

Abstract
Systems pharmacology aims to holistically understand mechanisms of drug actions to support drug discovery and clinical practice. Systems pharmacology modeling (SPM) is data driven. It integrates an exponentially growing amount of data at multiple scales (genetic, molecular, cellular, organismal, and environmental). The goal of SPM is to develop mechanistic or predictive multiscale models that are interpretable and actionable. The current explosions in genomics and other omics data, as well as the tremendous advances in big data technologies, have already enabled biologists to generate novel hypotheses and gain new knowledge through computational models of genome-wide, heterogeneous, and dynamic data sets. More work is needed to interpret and predict a drug response phenotype, which is dependent on many known and unknown factors. To gain a comprehensive understanding of drug actions, SPM requires close collaborations between domain experts from diverse fields and integration of heterogeneous models from biophysics, mathematics, statistics, machine learning, and semantic webs. This creates challenges in model management, model integration, model translation, and knowledge integration. In this review, we discuss several emergent issues in SPM and potential solutions using big data technology and analytics. The concurrent development of high-throughput techniques, cloud computing, data science, and the semantic web will likely allow SPM to be findable, accessible, interoperable, reusable, reliable, interpretable, and actionable.
Keywords: cloud computing, data science, machine learning, semantic web, computational modeling, systems biology, systems pharmacology modeling, NIH Commons
INTRODUCTION
Drug action is a complex process. A chemical, which can be synthesized, natural, or endogenous, starts its effect on biological systems through its interactions with biomolecules (proteins, DNAs, or RNAs), that is, its targets. In the case of most commonly investigated protein targets, many types of interactions are determined by protein conformational dynamics, from antagonism to partial agonism and inverse agonism, from biased signaling to allosteric modulation (1, 2). The change in the functional state of the biomolecule, which depends on the kinetics of ligand binding and unbinding and the thermodynamic transitions that ensue, will ultimately drive biological outcomes. Moreover, a chemical rarely binds to a single target. Multiple target binding (i.e., polypharmacology) is a common phenomenon (3). Even weak drug-target interactions can have a collectively strong effect on the physiological response of an organism (4). To understand how altering the functional state of multiple biomolecules changes the cellular milieu by regulating gene expression, signal transduction, and metabolism and ultimately modifies the physiological or pathological state of the individual, systems biology provides a means to model, simulate, and predict the phenotypic response of drug action (5). The fate of the drug molecule itself is also dependent on the activity and expression of transporters and metabolizing enzymes and the local physiological environment. Individual genetic and epigenetic variations and lifestyle choices such as diet add great diversity to the drug action. They may not only impose an effect on the thermodynamics and kinetics of drug binding as well as pharmacokinetics, but also rewire the biological network, thereby resulting in a dramatically different drug response. A reductionist view of drug action is too simple to explain and predict the drug phenotypic response to complex diseases. We need a holistic understanding of drug action in a background of diverse genetic, epigenetic, and environmental factors. These factors include conformational dynamics of drug-target interactions, emergent properties of biological systems, enzyme reactions, and physiology-based pharmacokinetics (6). Embracing the concept of multiscale systematic modeling of drug actions by integrating multiple omics data and biological mechanisms speaks to the emergence of a new discipline of systems pharmacology (6, 7). The fundamental features of systems pharmacology are (a) the development of actionable and interpretable mechanistic or predictive models through the integration of biological and clinical data at multiple temporal and spatial scales and (b) the use of the output from the model for generating novel hypotheses, discovering new biomedical knowledge, and supporting decision making in drug discovery and clinical practice. Systems pharmacology provides a promising avenue to gain a comprehensive and systematic view of drug action under the complex interplay of genetic, molecular, cellular, organismal, and environmental components. Such understanding may fill the current innovation gap in drug discovery (8).
Data-driven modeling plays a central role in systems pharmacology. Recent developments in high-throughput experiments have generated a huge amount of data across the multiple biological scales of the organism, across a wide range of timescales, and across multiple species. These data provide unprecedented opportunities for systems pharmacology but impose great challenges in data processing, management, sharing, and integration (9, 10). The rapid advances in cloud computing, big data technology, and data science offer an opportunity to clear the hurdles in data-driven modeling for systems pharmacology. The planned US National Strategic Computing Initiative will maximize the benefits of High Performance Computing. The development of the US National Institutes of Health (NIH) Data Science Commons (https://datascience.nih.gov/commons) will make biological data findable, accessible, interoperable, and reusable (11). These efforts will enhance significantly the availability and quality of biological data, thereby enhancing the capability of systems pharmacology modeling. Indeed, systems pharmacology models based on the integration of genome-wide, heterogeneous, and dynamic data sets have already shown promise in drug repurposing (12–14), predicting drug side effects (15–18), and developing combination therapy (19) and precision medicine (20).
With the explosion of mathematical and computational models for genomics, molecular dynamics (MD), biological networks, whole cells, tissues, organisms, and populations, we face new challenges. How can we manage these diverse models efficiently and effectively, including sharing, reuse, validation, reproducibility, access, and searching? How can we integrate diversified models that are from different resources, based on different methodologies, and at different temporal and spatial scales into a unified, potentially more powerful mechanistic or predictive model that captures the whole spectrum of drug actions? How can we translate mathematical languages or decipher black boxes representing these models into cause-and-effect relationships or simple rules that can be comprehended by biologists and clinicians, and integrate them with existing knowledge for automated reasoning and decision making? Addressing these challenges will no doubt facilitate harnessing big data for systems pharmacology and realize the full power of systems pharmacology in drug discovery and clinical practice. In this review, we discuss some of the unsolved issues in the management, integration, and translation of systems pharmacology models, and we propose possible solutions to them using big data technology and analytics. Researchers expect that parallel developments in high-throughput techniques, cloud computing, data science, and the semantic web will allow systems pharmacology models to be findable, accessible, interoperable, reusable, reliable, interpretable, and actionable.
MODEL MANAGEMENT
A comprehensive and systematic understanding of drug action requires the integration of diverse models from different data modalities (e.g., single-nucleotide variants, copy-number variations, methylations, proteomics, transcriptomics, metabolomics) across multiscales of cellular organization, from the atomic details of drug-target binding thermodynamics and kinetics to proteome-scale drug-target interactions, from the functional impact of mutations to emergent properties of biological networks, from cytochrome P450 enzyme reactions to physiology-based pharmacokinetics. They can be biophysics-based molecular models, machine learning models of molecular interactions, mathematical models of systems biology or pharmacokinetics, or, taking this to the extreme, connectome models of the human brain. Even in the same type of model, models can be significantly different. For example, an MD model of protein structure could be a Cα-represented coarse-grained elastic network model or an all-atomic conformational ensemble from microsecond MD simulations. A drug-target interaction model could be a graphic representation that abstracts each protein and drug as a single node and the interaction between them as an edge (21), whereas a drugome model includes three-dimensional structures of drug-target complexes (22). A systems biology model could be represented as a stoichiometric matrix and flux bounds (23), based on ordinary differential equations, or encoded as three-dimensional geometries and partial differential equations (24). Machine learning models could be inferred using different features and base learners. Furthermore, the models from different domains are often interleaved. For example, a drug-target binding/unbinding kinetics model could be a combination of an elastic network model and machine learning model (25). The diversity of models makes it a nontrivial task for scientists to discover, access, and reuse a model that is beyond their domain of expertise, as well as to integrate multiple models. Moreover, a model alone may not be sufficient for a real-world application; the model is often dependent on multiple data sets. Big data integration is an important topic in systems pharmacology that has been covered elsewhere (9, 10). Beyond the data challenge, models are coupled strongly with algorithms underlying the model, software that implements the algorithm to execute the model, and tools that process inputs and outputs. Software is often developed in different languages, compiled in different operating systems, and changed over time, making its interoperability, reuse, and reproducibility difficult (26). Moreover, software that is developed as part of a research program rather than a development process is rarely professional grade, robust, and easy to use. Innovative model management strategies for systems pharmacology, including but not limited to model storage, transfer, sharing, standardization, and validation, are urgently needed.
Model Storage, Transfer, and Sharing
With the exponential increase of biological data and computing power, the number of systems pharmacology models increases at an even faster pace. For example, understanding of drug binding thermodynamics and kinetics requires detailed study of the conformational dynamics of protein structures. Now it is possible to sample the conformational space of a protein structure at microsecond timescales and longer using MD simulations (27). MD generates millions of conformations for further analysis—ten times more than all the structures deposited in the Protein Data Bank (28) to date. Recent development of the Macromolecular Transmission Format for compact and accurate representation of biomolecular structural data may overcome storage and input-output hurdles in biomolecular structural modeling (http://mmtf.rcsb.org).
Heterogeneous network models of chemicals and proteins are very useful to predict genome-scale drug-target interactions. A network model of all chemicals in ChEMBL (29) may have millions of nodes and tens of millions of edges. Similarly, a network model for all sequences in UniProt (30) can quickly reach 100 million nodes. The number of edges will increase exponentially with the increase in nodes. The size of systems pharmacology models will impose a hurdle for model storage and transfer, making model sharing difficult. By taking advantage of the underlying properties of biological systems (e.g., redundancy), systems pharmacology models can be compressed without loss of information (31–34). Furthermore, big data storage and transfer technology, which has already gained substantial attention in genomics (35), can be applied to large-scale computational models in systems pharmacology.
Model Standardization
To make systems pharmacology modeling findable, accessible, interoperable, and reproducible, computational models must adhere to a common standard of representation and annotation, including a description of the execution and outcomes of the simulations. Much effort has been devoted to standardization in systems biology to facilitate collaboration (36, 37). Now nearly all modeling in systems biology follows the suggestion of the Minimal Information for Biological and Biomedical Investigations project (38). The minimum information required in the annotation of models (MIRIAM) provides guidelines to curate models (39). The MIRIAM registry proposes a connection between ontologies, model format, databases, and tools (40, 41). Ontologies have been developed to describe model structures and components, mathematical formulizations, and simulation algorithms (36). Several modeling formats have been proposed to encode systems biology models (36). Notably, the Pharmacometric Markup Language PharmML for the representation and exchange of pharmacometric models is under development (42). A recent development of the YAML metabolic modeling format enables version tracking and provides a flexible infrastructure for the distribution tracking and collaborative annotation of metabolic models (37).
The efforts made by the systems biology community should be extended to systems pharmacology, which is even more diversified than systems biology. It requires the development of new modeling ontologies, utilities, and visualizations in a specific domain, as well as protocols and tools that enable communication across domains. For example, to predict how drug inhibition of gene A and a mutation in gene B affect blood pressure collectively, we may need three models: a biophysical model to determine the strength of competitive inhibition of the drug on its target gene A, a machine learning model to predict the functional impact of mutations on gene B (e.g., neutral or deleterious), and a genome-scale metabolic model or a kinetic model that takes the outputs from drug binding and mutation models as inputs. The output of a biophysical model is usually in the form of a binding free energy. The output of a machine learning model of mutation could be the probability that the mutation is predicted to be deleterious. The input required for the stoichiometric or kinetic model is different: The stoichiometric model may need to constrain the flux corresponding to the reaction catalyzed by the target of the drug or harbored mutation, whereas the kinetic model may need to modify the kinetic parameters of the corresponding reaction. The integration of popular bioinformatics workflow systems such as Galaxy (43) with cloud computing (44) could be a powerful approach to linking diverse models together. However, existing workflow systems do not have sufficient semantic supports to represent and reproduce the communications between models. An ontology is needed to represent common molecular components and their interactions, which may have different representations in different models. For example, a gene can be represented by a structure of the encoded protein in the biophysical model but by a fragment of DNA sequence in the mutation model. Eventually they need to map to a variable in a mathematical model. Moreover, detailed descriptions of the experimental procedure (e.g., how to convert the binding free energy to a flux constraint) should be encoded in a way that makes it understandable to both machines and humans. Such efforts cannot be successful without an ecosystem that encourages collaborations. The NIH Data Science Commons is beginning to address this challenge (11).
Model Validation
A more serious problem in the application of systems pharmacology modeling is how to validate models, assess their accuracy and reliability on a new prediction, and define the scope of their application domains. Recent debates on the origins of inconsistency of constraint-based network modeling that describes a biological system by a set of constraints (e.g., mass balance, thermodynamics) highlight the difficulties in model validation (31, 45, 46). Constraint-based network modeling is a powerful tool in systems pharmacology. It has been applied to predict drug side effect profiles resulting from off-target binding (47), elucidate mechanisms of antibiotics (48), predict personalized drug responses (20), and identify drug targets and biomarkers (49). However, the application and reproducibility of constraint-based modeling are hampered by the lack of standard formats for network models and associated tools to parse the models (31, 37, 45, 46). A more fundamental issue is whether an exact arithmetic solver is required to achieve consistent results (31, 45, 46). The standardization efforts in systems biology discussed above may facilitate addressing these problems. The minimum information about a simulation experiment has been proposed to define unambiguously how to reproduce simulation results (50). Such information should be included in all types of models in systems pharmacology.
Machine learning–based big data analytics is playing an increasingly important role in systems pharmacology (10). Owing to the nature of pharmacological data that are often biased, incomplete, and heterogeneous, and our limited knowledge of biological systems and human physiology, the machine leaning models generalized using a specific algorithm and one particular set of data may not be applicable to a new case. Thus, the rigorous, on-the-fly validation of machine learning models is critical, particularly in a risk-sensitive domain (e.g., to determine if a cancer patient is sensitive to an experimental anticancer drug or if a lead compound should move into clinical trials, in which failure is costly). In this regard, the validation of a machine learning model for systems pharmacology using conventional techniques, such as cross-validation or a limited number of wet-lab experiments, is not sufficient, as such a model does not cover the whole pharmaceutical and physiological space and provides a only global performance measure. To define clearly the applicable domain of a model so that a nonexpert can have sufficient information to use the model wisely, new standards are needed for the future development and description of machine learning models for systems pharmacology. Firstly, the model should be evaluated constantly whenever new data become available. Models must be associated with computational tools that extract and format new data as well as metadata that describe the data on validation. Whenever feasible, the model can be retrained in the framework of active learning (51). Secondly, multiple evaluation metrics are needed, as different applications should be measured differently. For example, a ranking is sufficient for a web search but not for determining if a drug is effective in a patient. Finally, for each prediction, the reliability of the prediction should be estimated, as a new case may fall outside the generalized hypothesis space of a model that is based on biased and incomplete data. A case-based reasoning (CBR) framework may be useful to address this problem, as detailed below.
Model Access and Reusability
Even if systems pharmacology models and their associated data, algorithms, and software are well defined and validated, it may not be easy for a user to find the most suitable models unless all relevant documents and literatures are studied. Researchers have developed several databases to host computational models relevant to systems pharmacology, such as drug-target interaction models (52), constraint-based metabolic network models (53), and pharmacometrics models (54). However, several challenges still remain that hinder the accessibility and usability of systems pharmacology modeling. Firstly, data, software, and models are scattered around the Internet. Diverse systems pharmacology models have not been registered in a central place so that they can be easily accessed. Secondly, data, algorithms, and software that are used to build the model are often separated from each other. As discussed above, data and software that are used to develop systems pharmacology models are inseparable components of those models. Finally, there is no easy way to search for suitable models to use in a systems pharmacology modeling project.
The NIH Data Science Commons is building a shared virtual space that allows scientists to find, manage, share, use, and reuse digital objects (data, software, metadata, and workflows). It is a complex ecosystem including a computing environment, data sets, and software services and tools. The computing environment (e.g., cloud) supports access, use, and storage of digital objects. Publically available data sets need to adhere to Commons digital object compliance FAIR (findable, accessible, interoperable, and reproducible) principles. Software services and tools will enable scalable provisioning of compute resources; interoperability between digital objects within the Commons; discoverability of digital objects; sharing of digital objects between individuals or groups; access to and deployment of scientific analysis tools and pipeline workflows; and connectivity with other repositories, registries, and resources.
The concept of the Commons can include systems pharmacology models, each of which is a collection of model representation, data, software packages, and metadata that describe them. As shown in Figure 1, model, data, software, and metadata can be stored in different computers in a cloud computing environment. To make software reusable, big data technology such as Docker (https://www.docker.com/) can be used to wrap the software. Metadata are needed to standardize and characterize components of data, model, and software; define their interactions; and register the model system in the Commons.
Figure 1.
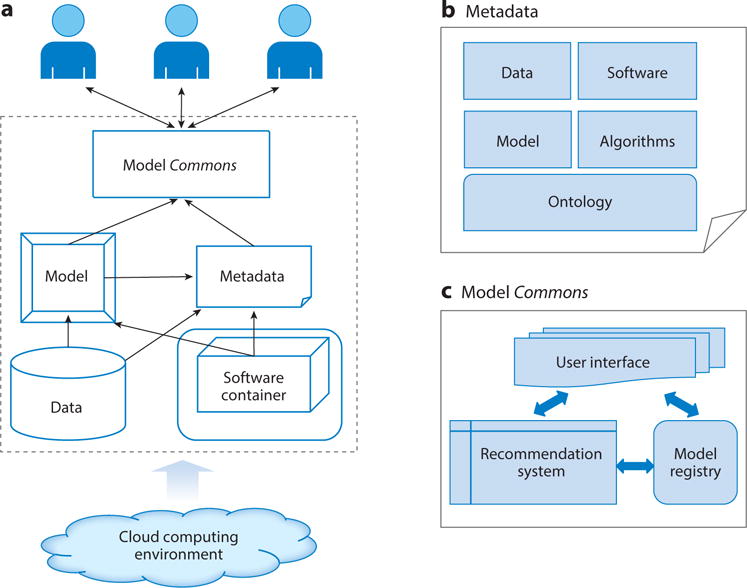
Scheme of a systems pharmacology model management system that adheres to Commons digital object compliance FAIR (findable, accessible, interoperable, and reusable) principles. (a) Architecture of model management systems. Models are linked with associated data sets, metadata, and software that are wrapped within a container and accessed through a model Commons. The whole system may be supported by a cloud computing environment. (b) Model metadata are built on ontologies, including information on the model itself, data, algorithms, and software. (c) The model Commons may need a recommendation system to rank the relevant models based on user requests in addition to a model registry and user interface.
To make models findable and accessible, a recommendation system similar to those used in Netflix or Amazon can be valuable. Each model can be associated with a set of features. These features may include the usage information of the model, description of embedded data and underlying algorithm in the model, summary of the software package used, and applications cited in the literature. Existing techniques in big data analytics, especially those for the recommendation system, can be applied to build the recommendation systems using the features associated with the model. Thus, the models can be ranked based on the user’s interest.
In summary, a systems pharmacology model is useful only when it can be used routinely by domain experts. The challenge in developing predictive models of drug action is highly complex and multidisciplinary. It is not likely to be overcome by any one group of researchers. Enabling scientists to reproduce and extend the work of others requires that the models and methods be distributed in a manner that is both accessible and usable. It requires close collaboration between model developers and end-users. The wise utilization of big data technology may facilitate establishment of an environment that fosters the development, dissemination, and effective use of systems pharmacology models, as well as the realization of community-based predictive modeling, a lofty goal in drug discovery (55).
MODEL INTEGRATION
The integration of multiple diverse models is critical for the success of systems pharmacology modeling. Firstly, investigators have recognized that multiscale modeling, from the atomic details of protein conformational dynamics to the emergent properties of biological networks, is needed for understanding drug actions comprehensively and holistically as well as developing precision medicine (6). The success of multiscale modeling depends on the mechanistic integration of molecular, network, tissue, organism, and populations models at several spatial and temporal scales. Owing to its complexity, a semantic integration that links data and information from diverse resources based on ontologies may be needed, as discussed in the next section. Secondly, on the same scale, multiple models can be combined by a plethora of machine learning techniques. The combination of multiple models will generally outperform a single model. However, there is no one-size-fits-all solution to achieve the optimal model combination in the context of systems pharmacology. Like the data integration in systems pharmacology (7), we refer to the model integration across different scales as vertical model integration but refer to model combination at the same scale as horizontal model integration. Finally, two of the fundamental questions in systems pharmacology are how to link in vitro drug potency to in vivo drug activity and how to extrapolate the drug response in animal models to that in humans. Solving these problems requires effective methods to reduce the transportability bias. Such bias arises when the population from which data are acquired is different from the one for which the inference is intended. Thus, the success of model integration in systems pharmacology depends on solutions to multiscale modeling (vertical model integration), horizontal model integration, and model transportability, as shown in Figure 2. The problem of multiscale modeling and model transportability in systems pharmacology (i.e., vertical integration) has been discussed elsewhere (10); here we focus on horizontal model integration.
Figure 2.
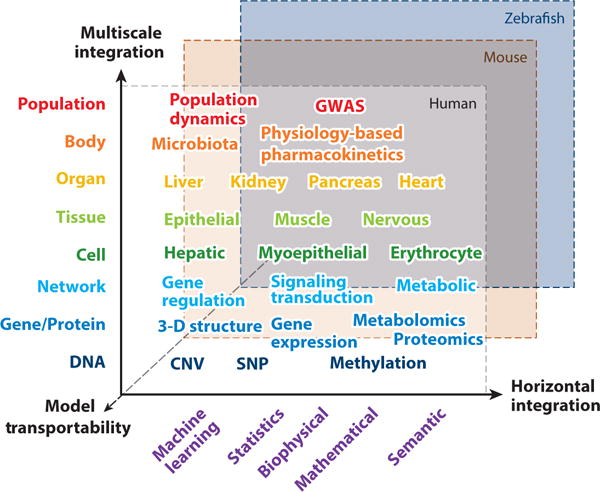
Model integration in systems pharmacology. Diverse models need to be integrated across multiple methodologies, multiple heterogeneous data sets, organismal hierarchy, and species (transportability). Abbreviations: CNV, copy-number variation; GWAS, genome-wide association studies; SNP, single-nucleotide polymorphisms.
Overview of Existing Techniques in Big Data Analytics for Horizontal Model Integration
Horizontal model integration can combine models that are built from different combinations of data sets, samples, features, and machine learning methods. Depending on the training data and the base method used, horizontal model integration can be cast as problems of ensemble learning, or data fusion in data science. Ensemble learning is a machine learning method that combines multiple predictive models to hopefully form a better model using the same base method, usually using a single data set. Alternatively, data fusion techniques use multiple data sets that are from different data modalities (a.k.a. views). For example, multiple genomics data sets such as those of mutations, copy-number variations (CNVs), methylation profiles, and gene expression profiles can all be used to develop predictive models for anticancer sensitivity. Each single data set is a modality or view.
Data fusion methods can be classified roughly into three categories. In the first category, a first-level model is built for each modality independently. Then these first-level models are combined by training a second-level model that uses the predictions of the first-level models as features (56) or via a meta-predictor that takes the majority votes or weights from the prediction of the first-level models; the meta-predictor has been widely used in chemoinformatics and bioinformatics (e.g., for predicting disease-associated mutations) (57). In the second category, a joint representation of multiple data sets is learned using deep neural networks (58). The third category combines different data sets based on their semantic relationships. The semantic relationship may include the similarity that corresponds naturally to different viewers or biological dependencies between views (e.g., the regulation of gene expression through DNA methylation). Data fusion methods have been applied to address many problems in systems pharmacology modeling. For example, a predictive model based on multikernel learning that combines kernels of genomics data sets is one of the best performers in the DREAM anticancer drug sensitivity challenge (59). The kernel is a similarity function between data points. Kernel-based matrix factorization that combines drug similarity and target similarity is a promising method to predict novel drug-target interactions (60). PARADIGM uses Bayesian network models to combine genetic variation, gene expression, and pathway information for gene enrichment analysis (61). The Bayesian network is a probabilistic graphic model that represents a set of biological measurements and their dependencies. Multitask learning that learns several related problems together at the same time using a shared representation of multiple data modalities has proved to be a valuable approach for inferring multitarget quantitative structure-activity relationship models for lead optimization (62). The advances in big data technology provide new opportunities for model combination. As different models in the combination can be trained independently, parallel and distributed computing models such as MapReduce will facilitate the implementation of horizontal model integration (63, 64). In spite of these advances, new strategies are needed to address inherent problems in systems pharmacology modeling.
Challenges in the Application of Big Data Analytics to Systems Pharmacology
When one is adapting existing techniques in big data analytics to model integration in systems pharmacology, several challenges remain owing to the inherent complexity of biological and clinical data. In addition to big volumes, pharmacological and clinical data are high dimensional, incomplete, biased, heterogeneous, dynamic, and noisy. Unlike big data in other domains such as social networks, imaging processing, and natural language, where there are huge numbers of samples, pharmacological and clinical data may be sparse but high dimensional. For example, in genome-wide association studies (GWAS), the sequencing data of a whole genome can have hundreds of gigabytes with hundreds of thousands of single-nucleotide polymorphisms. The sample size is typically in the range of hundreds of individual genomes. Although the total volume of data can be hundreds of terabytes, the number of variables is far larger than the number of samples. The One Million Genome Project cannot solve the under sampling problem completely owing to the diversity of disease phenotypes and pharmacogenomics profiles. The issue of how to handle extremely high-dimensional sparse data is an unsolved problem in big data analytics. For example, without integrating with other data, GWAS data alone cannot identify disease-associated mutations (65).
A second challenge is that existing data from pharmacology are often incomplete and biased. For example, only several thousand genes from multiple organisms have associated ligand binding information. Moreover, the number of associated ligands for each target is highly uneven, with many uncharacterized proteins playing important roles in drug action. Thirdly, in terms of heterogeneity, these data span the hierarchical organization of an organism (molecule, pathway, cell, tissue, organ, patient, and population), across a wide spectrum of timescales, and across multiple species. As mentioned above, multiscale modeling is required (6). Even in the same organism and at the same timescale of the same species, the data can be highly heterogeneous. For example, intertumor and intratumor heterogeneity have been observed ubiquitously (66). It is difficult to build a generalized machine learning model to predict anticancer drug sensitivity, as a new case can be unique and be out of the hypothesis space of trained models. Fourthly, the biological response to drug perturbation is dynamic. For example, cancer cells, bacteria, and viruses can evolve rapidly to gain drug resistance. Systems pharmacology modeling should take the dynamics of drug response into account. Finally, in terms of noise, systems pharmacology must not only consider the signal-to-noise ratio of the various experimental methods and data sets but also incorporate noise and stochasticity into its models, which are intrinsic properties of biological processes (67).
Conventional techniques for model integration in big data analytics are not sufficient to address the aforementioned challenges in systems pharmacology. In the case of ensemble learning, the most influential and practical methods that are based on random sampling of data points or features have limitations. They may be incapable of dealing with noisy data sets (68) or have difficulty in handing high-dimensional data. Moreover, the heterogeneity of samples may represent the underlying functional space of biological system (e.g., different tissue types, pathogenicity, races). The random sampling of data may not be the best strategy for ensemble learning to handle heterogeneous biological or clinical data. Several recently developed techniques may offer new solutions to adapting the ensemble learning to model integration in systems pharmacology. For example, researchers have proved that the minimization of sample intersections in the ensembled predictors will improve the performance of sampling (69). This implies that sampling may take advantage of the heterogeneity of data. Multiview ensemble learning is another technique that may facilitate systems pharmacology modeling using high-dimensional data (70). Here, randomly generated training sets (i.e., clustering or random feature set partitioning) are applied to select multiple subsets of features. Each subset of features is used as a view to train a model. An ensemble is constructed by the combination of these models. As discussed elsewhere (10), the incorporation of biological knowledge into data-driven modeling is critical to the process of systems pharmacology modeling. For example, protein-protein interaction networks may assist the optimal feature set partitioning.
A fundamental challenge in big data analytics is to discover unknowns outside the existing domain of knowledge. It is particularly difficult for systems pharmacology. As mentioned above, pharmacological and clinical data are often incomplete, biased, and heterogeneous. As a result, models built on these data cover only a portion of pharmacological and physiological space; that is, each model may be biased toward a certain knowledge domain. For example, only a portion of characterized druggable proteins have experimentally determined structures, and the pharmaceutical relevance of many protein structures is unknown. Models built on these divergent data sets are complementary but different. Existing paradigms for model integration may not be suitable. The underlying hypothesis of existing methods for model combination is that each predictive model can perform better than a random guess but not accurately enough to be useful. The existing algorithm can find the set of optimal weights that are based on observed data by randomly sampling the weight space. After training, the weight is fixed. It assumes that the high-weighted model always performs better than the low-weighted model. Both of these hypotheses do not hold in many cases of systems pharmacology, in which a model could be strong for one case but weak for another one. If the case is outside the knowledge space covered by all models, it is possible that all models fail. CBR may provide an alternative solution to integrate heterogeneous models in systems pharmacology. In the field of artificial intelligence, the CBR approach solves a new problem by adapting solutions to a previously similar problem (71). When one is applying CBR to model integration, it may work as follows: Firstly, old cases are clustered based on the similarity among them. Secondly, the performance of each model is evaluated for each case cluster. This generates a performance matrix. Then, given a new case, its similarity to each cluster is calculated. Finally, the models are weighted using the similarity of their associated clusters to the query case and the performance matrix. The model weight is case dependent in the CBR. The CBR strategy has been applied successfully to combine multiple protein-ligand interaction models for docking scoring and improved the performance of high-throughput screening significantly (72). The challenges for CBR are how to select relevant features and how to assess the similarity between cases. The combination of advances in data science and domain-specific knowledge may provide feasible solutions (10).
The ultimate goal of systems pharmacology modeling is to generate novel hypotheses for discovering new knowledge and supporting decision making in drug discovery and clinical practice. Followed by experimental validation, new biological knowledge can be discovered by validating or refuting the hypothesis. In turn, this will increase the coverage of knowledge space, thereby facilitating systems pharmacology modeling. It is more demanding to apply systems pharmacology modeling to support decision making in drug discovery and clinical practice, such as the prediction of anticancer drug efficacy for a particular patient. In such risk-sensitive domains, a reliable assessment of predictive modeling quality on an individual basis is essential. A fundamental assumption in machine learning is that the data sample on which an algorithm learns is representative of the complete data set to which the algorithm is applied. As a result, all methods proposed to address prediction reliability are tailored to generalize and may not apply to an individual case that may fall outside the space of the training data (73, 74). To assess the prediction reliability for a new case, it is critical to define the boundary of model space and to determine if the new case falls within the model space. The CBR paradigm may provide a solution to this problem. Another strategy is to incorporate the systems pharmacology modeling into existing biological and clinical knowledge, as discussed in the following section.
In summary, drug action is so complicated that any single model of systems pharmacology can touch just one part of the process. The information derived from any model can be biased, even misleading. It is critically important to clearly understand the scope of each model and to integrate diverse models—as many as possible—to gain a more comprehensive and reliable picture of the whole process of drug action. In spite of tremendous advances in data science, we still lack reliable and usable tools to integrate systems pharmacology models horizontally, vertically, and across species. More efforts are needed to develop new methods for model integration in systems pharmacology.
MODEL INTERPRETATION AND KNOWLEDGE INTEGRATION
The notable aim of systems pharmacology modeling is not only to maximize prediction accuracy but also to reveal the mechanism of drug action and to support decision making in drug discovery and clinical practice. Thus, it is necessary to have interpretable models, to integrate the models with existing knowledge, and to enable automated reasoning.
Systems pharmacology models can be assembled by either data-driven or mechanism-based approaches. These two approaches are often combined in systems pharmacology. Data-driven modeling—especially machine learning approaches—often generates a black box. Recent work may facilitate opening the black box of machine learning models. For example, Random Prism has been proposed as an alternative to the widely used Random Forest methods (75). The base learner of Random Prism is the Prism algorithm that learns a set of IF-THEN rules instead of trees. It may provide a better representation of knowledge, which cannot be encoded easily as a decision tree. In another case, sequence motifs are extracted from the output of kernel-based learning algorithms (76).
Although mechanistic-based models offer a more straightforward explanation of drug action, challenges still remain before fragmented biophysical or mathematical descriptions can be translated into unified biological knowledge. Firstly, computational models should be coupled with existing biological and clinical knowledge. The coupling will allow us to evaluate the model, generate new hypotheses, or identify knowledge gaps. Secondly, a mechanistic understanding of drug actions requires the combination of molecular, network, phenotype, and other models. However, these models are developed independently at different scales and using different modeling languages. Thus, integrating them into a unified model is not straightforward. Finally, mathematical languages used for modeling may not be comprehended easily by biologists and clinicians trying to establish causal relationships between genetic mutations, molecular interactions, and network modulations and pathophysiological processes and clinical outcomes. It is necessary to translate mathematical language into not only accessible human knowledge but also a machine-readable representation for automated reasoning. To address these challenges, effective knowledge representations of input-output relationships from systems pharmacology modeling, which can be understood by the researcher and read by machine, may be needed in addition to the development of ontologies that enable efficient communications between models, as discussed in the preceding section.
Using the Semantic Web for Knowledge Integration
The semantic web has promised to turn big data into linked and smart data and has emerged as a powerful technique for knowledge integration in systems biology (77) and health care (78). In the semantic web, knowledge is represented by the resource description framework (RDF) (http://www.w3.org/RDF/) in the form of subject-predicate-object triples. Domain knowledge is modeled as a graph of triples. The graph model is stored in RDF triple database management systems. The query language SPARQL has been developed to retrieve information from the RDF triple store. The Web Ontology Language (OWL) has been proposed to support database queries and rule-based technologies.
As semantic web technologies have matured, researchers have exploited them to link heterogeneous data sets into a unified knowledge base in systems biology. For example, BioGateway uses the semantic web to integrate the OBO foundry ontology, Gene Ontology, NCBI Taxonomy, and UniProt (79). eXframe provides a reusable framework for creating semantic web repositories of genomics experiments (80). Bio2RDF Release 2 links 19 data sets (81). Many of them are directly relevant to systems pharmacology modeling, such as the Comparative Toxicogenomics Database (82), DrugBank (83), Medical Subject Headings, National Drug Code Directory, Online Mendelian Inheritance in Man (84), or Pharmacogenomics Knowledge Base (85). Bio2RDF includes 57,850,248 unique subjects, 298,470,583 unique objects, 1,003 unique predicates, and 1,101,758,291 triples. In addition, the semantic web has been applied to manage electronic health records aiming to capture, standardize, integrate, describe, and disseminate health-related information (86–89). Researchers have proposed that a semantic data-driven environment is needed to address big data challenges in health care (78). The consolidation of semantic systems biology and semantic health care may provide new opportunities for GWAS, pharmacogenomics, and personalized medicine.
Integration of Systems Pharmacology Modeling with Biological and Clinical Knowledge
The efforts to apply the semantic web to systems biology and health care provide a solid foundation to advance the knowledge integration of systems pharmacology modeling. The incorporation of systems pharmacology modeling into a semantic, rich knowledge base may harness the power of systems pharmacology modeling in generating novel hypotheses and support decision making. It is important to transform the quantitative results from predictive models into logical descriptions or rules between biological entities. Then the logical relationships can be represented as RDF triples. The subject and the object are biological concepts (entities) such as genes. The predicate is the molecular interaction, functional association, or causal relationships between biological entities. For example, a predicted interaction between a chemical A and an agonist conformation of receptor B can be represented as A activates B (Figure 3). An ontology-driven uniformed concept mapping is needed to link genes, proteins, biological pathways, and phenotypes as well as their interactions from diverse models. With the uniformed concept mapping based on the ontologies and the translation of model outputs as an RDF triple, systems pharmacology models can be incorporated into existing semantic-based systems biology and health-care knowledge bases. In this way, software not only handles information to build inferences and test hypotheses but also generates computational and mathematical models that can be interpreted by humans. An example is shown in Figure 3. A knowledge graph has the following triples: Agonist and antagonist binding of peroxisome proliferator-activated receptor (PPAR) upregulate and downregulate the renin-angiotensin-aldosterone system (RAAS), respectively. The upregulation of RAAS increases blood pressure, whereas its downregulation decreases blood pressure. A computational model predicts that a drug binds the agonist or antagonist conformation of PPAR. The model result can be transferred to a triple in the knowledge base. Then the drug response phenotype (in this case, hypertension or hypotension) can be inferred.
Figure 3.
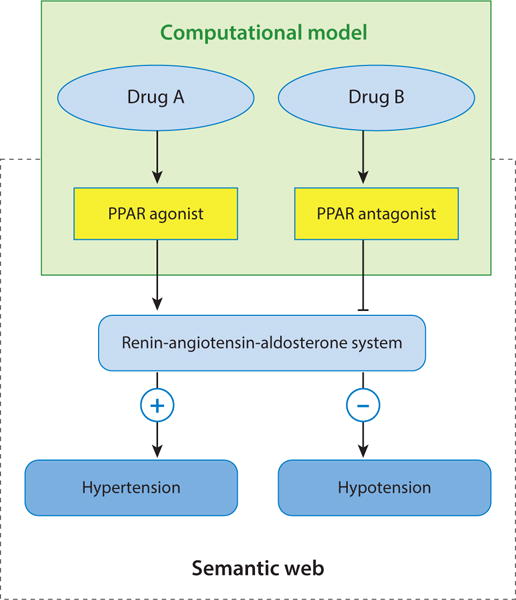
An example to integrate systems pharmacology modeling and the semantic web. The output of systems pharmacology models is translated into an RDF triple and associated with a knowledge base that is built on semantic web technology. The knowledge base will support automated model validation, reasoning, and decision making. Abbreviations: PPAR, peroxisome proliferator-activated receptor; RDF, resource description framework.
In summary, many systems pharmacology modeling approaches can provide only correlation rather than causality relationships between biological variables. One should take extreme caution when making decisions based on the correlation. For example, the correlation of a healthy heart with high levels of high-density lipoprotein (HDL) raises a great interest in developing HDL-targeted therapy for heart disease. However, the causal relationship between HDL and heart disease is unclear. It is not conclusive whether HDL causes a healthy heart or a healthy heart produces HDL (90). Such uncertainty may be the reason for several failed clinical trials in the development of drugs that increase the level of HDL. Beyond model integration, the integration of systems pharmacology modeling with existing biological and medical knowledge will be an important step toward the ultimate goal of using computational modeling to support decision making in drug discovery and clinical practice.
CONCLUSION
The conventional one drug–one target–one disease drug discovery process has been less successful in treating multigene, multifaceted, complex diseases. Systems pharmacology has emerged as a new discipline to tackle the current challenges in drug discovery. Systems pharmacology modeling uses diverse methodologies, integrates multiple omics data, crosses the hierarchy of an organism, spans a wide range of timescales, and addresses the uniqueness of the individual. Successful systems pharmacology modeling requires integrating multiple models to gain an integrated and comprehensive understanding of drug actions under diverse genetic and environmental conditions. Although data integration has already attracted tremendous attention in systems pharmacology, we face new challenges to enable systems pharmacology modeling to be findable, accessible, interoperable, reusable, reliable, interpretable, and actionable. Advances in big data technologies and data science may provide technical solutions to address these challenges. Beyond this, we need new business models— such as the NIH Commons—to prompt open science that is essential for systems pharmacology.
Acknowledgments
We appreciate the reviewers’ and editor’s careful review and constructive suggestions. We thank Dr. Philippe Youkharibache, Dr. Ying Zhang, and Dr. Zheng Zhao for their help in revising the manuscript. This research was supported by the National Library of Medicine of the National Institutes of Health under award number R01LM011986 (L.X.).
Glossary
- Drug action
how a drug interacts with and affects the human body; see agonist and antagonist, below
- Partial agonist
a drug or ligand that binds to a receptor but does not have the efficacy of a full agonist
- Inverse agonist
a drug or ligand that binds to the same receptor as an agonist but produces different effects
- Polypharmacology
designing drugs to work on many different targets, diseases, or both
- Semantic web
“[A] common framework that allows data to be shared and reused across application, enterprise, and community boundaries” (https://www.w3.org/RDF/FAQ)
- Machine learning
subfield of computer science; builds a model from an example data set of observations and makes predictions or decisions for a new data point
- Elastic network model
method to study macromolecular movements at short and long timescales; the macromolecule is represented as a mass-and-spring network
- Drugome
network with nodes represented by drugs and receptors and edges representing interactions between nodes; used to predict which drugs can be repurposed
- Heterogeneous models
models that use different data modalities or different algorithms
- Ontology
entities within a domain and their associated relationships; some ontologies are machine readable and actionable (e.g., the gene ontology)
- Stoichiometric model
see constraint-based metabolic network model
- Flux constraint
see constraint-based metabolic network model
- Constraint-based metabolic network model
shows metabolic pathways separated into metabolites, reactions, and enzymes, with each reaction constrained by each metabolite’s concentration, also known as flux or stoichiometry
- Active learning
special case of semisupervised machine learning; a learning algorithm queries the user (or another information source) interactively to obtain desired outputs at new data points
- Case-based reasoning (CBR)
a way to apply solutions from similar problems to the current problem
- Pharmacometrics model
model used to relate drugs and patients using pharmacology (pharmacokinetics and pharmacodynamics), physiology, and disease
- Vertical model integration
integrating models across different scales
- Horizontal model integration
integrating models on the same scale
- Ensemble learning
a machine learning method that averages over multiple models to establish one strong learner
- Data fusion
a technique that uses multiple data sets from different data modalities to build predictive models
- Sensitivity
true positive rate or recall, or the proportion of positives correctly classified; TP/(TP + FN), where TP = number of true positives and FN= number of false negatives
- Data modality
type of data that describes the problem of interest
- Multikernel learning
machine learning methods that use a predefined set of kernels and learn an optimal linear or nonlinear combination of kernels
- Kernel
a similarity function between data points
- Kernel-based matrix factorization
method to project data (i.e., drug compounds and target proteins in pharmacological models) into a subspace with lower dimensionality using kernel functions that estimate interactions (62)
- Multiview ensemble learning
combining models from multiple sources or views; each view is created by splitting features into subsets (70)
- Random Prism
Prism algorithm–based ensemble learning method that learns a set of IF-THEN rules
- Random Forest
an ensemble learning method that reduces overfitting to the training set by combining multiple decision trees
- Mechanistic-based model
a model that has a clear causal relationship between an object and its prediction
- RDF
resource description framework to model subject (resource), predicate (traits), and object expressions, known as an RDF triple
- Agonist
a drug or ligand that activates a receptor when bound
- Antagonist
a drug or ligand that blocks or limits the effects of an agonist
- Knowledge graph
multiple RDFs are linked as a graph
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
The Annual Review of Pharmacology and Toxicology is online at pharmtox.annualreviews.org
LITERATURE CITED
- 1.Kenakin T, Christopoulos A. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov. 2013;12:205–16. doi: 10.1038/nrd3954. [DOI] [PubMed] [Google Scholar]
- 2.Nussinov R, Tsai CJ, Csermely P. Allo-network drugs: harnessing allostery in cellular networks. Trends Pharmacol Sci. 2011;32:686–93. doi: 10.1016/j.tips.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xie L, Kinnings SL, Bourne PE. Novel computational approaches to polypharmacology as a means to define responses to individual drugs. Annu Rev Pharmacol Toxicol. 2012;52:361–79. doi: 10.1146/annurev-pharmtox-010611-134630. [DOI] [PubMed] [Google Scholar]
- 4.Xie L, Evangelidis T, Xie L, Bourne PE. Drug discovery using chemical systems biology: weak inhibition of multiple kinases may contribute to the anti-cancer effect of Nelfinavir. PLOS Comput Biol. 2011;7:e1002037. doi: 10.1371/journal.pcbi.1002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao S, Iyengar R. Systems pharmacology: network analysis to identify multiscale mechanisms of drug action. Annu Rev Pharmacol Toxicol. 2012;52:505–21. doi: 10.1146/annurev-pharmtox-010611-134520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xie L, Ge X, Tan H, Xie L, Zhang YL, et al. Towards structural systems pharmacology to study complex disease and personalized medicine. PLOS Comput Biol. 2014;10:e1003554. doi: 10.1371/journal.pcbi.1003554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sorger PK, Allerheiligen SRB, Abernethy DR, Altman RB, Brouwer KLR, et al. Quantitative and systems pharmacology in the post-genomic era: new approaches to discovering drugs and understanding therapeutic mechanisms. White Pap. QSP Workshop Group, Natl. Inst. Health; Bethesda, MD: 2011. https://www.nigms.nih.gov/Training/Documents/SystemsPharmaWPSorger2011.pdf. [Google Scholar]
- 8.Zerhouni EA. Turning the Titanic. Sci Transl Med. 2014;6:221ed2. doi: 10.1126/scitranslmed.3008294. [DOI] [PubMed] [Google Scholar]
- 9.Ma’ayan A, Rouillard AD, Clark NR, Wang Z, Duan Q, Kou Y. Lean Big Data integration in systems biology and systems pharmacology. Trends Pharmacol Sci. 2014;35:450–60. doi: 10.1016/j.tips.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hart T, Xie L. Providing data science support for systems pharmacology and its implications to drug discovery. Expert Opin Drug Discov. 2015;11:241–56. doi: 10.1517/17460441.2016.1135126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bourne PE, Lorsch JR, Green ED. Perspective: sustaining the big-data ecosystem. Nature. 2015;527:S16–17. doi: 10.1038/527S16a. [DOI] [PubMed] [Google Scholar]
- 12.Kinnings SL, Liu N, Buchmeier N, Tonge PJ, Xie L, Bourne PE. Drug discovery using chemical systems biology: repositioning the safe medicine Comtan to treat multi-drug and extensively drug resistant tuberculosis. PLOS Comput Biol. 2009;5:e1000423. doi: 10.1371/journal.pcbi.1000423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ng C, Hauptman R, Zhang Y, Bourne PE, Xie L. Anti-infectious drug repurposing using an integrated chemical genomics and structural systems biology approach. Proc Pac Symp Biocomput, Kohala Coast, Hawaii, Jan 3–7. 2014:136–47. [PMC free article] [PubMed] [Google Scholar]
- 14.Ho Sui SJ, Lo R, Fernandes AR, Caulfield MDG, Lerman JA, et al. Raloxifene attenuates Pseudomonas aeruginosa pyocyanin production and virulence. Int J Antimicrob Agents. 2012;40:246–51. doi: 10.1016/j.ijantimicag.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bai JPF, Fontana RJ, Price ND, Sangar V. Systems pharmacology modeling: an approach to improving drug safety. Biopharm Drug Dispos. 2014;35:1–14. doi: 10.1002/bdd.1871. [DOI] [PubMed] [Google Scholar]
- 16.Berger SI, Ma’ayan A, Iyengar R. Systems pharmacology of arrhythmias. Sci Signaling. 2010;3:ra30. doi: 10.1126/scisignal.2000723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berger SI, Iyengar R. Role of systems pharmacology in understanding drug adverse events. Wiley Interdiscip Rev Syst Biol Med. 2011;3:129–35. doi: 10.1002/wsbm.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xie L, Li J, Xie L, Bourne PE. Drug discovery using chemical systems biology: identification of the protein-ligand binding network to explain the side effects of CETP inhibitors. PLOS Comput Biol. 2009;5:e1000387. doi: 10.1371/journal.pcbi.1000387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao S, Nishimura T, Chen Y, Azeloglu EU, Gottesman O, et al. Systems pharmacology of adverse event mitigation by drug combinations. Sci Transl Med. 2013;5:206ra140. doi: 10.1126/scitranslmed.3006548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bordbar A, McCloskey D, Zielinski DC, Sonnenschein N, Jamshidi N, Palsson BO. Personalized whole-cell kinetic models of metabolism for discovery in genomics and pharmacodynamics. Cell Syst. 2015;1:283–92. doi: 10.1016/j.cels.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 21.Yildirim MA, Goh KI, Cusick ME, Barabasi AL, Vidal M. Drug-target network. Nat Biotechnol. 2007;25:1119–26. doi: 10.1038/nbt1338. [DOI] [PubMed] [Google Scholar]
- 22.Kinnings SL, Xie L, Fung K, Xie L, Bourne PE. The Mycobacterium tuberculosis drugome and its polypharmacological implications. PLOS Comput Biol. 2010;6:e100976. doi: 10.1371/journal.pcbi.1000976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bordbar A, Monk JM, King ZA, Palsson BO. Constraint-based models predict metabolic and associated cellular functions. Nat Rev Genet. 2014;15:107–20. doi: 10.1038/nrg3643. [DOI] [PubMed] [Google Scholar]
- 24.Moraru II, Schaff JC, Slepchenko BM, Blinov ML, Morgan F, et al. Virtual cell modelling and simulation software environment. IET Syst Biol. 2008;2:352–62. doi: 10.1049/iet-syb:20080102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiu SH, Xie L. Toward high-throughput predictive modeling of protein binding/unbinding kinetics. 2015 doi: 10.1021/acs.jcim.5b00632. bioRxiv:10.1101/024513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garijo D, Kinnings SL, Xie L, Xie L, Zhang YL, et al. Quantifying reproducibility in computational biology: the case of the tuberculosis drugome. PLOS ONE. 2013;8:e80278. doi: 10.1371/journal.pone.0080278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klepeis JL, Lindorff-Larsen K, Dror RO, Shaw DE. Long-timescale molecular dynamics simulations of protein structure and function. Curr Opin Struct Biol. 2009;19:120–27. doi: 10.1016/j.sbi.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 28.Deshpande N, Addess KJ, Bluhm WF, Merino-Ott JC, Townsend-Merino W, et al. The RCSB Protein Data Bank: a redesigned query system and relational database based on the mmCIF schema. Nucleic Acids Res. 2005;33:D233–37. doi: 10.1093/nar/gki057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40:D1100–7. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bairoch A, Apweiler R, Wu CH, Barker WC, Boeckmann B, et al. The Universal Protein Resource (UniProt) Nucleic Acids Res. 2005;33:D154–59. doi: 10.1093/nar/gki070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chindelevitch L, Trigg J, Regev A, Berger B. An exact arithmetic toolbox for a consistent and reproducible structural analysis of metabolic network models. Nat Commun. 2014;5:4893. doi: 10.1038/ncomms5893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Daniels NM, Gallant A, Peng J, Cowen LJ, Baym M, Berger B. Compressive genomics for protein databases. Bioinformatics. 2013;29:i283–90. doi: 10.1093/bioinformatics/btt214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loh PR, Baym M, Berger B. Compressive genomics. Nat Biotechnol. 2012;30:627–30. doi: 10.1038/nbt.2241. [DOI] [PubMed] [Google Scholar]
- 34.Yu YW, Daniels NM, Danko DC, Berger B. Entropy-scaling search of massive biological data. Cell Syst. 2015;1:130–40. doi: 10.1016/j.cels.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feltus FA, Breen JR, III, Deng J, Izard RS, Konger CA, et al. The widening gulf between genomics data generation and consumption: a practical guide to big data transfer technology. Bioinform Biol Insights. 2015;2015(Suppl. 1):9–19. doi: 10.4137/BBI.S28988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Drager A, Palsson BO. Improving collaboration by standardization efforts in systems biology. Front Bioeng Biotechnol. 2014;2:61. doi: 10.3389/fbioe.2014.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steffensen JL, Dufault-Thompson K, Zhang Y. PSAMM: a Portable System for the Analysis of Metabolic Models. PLOS Comput Biol. 2016;12:e1004732. doi: 10.1371/journal.pcbi.1004732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taylor CF, Field D, Sansone SA, Aerts J, Apweiler R, et al. Promoting coherent minimum reporting guidelines for biological and biomedical investigations: the MIBBI project. Nat Biotechnol. 2008;26:889–96. doi: 10.1038/nbt.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Le Novere N, Finney A, Hucka M, Bhalla US, Campagne F, et al. Minimum information requested in the annotation of biochemical models (MIRIAM) Nat Biotechnol. 2005;23:1509–15. doi: 10.1038/nbt1156. [DOI] [PubMed] [Google Scholar]
- 40.Juty N, Le Novère N, Laibe C. Identifiers.org and MIRIAM Registry: community resources to provide persistent identification. Nucleic Acids Res. 2012;40:D580–86. doi: 10.1093/nar/gkr1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laibe C, Le Novère N. MIRIAM Resources: tools to generate and resolve robust cross-references in Systems Biology. BMC Syst Biol. 2007;1:58. doi: 10.1186/1752-0509-1-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Swat MJ, Moodie S, Wimalaratne SM, Kristensen NR, Lavielle M, et al. Pharmacometrics Markup Language (PharmML): opening new perspectives for model exchange in drug development. CPT Pharmacomet Syst Pharmacol. 2015;4:316–19. doi: 10.1002/psp4.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goecks J, Nekrutenko A, Taylor J. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Torreno O, Krieger MT, Heinzlreiter P, Trelles O. Pairwise genome comparison workflow in the cloud using Galaxy. Procedia Comput Sci. 2015;51:2864–68. [Google Scholar]
- 45.Ebrahim A, Almaas E, Bauer E, Bordbar A, Burgard AP, et al. Do genome-scale models need exact solvers or clearer standards? Mol Syst Biol. 2015;11:831. doi: 10.15252/msb.20156157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chindelevitch L, Trigg J, Regev A, Berger B. Reply to “Do genome-scale models need exact solvers or clearer standards?”. Mol Syst Biol. 2015;11:830. doi: 10.15252/msb.20156548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chang RL, Xie L, Xie L, Bourne PE, Palsson B. Drug off-target effects predicted using structural analysis in the context of a metabolic network model. PLOS Comput Biol. 2010;6:e1000938. doi: 10.1371/journal.pcbi.1000938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang RL, Xie L, Bourne PE, Palsson BO. Antibacterial mechanisms identified through structural systems pharmacology. BMC Syst Biol. 2013;7:102. doi: 10.1186/1752-0509-7-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jerby L, Ruppin E. Predicting drug targets and biomarkers of cancer via genome-scale metabolic modeling. Clin Cancer Res. 2012;18:5572–84. doi: 10.1158/1078-0432.CCR-12-1856. [DOI] [PubMed] [Google Scholar]
- 50.Waltemath D, Adams R, Beard DA, Bergmann FT, Bhalla US, et al. Minimum Information About a Simulation Experiment (MIASE) PLOS Comput Biol. 2011;7:e1001122. doi: 10.1371/journal.pcbi.1001122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Warmuth MK, Liao J, Rätsch G, Mathieson M, Putta S, Lemmen C. Active learning with support vector machines in the drug discovery process. J Chem Inf Comput Sci. 2003;43:667–73. doi: 10.1021/ci025620t. [DOI] [PubMed] [Google Scholar]
- 52.Wang C, Hu G, Wang K, Brylinski M, Xie L, Kurgan L. PDID: database of molecular-level putative protein–drug interactions in the structural human proteome. Bioinformatics. 2015;32:579–86. doi: 10.1093/bioinformatics/btv597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.King ZA, Lu J, Drager A, Miller P, Federowicz S, et al. BiGG Models: a platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res. 2016;44:D515–22. doi: 10.1093/nar/gkv1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harnisch L, Matthews I, Chard J, Karlsson MO. Drug and disease model resources: a consortium to create standards and tools to enhance model-based drug development. CPT Pharmacomet Syst Pharmacol. 2013;2:e34. doi: 10.1038/psp.2013.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Derry JM, Mangravite LM, Suver C, Furia MD, Henderson D, et al. Developing predictive molecular maps of human disease through community-based modeling. Nat Genet. 2012;44:127–30. doi: 10.1038/ng.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Breiman L. Stacked regressions. Mach Learn. 1996;24:49–64. [Google Scholar]
- 57.Capriotti E, Altman RB, Bromberg Y. Collective judgment predicts disease-associated single nucleotide variants. BMC Genom. 2013;14(Suppl. 3):S2. doi: 10.1186/1471-2164-14-S3-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ngiam J, Khosla A, Kim M, Nam J, Lee H, Ng AY. Multimodal deep learning. Proc 28th Int Conf Mach Learn (ICML-11) 2011:689–96. [Google Scholar]
- 59.Costello JC, Heiser LM, Georgii E, Gonen M, Menden MP, et al. A community effort to assess and improve drug sensitivity prediction algorithms. Nat Biotechnol. 2014;32:1202–12. doi: 10.1038/nbt.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gonen M. Predicting drug-target interactions from chemical and genomic kernels using Bayesian matrix factorization. Bioinformatics. 2012;28:2304–10. doi: 10.1093/bioinformatics/bts360. [DOI] [PubMed] [Google Scholar]
- 61.Ng S, Collisson EA, Sokolov A, Goldstein T, Gonzalez-Perez A, et al. PARADIGM-SHIFT predicts the function of mutations in multiple cancers using pathway impact analysis. Bioinformatics. 2012;28:i640–46. doi: 10.1093/bioinformatics/bts402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rosenbaum L, Dorr A, Bauer MR, Boeckler FM, Zell A. Inferring multi-target QSAR models with taxonomy-based multi-task learning. J Cheminformatics. 2013;5:33. doi: 10.1186/1758-2946-5-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Panda B, Herbach JS, Basu S, Bayardo RJ. PLANET: massively parallel learning of tree ensembles with MapReduce. Proc. VLDB ’09; Aug. 24–28; Lyon, Fr.. 2009. pp. 1426–37. [Google Scholar]
- 64.Palit I, Reddy CK. Scalable and parallel boosting with MapReduce. Knowl Data Eng IEEE Trans. 2012;24:1904–16. [Google Scholar]
- 65.Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, et al. Schizophrenia risk from complex variation of complement component 4. Nature. 2016;530:177–83. doi: 10.1038/nature16549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McGranahan N, Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell. 2015;27:15–26. doi: 10.1016/j.ccell.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 67.Tsimring LS. Noise in biology. Rep Prog Phys. 2014;77:026601. doi: 10.1088/0034-4885/77/2/026601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Long PM, Servedio RA. Random classification noise defeats all convex potential boosters. Mach Learn. 2010;78:287–304. [Google Scholar]
- 69.Papakonstantinou PA, Xu J, Cao Z. Bagging by design (on the suboptimality of bagging) Proc Twenty-Eighth AAAI Conf Artif Intell. 2014:2041–47. [Google Scholar]
- 70.Kumar V, Minz S. Multi-view ensemble learning: an optimal feature set partitioning for high-dimensional data classification. Knowledge Inf Syst. 2015;2015:1–59. [Google Scholar]
- 71.Aamodt A, Plaza E. Case-based reasoning: foundational issues, methodological variations, and system approaches. Artif Intell Commun. 1994;7:39–59. [Google Scholar]
- 72.Epstein SL, Yun X, Xie L. Multi-agent, multi-case-based reasoning. In: Diáz-Agudo B, Watson I, editors. Case-Based Reasoning Research and Development: International Conference on Case-Based Reasoning 2012. Berlin: Springer; 2012. pp. 74–88. [Google Scholar]
- 73.Kononenko I, Štrumbelj E, Bosnic Z, Pevec D, Kukar M, Robnik-Šikonja M. Explanation and reliability of individual predictions. Informatica. 2013;37:41–48. [Google Scholar]
- 74.Kukar M. Transductive reliability estimation for individual classifications. In: Dai H, Liu JN, Smirnov E, editors. Machine Learning and Data Mining: Reliable Knowledge Discovery. Cham, Switz: Springer; 2012. pp. 3–27. [Google Scholar]
- 75.Stahl F, Bramer M. Random Prism: a noise-tolerant alternative to Random Forests. Expert Syst. 2014;31:411–20. [Google Scholar]
- 76.Vidovic MM-C, Görnitz N, Müller K-R, Rätsch G, Kloft M. Opening the black box: revealing interpretable sequence motifs in kernel-based learning algorithms. In: Appice A, Rodrigues PP, Costa VS, Gama J, Jorge A, Soares C, editors. Lecture Notes in Computer Science, Vol. 9285: Machine Learning and Knowledge Discovery in Databases. Cham, Switz: Springer; 2015. pp. 137–53. [Google Scholar]
- 77.Antezana E, Mironov V, Kuiper M. The emergence of semantic systems biology. N Biotechnol. 2013;30:286–90. doi: 10.1016/j.nbt.2012.11.008. [DOI] [PubMed] [Google Scholar]
- 78.Panahiazar M, Taslimitehrani V, Jadhav A, Pathak J. Empowering personalized medicine with big data and semantic web technology: promises, challenges, and use cases. Proc Big Data, 2014 IEEE Int Conf. 2014:790–95. doi: 10.1109/BigData.2014.7004307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Antezana E, Blonde W, Egana M, Rutherford A, Stevens R, et al. BioGateway: a semantic systems biology tool for the life sciences. BMC Bioinform. 2009;10(Suppl. 10):S11. doi: 10.1186/1471-2105-10-S10-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Merrill E, Corlosquet S, Ciccarese P, Clark T, Das S. Semantic Web repositories for genomics data using the eXframe platform. J Biomed Semantics. 2014;5:1. doi: 10.1186/2041-1480-5-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Callahan A, Cruz-Toledo J, Ansell P, Dumontier M. Bio2RDF release 2: improved coverage, interoperability and provenance of life science linked data. In: Cimiano P, Corcho O, Presutti V, Hollink L, Rudolph S, editors. Lecture Notes in Computer Science, Vol. 7882: The Semantic Web: Semantics and Big Data. Cham, Switz: Springer; 2013. pp. 200–12. [Google Scholar]
- 82.Davis AP, Murphy CG, Johnson R, Lay JM, Lennon-Hopkins K, et al. The Comparative Toxicogenomics Database: update 2013. Nucleic Acids Res. 2013;41:D1104–14. doi: 10.1093/nar/gks994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wishart DS, Knox C, Guo AC, Cheng D, Shrivastava S, et al. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008;36:D901–6. doi: 10.1093/nar/gkm958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33:D514–17. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Thorn CF, Klein TE, Altman RB. PharmGKB: the pharmacogenetics and pharmacogenomics knowledge base. Methods Mol Biol. 2005;311:179–91. doi: 10.1385/1-59259-957-5:179. [DOI] [PubMed] [Google Scholar]
- 86.Robles M, Fernández-Breis JT, Maldonado JA, Moner D, Martínez-Costa C, et al. ResearchEHR: use of semantic web technologies and archetypes for the description of EHRs. Stud Health Technol Inform. 2010;155:129. [PubMed] [Google Scholar]
- 87.Tao C, Pathak J, Welch SR, Bouamrane M-M, Huff SM, Chute CG. Toward semantic web based knowledge representation and extraction from electronic health records. Proc Int Workshop Manag Interoperability Complex Health Syst. 2011:75–78. [Google Scholar]
- 88.Lozano-Rubí R, Pastor X, Lozano E. OWLing clinical data repositories with the ontology web language. JMIR Med Inform. 2014;2:e14. doi: 10.2196/medinform.3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pathak J, Kiefer RC, Bielinski SJ, Chute CG. Applying semantic web technologies for phenome-wide scan using an electronic health record linked Biobank. J Biomed Semantics. 2012;3:10. doi: 10.1186/2041-1480-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rader DJ, Hovingh GK. HDL and cardiovascular disease. Lancet. 2014;384:618–25. doi: 10.1016/S0140-6736(14)61217-4. [DOI] [PubMed] [Google Scholar]