identification and metabolic stability experiments in the S9 fraction of mouse liver tissue confirmed that the parent drugs 1–4 and their respective alkynylated probes 5–8 form analogous metabolites, including glutathione (GSH) adducts of these metabolites25, and display similar overall metabolic stability (Figure S1B and Table S1).
Figure 1.

Structures and initial profiling of the reactivity of chemical proteomic probes for hepatoxic drugs. (A) Structures of the parent hepatotoxic drugs (1–4) and their corresponding alkyne-modified (clickable) probes (5–8). The alkyne functionality was incorporated at sites intended to minimize interference with the known routes of metabolism of the drugs; see Figure S1 for more details. (B) In vitro reactivity profiles for probes 5–8 in mouse liver lysates with or without NADPH. Freshly prepared mouse liver proteome (2 mg/mL) was treated with DMSO or indicated probes (0.1–50 μM, 2 h, room temperature), and protein reactivity events analyzed by CuAAC with an azide-rhodamine tag followed by SDS-PAGE and in-gel fluorescence scanning (fluorescent gels shown in grayscale). See Figure S2A for a lower intensity image of the gel. (C) Schematic for chemical proteomic workflow used to profile the in vivo reactivity of probes 5–8. Following treatment with vehicle or probes (i.p., 2 h), mice were sacrificed and liver proteomes prepared and analyzed for protein reactivity events as describes in part B. (D) Concentration-dependent reactivity of mouse liver soluble (left) and membrane (right) proteomes following treatment of mice with vehicle or probes 5–8 (i.p., 2 h) at the indicated doses. The dosing range for probe 8 was extended into the acute hepatoxic range for acetaminophen (300 mg/kg). (E) Proteomic reactivity of five tissues from mice treated with vehicle or probes 5–8 (i.p., 90 mg/kg, 2 h). Data shown are for soluble proteomes; see Figure S2B for membrane proteome reactivity data. (F) Proteomic reactivity of liver from mice treated for the indicated number of days with vehicle or probes 5–7 (30 mg/kg, i.p., once daily). Data shown are for membrane proteomes; see Figure S2C for soluble proteome reactivity data.
We initially evaluated the in vitro proteomic reactivity of 5–8 by exposing the probes (0.1 – 50 μM) to mouse liver homogenates in the presence or absence of NADPH (a cofactor for CYP enzymes) for 2 h, followed by CuAAC coupling to an azide-rhodamine reporter tag, SDS-PAGE, and in-gel fluorescence scanning. Each probe displayed a distinct concentration-dependent proteomic reactivity profile that was much stronger in the presence of NADPH (Figures 1B and S2A), consistent with metabolic activation being mediated, as expected, by CYP enzymes. We next examined the in vivo proteomic reactivity of 5–8 by treating mice with the probes (10–90 mg/kg, i.p., with probe 8 also being evaluated at 200 and 300 mg/kg) for 2 h, followed by isolation of liver tissue and gel-based analysis of probe-modified proteins (Figure 1C). Each probe produced substantial, dose-dependent protein reactivity in mouse liver, and, as was observed in vitro, the profiles of modified proteins differed for each probe in vivo (Figure 1D). The overall extent of proteomic reactivity also differed markedly for probes in vitro versus in vivo. The troglitazone (5) and tienilic acid (7) probes, for instance, showed strong protein reactivity in liver homogenates (Figure 1B), but more tempered protein labeling profiles in vivo (Figure 1D). In contrast, the acetaminophen probe (8) exhibited extensive protein labeling in vivo (Figure 1D), but limited in vitro reactivity in liver homogenates (Figure 1B). Probes 5–8 showed very limited in vivo proteomic reactivity in other mouse tissues (Figures 1E and S2B), consistent with the predominant expression of CYPs in liver and the corresponding CYP-generated metabolites being restricted in reactivity to proteins in this tissue. One exception was a probe 5-labeled 70 kDa protein that appeared in every tissue, which we later confirmed by MS analysis to be serum albumin (see below), an abundant protein that is produced in liver and then secreted into blood, likely accounting for its detection in other tissues.
We also determined if probes 5–7 showed cumulative protein reactivity in liver following multi-day dosing in mice. Mice were treated with probes or vehicle once-per-day (30 mg/kg, i.p.) for one, three, or seven days and then sacrificed and liver proteomes analyzed for protein reactivity by gel-based analysis. Each probe showed evidence of cumulative protein labeling over the seven-day treatment period, with particularly striking increases in liver protein reactivity being observed for probes 5 and 6 (Figures 1F and S2C).
Having found by gel-based profiling that clickable analogues of hepatoxic drugs provide probes for visualizing reactive metabolite-protein interactions in liver both in vitro and in vivo, we next focused on identifying the probe-modified proteins using quantitative MS methods.
Identification of protein targets of reactive metabolites
To identify proteins modified by probes 5–8 in vivo, we treated pairs of mice with a probe or the corresponding parent drug under acute (8 or 4, 200 mg/kg, 2 h) or subchronic (5–7 or 1–3, 30 mg/kg, 7 days, q.d) dosing conditions. Comparator mice were treated with parent drug (rather than vehicle) to control for potential drug-induced gene expression in liver tissue. Probe-modified proteins were then identified from liver tissue of mice by CuAAC coupling to an azide-biotin tag, streptavidin enrichment, and quantitative LC-MS using amine reductive-dimethylation (ReDiMe) chemistry26 to incorporate isotopically ‘heavy’ or ‘light’ methyl groups into the tryptic peptides of proteins from probe- versus control-treated mice, respectively (Figure 2A). Targets of each probe were defined as proteins showing four-fold or higher ReDiMe ratios (or R values) in probe- vs parent drug-treated mice (Figure 2B, Figure S3A–C and Tables S2 and S3; n = 2/group). Under the tested conditions, and generally consistent with the gel-based profiles of protein labeling events in mouse liver (Figure 1D, F), probes 5, 6, and 8 exhibited similarly high proteomic reactivity (~50–80 targets each), while probe 7 displayed fewer protein modification events (9 targets) (Figure 2C and Table S2).
Figure 2.

Quantitative MS-based characterization of the in vivo proteome reactivity of hepatotoxic drugs. (A) Schematic of the workflow for quantitative MS-based proteomic experiments to identify mouse liver proteins modified by probes 5–8 in vivo. Mice were treated with the probes 5–8 or the corresponding parent drugs 1–4 using acute (8 or 4, 200 mg/kg, 2 h) or subchronic (5–7 or 1–3, 30 mg/kg, 7 days, q.d) dosing conditions. Following sacrifice of animals, liver proteomes were prepared and probe-modified proteins conjugated by CuAAC to an azide-biotin tag, enriched using streptavidin, digested on-bead with trypsin, and the resulting tryptic peptides isotopically labeled at N-terminal and lysine ε-amino groups by reductive dimethylation (ReDiMe) with “heavy” (D2C13O, probe-treated sample) or “light” (H2CO, control sample) formaldehyde. LC-MS/MS analysis of the combined heavy and light samples enabled identification and quantification of proteins based on MS2 and MS1 signals, respectively. (B) Representative ReDiMe ratio plot for proteins identified from mice treated with probe 8 (heavy) versus drug 4 (light, control). Proteins with heavy/light ratios (or R values) ≥ 4 (dashed red line) were considered covalent targets of probe 8. See Figure S3 for the corresponding ReDiMe ratio plots for probes 5–7. (C) Bar graph depicting the number of protein targets for the indicated probes in mouse liver. (D) Left bar, stacked bar graph showing the aggregate number of protein targets of probes 5–8 and the fraction of these targets enriched by one or more probes. Right bar, fraction of protein targets of probes 5–8 representing proteins previously identified as targets of chemically reactive metabolites in mice, as assessed by presence in the Target Protein Database (TPDB; http://targetprotein.res.ku.edu). (E) Stacked bar graph depicting the shared and unique protein targets of probes 5–8 in mouse liver. (F) Bar graph comparing the percentage of proteins containing an annotated functional cysteine residue in the Uniprot Knowledgebase for all mouse proteins (3%) versus proteins identified as targets of probes 5–8 (15%). (G) Stacked bar depicting the percentage of protein targets of probe 5–8 for which orthologous human proteins were identified (combined red and blue sections) and found to be targets of cysteine-directed fragment electrophiles (blue section) in a recent chemical proteomic study28. (H) Bar graph depicting the percentage of protein targets of probes 5, 6, and 8 that are involved in the indicated primary metabolic processes, according to the PANTHER classification system (http://pantherdb.org). The protein targets were first classified by biological process (Figure S4A), which revealed a major subset of targets (~60% for all combined probes) classified as metabolic. These metabolic targets were further categorized by metabolic process, and the majority (~70% for all combined probes) were listed as primary metabolic (Figure S4B). The data in panel H represent further classification of these metabolic targets into subcategories of primary metabolic processes.
We next examined the overlap across the target profiles of the tested hepatotoxic drugs. Here, we took caution to minimize the potential for stochastic protein detection by LC-MS to alter our estimates of unique vs overlapping targets for probes 5–8. Accordingly, we required that a protein was quantified in data sets from at least three of the four tested probes, or was strongly and consistently enriched by a single probe (≥ 10 total quantified peptides with detection in both replicate experiments), to be part of the target overlap analysis. Of the 152 aggregate targets of probes 5–8, 126 met this requirement, and, notably, 70% (91) of these proteins were enriched by only a single probe, with the remaining ~30% of proteins being mostly enriched by two probes (30 proteins) along with a handful (5 proteins) enriched by three probes (Figure 2D). Only a handful of the 152 targets of probes 5–8 are found in a reactive metabolite target protein database (TPDB; http://targetprotein.res.ku.edu/)27 (Figure 2D), which has mostly been assembled from studies performed with radiolabeled drugs. A deeper analysis of target overlap revealed that probes 6 and 8 shared many targets, while the target profile of probe 5 consisted mostly of unique targets (Figure 2E), including serum albumin (Figure S3), matching the ~70 kDa probe 5-labeled protein detected in most tissues (Figure 1E). Even though probe 7 only enriched nine targets, some of these proteins were unique to this probe (Figure 2E).
Functional classification of protein targets of reactive metabolites
A unifying theme for the targets across all reactive metabolite probes was an enrichment for proteins containing functional (seleno)cysteine residues, including those annotated in the UniprotKB as active-site, redox-active disulfide, or post-translationally modified (Figure 2F). A substantial number of the probe targets also showed evidence of covalent ligandability in a recent proteome-wide assessment of cysteine-directed fragment electrophiles using competitive profiling with a broad-spectrum iodoacetamide (IA)-alkyne probe28 (Figure 2G). These findings are consistent with previous work demonstrating that i) reactive metabolites of drugs, including several of the drugs tested herein, have a propensity to covalently modify cysteine residues23, 24, 29; and ii) functional cysteines have, in general, higher intrinsic reactivity and consequently greater potential for covalent ligand modification compared to other cysteines in the proteome28, 30. We also note that the targets of reactive metabolite probes included several proteins with conserved, functional selenocysteine residues; for instance, six selenoproteins (TXNRD1, VIMP, SEPHS2, SELT, TXNRD3, and SELO) were enriched by probe 8, including thioredoxin reductase 1 (TXNRD1), which was recently described as a covalent target of reactive metabolites of acetaminophen31.
We next asked whether the targets of reactive metabolite probes showed enrichment for specific biological processes using the PANTHER classification system32, which revealed that the vast majority of probe targets were involved in primary metabolic processes (Figure S4A, B). This output may reflect enrichment for cysteine-dependent proteins within metabolic processes or simply attributed to the high metabolic composition of the liver proteome. Further inspection revealed that individual probes enriched proteins from different primary metabolic pathways. Probe 5, for instance, showed a striking preference for proteins involved in lipid or steroid metabolism, while probes 6 and 8 exhibited greater enrichment of proteins involved in amino acid and protein metabolism (Figure 2H). These results suggest that the features of troglitazone that promote interactions with its primary targets – the lipid-activated nuclear receptors PPARs – may also endow the metabolic products of this drug with preferential binding to and reactivity with other lipid-interacting proteins.
We finally asked if the chemical proteomic analysis of reactive metabolite targets could be extended to freshly isolated human hepatocytes using probe 5 as a case study. Primary human hepatocytes were treated in situ with probe 5 (20 μM, 4 h) or compound 1, followed by CuAAC coupling to an azide-Rh tag, SDS-PAGE, and in-gel fluorescence scanning, which revealed extensive proteomic reactivity for probe 5 (Figure 3A). We followed up this gel-based study with a quantitative LC-MS (RediMe) analysis, which identified > 200 protein targets of probe 5 (R value ≥ 4) in human hepatocytes (Figure 3B). The number of proteins targets identified for probe 5 in suspended primary human hepatocytes was substantially larger than that found in liver from mice treated with this probe (> 200 vs ~50), which could reflect that the concentration of probe used in the human hepatocyte treatments (20 μM) exceeded the probe exposure in vivo or that the metabolic activation of probe 5 was more efficient in human hepatocytes. Regardless, we observed good consistency in targets identified in both systems, as orthologues and homologues of many mouse protein targets were enriched by probe 5 in human hepatocytes (Figure 3C). Also, as was observed in mouse liver tissue, the targets of probe 5 in human hepatocytes showed substantial enrichment for proteins involved in in lipid and steroid metabolism (Figures 3D and S4C, D).
Figure 3.

Characterization of the proteomic reactivity of the troglitazone probe 5 in human hepatocytes. (A) Analysis of the membrane and soluble proteomic reactivity of primary human hepatocytes treated with vehicle (DMSO) or probe 5 (20 μM, 2 h). (B) A ReDiMe ratio plot for proteins enriched and identified from human hepatocytes treated with probe 5 (heavy) versus DMSO control (light) corresponding probe/control (heavy/light isotopic) ratios. Proteins with R values ≥ 4 (dashed red line) were considered covalent targets of probe 5. (C) Venn diagram demonstrating the overlap of orthologous or closely related proteins found to be targets of probe 5 in both mouse liver and human hepatocytes. (D) Bar graph depicting the percentage of protein targets of probes 5, 6, and 8 from human hepatocytes that are involved in the indicated primary metabolic processes, according to the PANTHER classification system. The protein targets were first classified by biological process, which revealed a major subset (~65%) classified as metabolic (Figure S4C). Further categorization indicated that 75% of the metabolic targets were involved in primary metabolic processes (Figure S4D). The data in panel D represents further classification of these metabolic targets into sub-categories of primary metabolic processes.
Assessing the stoichiometry of reactive metabolite-protein interactions in vivo
While covalent protein binding may contribute to drug toxicity by creating immunogenic adducts, it is also possible that reactive metabolites perturb the specific functions of individual proteins, especially if metabolite reactions occur at high stoichiometry with these proteins. We therefore sought to complement our chemical proteomic analysis of proteins modified by reactive metabolites with determination of the fractional engagement of these modifications events in vivo. As a case study, we evaluated the hepatotoxin, acetaminophen (4).
Our original enrichment experiments identified 82 targets of the acetaminophen probe 8 in mouse liver (Figure 2C). We next assessed the fractional engagement of these events by treating mice with a dose range of 4 (200–600 mg/kg, 2 h) followed by probe 8 (100 mg/kg, 1 h). Gel-based analysis of these “competition” profiling experiments revealed that 4 produced a concentration-dependent blockade of probe 8-labeling for a subset of proteins in the liver proteome (Figure 4A). We identified these 4-competed proteins by quantitative MS-based proteomics of liver tissue from mice treated with 4 (500 mg/kg, 2 h) or vehicle followed by probe 8 (100 mg/kg, 1 h) (Figure 4B, where high-occupancy targets of 4 were defined as proteins showing R values > 3 for vehicle/4 comparisons). The high-engagement protein targets of 4 included several selenoproteins and proteins that use conserved cysteine residues for catalysis (Figure 4B).
Figure 4.

Characterization of high-occupancy protein reactivity events for acetaminophen (4) in mouse liver. (A) Gel-based profile of competitive blockade of probe 8 reactivity with mouse liver proteins by acetaminophen (4). Mice were treated with vehicle or 4 at the indicated dose (2 h, i.p.), followed by treatment with the probe 8 (100 mg/kg, 1 h, i.p.). Animals were then sacrificed and liver soluble and membrane protein fractions conjugated to rhodamine-azide by CuAAC and analyzed by SDS-PAGE and in-gel fluorescence scanning. Arrows mark proteins for which probe 8 reactivity was competed by pretreatment with 4. (B) A ReDiMe ratio plot for proteins identified from liver tissue of mice treated with 4 (500 mg/kg, 2 h, i.p.) (light) or vehicle (heavy), followed by probe 8 (100 mg/kg, 1 h, i.p.). Proteins with R values ≥ 3 (dashed red line) were considered to be competitively blocked in reactivity with probe 8 and designated as high-occupancy targets of 4 (shown in the magnified region on the right). High-occupancy targets containing catalytic cysteine or selenocysteine residues are shown in red and blue, respectively.
The acetaminophen isomer N-acetyl-m-aminophenol (AMAP, 9; Figure 5A) does not show hepatotoxicity in mice, even though both 4 and 9 have been reported to exhibit similar overall covalent protein binding in rodent liver as assessed by radiolabeled protein binding assays33. We first compared the protein reactivity profiles of liver proteomes from mice treated with 8 or 1025 (an alkyne probe analogue of 9; Figure 5A) (10–300 mg/kg, 2 h), which revealed that the magnitude of covalent reactivity of probe 8 greatly exceeded that of probe 10 (Figure 5B). Because previous studies have indicated that the protein adducts formed by 9 may be less stable under conditions of gel electrophoresis compared to the protein adducts of 433, we next assessed the fractional engagement of 8-labeled liver proteins by 9 in vivo (400 mg/kg, 2 h) using quantitative (ReDiMe) MS-based proteomics, which revealed that 9 shared several high-occupancy targets with 4 (Table S2). We then directly compared 4 versus 9 for their respective competitive blockade of protein labeling by 8 in vivo. In these experiments, mice were treated with 4 or 9 (400 mg/kg, 2 h) followed by 8 (100 mg/kg, 1 h) and their liver tissues compared by quantitative (ReDiMe) MS-based proteomics. Plotting the R values (Log2) for 9-treated/4-treated mice (where 0 = no differences; > 1 = preferentially competed by 4; < 1 = preferentially competed by 9) revealed that fourteen proteins were preferentially competed by 4, while only two targets were better competed by 9 (Figure 5C). Of the fourteen proteins preferentially targeted by 4, six were selenoproteins (Figure 5C).
Figure 5.

Comparative proteomic reactivity profiling of acetaminophen and its less toxic analogue AMAP. (A) Structures of AMAP (9) and the alkyne-modified probe analogue 10. (B) Comparative gel-based analysis of the mouse liver proteomic reactivity of probes 8 and 10 in vivo. Mice were treated with the indicated probe and dose (10–300 mg/kg, 2 h, i.p.), sacrificed, and liver soluble and membrane protein fractions conjugated to rhodamine-azide by CuAAC and analyzed by SDS-PAGE and in-gel fluorescence scanning. (C) A ReDiMe ratio plot for proteins identified from liver tissue of mice treated wwith 4 or 9 (400 mg/kg, 2 h, i.p.), followed by 8 (100 mg/kg, 1 h, i.p.). Dashed red lines designated proteins with log2 R values > 1 (preferentially competed by 4) or < 1 (preferentially competed by 9). Six of the fourteen proteins preferentially competed by 4 were selenocysteine-containing proteins (shown in blue).
These data, taken together, indicate that acetaminophen (4) modifies with high engagement a select subset of liver proteins, many of which have conserved, functional (seleno)cysteines and are more substantially modified by 4 over its less toxic analogue 9.
Quantitative profiling of (seleno)cysteine residues adducted by acetaminophen
The prevalence of high-engagement targets of 4 with conserved, functional (seleno)cysteine residues motivated us to examine whether these and possibly other functional (seleno)cysteine residues were directly modified by 4 in vivo. In this regard, cysteine and, more recently, selenocysteine residues31, due to their intrinsic nucleophilicity, are considered major sites for modification by reactive metabolites. We used a quantitative chemical proteomic method termed competitive isoTOP-ABPP28, 34 to compare the reactivity of (seleno)cysteine residues in liver proteomes from mice treated with 4 (500 mg/kg, 2 h) or vehicle (Figure S5). Two variants of the isoTOP-ABPP protocol were employed – 1) the established method, where liver proteomic lysates from 4-treated mice was exposed to an IA-alkyne probe; and 2) an in vivo profiling variant, where 4-treated mice were administered 8 (100 mg/kg, 1 h). Combining data from both experiments, we found that treatment of mice with 4 produced a substantial reduction in reactivity (R value ≥ 3 for vehicle/4 comparisons) for 111 (seleno)cysteine residues in mouse liver (Figures S6A). Several of these residues were functional (seleno)cysteines (Table 1 and Figure S6B), a subset of which also corresponded to the high-engagement targets of 4 identified in whole protein enrichment experiments performed with probe 8 (e.g., Selt, Selo, Txnrd1), while others were functional (seleno)cysteines in mechanistically related proteins that were either not detected or not competed by 4 in probe 8 experiments (e.g., additional selenoproteins, aldehyde dehydrogenases (ALDHs)) (compare Table 1 and Figure 4B). The instances where 4-competed (seleno)cysteine residues were found on proteins that were not themselves identified as high-engagement targets of 4 in whole protein profiling experiments with probe 8 (see entries 11 – 32 in Table 1) may reflect cases where the isoTOP-ABPP method exhibited superior sensitivity compared to whole protein enrichment experiments or where multiple sites of 8-reactivity occurred on the same protein, only one of which was substantially competed by 4. Conversely, the failure to detect 4-competed (seleno)cysteines for some of the high-occupancy proteins of 4 denoted in Figure 4B may reflect cases where the tryptic peptides containing 4-competed (seleno)cysteines were not detected by LC-MS, or where other non-cysteine adducts were formed on proteins by reactive metabolites of 4. More curiously, however, were rare, but clear examples of proteins identified as high-engagement targets of 4 in competition experiments with probe 8, as determined by either whole protein capture or isoTOP-ABPP, where the corresponding functional cysteine was observed, but not competed by 4 in isoTOP-ABPP experiments performed with the IA-alkyne probe (e.g., C32 of GSTO1, Table 1). This result may indicate that some of the adducts formed by 4 with functional cysteines are unstable such that they are lost during the processing of liver tissue prior to in vitro analysis by isoTOP-ABPP.
Table 1.
High-occupancy protein targets of acetaminophen 4 in mouse liver as determined by quantitative MS-based proteomics.
| Entry | Uniprot ID | Gene name | probe 8 enriched, whole protein | 4 competed, whole protein analysis | 4 competed, peptide analysis | Competed residue(s), peptide |
|---|---|---|---|---|---|---|
| 1 | Q9CXT8 | Pmpcb | 20 | 20 | 20 | C248* |
| 2 | Q9D172 | D10Jhu81e | 20 | 19.8 | 20 | C174# |
| 3 | P17751 | Tpi1 | 20 | 10.8 | 13*, 20# | C71*# |
| 4 | Q9DBC0 | Selo^ | 10 | 20 | 18*, 20# | U665*# |
| 5 | O09131 | Gsto1^ | 20 | 8.0 | 1.2*, 20# | C32*# |
| 6 | Q9JMH6 | Txnrd1^ | 20 | 20 | 5.5*, 6.5# | U612*# |
| 7 | P62342 | Selt^ | 20 | 20 | 4.3 | U49* |
| 8 | O09172 | Gclm | 12 | 4.9 | 3.8 | C35* |
| 9 | Q9CRB3 | Urah | 20 | 3.7 | 2.8*, 6.4# | C35*# |
| 10 | Q8BRK8 | Prkaa2 | 5.7 | 3.1 | 4.0 | C413* |
| 11 | Q791V5 | Mtch2 | 6.6 | NC | 3.4, 5.5 | C296*, C56* |
| 12 | Q8VHE0 | Sec63 | 8.8 | NC | 7.5*, 20# | C295*# |
| 13 | Q8BUV3 | Gphn | 9.2 | NC | 3.9*, 14# | C452*# |
| 14 | Q8VCF0 | Mavs | 20 | NC | 4.8 | C470* |
| 15 | Q99LX0 | Park7^ | 11 | NC | 4.4 | C106* |
| 16 | Q9DBA6 | Tysnd1 | 5.6 | NC | 3.6 | C106/C110* |
| 17 | E9Q3C1 | C2cd2 | 20 | NC | 3.6 | C408* |
| 18 | Q9WV55 | Vapa | 4.3 | NC | 3.5 | C60* |
| 19 | O08573 | Lgals9 | 4.8 | NC | 3.4 | C138* |
| 20 | P42227 | Stat3^ | 5.8 | NC | 3.2 | C259* |
| 21 | Q922Q1 | Marc2 | 4.4 | NC | 2.4*, 4.0# | C74*# |
| 22 | O70252 | Hmox2^ | 20 | ND | 4.4 | C281* |
| 23 | Q9JLJ1 | Selk^ | ND | ND | 7.9 | U497* |
| 24 | Q8VC19 | Alas1^ | ND | ND | 5.6 | C108* |
| 25 | O08997 | Atox1^ | ND | ND | 4.8 | C12/C15* |
| 26 | P97821 | Ctsc^ | ND | ND | 3.4 | C254/C257* |
| 27 | Q9JLC3 | Msrb1^ | ND | ND | 3.4 | U95* |
| 28 | G5E860 | Usp16^ | ND | ND | 3.3 | C204* |
| 29 | Q8BH00 | Aldh8a1^ | 1.9 | 1.5 | 11*, 12# | C287/289*# |
| 30 | Q9JLT4 | Txnrd2^ | 1.8 | 1.6 | 3.5*, 4# | U497*# |
| 31 | Q9JLJ2 | Aldh9a1^ | 1.7 | 1 | 3.9 | C288/289* |
| 32 | P47738 | Aldh2^ | 1.5 | 0.9 | 3.8*, 3.5# | C320-322*# |
| 33 | Q9BCZ4 | Vimp^ | 20 | 20 | ND | |
| 34 | Q9ESW8 | Pgpep1^ | 20 | 20 | ND | |
| 35 | Q8CDN6 | Txnl1^ | 20 | 4.7 | ND | |
| 36 | E9Q137 | Tex264^ | 7.1 | 3.5 | ND |
contains conserved functional (seleno)cysteine residue
NC, not competed
ND, not detected
Measured by competition with IA alkyne probe (in vitro treatment)
Measured by competition with probe 8 (in vivo treatment)
These results provide further evidence that acetaminophen (4) generates high-engagement adducts with functional (seleno)cysteines in several liver proteins in vivo, and also reveal complementary features of data obtained by whole protein enrichment (e.g., competition with probe 8) versus site-specific (e.g., isoTOP-ABPP) chemical proteomic experiments.
Discussion
Despite the prevalence of drug-induced liver injury (DILI), our understanding of its mechanistic basis is limited. It is generally appreciated that reactive drug metabolites may be a root cause of DILI, and several methods have accordingly been introduced to identify proteins that are covalently modified by drugs in the liver, including the use of radiolabeled drugs12 and antibodies that enrich specific drug-protein adducts35. These methods have succeeded in providing a growing inventory of liver proteins targeted by reactive drug metabolites; but, this inventory is likely quite incomplete, given that different studies performed on the same drug often identify virtually non-overlapping lists of protein targets14. Thus, fundamental questions about reactive drug metabolism remain largely unanswered, such as - i) what is the shared versus unique protein target landscape for reactive metabolites generated from different drugs in vivo; and ii) what is the stoichiometry of modification, or fractional engagement, of targets by reactive drug metabolites in vivo? We surmised that a chemical proteomic strategy leveraging bioorthogonal reactivity by incorporating alkyne groups at sites on drugs that minimally perturb metabolism might offer a complementary and potentially superior route for comprehensively visualizing, enriching, and mapping of protein targets of reactive metabolites in vivo. While incorporation of a bioorthogonal handle into drugs could affect their metabolism and corresponding proteomic reactivity, we found that the major glutathione adducts of the drugs tested herein were also observed for the corresponding alkyne probes when evaluated in liver S9 fractions (as previously reported for probe 825, Figure S1B). Additionally, if metabolism occurred on the alkyne itself, the metabolic products should not contribute to our chemical proteomic results, since CuAAC coupling to azide reporter tags would have been blocked. We should also mention that alkynes, in particular, aryl alkynes, can serve as mechanism-based inhibitors of CYPs36 and have even served as the basis for CYP-directed ABPP probes37, and it is therefore important to consider these factors when identifying locations for alkyne placement or, alternatively, select azides as the latent affinity handle. It may also prove difficult to incorporate an alkyne or azide group into some drugs at sites that do not perturb metabolism or interactions with potential protein targets.
Several potentially general conclusions can be drawn from our studies. First, the drugs examined herein showed markedly different protein reactivity profiles in vitro versus in vivo. This was particularly clear for probes 5 (troglitazone) and 8 (acetaminophen), which showed substantially greater proteomic reactivity in liver homogenates and in vivo, respectively (e.g., compare Figures 1B and 1D). Such cases may indicate different routes for, or extents of, drug metabolism in liver homogenates versus in vivo. Second, individual drugs modified distinct sets of liver proteins in vivo, with only a modest fraction of proteins being targeted by two or more of the tested drugs (Figure 2D). The unique profiles of drug modification were not a technical artifact of stochastic protein detection in MS-based proteomic experiments because the proteins under comparison were quantified in multiple (often all) data sets, but only showed enrichment by one, or more rarely two or three probes. Of particular interest was the preferential enrichment of proteins involved in lipid metabolism by the troglitazone probe 5, which was observed in both mouse liver (Figure 2H) and human hepatocytes (Figure 3D). Considering that the primary target of troglitazone is the lipid receptor PPARγ, our proteomic data suggest that the reactive metabolites of troglitazone retain preferred interactions with proteins that bind lipids. In as much as these proteins participate in shared or convergent pathways, their concurrent modification by troglitazone metabolites may lead to lipid network dysregulation that contributes to some of the established features of troglitazone-induced toxicity, including perturbation of bile acid homeostasis, inhibition of fatty acid oxidation, and mitochondrial toxicity38, 39. The chemical proteomic methods described herein may also facilitate the comparative mapping of drug targets across species to enhance our understanding of species-specific drug metabolism and associated DILI events. Finally, our findings comparing acetaminophen versus the less hepatotoxic analogue AMAP emphasize the potential importance of the stoichiometry of protein modification by drugs, as the overall target landscape for the two compounds was similar, but APAP exhibited higher relative engagement for most of these shared targets (Figure 5C), possibly reflecting differences in the corresponding amounts or intrinsic reactivities of metabolites generated from each drug in vivo40. That several of the protein targets of acetaminophen were modified on functional (seleno)cysteine residues underscores the possible deleterious impact of high fractional engagement of these residues. A recent chemical proteomic analysis also identified thiolases (e.g., ACAT2) and aldehyde dehydrogenases (e.g., ALDH9A1) as targets of the herbicide glyphosphate in mouse liver41, possibly indicating that these classes of cysteine-dependent enzymes are frequent sites of action for reactive metabolites.
The hepatotoxicity of acetaminophen has been shown to require apoptosis signaling kinase 1 (ASK1), which activates downstream c-jun-N-terminal kinase (JNK) to promote acetaminophen-induced cell death in mouse liver and human hepatocytes42. We were accordingly intrigued to identify several high-occupancy targets of acetaminophen for which loss of activity stimulates stress signaling through ASK1. As outlined in Figure 6, these proteins include multiple thioredoxin reductases, two of which we confirmed were targeted at their key redox center (TXNRD1, TXNRD2). Pharmacological blockade of thioredoxin reductases is thought to promote apoptosis by depletion of reduced thioredoxin43, 44, which is a direct negative regulator of ASK145. We also identified multiple high-occupancy selenoprotein targets of acetaminophen (SELK, VIMP; Figures 4B and 5C and Table 1) that are critical components of the ER-associated degradation machinery46, 47, the perturbation of which can also lead to ASK1 activation48. Another negative regulator of ASK1 activation49 PARK7, or DJ-1, was targeted by acetaminophen at a key active-site residue (Table 1) and has been recently characterized as a glutathione-independent glyoxalase/protein deglycase critical for oxidative stress resistance50, 51. Taken together, our chemical proteomic data thus offer potential mechanistic insights into the biochemical stress response pathways that are perturbed by acetaminophen treatment, and lead to activation of ASK1 to promote hepatotoxicity. That proteins in some of these pathways were also discovered herein as targets of additional hepatotoxic drugs (e.g., clozapine) points to potentially common sites of action for mediating DILI.
Figure 6.

Schematic depicting pathways containing high-occupancy protein targets of acetaminophen (4), including redox homeostasis (e.g., Txnrd1, Txnrd2, Msrb1) and ER-associated degradation (ERAD) (Park7, Selk, Timp) pathways, the disruption of which stimulates stress signaling by activation of the apoptosis signaling kinase 1 (ASK1). Signaling through ASK1, followed by activation of JNK, is required for acetaminophen-induced cell death42.
Experimental Methods
Troglitazone, clozapine, tienilic acid, and acetaminophen were purchased from Tocris Biosciences, Enzo Life Sciences, Sigma-Aldrich, and Sigma-Aldrich, respectively, and used without further purification. Other compounds and probes were synthesized and routes and analytical characterization are described in Supplementary Material. Mouse liver S9 fraction was obtained as an equal mix of male and female, samples from Xenotech, Lenexa, KS. All mouse studies were performed following protocols that received approval from The Scripps Research Institute–Institutional Animal Care and Use Committee office.
See Supplementary Methods sections for detailed experimental procedures.
Supplementary Material
Acknowledgments
This work was supported by the NIH (CA087660) and Pfizer Inc. We thank B. Huang, Y. Liu, and A. Yu (WuXi AppTec) for the design and execution of the synthetic routes to probes 5 and 7.
Footnotes
Supporting Information Available: This material is available free of charge via the Internet.
References
- 1.Bouvy JC, De Bruin ML, Koopmanschap MA. Epidemiology of adverse drug reactions in Europe: a review of recent observational studies. Drug Saf. 2015;38:437–453. doi: 10.1007/s40264-015-0281-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miguel A, Azevedo LF, Araujo M, Pereira AC. Frequency of adverse drug reactions in hospitalized patients: a systematic review and meta-analysis. Pharmacoepidemiol Drug Saf. 2012;21:1139–1154. doi: 10.1002/pds.3309. [DOI] [PubMed] [Google Scholar]
- 3.Leung L, Kalgutkar AS, Obach RS. Metabolic activation in drug-induced liver injury. Drug Metabolism Reviews. 2012;44:18–33. doi: 10.3109/03602532.2011.605791. [DOI] [PubMed] [Google Scholar]
- 4.Licata A. Adverse drug reactions and organ damage: The liver. Eur J Intern Med. 2016;28:9–16. doi: 10.1016/j.ejim.2015.12.017. [DOI] [PubMed] [Google Scholar]
- 5.Kaplowitz N. Idiosyncratic drug hepatotoxicity. Nat Rev Drug Discov. 2005;4:489–499. doi: 10.1038/nrd1750. [DOI] [PubMed] [Google Scholar]
- 6.Park BK, Boobis A, Clarke S, Goldring CEP, Jones D, Kenna JG, Lambert C, Laverty HG, Naisbitt DJ, Nelson S, Nicoll-Griffith DA, Obach RS, Routledge P, Smith DA, Tweedie DJ, Vermeulen N, Williams DP, Wilson ID, Baillie TA. Managing the challenge of chemically reactive metabolites in drug development. Nat Rev Drug Discov. 2011;10:292–306. doi: 10.1038/nrd3408. [DOI] [PubMed] [Google Scholar]
- 7.Uetrecht J, Naisbitt DJ. Idiosyncratic Adverse Drug Reactions: Current Concepts. Pharmacol Rev. 2013;65:779–808. doi: 10.1124/pr.113.007450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stepan AF, Walker DP, Bauman J, Price DA, Baillie TA, Kalgutkar AS, Aleo MD. Structural alert/reactive metabolite concept as applied in medicinal chemistry to mitigate the risk of idiosyncratic drug toxicity: a perspective based on the critical examination of trends in the top 200 drugs marketed in the United States. Chem Res Toxicol. 2011;24:1345–1410. doi: 10.1021/tx200168d. [DOI] [PubMed] [Google Scholar]
- 9.Adams DH, Ju C, Ramaiah SK, Uetrecht J, Jaeschke H. Mechanisms of immune-mediated liver injury. Toxicol Sci. 2010;115:307–321. doi: 10.1093/toxsci/kfq009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dalvie D, Kalgutkar AS, Chen W. Practical approaches to resolving reactive metabolite liabilities in early discovery. Drug Metab Rev. 2015;47:56–70. doi: 10.3109/03602532.2014.984813. [DOI] [PubMed] [Google Scholar]
- 11.Liebler DC. Protein damage by reactive electrophiles: targets and consequences. Chem Res Toxicol. 2008;21:117–128. doi: 10.1021/tx700235t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou S. Separation and Detection Methods for Covalent Drug-Protein Adducts. J Chromatog B. 2003;797 doi: 10.1016/s1570-0232(03)00399-4. [DOI] [PubMed] [Google Scholar]
- 13.Qiu Y, Burlingame AL, Benet LZ. Identification of the Hepatic Protein Targets of Reactive Metabolites of Acetaminophen in Vivo in Mice Using Two-Dimensional Gel Electrophoresis and Mass Spectrometry. J Biol Chem. 1998;273 doi: 10.1074/jbc.273.28.17940. [DOI] [PubMed] [Google Scholar]
- 14.Koen YM, Sarma D, Williams TD, Galeva NA, Obach RS, Hanzlik RP. Identification of Protein Targets of Reactive Metabolites of Tienilic Acid in Human Hepatocytes. Chem Res Toxicol. 2012;25:1145–1154. doi: 10.1021/tx300103j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lanning BR, Whitby LR, Dix MM, Douhan J, Gilbert AM, Hett EC, Johnson TO, Joslyn C, Kath JC, Niessen S, Roberts LR, Schnute ME, Wang C, Hulce JJ, Wei B, Whiteley LO, Hayward MM, Cravatt BF. A road map to evaluate the proteome-wide selectivity of covalent kinase inhibitors. Nat Chem Biol. 2014;10:760–767. doi: 10.1038/nchembio.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Medina-Cleghorn D, Bateman LA, Ford B, Heslin A, Fisher KJ, Dalvie ED, Nomura DK. Mapping Proteome-Wide Targets of Environmental Chemicals Using Reactivity-Based Chemoproteomic Platforms. Chem Biol. 2015;22:1394–1405. doi: 10.1016/j.chembiol.2015.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wright MH, Sieber SA. Chemical proteomics approaches for identifying the cellular targets of natural products. Nat Prod Rep. 2016;33:681–708. doi: 10.1039/c6np00001k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew Chem Int Ed Engl. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 19.Speers AE, Adam GC, Cravatt BF. Activity-based protein profiling in vivo using a copper(i)-catalyzed azide-alkyne [3 + 2] cycloaddition. J Am Chem Soc. 2003;125:4686–4687. doi: 10.1021/ja034490h. [DOI] [PubMed] [Google Scholar]
- 20.Ong SE, Mann M. Mass spectrometry-based proteomics turns quantitative. Nat Chem Biol. 2005;1:252–262. doi: 10.1038/nchembio736. [DOI] [PubMed] [Google Scholar]
- 21.Hinson JA, Roberts DW, James LP. Mechanisms of acetaminophen-induced liver necrosis. Handb Exp Pharmacol. 2010:369–405. doi: 10.1007/978-3-642-00663-0_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kassahun K, Pearson PG, Tang W, McIntosh I, Leung K, Elmore C, Dean D, Wang R, Doss G, Baillie TA. Studies on the metabolism of troglitazone to reactive intermediates in vitro and in vivo. Evidence for novel biotransformation pathways involving quinone methide formation and thiazolidinedione ring scission. Chem Res Toxicol. 2001;14:62–70. doi: 10.1021/tx000180q. [DOI] [PubMed] [Google Scholar]
- 23.Pirmohamed M, Williams D, Madden S, Templeton E, Park BK. Metabolism and Bioactivation of Clozapine by Human Liver in-Vitro. J Pharmacol Exp Ther. 1995;272:984–990. [PubMed] [Google Scholar]
- 24.Dansette PM, Amar C, Smith C, Pons C, Mansuy D. Oxidative activation of the thiophene ring by hepatic enzymes. Hydroxylation and formation of electrophilic metabolites during metabolism of tienilic acid and its isomer by rat liver microsomes. Biochem Pharmacol. 1990;39:911–918. doi: 10.1016/0006-2952(90)90207-2. [DOI] [PubMed] [Google Scholar]
- 25.Koen YM, Liu K, Shinogle H, Williams TD, Hanzlik RP. Comparative Toxicity and Metabolism of N-Acyl Homologues of Acetaminophen and Its Isomer 3′-Hydroxyacetanilide. Chem Res Toxicol. 2016;29:1857–1864. doi: 10.1021/acs.chemrestox.6b00270. [DOI] [PubMed] [Google Scholar]
- 26.Wilson-Grady JT, Haas W, Gygi SP. Quantitative comparison of the fasted and re-fed mouse liver phosphoproteomes using lower pH reductive dimethylation. Methods. 2013;61:277–286. doi: 10.1016/j.ymeth.2013.03.031. [DOI] [PubMed] [Google Scholar]
- 27.Hanzlik RP, Koen YM, Fang J. Bioinformatic analysis of 302 reactive metabolite target proteins. Which ones are important for cell death? Toxicol Sci. 2013;135:390–401. doi: 10.1093/toxsci/kft166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Backus KM, Correia BE, Lum KM, Forli S, Horning BD, Gonzalez-Paez GE, Chatterjee S, Lanning BR, Teijaro JR, Olson AJ, Wolan DW, Cravatt BF. Proteome-wide covalent ligand discovery in native biological systems. Nature. 2016;534:570–574. doi: 10.1038/nature18002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Streeter AJ, Dahlin DC, Nelson SD, Baillie TA. The covalent binding of acetaminophen to protein. Evidence for cysteine residues as major sites of arylation in vitro. Chem Biol Interact. 1984;48:349–366. doi: 10.1016/0009-2797(84)90145-5. [DOI] [PubMed] [Google Scholar]
- 30.Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MB, Bachovchin DA, Mowen K, Baker D, Cravatt BF. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature. 2010;468:790–795. doi: 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jan YH, Heck DE, Dragomir AC, Gardner CR, Laskin DL, Laskin JD. Acetaminophen Reactive Intermediates Target Hepatic Thioredoxin Reductase. Chem Res Toxicol. 2014;27:882–894. doi: 10.1021/tx5000443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mi H, Poudel S, Muruganujan A, Casagrande JT, Thomas PD. PANTHER version 10: expanded protein families and functions, and analysis tools. Nucleic Acids Res. 2016;44:D336–342. doi: 10.1093/nar/gkv1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Myers TG, Dietz EC, Anderson NL, Khairallah EA, Cohen SD, Nelson SD. A comparative study of mouse liver proteins arylated by reactive metabolites of acetaminophen and its nonhepatotoxic regioisomer, 3′-hydroxyacetanilide. Chem Res Toxicol. 1995;8:403–413. doi: 10.1021/tx00045a012. [DOI] [PubMed] [Google Scholar]
- 34.Wang C, Weerapana E, Blewett MM, Cravatt BF. A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles. Nat Methods. 2014;11:79–85. doi: 10.1038/nmeth.2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pumford NR, Halmes NC, Hinson JA. Covalent binding of xenobiotics to specific proteins in the liver. Drug Metab Rev. 1997;29:39–57. doi: 10.3109/03602539709037572. [DOI] [PubMed] [Google Scholar]
- 36.Hollenberg PF, Kent UM, Bumpus NN. Mechanism-based inactivation of human cytochromes p450s: experimental characterization, reactive intermediates, and clinical implications. Chem Res Toxicol. 2008;21:189–205. doi: 10.1021/tx7002504. [DOI] [PubMed] [Google Scholar]
- 37.Wright AT, Song JD, Cravatt BF. A suite of activity-based probes for human cytochrome P450 enzymes. J Am Chem Soc. 2009;131:10692–10700. doi: 10.1021/ja9037609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Begriche K, Massart J, Robin MA, Borgne-Sanchez A, Fromenty B. Drug-induced toxicity on mitochondria and lipid metabolism: mechanistic diversity and deleterious consequences for the liver. J Hepatol. 2011;54:773–794. doi: 10.1016/j.jhep.2010.11.006. [DOI] [PubMed] [Google Scholar]
- 39.Yang K, Woodhead JL, Watkins PB, Howell BA, Brouwer KL. Systems pharmacology modeling predicts delayed presentation and species differences in bile acid-mediated troglitazone hepatotoxicity. Clin Pharmacol Ther. 2014;96:589–598. doi: 10.1038/clpt.2014.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Howell BA, Siler SQ, Watkins PB. Use of a systems model of drug-induced liver injury (DILIsym((R))) to elucidate the mechanistic differences between acetaminophen and its less-toxic isomer, AMAP, in mice. Toxicol Lett. 2014;226:163–172. doi: 10.1016/j.toxlet.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 41.Ford B, Bateman LA, Gutierrez-Palominos L, Park R, Nomura DK. Mapping Proteome-wide Targets of Glyphosate in Mice. Cell Chem Biol. 2017;24:133–140. doi: 10.1016/j.chembiol.2016.12.013. [DOI] [PubMed] [Google Scholar]
- 42.Xie Y, Ramachandran A, Breckenridge DG, Liles JT, Lebofsky M, Farhood A, Jaeschke H. Inhibitor of apoptosis signal-regulating kinase 1 protects against acetaminophen-induced liver injury. Toxicol Appl Pharmacol. 2015;286:1–9. doi: 10.1016/j.taap.2015.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang J, Li Y, Duan D, Yao J, Gao K, Fang J. Inhibition of thioredoxin reductase by alantolactone prompts oxidative stress-mediated apoptosis of HeLa cells. Biochem Pharmacol. 2016;102:34–44. doi: 10.1016/j.bcp.2015.12.004. [DOI] [PubMed] [Google Scholar]
- 44.Duan D, Zhang J, Yao J, Liu Y, Fang J. Targeting Thioredoxin Reductase by Parthenolide Contributes to Inducing Apoptosis of HeLa Cells. J Biol Chem. 2016 doi: 10.1074/jbc.M115.700591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee JH, Park KJ, Jang JK, Jeon YH, Ko KY, Kwon JH, Lee SR, Kim IY. Selenoprotein S-dependent Selenoprotein K Binding to p97(VCP) Protein Is Essential for Endoplasmic Reticulum-associated Degradation. J Biol Chem. 2015;290:29941–29952. doi: 10.1074/jbc.M115.680215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shchedrina VA, Everley RA, Zhang Y, Gygi SP, Hatfield DL, Gladyshev VN. Selenoprotein K binds multiprotein complexes and is involved in the regulation of endoplasmic reticulum homeostasis. J Biol Chem. 2011;286:42937–42948. doi: 10.1074/jbc.M111.310920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol. 2015;10:173–194. doi: 10.1146/annurev-pathol-012513-104649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Raninga PV, Trapani GD, Tonissen KF. Cross Talk between Two Antioxidant Systems, Thioredoxin and DJ-1: Consequences for Cancer. Oncoscience. 2014;1:95–110. doi: 10.18632/oncoscience.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Richarme G, Mihoub M, Dairou J, Bui LC, Leger T, Lamouri A. Parkinsonism-associated protein DJ-1/Park7 is a major protein deglycase that repairs methylglyoxal- and glyoxal-glycated cysteine, arginine, and lysine residues. J Biol Chem. 2015;290:1885–1897. doi: 10.1074/jbc.M114.597815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bankapalli K, Saladi S, Awadia SS, Goswami AV, Samaddar M, D’Silva P. Robust glyoxalase activity of Hsp31, a ThiJ/DJ-1/PfpI family member protein, is critical for oxidative stress resistance in Saccharomyces cerevisiae. J Biol Chem. 2015;290:26491–26507. doi: 10.1074/jbc.M115.673624. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.