

Abstract
Ras/Rap1-specific endopeptidase (RRSP) is a cytotoxic effector domain of Multifunctional-autoprocessing repeats-in-toxins (MARTX) toxin of highly virulent strains of Vibrio vulnificus. RRSP blocks RAS-MAPK kinase signaling by cleaving Ras and Rap1 within the Switch I region between Y32 and D33. Although the RRSP processing site is highly conserved among small GTPases, only Ras and Rap1 have been identified as proteolytic substrates. Here we report that Y32-D33 residues at the scissile bond play an important role for RRSP substrate recognition, while nucleotide state of Ras has only a minimal effect. In addition, substrate specificity is generated by residues across the entire Switch I region. Indeed, swapping the Ras Switch I region into either RalA or RhoA, GTPases that are not recognized by RRSP, generated chimeras that are substrates of RRSP. However, a difference in processing efficiency of the Ras Switch I in the context of Ras, RalA, or RhoA indicates that protein regions outside the Ras Switch I also contribute to efficient RRSP substrate recognition. Moreover, we show that synthetic peptides corresponding to the Ras and Rap1, but not RalA, Switch I regions are cleaved by RRSP demonstrating sequence specific substrate recognition. In conclusion, this work demonstrates that the GTPase recognition of RRSP is nucleotide independent, and is mainly driven by the Ras and Rap1 Switch I loop and also influenced by additional protein-protein interactions, increasing the substrate specificity of RRSP.
Keywords: bacterial toxin, Ras protein, Ras-related protein 1, GTPase KRas, bacterial pathogenesis, RRSP, endopeptidase, substrate specificity
TEXT Vibrio vulnificus is a motile Gram-negative foodborne pathogen that can cause life-threatening infections. Ingestion of contaminated seafood or the exposure of open wounds to water harboring V. vulnificus can result in fatal septicemia, gastroenteritis and necrotizing fasciitis. The mortality rate of human subjects with liver damage or compromised immune systems can be greater than 50%1, 2. The severity of V. vulnificus infection is due to the secretion of the cytolytic pore-forming V. vulnificus cytolysin (VVC) and the multifunctional autoprocessing repeats-in-toxin (MARTXVv) toxin, which together diminish the innate immune response and induce tissue necrosis at the site of infection, helping the bacterial to invade and spread systemically3–10. In particular, MARTXVv has been recognized as the most significant virulence factor for V. vulnificus pathogenesis5–9, 11, 12.
MARTX toxins are large protein toxins (3500–5300 aa) secreted by Gram-negative bacteria13. MARTX toxins may carry up to five different enzymatically active effector domains that are delivered into the eukaryotic cytosol across the plasma membrane13. Once in the cytosol, the MARTX cysteine protease domain (CPD) binds inositol hexakisphosphate, initiating autoprocessing of the toxin at leucine residues located in unstructured regions that link the effector domains14. When released in target cells, each effector domain has a unique enzymatic function, resulting in cytopathicity or cytotoxicity15–20.
The toxin effector designated Domain of Unknown Function in position 5 (DUF5) is carried by some, but not all, V. vulnificus MARTXVv toxins21. Notably, a V. vulnificus strain producing a MARTXVv toxin with DUF5 shows about 50-fold higher LD50 in mice than strains producing a toxin without DUF521. In addition, delivery of DUF5 alone into cells results in cytotoxicity characterized by cell rounding and lack of cell proliferation15, 22. In order to understand how DUF5 dramatically increases the virulence potential of V. vulnificus, we recently solved its molecular mechanism within cells. DUF5 directly inhibits the mitogen-activated protein kinases pathway (MAPK) by proteolytically processing the small guanosine triphosphatase (GTPase) Ras, thereby preventing the phosphorylation of extracellular signal–regulated kinases (ERK) and blocking cell proliferation22.
Ras is the representative protein of the small GTPase superfamily that includes more than 150 proteins separated into five families: Ras, Rho, Rab, Arf, and Ran23. These small GTPases control various cell processes by alternating conformational states finely regulated by guanine-nucleotide-exchange factors (GEFs), which promote formation of the GTP-bound active state, and by GTPase-activating proteins (GAPs), which accelerate the intrinsic GTPase activity to promote formation of the GDP-bound inactive state23–25. In particular, the conformation of the Switch I region (residues 30–38 in Ras) in the GTP-bound state is essential for binding of effectors and activation of downstream signaling cascades26–28. DUF5 was found to cleave Ras between Y32 and D33 within the Switch I region22.
There are three human RAS genes (NRas, HRas and KRas) that encode four Ras proteins: NRas, HRas, and two isoforms of KRas, of which KRas4B is the predominant splice variant expressed in many tissues 29. The primary sequence of the four Ras proteins is highly conserved (85–87% amino acid sequence identity) and they share many structural and biochemical properties. The N-terminal G domain (residues 5–166) is involved in the nucleotide binding and GTP hydrolysis including the highly conserved Switch I region (Fig. 2). The hypervariable regions (HVRs) at the C-termini are subject to different post-translational modifications that direct Ras isoforms to different subcellular locations23, 30. In our previous study, we found that DUF5 cleaves full-length KRas, NRas and HRas equally well, demonstrating that the different C-terminal HVRs among Ras isoforms are not involved in substrate recognition by the endopeptidase22.
Figure 2.

Scissile bond residues in KRas Y32 are important for RRSP specificity. A–C SDS-PAGE analysis of in vitro cleavage products of 10 μM rKRas as indicated incubated for 10 min at 37° C with and without 10 μM rRRSP. B–D Time course experiment to assess RRSP activity for rKRas as indicated. Each curve represents the average and standard deviation of three independent biological replicates. WT KRas curve is the same in panel B and D because experiments were conducted simultaneously.
The Ras subfamily, in addition to KRas, HRas, and NRas, includes other similar GTPases like Ras-related proteins 1 and 2 (Rap1 and Rap2), as well as Ral, Rheb, and Rit proteins. These other subfamily members regulate many cellular functions including cell proliferation, differentiation and survival, cell adhesion, mTOR signaling, and neuronal differentiation31–34. Among several small GTPases tested belonging to this Ras subfamily, DUF5 was found to efficiently cleave only Rap1 in addition to Ras22, and did not cleave RalA, RheB2, or Rit222. The endopeptidase also did not cleave GTPases from other small GTPase subfamilies, including RhoA, which regulates cytoskeleton assembly. Since DUF5 exclusively cleaves the small GTPases Ras and Rap1, this effector domain was renamed Ras/Rap1-specific endopeptidase (RRSP).
Ras itself is the central node in the cell that coordinates extracellular signals to ultimately regulate expression of genes for cell proliferation, differentiation, and survival through its control of the MAPK signaling cascade35. Rap1 mainly controls establishment of cell polarity, activation of integrin-mediated cell adhesion and the regulation of cell–cell contacts31, 36, 37. Interestingly, both Ras and Rap1 control signal transduction in response to bacterial infection, likely accounting for why they are targeted by this bacterial toxin38, 39, although the biochemical basis for the substrate specificity has not been previously investigated.
In this study, we demonstrate the Switch I sequence confers specificity of RRSP for Ras and Rap1. RRSP is able to cleave Ras and Rap1 Switch I peptides corresponding to residues comprised between 28 and 38, but not a peptide from the closely related RalA. In addition, swapping the Ras Switch I sequence into RalA and RhoA converts these non-substrate GTPases into substrates. Differences in the efficiency of processing of these chimeras reveal a contribution of other regions of the substrates to the recognition. The contribution is unlikely to be related to nucleotide binding since RRSP cleaves both GTP- and GDP-bound Ras. Overall, we show that the substrate specificity of RRSP for Ras and Rap1 is mainly driven by sequence specific recognition of the Switch I sequence of Ras and Rap1 with regions outside of the Switch I region contributing to processing efficiency.
EXPERIMENTAL PROCEDURES
Materials
E. coli TOP10 cells were obtained from Life Technologies and E. coli BL21(DE3)Magic and pMCSG7 bacterial expression vector were provided by Andrzej Joachimiak (Argonne National Laboratory, IL). Proteins were expressed in the kanamycin-resistant BL21(DE3)(pMagic) E. coli strain. The pMagic plasmid provides the rare codon tRNA gene argU (40 and A. Joachimiak, personal communication). Recombinant KRas-Tagless (2–185) proteins loaded with GDP and GppNHp were provided by Bindu Lakshman and Andrew Stephen (Frederick National Laboratories for Cancer Research, MD). The amount of nucleotide bound to KRas was detected by high-performance liquid chromatography and the elution times were compared to a standard curve of either GDP/GppNHp. Common reagents were obtained from Sigma-Aldrich, Fisher or VWR. SspI restriction enzyme and Gibson Assembly® Master Mix were obtained from New England Biolabs. Custom DNA oligonucleotide primers and GBlocks™ as listed in Table S1 were purchased from Integrated DNA Technologies (Coralville, IA). Plasmids were purified by using Epoch spin columns or Qiagen Midi Prep kit according to the manufacturer’s recommended protocol. Plasmids were introduced into E. coli. BL21(DE3)(pMagic) by electroporation. Recombinant proteins were purified using ÄKTA Xpress Purifier system (GE Healthcare Bio-Sciences, Pittsburgh, PA). Prepacked columns with Nickel Sepharose resin (Ni-NTA) HisTrap column for affinity chromatography or Superdex 200 (26/60) for size exclusion chromatography (SEC) were purchased from GE Healthcare Bio-Sciences.
Expression and purification of RRSP
The N-terminal 6xHis tagged RRSP was purified over a Ni-NTA HisTrap column followed by SEC using the ÄKTA protein purification system as previously described22.
Cloning, expression and purification of small GTPase
DNA sequences corresponding to RalA (NP_005393.2) and RhoA (NP_001300870.1) were amplified from templates previously described22, using primers designed for Gibson Assembly® (New England Biolabs). PCR products were cloned into the pMCSG7 expression vector previously digested with SspI. The Y32A, D33A and D33E mutations were introduced by site-directed mutagenesis using the pMCSG7-KRas vector as a template. Custom gBlocks™ designed to introduce the modified KRas, RalA and RhoA Switch I sequences were assembled into the SspI-digested pMCSG7 vector using Gibson Assembly®, following the manufacturer’s recommended protocol. Plasmids were confirmed to be accurate by DNA sequencing and then transformed into BL21(DE3)Magic cells. All small GTPases were purified as described above for rRRSP, except all buffers were adjusted to pH 7.5 and contained 10 mM MgCl2.
In vitro RRSP cleavage assay
Small GTPases were incubated with rRRSP proteins at equimolar concentrations (10 μM) in 10 mM Tris pH 7.5, 500 mM NaCl, 10 mM MgCl2 at 37°C for 10 min. The time course cleavage assay the reaction was conducted at 10 min and 30 min, 1, 2, 4, and 8 hours; in addition, for rKRas and rRalA:S1:Ras the cleavage reaction was monitored even at shorter time points such as 1, 3 and 5 min. The time course cleavage assay with tagless GDP-rKRas and also GppNHp-rKRas was performed using 10 μM of GTPases and 10 nM of RRSP and monitored after 2, 4, 8, 16, 32 and 64 min. Reactions were stopped by adding 6X Laemmli sample buffer and incubating the sample at 90°C for 5 min. Proteins were separated on 18% SDS–polyacrylamide gels and visualized using Coomassie stain.
SDS-PAGE analysis and quantification
Cleavage products of GTPases were quantified from scanned SDS–polyacrylamide gels using NIH ImageJ 1.64. The percentage of cleavage product was calculated by the following formula:
where cleaved GTPase is the band running at 18 KDa. Data were fit in GraphPad Prism 6.0 to a one-phase exponential association curve to determine the kobs. Each point of the time course cleavage assay represents the average and standard deviation of results from three independent experiments.
Peptide synthesis and in vitro peptide cleavage assay
Peptides corresponding to Switch I sequence of Ras (FVDEYDPTIE), Rap1 (FVEKYDPTIE), Ral (FVEDYEPTKA) and Ras Switch I Y32A/D33A (FVDEAAPTIE) were synthesized by fast flow Fmoc solid phase peptide synthesis on H-Rink Amide-ChemMatrix resin41. Each peptide was purified using an Agilent Zorbax 300SB-C18 column (5 μm, 9.4 x 250mm) at 7 mL/min and 1–31% acetonitrile (0.1% TFA) over 60 minutes. 10 μM of each peptide was then incubated in the presence or absence of RRSP (10 μM) in 20 mM Tris pH 7.5 with 150 mM NaCl in a 37°C water bath. Time points were pulled at 0, 1, 2, 5, and 8 hours, quenched with 1:1 water (0.1% TFA):acetonitrile (0.1%TFA), and analyzed by Agilent 6520 ESI-Q-TOF mass spectrometer with an Agilent Zorbax 300SB-C3 column (5 μm, 2.1 x 150 mm) at 0.8 mL/min and 1–41% acetonitrile (0.1% formic acid) over 8 minutes.
RESULTS
Y32 and D33 residues of Ras Switch I are important for the recognition by RRSP
RRSP was previously shown22 to effectively process Ras isoforms (H/N/K) and Rap1 within the Switch I loop; the scissile bond in Ras and Rap1 was identified between Y32 and D33 (Fig. 1).
Figure 1.

Switch I sequence alignment and RRSP cleavage site. A Alignment of amino acid sequences of the Switch I sequence of several small GTPases by Clustal Omega and ESPript 3 62. The black arrow between Y32 and D33 indicates the RRSP processing site. The RRSP protein substrates (Ras/Rap1) are located above the black dashed line. Identical amino acid residues are highlighted in red. The accession numbers of amino acid sequences were obtained from NCBI: KRas (NP_004976), HRas (NP_001123914), NRas (NP_002515), Rap1A (CAA00804), Rap1B (CAA00805), RalA (NP_005393), RalB (AAA60250), RhoA (NP_001300870) and RhoB (NP_004031). B Crystal structure of KRas-GDP (PDB 4OBE 63) prepared using PyMOL Molecular Graphics System, Version 1.8 (Schrödinger, LLC). Switch I region is colored in blue and the amino acid Y32 and D33 next to RRSP processing site are shown as stick models.
To define the impact of the scissile pair amino acids on RRSP activity, full-length recombinant KRas4B (rKRas) was purified, mixed at equimolar concentration with rRRSP, incubated at 37°C, and the reaction products were analyzed by SDS-PAGE. As shown previously, rKRas is efficiently cleaved by rRRSP within 10 minutes reducing the electrophoretic mobility of rKRas by ~5 kDa 22. rKRas variants with Y32A and D33A amino acid substitutions were also generated and purified. About 50% of rKRas Y32A and D33A variants were cleaved, indicating that both residues contribute to RRSP specificity, but neither is essential (Fig. 2A). By contrast, a rKRas variant with both Y32A and D33A mutations was found to be resistant to RRSP processing, demonstrating an additive effect of both Y32 and D33 on RRSP specificity (Fig. 2A).
To quantify the processing defect, rKRas and rKRas Y32A, D33A and Y32A/D33A mutant variants were each tested as substrates in a time course experiment. As shown in Fig. 1B and Table 1, the catalytic rates of rRRSP for rKRas Y32A (rRRSP kobs 0.031 min−1) and D33A (rRRSP kobs 0.017 min−1) were slower than for rKRas (rRRSP kobs 2.35 min−1), although complete cleavage of both did occur after 4 h. By contrast, rKRas Y32A/D33A presented no cleavage products at any time points (Fig. 2B).
Table 1.
Catalytic activity of RRSP (kobs) for different substrates.
| Substrate | RRSP kobs (min−1) | RRSP activity reduction (fold-change) |
|---|---|---|
| rKRas | 2.35 ± 0.14 | - |
| rKRas Y32A | 0.031 ± 0.0054 | 75.8 |
| rKRas D33A | 0.017 ± 0.0019 | 138 |
| rKRas Y32F | 0.094 ± 0.0079 | 25.0 |
| rKRas Y32A/D33A | undetectable | - |
| rKRas Y32F/D33A | 0.00073 ± 0.0014 | 2350 |
| rRalA | undetectable | - |
| rKRas:S1:Ral | 0.0041 ± 0.0027 | 573 |
| rRalA:S1:Ras | 0.91 ± 0.095 | 2.58 |
| rRhoA | undetectable | - |
| rKRas:S1:Rho | undetectable | - |
| rRhoA:S1:Ras | 0.0049 ± 0.0074 | 479 |
The loss of activity of RRSP for rKRas Y32A/D33A could be due to either dramatically decreased substrate specificity or the direct involvement of both Y32 and D33 to the cleavage mechanism of RRSP. To define the role of the phenol group of Y32 or the carboxyl group of D33 for RRSP activity, rKRas Y32F and rKRas Y32F/D33A were produced and tested with rRRSP at equimolar concentration. Recombinant rKRas Y32F, showed partial cleavage after 10 minutes, reaching a complete cleavage after 4 hours incubation, similar to the results obtained with the Y32A variant (Fig. 2C). In addition, RRSP kobs with rKRas Y32F as substrate was 0.094 min−1 , about 25-fold lower than for rKRas (Table 1), indicating that the reduced RRSP activity for both rKRas Y32A and Y32F was due to the absence of the hydroxyl group. Thus, while the presence of the hydroxyl group is not essential for catalysis, and this group might play an important role for the interaction between RRSP and the Switch I of Ras.
However, rKRas Y32F/D33A showed almost 35% cleavage only after 8 hours and RRSP catalytic activity was 0.00073 min−1 (Fig. 2C,D and Table 1). This further suggests that the phenyl group of Y32F helps in the recognition of the Switch I by RRSP, but the absence of the carbonyl group in position 33 dramatically reduces the cleavage of the Switch I. Overall, these results show that residues Y32 and D33 play a crucial role for RRSP substrate recognition and interaction, but are not fundamental for RRSP catalysis.
Residues downstream of Y32 are partially involved in RRSP substrate recognition
It was also previously shown22 that RRSP does not cleave RalA, a close relative of Ras and Rap in the Ras subfamily (Fig. 1). If Y32-D33 amino acids are the sole contributors to RRSP specificity, it seems unlikely that RRSP cannot cleave RalA, which has a conservative Y-E amino acid flanking its potential Switch 1 scissile bond (Fig. 1). Thus, recombinant mutant protein rKRas D33E was purified and tested to assess whether RRSP can cleave a Y-E scissile bond. As shown in Fig. 3B rRRSP cleaved almost 90% of rKRas D33E in 10 minutes, demonstrating that the presence of E44 within the RalA Switch I does not explain why RalA is not a substrate of RRSP.
Figure 3.
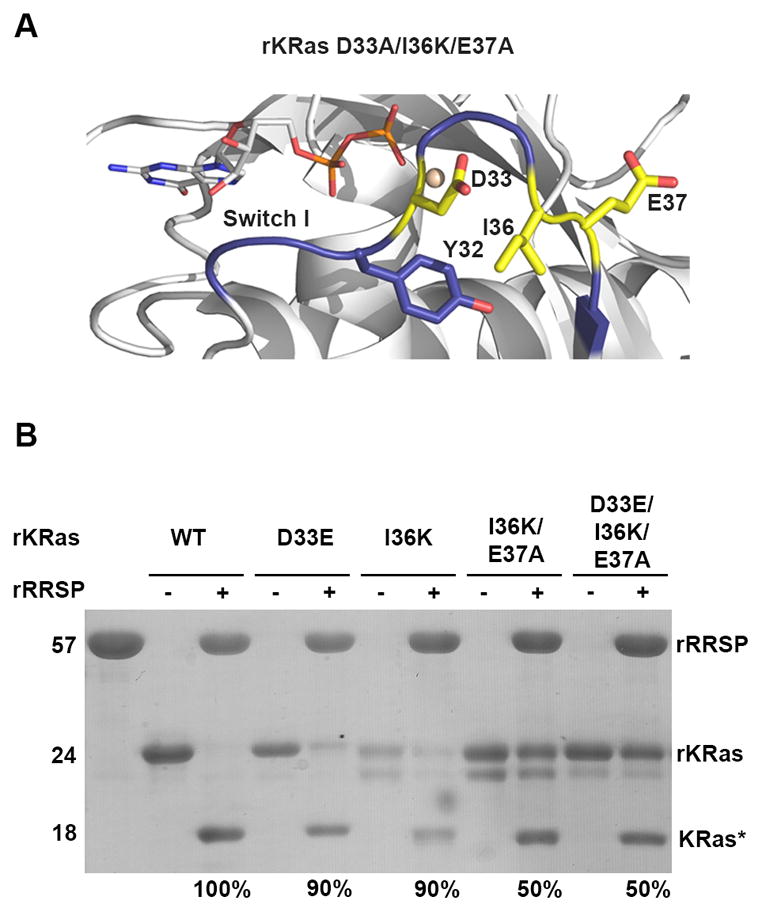
Residues downstream of Y32 are important but not essential for RRSP substrate recognition. A Illustration of the residues of the Ras Switch I region substituted with amino acid corresponding to Ral Switch I. KRas-GDP switch I is colored blue and residues substituted with RalA Switch I amino acids are colored yellow and shown as stick models. B SDS-PAGE analysis of in vitro cleavage products of 10 μM rKRas mutants incubated for 10 minutes at 37°C with and without 10 μM rRRSP, representing at least 3 independent experiments.
RalA Switch I amino acids upstream of the scissile bond are highly conserved with Ras or Rap1, while downstream of D33 the RalA Switch I has K47 and A48 instead of the Ras or Rap1 I36 and E37. These amino acid differences change the overall charge downstream of the scissile bond (Fig. 1). To evaluate their contribution to RRSP substrate specificity, RalA Switch I residues were introduced in the sequence of the rKRas Switch I region, generating the protein variant rKRas I36K and the double mutant protein rKRas I36K/E37A (Fig. 3A). Purified rKRas I36K and rKRas I36K/E37A were each incubated for 10 minutes with RRSP. About 90% of rKRas I36K was cleaved by RRSP, showing that the mutation I36K did not affect the interaction between KRas and RRSP (Fig. 3B). However, rKRas I36K/E37A showed only about 50% of cleaved product, suggesting that there was an effect when both residues were mutated, probably due to the change of the net charge from negative (I36-E37) to positive (K36-A37) (Fig. 3B). Moreover, rKRas D33E/I36K/E37A protein was also purified and tested with RRSP and similar to rKRas I36K/E37A, this protein was also cleaved about 50% (Fig. 3B). Taken together, these results suggest Switch I residues downstream of the scissile bond are important for RRSP specificity, but are not entirely essential.
The total sequence of Ras Switch I is necessary for RRSP specificity
To further investigate the contribution of the entire Switch 1, the KRas Switch I sequence between 30–37 residues was entirely replaced with corresponding amino acids of non-substrate RalA Switch I (residues 41–48), creating rKRas D30E/E31D/D33E/I36K/E37A (rKRas:S1:Ral) (Fig. 4A,B). In 10 min, neither rRalA nor rKRas:S1:Ral was efficiently processed by RRSP (Fig. 4C), indicating that it is the RalA Switch I sequence that is poorly recognized by RRSP. When quantified, the change of Ras Switch I to RalA Switch I decreased the kobs of RRSP from 2.35 min−1 (KRas) to 0.0041 min−1 (KRas:S1:Ral), a reduction of 573-fold (Fig. 4D and Table 1). This resulted in about 40% processing of rKRas:S1:Ral after 8 h (Fig. 4C). However, this residual cleavage of rKRas:S1:Ral compared to a complete absence of cleavage of rRalA reveals that while RRSP can at very low efficiency cleave the RalA Switch I in vitro, this occurs only when appropriately presented to RRSP in the context of the KRas protein, suggesting there may be a contribution of the backbone to RRSP substrate recognition.
Figure 4.

Ras Switch I drives the specificity of RRSP. A Superimposition of crystal structures of KRas-GDP (blue) (PDB 4OBE 63) and RalA-GDP (yellow) (1U8Z 64). Switch I residues are shown as stick models and annotated accordingly to their positions. B Scheme of KRas/RalA chimera constructs designed, expressed and purified. C SDS-PAGE analysis of in vitro cleavage products of 10 μM of rKRas, rRalA, KRas:S1:RalA and RalA:S1:Ras incubated for 10 min at 37°C with and without 10 μM rRRSP. D Time course experiment to assess RRSP activity for recombinant protein substrates, as described for Fig. 2.
RalA but not RhoA backbone interactions contribute to processing by RRSP
To further explore the potential contribution of sequences outside of the Switch I to RRSP substrate specificity, an comparable chimera mimicking the Ras Switch I sequence (rRalA:S1:Ras) was also generated by substituting RalA residues 41–48 with corresponding residues of Ras residues 30–37 ((E41D/D42E/E44D/K47I/A48E)(Fig. 4A,B). Remarkably, rRalA:S1:Ras was efficiently cleaved in 10 minutes, similar to rKRas, demonstrating that swapping the entire Ras Switch I into a non-substrate GTPase converted it to a suitable substrate for RRSP (Fig. 4C). A time course experiment revealed that rRRSP had about 2.6-fold reduced catalytic activity (kobs) for the Ras Switch I when presented by RalA (rRalA:S1:Ras) compared to the natural rKRas substrate (Fig. 4D, Table 1). Thus, a RalA backbone has only a minimal effect on processing efficiency and further confirms that rRalA is not cleaved due its Switch I sequence, not due to the backbone interactions with RRSP.
RalA is a close relative of Ras within the Ras subfamily perhaps accounting for its ability to serve as a presenter of the Ras Switch I to RRSP if backbone interactions impart some specificity of RRSP for Ras and Rap1. To further test the importance of Ras Switch I compared to the backbone, the RhoA Switch I sequence was swapped with the Ras Switch I sequence (RhoA:S1:Ras) (Fig. 5A). As previously shown22, rRhoA was not cleaved by RRSP. Furthermore, rKRas:S1:Rho was not cleaved, indicating that RRSP does not recognize RhoA Switch I even when presented by KRas. By contrast, when the corresponding amino acid positions of RhoA Switch I (residues 31–41) were substituted into KRas (Fig. 5A) RRSP processed rRhoA:S1:Ras about 25% after 10 minutes (Fig. 5B). Interestingly, this shows that compared to rRalA:S1:Ras, which was effectively cleaved in 10 minutes, RRSP is less able to recognize the Ras Switch I when presented in the context of RhoA. Indeed, in a time course experiment, rRhoA:S1:Ras was slowly processed, reaching 90% of cleavage product formation only after 8 hours (Fig. 5C and Table 1). In addition, RRSP for rRhoA:S1:Ras (kobs 0.0049 min−1), was ~185-fold and ~480-fold lower than rRalA:S1:Ras and rKRas, respectively. The partial cleavage of rRhoA:S1:Ras suggests that the RhoA structural region outside of Switch I does not contribute to RRSP specificity, while the more closely related GTPase RalA does provide these interactions. Thus, RRSP substrate recognition is predominantly driven by the Switch I sequence but the efficiency of processing is also influenced by additional protein-protein interactions.
Figure 5.

RRSP cleaves RhoA with Ras Switch I. A Scheme of KRas/RhoA chimera constructs designed, expressed and purified. B SDS-PAGE analysis of in vitro cleavage products of 10 μM of rKRas, rRhoA, KRas:S1:Rho and RhoA:S1:Ras incubated for 10 min at 37°C with and without 10 μM rRRSP. C Time course experiment to assess RRSP activity for recombinant protein substrates, as described for Fig. 2.
RRSP cleaves Ras in the active (GTP-bound) and inactive (GDP-bound) states
The structural conformation of Switch I when bound to nucleotide is known to influence the recognition of downstream effectors and bacterial toxins26, 42, 43. In particular, structural studies of Ras revealed residues Y32 and D33 dramatically change their orientation, depending on whether Ras is bound to GDP or GTP (Fig. S1). To define whether RRSP specificity is nucleotide dependent explaining in part the sequence specificity of Switch I, rKRas proteins loaded with GDP and GppNHp, a non-hydrolysable GTP analog were used. Both GDP- and GppNHp rKRas were mixed at different molar ratios with increasing amounts of recombinant RRSP (rRRSP). At equimolar ratio GDP-rKRas and GppNHp-rKRas were efficiently cleaved by rRRSP (Fig. 6A-inset). Also, at molar ratio 1:10 and 1:100 (rRRSP:rKRas), rRRSP cleaved both GppNHp- and GDP-bound KRas (Fig. 6A).
Figure 6.

RRSP cleaves Ras in the active (GTP-bound) and inactive (GDP-bound) states. A In vitro cleavage of 10 μM rKRas with 10 μM rRRSP (inset) or concentration indicated. B Time course experiment to assess RRSP activity for GDP-rKRas and GppNHp-rKRas as indicated. Each curve represents the average and standard deviation of three independent biological replicates. Each curve present in A and B represents the average and standard deviation of three independent biological replicates.
At molar ratio 1:1000 partial processing was observed of about 80% for GppNHp-rKRas and only 50% for GDP-rKRas (Fig. 6A), indicating a slight preference for GppNHP-KRas as substrate. To quantify this alight difference at molar ratio 1:1000, a time course experiment was conducted to determine the cleavage rates of RRSP for both nucleotide-bound forms. For a reaction with only 0.1 μM rRRSP, GppNHp-rKRas was processed 3-fold faster than GDP-rKRas, although after one hour both rKRas forms were completely cleaved. Thus, the nucleotide state and the orientation of Y32 and D33 on Switch I had only a minor effect on rRRSP cleavage kinetics, demonstrating that RRSP is able to process both GDP- and GTP-bound forms.
RRSP cleaves peptides corresponding to Ras and Rap1 Switch I
All total, our data suggest that RRSP has sequence specificity for the Ras and Rap1 Switch I with GTPase backbone and bound nucleotide contributing to only efficiency. If this is the case, then a peptide alone should be processed by RRSP without a backbone or nucleotide. Peptides corresponding to the Switch I (residues 28–38) of Ras and Rap1 (peptides Ras-S1 and Rap1-S1, respectively) were synthesized and incubated with recombinant RRSP (rRRSP). Each peptide was incubated at equimolar ratio with 10 μM rRRSP at 37°C, the reactions were quenched after 0, 1, 2, 5 and 8 h and the cleavage products were analyzed by liquid chromatography-mass spectrometry (LC-MS). Both Ras-S1 and Rap1-S1 were completely processed after 2 h incubation with rRRSP. The reaction products of Ras-S1 in the presence of rRRSP revealed the formation of a smaller peptide (Fig. 7A, peak 1) with an observed mass of 671.3 Da, corresponding to the N-terminus cleavage product peptide FVDEY. Similarly, a product with observed mass of 684.3 Da (FVEKY) was detected for Rap1-S1 when incubated with rRRSP (Fig. 7B, peak 3). Since RalA Switch I is not a substrate of RRSP (Fig. 5A) a peptide corresponding to Ral Switch I (Ral-S1) was also tested as a substrate. As expected, no cleavage product was detected for Ral-S1 even after 8 hours at 37°C (Fig. 7C, peak 5). Further, exchange of the Ras scissle bond residues to Y32A/D33A generated a non-cleavable peptide. Thus, sequences that are not substrates in the context of intact Ras are also not substrates as peptides. This demonstrates that amino acid differences in the Switch I are sufficient to determine RRSP specificity, discriminating between substrate and non-substrate GTPases. However, the lower efficiency of peptide processing compared to processing of the whole GTPase proteins underscores that the nature of the GTPases and its nucleotide state can contribute to efficiency.
Figure 7.

RRSP cleaves Ras and Rap1 Switch I peptides. In vitro cleavage assay using LC-MS of Switch I peptides was performed at equimolar concentration with rRRSP (10 μM) at different time points. A Results of Ras Switch I peptide (Ras-S1); B Results of Rap1 Switch I peptide (Rap1-S1); C Results of Ral Switch I peptide (Ral-S1). Upper and middle panels show the retention times of the cleavage products Switch I peptides incubated with and without rRRSP. Incubation times are color-coded: time zero (black), 1 h (red), 2 h (green), 4 h (violet) and 8 h (purple). Lower panels indicate the observed and calculated monoisotopic mass values for each Switch I peptides and their cleavage product. D In vitro cleavage assay of Ras Y32A/D33A Switch I peptide was performed at equimolar concentration with 10 μM rRRSP for time indicated.
DISCUSSION
Small GTPases regulate a variety of cellular mechanisms that span from cell proliferation and differentiation to vesicle trafficking and apoptosis. Because of their central role in major cellular pathways, small GTPases are naturally involved in cell defense mechanisms of the immune system, in particular in the activation of the innate immune response against external pathogens39, 44. Host cells use pattern recognition receptors, such as Toll-like receptors (TLRs), to sense pathogen-associated molecular patterns and to activate the immune system. Bacteria are recognized through TLR2, TLR4 and TLR5 by binding lipoproteins, lipopolysaccharide and flagellin, respectively44. This activates the innate and adaptive immune responses, eliminating the pathogens and establishing protective immunity against them. However, bacterial pathogens, through the use of secreted toxins, have developed specific ways to destroy the eukaryotic cell cytoskeleton and to reduce the host response against bacterial infection by inhibiting or completely inactivating the small GTPases45. Among these toxins, some have broad specificity targeting many small GTPases, while others show more limited substrate preference45, 46.
RRSP was recently identified as the first member of an uncharacterized endopeptidase family able to selectively cleave the Switch I of Ras and Rap1 small GTPases, blocking Ras-MAPK signaling22. In this study, we found that the Switch I of either Ras or Rap1 was the minimal sequence required for RRSP recognition. The final demonstration of cleavage of Ras-S1 or Rap1-S1 peptides by RRSP demonstrated that the primary sequence of Switch I between residues 28–38 was sufficient for RRSP to discriminate substrates and identify the scissile bond between Y32 and D33. Since the Switch I sequence is directly involved in the interaction with downstream effectors, the specific recognition of RRSP to this region might indicate similar surface contact. In fact, Ras Switch I residues 30 and 31 are known to be critical for the specific binding to BRaf, a downstream effector of Ras35, 47. Interestingly, changing residues 30 and 31 in Rap1 to those found in the corresponding positions of Ras lead to favorable interaction between Rap1 and BRaf in vitro26. We found that Y32 and D33 play important roles in RRSP activity, as their substitution with alanine residues entirely inhibits the cleavage of Switch I. Although Y32 and D33 at the scissile bond are highly but not completely conserved among other GTPases, RRSP has been shown to cleave only Ras and Rap122. Indeed, the scissile pair does not fully account for substrate specificity, as the Y-E bond found in RalA was a suitable bond when substituted into Ras. In addition, swapping individual residues found in RalA into Ras failed to identify a single residue that would result in complete RRSP loss of activity. Rather, we found that it is the whole Switch I primary sequence, comprised of residues 28–38 that defines the RRSP specificity.
This recognition of the Switch I sequence is a distinct strategy compared to other toxins that target the Switch I of GTPases. The well-characterized toxins TcdA and TcdB of the pathogen Clostridium difficile use UDP-glucose as the sugar donor to mono-O-glucosylate Rho proteins, including RhoA, RhoB, RhoC, RhoG, Rac1 and Cdc4248–50. However, recently Zaiser et al using a proteomic approach have found that TcdA and TcdB also glucosylate Ras proteins (K/H/N) and other Ras subfamily proteins like Rap and Ral51. Clostridium sordellii Lethal Toxin (LT or TcsL) uses mono-O-glucosylation to modify GTPases from both the Rho proteins, like Rac1 and Ras subfamily proteins, including Rac, Ras, Rap and Ral43, 52. The TpeL toxin of Clostridium perfringens is more stringent in its recognition and targets Ras proteins by mono-O-GlcNAcylation using UDP-N-acetylglucosamine (UDP-GlcNAc) as the sugar donor53. All these clostridial toxins mono-O-glucosylate the conserved threonine in the Switch I (T35 or T37 of Rho or Ras proteins, respectively). Glucosylation of T35 and T37 impair GTPase activity, blocking their signaling functions54–56. Since the targeted threonine residue within the Switch I is 100% conserved across all GTPases23, sequence specificity of the modified residue cannot account for the substrate preference of these clostridial toxins.
Instead, for these other toxins, specificity is driven in part by conformation change due to the bound nucleotide. LT preferentially modifies Rho and Ras in GDP-from, as the target hydroxyl group of the threonine residue directly coordinates the magnesium cation and binds the γ-phosphate of the GTP43. By contrast, PaTox of Photorhabdus asymbiotica GlcNAcylation of GDP-loaded RhoA is poor while GTP-RhoA is efficiently modified in the Switch I. Interestingly, PaTox mono-O-glucosylates Y34 of RhoA, the equivalent residue Y32 of Ras, indicating that Y34 is in a favorable and exposed conformation to be modified when RhoA is in its active form42. This precedence established for the glucosyltransferase toxins to discriminate GTP- and GDP-bound forms of target GTPases is found to be quite different for RRSP. In this work, we showed that RRSP efficiently cleaved both GDP- and GTP-KRas, demonstrating that differences in the orientations of Ras Y32 due to nucleotide binding had only a minor 2.8-fold effect on Switch I recognition. In this way, RRSP is more similar to the Clostridium botulinum C3 exoenzyme, which catalyzes the ADP-ribosylation of N41 of RhoA/B/C. C3 is capable of modifying both GTP- and GDP-bound forms of Rho proteins. Similarly to RRSP, the nucleotide occupancy does not affect the enzyme recognition but only the kinetics57. However, since the nucleotide state of any small GTPase is crucial for association withs downstream effectors or guanosine nucleotide dissociation inhibitors in the cell, it is possible RRSP might have different substrate accessibility in the cellular environment due to binding of other proteins.
Among the many toxins that target small GTPases, substrate specificity is also dictated by interactions outside of the Switch I region, but structurally nearby. For TcdB and LT, a single amino acid modification within the Switch II region of RhoB and RhoD was sufficient to change the target specificity of both toxins58. The adenyltransferase IbpA of Histophilus somni also targets the Switch I, by catalyzing the addition of adenosine monophosphate to Y34 of RhoA and to Y32 of both Cdc42 and Rac159. Similarly to TcdB and LT, IbpA also recognizes Rho-family GTPases by binding to the Switch II region60. Moreover, C3 exoenzyme from C. botulinum, which specifically modifies RhoA/B/C but not other members of either Rho or Ras subfamilies, recognizes Switch II and interswitch regions in addition to Switch I61.
Unlike these other toxins, the specificity of RRSP is largely driven by the sequence of Switch I as shown when the entire Ras Switch I was exchanged into RalA, RalA became a substrate for RRSP and the rate of processing decreased only slightly. Yet, when exchanged into RhoA, the processing rate declined dramatically. This suggests that, in addition to a different presentation of the Ras Switch I by RhoA, there is a contribution of the protein backbone to substrate recognition. However, this result also indicates that the Ras Switch I is strictly required for substrate specificity. Notably, the complete cleavage of both Ras-S1 and Rap-S1 peptides by RRSP required two hours, while ten minutes was sufficient for cleavage of intact KRas protein. These data further demonstrate that although the sequence recognition of the Switch I region is mainly responsible for the RRSP specificity, possible additional interactions with the whole GTPase structure increases the recognition and efficiency of RRSP. Indeed, the next question that will need to be addressed is how these other regions of Ras and Rap1 outside the Switch I can contribute to a second level of specificity.
Supplementary Material
Acknowledgments
Funding Sources
This work was supported by the PanCan/FNCLR KRas Fellowship (to M.B); a National Science Foundation Graduate Research Fellowship (to A.E.R.); an MIT/NIGMS Interdepartmental Biotechnology Training Program Award T32GM008334 (to A.R.L.), Massachusetts Institute of Technology start-up fund, a Damon Runyon Cancer Research Foundation Award, and the Sontag Foundation Distinguished Scientist Award (to B.L.P.); and the Northwestern Catalyst Fund and National Institute of Allergy and Infectious Diseases awards R01AI092825 and R01AI098369 (to K.J.F.S).
We thank Bindu Lakshman, Peter Frank and Dr. Andrew Stephen for providing GDP-KRas and GppNHp-KRas (NCI-RAS Initiative, Frederick National Laboratory for Cancer Research). We thank Prof. Andrzej Joachimiak for the gift of the BL21(DE3)(pMagic) and pMCSG7. We also acknowledge Jennifer Wong for the provided technical assistance and Wayne Anderson and Vadim Gaponenko for their imput. The Center of Structural Genomics of Infectious Diseases (CSGID) for the provided access to AKTA Xpress purifier and the Genomics Core Facility at Northwestern University for sequence confirmation of plasmids.
ABBREVIATIONS
- RRSP
Ras/Rap1-specific endopeptidase
- MARTX
Multifunctional-autoprocessing repeats-in-toxins
- MAPK
mitogen-activated protein kinases pathway
- GTPase
guanosine triphosphatase
Footnotes
Author Contributions
M.B. conceived and conducted most of the experiments, analyzed the data and coordinated the study. Z.L. A.R.L. A.E.R., and B.L.P. produced the synthetic peptides, conducted in vitro cleavage studies, and analyzed data. M.B. and K.J.F.S. wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Notes
M.B. and K.J.F.S. hold a patent application covering RRSP application. The remaining authors declare no competing financial interest.
References
- 1.Jones MK, Oliver JD. Vibrio vulnificus: disease and pathogenesis. Infect Immun. 2009;77:1723–1733. doi: 10.1128/IAI.01046-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Daniels NA. Vibrio vulnificus oysters: pearls and perils. Clin Infect Dis. 2011;52:788–792. doi: 10.1093/cid/ciq251. [DOI] [PubMed] [Google Scholar]
- 3.Fan JJ, Shao CP, Ho YC, Yu CK, Hor LI. Isolation and characterization of a Vibrio vulnificus mutant deficient in both extracellular metalloprotease and cytolysin. Infect Immun. 2001;69:5943–5948. doi: 10.1128/IAI.69.9.5943-5948.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gavin HE, Beubier NT, Satchell KJ. The effector domain region of the Vibrio vulnificus MARTX toxin confers biphasic epithelial barrier disruption and is essential for systemic spread from the intestine. PLoS Pathog. 2017;13:e1006119. doi: 10.1371/journal.ppat.1006119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jeong HG, Satchell KJ. Additive function of Vibrio vulnificus MARTX(Vv) and VvhA cytolysins promotes rapid growth and epithelial tissue necrosis during intestinal infection. PLoS Pathog. 2012;8:e1002581. doi: 10.1371/journal.ppat.1002581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim YR, Lee SE, Kook H, Yeom JA, Na HS, Kim SY, Chung SS, Choy HE, Rhee JH. Vibrio vulnificus RTX toxin kills host cells only after contact of the bacteria with host cells. Cell Microbiol. 2008;10:848–862. doi: 10.1111/j.1462-5822.2007.01088.x. [DOI] [PubMed] [Google Scholar]
- 7.Lee JH, Kim MW, Kim BS, Kim SM, Lee BC, Kim TS, Choi SH. Identification and characterization of the Vibrio vulnificus rtxA essential for cytotoxicity in vitro and virulence in mice. J Microbiol. 2007;45:146–152. [PubMed] [Google Scholar]
- 8.Liu M, Alice AF, Naka H, Crosa JH. The HlyU protein is a positive regulator of rtxA1, a gene responsible for cytotoxicity and virulence in the human pathogen Vibrio vulnificus. Infect Immun. 2007;75:3282–3289. doi: 10.1128/IAI.00045-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lo HR, Lin JH, Chen YH, Chen CL, Shao CP, Lai YC, Hor LI. RTX toxin enhances the survival of Vibrio vulnificus during infection by protecting the organism from phagocytosis. J Infect Dis. 2011;203:1866–1874. doi: 10.1093/infdis/jir070. [DOI] [PubMed] [Google Scholar]
- 10.Wright AC, Morris JG., Jr The extracellular cytolysin of Vibrio vulnificus: inactivation and relationship to virulence in mice. Infect Immun. 1991;59:192–197. doi: 10.1128/iai.59.1.192-197.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee CT, Pajuelo D, Llorens A, Chen YH, Leiro JM, Padros F, Hor LI, Amaro C. MARTX of Vibrio vulnificus biotype 2 is a virulence and survival factor. Environ Microbiol. 2013;15:419–432. doi: 10.1111/j.1462-2920.2012.02854.x. [DOI] [PubMed] [Google Scholar]
- 12.Ziolo KJ, Jeong HG, Kwak JS, Yang S, Lavker RM, Satchell KJ. Vibrio vulnificus biotype 3 multifunctional autoprocessing RTX toxin is an adenylate cyclase toxin essential for virulence in mice. Infect Immun. 2014;82:2148–2157. doi: 10.1128/IAI.00017-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Satchell KJ. Structure and function of MARTX toxins and other large repetitive RTX proteins. Annu Rev Microbiol. 2011;65:71–90. doi: 10.1146/annurev-micro-090110-102943. [DOI] [PubMed] [Google Scholar]
- 14.Prochazkova K, Shuvalova LA, Minasov G, Voburka Z, Anderson WF, Satchell KJ. Structural and molecular mechanism for autoprocessing of MARTX toxin of Vibrio cholerae at multiple sites. J Biol Chem. 2009;284:26557–26568. doi: 10.1074/jbc.M109.025510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Antic I, Biancucci M, Satchell KJ. Cytotoxicity of the Vibrio vulnificus MARTX toxin effector DUF5 is linked to the C2A subdomain. Proteins. 2014;82:2643–2656. doi: 10.1002/prot.24628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agarwal S, Kim H, Chan RB, Agarwal S, Williamson R, Cho W, Paolo GD, Satchell KJ. Autophagy and endosomal trafficking inhibition by Vibrio cholerae MARTX toxin phosphatidylinositol-3-phosphate-specific phospholipase A1 activity. Nat Commun. 2015;6:8745. doi: 10.1038/ncomms9745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Agarwal S, Zhu Y, Gius DR, Satchell KJ. The Makes Caterpillars Floppy (MCF)-Like domain of Vibrio vulnificus induces mitochondrion-mediated apoptosis. Infect Immun. 2015;83:4392–4403. doi: 10.1128/IAI.00570-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahrens S, Geissler B, Satchell KJ. Identification of a His-Asp-Cys catalytic triad essential for function of the Rho inactivation domain (RID) of Vibrio cholerae MARTX toxin. J Biol Chem. 2013;288:1397–1408. doi: 10.1074/jbc.M112.396309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kudryashov DS, Cordero CL, Reisler E, Satchell KJ. Characterization of the enzymatic activity of the actin cross-linking domain from the Vibrio cholerae MARTX Vc toxin. J Biol Chem. 2008;283:445–452. doi: 10.1074/jbc.M703910200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim BS, Satchell KJ. MARTX effector cross kingdom activation by Golgi-associated ADP-ribosylation factors. Cell Microbiol. 2016;18:1078–1093. doi: 10.1111/cmi.12568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kwak JS, Jeong HG, Satchell KJ. Vibrio vulnificus rtxA1 gene recombination generates toxin variants with altered potency during intestinal infection. Proc Natl Acad Sci USA. 2011;108:1645–1650. doi: 10.1073/pnas.1014339108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Antic I, Biancucci M, Zhu Y, Gius DR, Satchell KJ. Site-specific processing of Ras and Rap1 Switch I by a MARTX toxin effector domain. Nat Commun. 2015;6:7396. doi: 10.1038/ncomms8396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wennerberg K, Rossman KL, Der CJ. The Ras superfamily at a glance. J Cell Sci. 2005;118:843–846. doi: 10.1242/jcs.01660. [DOI] [PubMed] [Google Scholar]
- 24.Milburn MV, Tong L, deVos AM, Brunger A, Yamaizumi Z, Nishimura S, Kim SH. Molecular switch for signal transduction: structural differences between active and inactive forms of protooncogenic ras proteins. Science. 1990;247:939–945. doi: 10.1126/science.2406906. [DOI] [PubMed] [Google Scholar]
- 25.Wittinghofer A, Pai EF. The structure of Ras protein: a model for a universal molecular switch. Trends Biol Sci. 1991;16:382–387. doi: 10.1016/0968-0004(91)90156-p. [DOI] [PubMed] [Google Scholar]
- 26.Fetics SK, Guterres H, Kearney BM, Buhrman G, Ma B, Nussinov R, Mattos C. Allosteric effects of the oncogenic RasQ61L mutant on Raf-RBD. Structure. 2015;23:505–516. doi: 10.1016/j.str.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Herrmann C. Ras-effector interactions: after one decade. Curr Opin Struct Biol. 2003;13:122–129. doi: 10.1016/s0959-440x(02)00007-6. [DOI] [PubMed] [Google Scholar]
- 28.Herrmann C, Ahmadian MR, Hofmann F, Just I. Functional consequences of monoglucosylation of Ha-Ras at effector domain amino acid threonine 35. J Biol Chem. 1998;273:16134–16139. doi: 10.1074/jbc.273.26.16134. [DOI] [PubMed] [Google Scholar]
- 29.Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci. 2016;129:1287–1292. doi: 10.1242/jcs.182873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ahearn IM, Haigis K, Bar-Sagi D, Philips MR. Regulating the regulator: post-translational modification of RAS. Nat Rev Mol Cell Biol. 2012;13:39–51. doi: 10.1038/nrm3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bos JL, de Rooij J, Reedquist KA. Rap1 signalling: adhering to new models. Nat Rev Mol Cell Biol. 2001;2:369–377. doi: 10.1038/35073073. [DOI] [PubMed] [Google Scholar]
- 32.Dibble CC, Cantley LC. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015;25:545–555. doi: 10.1016/j.tcb.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hazelett CC, Sheff D, Yeaman C. RalA and RalB differentially regulate development of epithelial tight junctions. Mol Biol Cell. 2011;22:4787–4800. doi: 10.1091/mbc.E11-07-0657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi GX, Cai W, Andres DA. Rit subfamily small GTPases: regulators in neuronal differentiation and survival. Cell Signal. 2013;25:2060–2068. doi: 10.1016/j.cellsig.2013.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3:459–465. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- 36.Shimonaka M, Katagiri K, Nakayama T, Fujita N, Tsuruo T, Yoshie O, Kinashi T. Rap1 translates chemokine signals to integrin activation, cell polarization, and motility across vascular endothelium under flow. J Cell Biol. 2003;161:417–427. doi: 10.1083/jcb.200301133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Price LS, Hajdo-Milasinovic A, Zhao J, Zwartkruis FJ, Collard JG, Bos JL. Rap1 regulates E-cadherin-mediated cell-cell adhesion. J Biol Chem. 2004;279:35127–35132. doi: 10.1074/jbc.M404917200. [DOI] [PubMed] [Google Scholar]
- 38.David MD, Cochrane CL, Duncan SK, Schrader JW. Pure lipopolysaccharide or synthetic lipid a induces activation of p21Ras in primary macrophages through a pathway dependent on Src family kinases and PI3K. J Immunol. 2005;175:8236–8241. doi: 10.4049/jimmunol.175.12.8236. [DOI] [PubMed] [Google Scholar]
- 39.Scheele JS, Marks RE, Boss GR. Signaling by small GTPases in the immune system. Immunol Rev. 2007;218:92–101. doi: 10.1111/j.1600-065X.2007.00530.x. [DOI] [PubMed] [Google Scholar]
- 40.Wu N, Christendat D, Dharamsi A, Pai EF. Purification, crystallization and preliminary X-ray study of orotidine 5′-monophosphate decarboxylase. Acta Crystallogr D Biol Crystallogr. 2000;56:912–914. doi: 10.1107/s090744490000576x. [DOI] [PubMed] [Google Scholar]
- 41.Simon MD, Heider PL, Adamo A, Vinogradov AA, Mong SK, Li X, Berger T, Policarpo RL, Zhang C, Zou Y, Liao X, Spokoyny AM, Jensen KF, Pentelute BL. Rapid flow-based peptide synthesis. Chembiochem. 2014;15:713–720. doi: 10.1002/cbic.201300796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jank T, Bogdanovic X, Wirth C, Haaf E, Spoerner M, Bohmer KE, Steinemann M, Orth JH, Kalbitzer HR, Warscheid B, Hunte C, Aktories K. A bacterial toxin catalyzing tyrosine glycosylation of Rho and deamidation of Gq and Gi proteins. Nat Struct Mol Biol. 2013;20:1273–1280. doi: 10.1038/nsmb.2688. [DOI] [PubMed] [Google Scholar]
- 43.Just I, Selzer J, Hofmann F, Green GA, Aktories K. Inactivation of Ras by Clostridium sordellii lethal toxin-catalyzed glucosylation. J Biol Chem. 1996;271:10149–10153. doi: 10.1074/jbc.271.17.10149. [DOI] [PubMed] [Google Scholar]
- 44.Diacovich L, Gorvel JP. Bacterial manipulation of innate immunity to promote infection. Nat Rev Microbiol. 2010;8:117–128. doi: 10.1038/nrmicro2295. [DOI] [PubMed] [Google Scholar]
- 45.Aktories K. Bacterial protein toxins that modify host regulatory GTPases. Nat Rev Microbiol. 2011;9:487–498. doi: 10.1038/nrmicro2592. [DOI] [PubMed] [Google Scholar]
- 46.Lemichez E, Aktories K. Hijacking of Rho GTPases during bacterial infection. Exp Cell Res. 2013;319:2329–2336. doi: 10.1016/j.yexcr.2013.04.021. [DOI] [PubMed] [Google Scholar]
- 47.Nassar N, Horn G, Herrmann C, Block C, Janknecht R, Wittinghofer A. Ras/Rap effector specificity determined by charge reversal. Nat Struct Biol. 1996;3:723–729. doi: 10.1038/nsb0896-723. [DOI] [PubMed] [Google Scholar]
- 48.Just I, Selzer J, Wilm M, von Eichel-Streiber C, Mann M, Aktories K. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature. 1995;375:500–503. doi: 10.1038/375500a0. [DOI] [PubMed] [Google Scholar]
- 49.Just I, Wilm M, Selzer J, Rex G, von Eichel-Streiber C, Mann M, Aktories K. The enterotoxin from Clostridium difficile (ToxA) monoglucosylates the Rho proteins. J Biol Chem. 1995;270:13932–13936. doi: 10.1074/jbc.270.23.13932. [DOI] [PubMed] [Google Scholar]
- 50.Genth H, Dreger SC, Huelsenbeck J, Just I. Clostridium difficile toxins: more than mere inhibitors of Rho proteins. Int J Biochem Cell Biol. 2008;40:592–597. doi: 10.1016/j.biocel.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 51.Zeiser J, Gerhard R, Just I, Pich A. Substrate specificity of clostridial glucosylating toxins and their function on colonocytes analyzed by proteomics techniques. J Proteome Res. 2013;12:1604–1618. doi: 10.1021/pr300973q. [DOI] [PubMed] [Google Scholar]
- 52.Popoff MR, Chaves-Olarte E, Lemichez E, von Eichel-Streiber C, Thelestam M, Chardin P, Cussac D, Antonny B, Chavrier P, Flatau G, Giry M, de Gunzburg J, Boquet P. Ras, Rap, and Rac small GTP-binding proteins are targets for Clostridium sordellii lethal toxin glucosylation. J Biol Chem. 1996;271:10217–10224. doi: 10.1074/jbc.271.17.10217. [DOI] [PubMed] [Google Scholar]
- 53.Guttenberg G, Hornei S, Jank T, Schwan C, Lu W, Einsle O, Papatheodorou P, Aktories K. Molecular characteristics of Clostridium perfringens TpeL toxin and consequences of mono-O-GlcNAcylation of Ras in living cells. J Biol Chem. 2012;287:24929–24940. doi: 10.1074/jbc.M112.347773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sehr P, Joseph G, Genth H, Just I, Pick E, Aktories K. Glucosylation and ADP ribosylation of Rho proteins: effects on nucleotide binding, GTPase activity, and effector coupling. Biochemistry. 1998;37:5296–5304. doi: 10.1021/bi972592c. [DOI] [PubMed] [Google Scholar]
- 55.Barth N, Ziegler A, Himmelmann GW, Coners H, Wabitsch M, Hennighausen K, Mayer H, Remschmidt H, Schafer H, Hebebrand J. Significant weight gains in a clinical sample of obese children and adolescents between 1985 and 1995. Int J Obes Relat Metab Disord. 1997;21:122–126. doi: 10.1038/sj.ijo.0800376. [DOI] [PubMed] [Google Scholar]
- 56.Pai EF, Krengel U, Petsko GA, Goody RS, Kabsch W, Wittinghofer A. Refined crystal-structure of the triphosphate conformation of H-Ras p21 at 1.35 Å resolution - implications for the mechanism of GTP hydrolysis. EMBO J. 1990;9:2351–2359. doi: 10.1002/j.1460-2075.1990.tb07409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wilde C, Genth H, Aktories K, Just I. Recognition of RhoA by Clostridium botulinum C3 exoenzyme. J Biol Chem. 2000;275:16478–16483. doi: 10.1074/jbc.M910362199. [DOI] [PubMed] [Google Scholar]
- 58.Jank T, Pack U, Giesemann T, Schmidt G, Aktories K. Exchange of a single amino acid switches the substrate properties of RhoA and RhoD toward glucosylating and transglutaminating toxins. J Biol Chem. 2006;281:19527–19535. doi: 10.1074/jbc.M600863200. [DOI] [PubMed] [Google Scholar]
- 59.Zekarias B, Mattoo S, Worby C, Lehmann J, Rosenbusch RF, Corbeil LB. Histophilus somni IbpA DR2/Fic in virulence and immunoprotection at the natural host alveolar epithelial barrier. Infect Immun. 2010;78:1850–1858. doi: 10.1128/IAI.01277-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xiao J, Worby CA, Mattoo S, Sankaran B, Dixon JE. Structural basis of Fic-mediated adenylylation. Nat Struct Mol Biol. 2010;17:1004–1010. doi: 10.1038/nsmb.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Toda A, Tsurumura T, Yoshida T, Tsumori Y, Tsuge H. Rho GTPase recognition by C3 exoenzyme based on C3-RhoA complex structure. J Biol Chem. 2015;290:19423–19432. doi: 10.1074/jbc.M115.653220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42:W320–324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hunter JC, Gurbani D, Ficarro SB, Carrasco MA, Lim SM, Choi HG, Xie T, Marto JA, Chen Z, Gray NS, Westover KD. In situ selectivity profiling and crystal structure of SML-8-73-1, an active site inhibitor of oncogenic K-Ras G12C. Proc Natl Acad Sci USA. 2014;111:8895–8900. doi: 10.1073/pnas.1404639111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nicely NI, Kosak J, de Serrano V, Mattos C. Crystal structures of Ral-GppNHp and Ral-GDP reveal two binding sites that are also present in Ras and Rap. Structure. 2004;12:2025–2036. doi: 10.1016/j.str.2004.08.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.