

Abstract
The progesterone receptor (PR) regulates transcriptional programs that drive proliferation, survival, and stem cell phenotypes. Although the role of native progesterone in the development of breast cancer remains controversial, PR clearly alters the transcriptome in breast tumors. This study identifies a class of genes, interferon-stimulated genes (ISGs), potently downregulated by ligand-activated PR which have not been previously shown to be regulated by PR. Progestin-dependent transcriptional repression of ISGs was observed in breast cancer cell line models and human breast tumors. Ligand-independent regulation of ISGs was also observed, as basal transcript levels were markedly higher in cells with PR knockdown. PR repressed ISG transcription in response to interferon treatment, the canonical mechanism through which these genes are activated. Liganded PR is robustly recruited to enhancer regions of ISGs, and ISG transcriptional repression is dependent upon PR’s ability to bind DNA. In response to PR activation, key regulatory transcription factors that are required for interferon-activated ISG transcription, STAT2 and IRF9, exhibit impaired recruitment to ISG promoter regions, correlating with PR/ligand-dependent ISG transcriptional repression. Interferon activation is a critical early step in nascent tumor recognition and destruction through immune surveillance. As the large majority of breast tumors are PR-positive at the time of diagnosis, PR-dependent downregulation of interferon signaling may be a mechanism through which early PR-positive breast tumors evade the immune system and develop into clinically relevant tumors.
Implications: This study highlights a novel transcriptional mechanism through which PR drives breast cancer development and potentially evades the immune system.
Keywords: progesterone receptor, transcription, interferon-stimulated genes
Introduction
There is an emerging role for the ovarian steroid hormone, progesterone, and its receptor, the progesterone receptor (PR), in the development of breast cancer (1–3). Clinical data have shown increased breast cancer incidence in women taking post-menopausal hormone replacement therapy (HRT) whose combined regimens included estrogen and progestins; this increased risk was not present in women taking estrogen-only HRT (4). Although much debate continues regarding the role for native progesterone in breast cancer development, it is clear that PR, alone or in combination with the estrogen receptor (ER), affects the transcriptional landscape of breast cancer (5–8).
PR is a ligand-activated transcription factor that binds DNA either directly through progesterone response elements (PREs), or indirectly through tethering interactions with other transcription factors. These interactions, together with the recruitment of transcriptional co-regulators, lead to transcriptional activation or repression of PR target genes. In the breast, these PR-dependent gene programs can drive proliferation, cell survival, and mammary stem cell self-renewal (reviewed in (3)). The mechanisms for PR-dependent transcriptional activation have been well studied, while PR-mediated transcriptional repression, especially direct repression in response to ligand, remains less understood.
Interferon signaling is a critical response of the innate immune system, which typically occurs following pathogen detection. Following interferon (IFN; types I–III) production and binding to their cognate receptors, a signaling cascade mediated by JAK/STAT (Janus Activated Kinase/Signal Transducer and Activator of Transcription) is initiated, culminating in a transcriptional response whose gene products aid the cell in responding to a pathogenic threat. The genes activated in response to interferons are collectively called interferon-stimulated genes, or ISGs (reviewed in (9)). In response to type I interferons, such as IFN-alpha (IFNα), a heterodimeric receptor (IFNAR1 [interferon-alpha receptor 1] and IFNAR2) complex is activated/auto-phosphorylated in response to ligand, promoting JAK1/Tyrosine Kinase 2 (TYK2)-dependent phosphorylation of STAT1 and STAT2. Phosphorylated STAT1 and STAT2, together with IFN-regulatory factor 9 (IRF9), form a transcriptional complex referred to as IFN-stimulated gene factor 3 (ISGF3) which binds to DNA-sequences within ISG promoter regions, referred to as IFN-stimulated response elements (ISREs). Binding of ISGF3 to ISREs leads to transcription of ISGs, preparing the cell with a diverse response to viral infection. Interferon-signaling activation and ISG expression has also been detected in human tumors, independent of viral infection. The role of interferon signaling in human tumors is not well known, and remains an area of intense research (10). The cancer immunoediting hypothesis postulates that IFN-signaling is an early step required for immune-recognition and subsequent destruction of nascent tumors by immune modulatory cells (11). Alterations in immune surveillance, recognition, and destruction may have dramatic effects contributing to the development of clinically overt tumors. This effect appears to be most prominent in the early stages of tumor development. The opposite phenotype is seen in advanced, therapy-resistant tumors: evidence of interferon signaling (high ISG levels) is associated with a range of tumor types, including breast, that have escaped chemotherapy, radiation, or immunotherapy (12). Herein, we present evidence of a novel role for PR in mediating transcriptional repression of ISGs. This newly described activity of PR has significant clinical implications for immune evasion and development of PR-positive human breast tumors.
Materials and Methods
Cell lines and constructs
T47D-co, T47D-Y, T47D-YB, T47D-YA, and Hela-PR cells have been previously described (13, 14) and were a generous gift of Dr. Carol Lange (University of Minnesota). T47D cells (unmodified) were obtained from ATCC, and cultured as recommended. Cell line authentication is currently underway. MDA-MB-231 cells were maintained in Minimum Essential Media (MEM; CellGro) supplemented with 5% FBS, 1% Penicillin/Streptomycin, 1% non-essential amino acids, and 6 ng/ml insulin (cMEM). BT549 cells were maintained in RPMI-1640 Media (CellGro) supplemented with 10% FBS, 1% Penicillin/Streptomycin, 1% non-essential amino acids, and 6 ng/ml insulin. The PR-mDBD construct was a generous gift of Dr. Kathryn Horwitz (University of Colorado). Cells were treated with the following reagents (when applicable): R5020 (10nM; Sigma), human recombinant interferon-alpha (Sigma-Aldrich, SPR4594).
Gene Set Enrichment Analysis
Gene set enrichment analysis (GSEA) was performed using the javaGSEA desktop software; the c2 Molecular Signatures Database (MSigDB) version 5.2 was queried (15, 16). Dataset files (GEO accession number:GSE46850) were developed based on normalized Illumina expression intensities from cells that constitutively express wt PR-B (T47D-YB cells), as published in (13). Specifically, the log2 transformed expression values were compared for two phenotypes: T47D-YB EtOH (vehicle) and T47D-YB R5020. GSEA was executed using the default settings, except the permutation type was set to Gene_set with 1000 permutations, and the metric for ranking genes was set to Diff_of_Classes, because normalized expression data was log2 transformed. Leading Edge analysis was performed on the 54 gene sets from MSigDB c2 analysis (PR-B EtOH vs R5020) that achieved FDR values ≤ 0.05.
Tumor explants
Tumor explant processing and treatments were done as previously described in (8). Raw FASTQ data was re-analyzed using the VIPER RNA-seq pipeline (https://bitbucket.org/cfce/viper/) at Center for Functional Cancer Epigenetics (CFCE) at the Dana-Farber Cancer Institute. For visualizing gene expression data, we used heatmap.2 function of gplots package in R programming language.
Immunoblotting
Immunoblotting/Western blotting was performed as previously described (13, 14). Membranes were probed with primary antibodies recognizing total PR (Santa Cruz Biotechnology, sc-7208 or ThermoScientific, MS-298-P), IRF9 (Cell Signaling, 76684), IFIT1 (Cell Signaling, 14769), IFIT3 (Santa Cruz Biotechnology, sc-393512), OAS1 (Cell Signaling, 14498), and ERK (Cell Signaling, 9102). All Western blotting experiments were performed in triplicate, and representative experiments are shown.
siRNA/shRNA
ON-TARGETplus SMARTpool (a pool of 4 individual siRNAs) designed to target human PR was purchased from Dharmacon (L-003433-00), as were Non-silencing (NS) siRNA controls (D-001810-01-05) and the suggested DharmaFect 1 Transfection Reagent (T-2001-03). For siRNA experiments, the cells were plated at a density of 3×105 cells per well in a 6-well plate. 24 hours following plating, T47D-co cells were transfected with 25nM NS or PR siRNA per manufacturer’s siRNA transfection protocol. At 48 hours post-transfection, the cells were starved in serum/phenol red-free IMEM (Gibco, A10488-01). At 72hr post-transfection the cells were treated with EtOH or 10nM R5020 for 6hr. Subsequent RNA isolation and qPCR experiments were carried out and analyzed as described below. PR shRNA knockdown cells were created using viral particles (GE/Dharmacon) targeting three different regions of human PR. Viral transduction protocol was followed as per manufacturer’s instructions. Transduced, stable cell line pools expressing NS or PR shRNA were created in T47D-co or T47D-YB cells following 14 days of selection in 2.5 ug/ml Puromycin (MP Biomedicals). Target si/shRNA sequences are listed in Supplementary Table 1.
Luciferase Transcription Assays
pGL4.45[luc2P/ISRE/Hygro] luciferase construct (Promega) was stably integrated in HeLa and HeLa-PR cells using Hygromycin selection. Luciferase assays were performed as previously described (17) using the Dual Luciferase Reporter Assay (Promega). Starved cells were treated for 18hr with 10nM R5020 and/or 10,000IU/ml interferon-alpha.
Real-Time Quantitative PCR (qPCR)
RNA isolation, cDNA creation, and qPCR were performed as previously described (13, 14), with modifications noted here and in the Figure legends. qPCR was performed using the Faststart Essential DNA Green Master (Roche) on a Roche LightCycler96. Relative concentrations were quantified using the LightCycler96 (Roche, Software 1.1, Absolute Quantification Analysis), using a 6-point standard curve. Primer sets are listed in Supplementary Table 1. Relevant genomic sequence information is based on GR37 hg19.
ChIP assays
ChIP was performed using the ChIP-IT Express Kit (Active Motif) according to manufacturer’s instructions using sonication for chromatin shearing. Lysates were immunoprecipitated (IP) overnight (18hr) with the following antibodies: PR (Santa Cruz Biotechnology, sc-7208), STAT2 (Santa Cruz, sc-476), IRF9 (Santa Cruz Biotechnology, sc-10793), H3K27ac1 (Cell Signaling, 8173), or an equal amount of negative control mouse or rabbit IgG. Resulting DNA was analyzed using qPCR as described above, and data is represented as a percentage of input DNA. In silico analysis was performed using MatInspector (Genomatix) to identify potential PRE-binding sites. Primer sets are listed in Supplementary Table 1.
Statistics
Statistical significance for all experiments was determined using an unpaired Student’s t-test, unless otherwise specified. A p value ≤ 0.05 is considered statistically significant. The Delta method was used to calculate standard deviation for the ratio of two variables using their individual standard deviations, as seen when plotting fold relative RNA expression data between two treatment groups/cell lines (18).
Results
Interferon gene sets enriched in PR-regulated transcriptional data set
Gene Set Enrichment Analysis (GSEA) is a powerful computational tool for comparing gene expression datasets of interest (i.e. genes regulated by PR) to published gene sets culled from the literature (15, 16). Using GSEA, we analyzed our previously published microarray dataset from ligand (synthetic progestin; R5020)-treated T47D breast cancer cells stably expressing full length PR (GEO:GSE46850) (13). GSEA revealed that multiple interferon-related gene sets (examples shown in Fig 1A) were significantly enriched in the absence of ligand; this enrichment was lost in cells treated with ligand, representing a negative correlation between ligand treatment and enrichment with interferon-related gene sets. Nearly 20% of the top significantly-regulated gene sets (54 sets with a false discovery rate [FDR] ≤ 0.05) are interferon-related gene sets (select sets shown in Fig 1B). As enrichment in these gene sets is lost in progestin-treated cells, these data imply that interferon gene programs are repressed/lost in response to ligand. Leading Edge (LE) analysis is a component of GSEA that allows one to identify individual genes that are present in multiple highly significant gene sets (i.e. core genes that drive the enrichment of a particular gene set). LE analysis identified multiple genes that are transcriptional targets of interferon-signaling pathways (i.e. IFITs, IRF7, OAS1/2, STAT1 and MX2) whose regulation is lost in ligand-treated cells (Fig 1C). These genes, classically activated by interferons, are collectively referred to as interferon-stimulated genes (ISGs). Cumulatively, these computational data suggest that interferon gene programs are negatively regulated by progestins.
Figure 1. Interferon signaling enriched in PR data set.
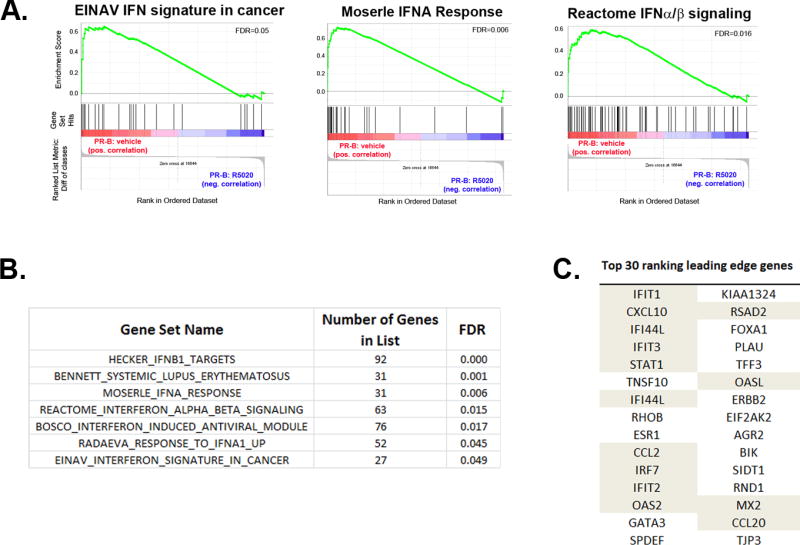
(A). GSEA comparing gene expression datasets obtained using the Illumina Microarray data set published in Hagan et al, 2013. Vehicle vs R5020 (synthetic PR ligand) are compared in T47D cells stably expressing full length PR. Select gene sets are shown from the C2 MSigDB. (B). Summary of interferon-related gene sets that are included in the most significant (FDR < 0.05; 54 gene sets in total) gene sets from MSigDB C2 GSEA analysis. (C). Top 30 ranking genes as identified using Leading Edge (LE) analysis on 54 gene sets referred to in (B). Shading denotes genes that are interferon-signaling related and/or interferon-stimulated genes (ISGs).
ISGs are downregulated by progestins in human breast cancer
We used a human tumor-explant model, previously described by Singhal et al (8), to determine if progestin-dependent ISG downregulation occurs in human breast tumors. Sliced portions of ER/PR-positive tumors from eight patients were obtained during surgical breast cancer resection. These tumor samples were grown in an ex vivo culture system in media containing vehicle (EtOH) or R5020 (24–48hr). RNA-seq was performed on cultures of each tumor sample; these data were analyzed for ISG repression following R5020 treatment. The heatmap in Figure 2 shows fold expression (R5020/vehicle), for RNA-seq data obtained from all eight patients, for select ISGs. There is a clear trend towards transcriptional repression for ISGs in patient ex vivo tumor explants. These data indicate that ISGs are regulated by progestins not only in human breast cancer cell lines (Fig 1), but, importantly, in human breast tumors as well (Fig 2).
Figure 2. Progestin-dependent ISG repression in human breast tumors.
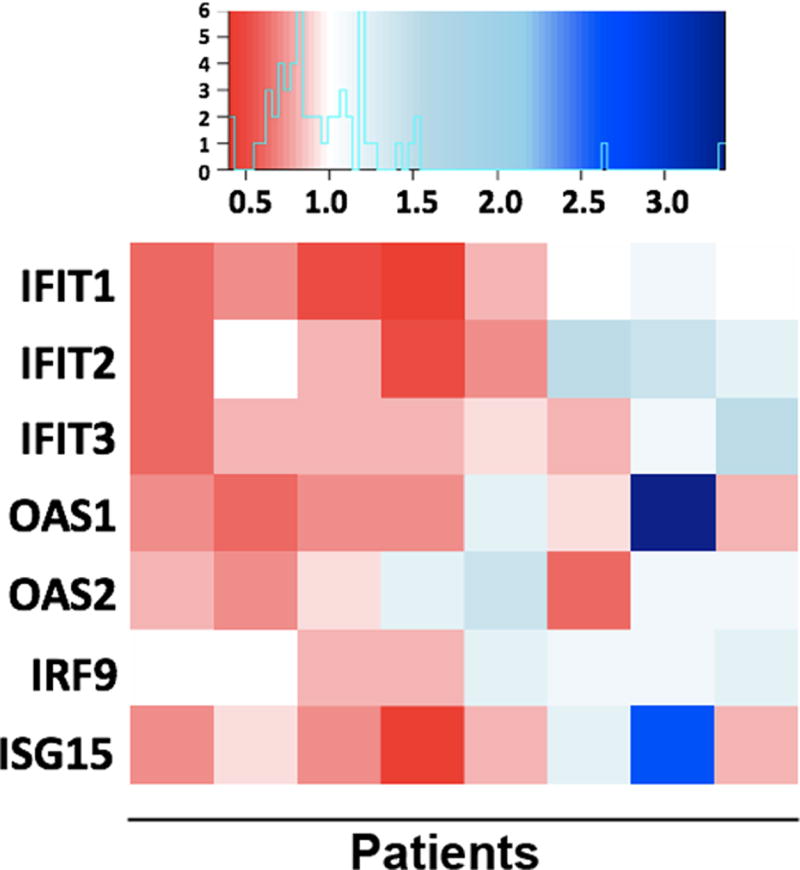
Human breast tumors were grown ex vivo for 24–48hr in media containing vehicle or 10nM R5020, followed by RNA-seq. Heat map represents fold RNA expression values for R5020/vehicle for seven ISGs, in each of eight patient samples. Red shading indicates downregulation of RNA expression in R5020-treated tumors, blue represents gene upregulation in response to R5020 treatment, white represents no change.
ISG repression is PR-dependent
To further characterize the mechanism through which progestins regulate ISG expression, we studied progestin-dependent ISG repression in multiple breast cancer cell line models. A brief note about our most frequently used model system: PR is an important target gene of ER and, as such, PR expression is regulated by estrogen in most tissues (19, 20). In order to differentiate between the effects of ER/estrogen and PR/progesterone, our laboratory uses PR-positive (T47D-co) and PR-null (T47D-Y) variants of the ER/PR+ breast cancer cell line, T47D (21). T47D-co cells endogenously express both isoforms of PR, PR-A and PR-B, without the need for exogenously added estrogen, allowing us to study the function of PR without the confounding effects of estrogen. T47D-Y cells can also be used to reintroduce single isoform variants or mutants of PR, such as PR-A (T47D-YA cells) and PR-B (T47D-YB cells) or DNA-binding mutant PR (T47D-PRB-mDBD). We have published extensively using these cell line models to define isoform- and phosphorylation-specific PR gene regulation and protein-protein interactions (13, 14, 22–24), and this cell line model remains a powerful and well-established system for studying PR activity (5).
To characterize regulation of ISGs by progestins, we analyzed RNA levels of ISGs selected from the LE gene list in T47D-co breast cancer cells. T47D-co cells were treated for 6hr with synthetic progestins (R5020 or medroxyprogesterone acetate [MPA]), native progesterone, or vehicle (EtOH); isolated RNA was used for quantitative real-time PCR (qPCR) analysis. For the ISGs we assayed, six of which are shown in Figure 3A, expression was transcriptionally repressed (2- to 10-fold) by all progestins, both synthetic and native. Individual gene validation (such as those shown in Fig 3A, and data not shown) and microarray data mining (13) showed that this transcriptional repression is conserved for a large cohort of ISGs. Progestin-dependent ISG transcriptional downregulation can be mediated via both isoforms of PR, as T47D-Y cells stably expressing either PR-B or PR-A can both repress ISG RNA levels, although PR-B appears to have greater transcriptional repressor activity on the ISGs assayed (Fig S1A). Additionally, because the T47D-co cells are unique in that exogenous estrogen is not needed for PR expression, we verified that ISG transcripts were repressed following progestin treatment in unmodified, parental T47D cells grown in an estrogenic environment (T47D-ATCC; Fig S1B). To determine if repression of ISGs by PR is cell-type specific, we stably expressed PR-B in HeLa cells (normally PR-null) and observed ISG transcriptional repression in response to progestin treatment (Fig S1C). Finally, this transcriptional repression was associated with a concomitant decrease in ISG protein levels. ISG proteins were assayed from T47D-co breast cancer cells treated with ligand (R5020; 18hr); decreased protein levels were observed for all ISGs assayed via Western blotting (Fig 3B). Cumulatively, these data suggest that ligand-activated PR promotes the downregulation of ISG RNA and protein levels.
Figure 3. ISGs are repressed by ligand-activated PR.
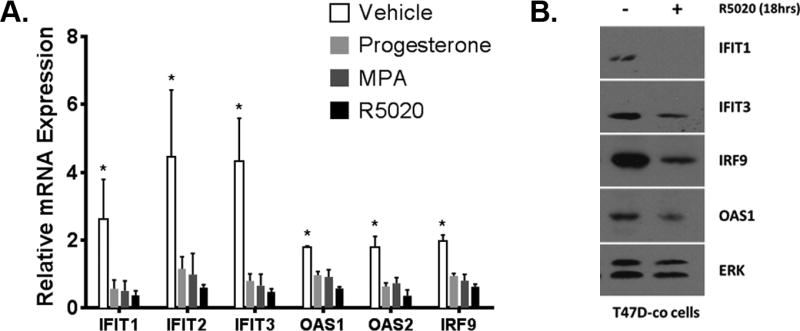
(A). T47D PR-positive breast cancer cells (T47D-co) were starved for 18hr in serum-free media, followed by treatment with 10nM R5020, 10nM medroxyprogesterone acetate (MPA), 100nM native progesterone, or vehicle for 6hr. Isolated RNA was analyzed for select genes. Gene values were normalized to an internal control (β-actin). Error bars represent standard deviation (SD) between biological triplicates. Asterisks represent statistical significance between the vehicle treated groups and all treatment groups (R5020, MPA, and progesterone); p < 0.05, as determined using an unpaired Student’s t-test. This experiment was performed in triplicate, and a representative experiment is shown here. (B). T47D-co cells were starved for 18hr in serum-free media and then treated with 10nM R5020 or vehicle for 18hr. Protein lysates were analyzed via Western blotting. ERK represents the loading control.
To determine whether ISG transcriptional repression following progestin treatment is dependent upon PR, we assayed ISG transcript levels in cells where PR expression was knocked-down using a pool of four siRNAs. T47D-co cells transiently transfected with non-silencing (NS) or PR siRNA were treated with vehicle or R5020 for 6hr, and RNA was analyzed using qPCR. ISG transcriptional repression in response to ligand was lost in cells with PR knockdown (Fig 4A; PR knockdown efficiency shown in Fig 4B). These data were repeated in cells stably expressing PR shRNA (Fig S2). Surprisingly, basal levels (vehicle treated, in the absence of progestin) of ISGs were markedly higher in cells lacking PR (compare vehicle bars between NS and PR siRNA for each gene shown in Fig 4A). Basal levels of select ISGs were increased 1.5 to 3.5-fold when PR expression was knocked-down using siRNA (Fig 4C). These data indicate that PR has ligand-independent functions that appear to maintain low levels of ISG expression. Moreover, when ISG expression was assayed in two PR-negative (triple negative, lacking ER/PR/HER2) breast cancer cell lines, we saw a similar phenotype: no ISG repression in response to ligand and high basal levels of ISGs (Fig S3). Together, these data indicate that PR attenuates ISG expression, even in the absence of ligand, suggesting a concerted biological program aimed at PR-dependent down-regulation of ISGs.
Figure 4. ISG repression is PR-dependent.
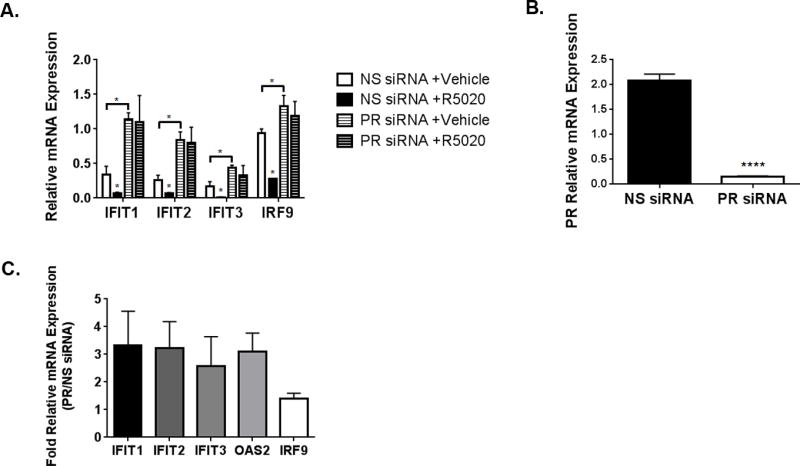
(A). T47D-co cells were transfected with non-silencing (NS) or a pool of four PR siRNAs. 48hr following siRNA transfection, cells were starved for 18hr, followed by 10nM R5020 or vehicle for 6hr. Isolated RNA was analyzed for select genes. Gene values were normalized to an internal control (β-actin). Error bars represent SD between biological triplicates. Asterisks represent statistical significance between the vehicle and R5020-treated groups; p < 0.05, as determined using an unpaired Student’s t-test. This experiment was performed in triplicate, and a representative experiment is shown here. (B). PR knockdown efficiency by siRNA was determined using relative PR levels between the NS and PR siRNA transfected cells. Asterisks represent statistical significance; p < 0.0001. (C). Fold increases in basal (no ligand) ISG expression between PR and NS siRNA transfected cells. Error bars represent the SD for the ratio of two variables using their individual standard deviations, calculated using the Delta method.
Interferon-activation of ISGs is repressed by ligand-activated PR
Most typically, ISGs are activated in response to interferons in specialized cell types of the innate immune system, such as dendritic cells. However, all cell types have the capacity to produce and respond to interferons (including fibroblasts and epithelial cells), as long as the proper receptors (IFNARs) and transcriptional machinery are intact (10). Although our T47D-co cells do not secrete endogenous interferons (measured using highly-sensitive ELISA assays designed to detect picomolar concentrations; data not shown), they retain their response to exogenous treatment with IFNα by transcriptionally upregulating ISGs (Fig 5). Interestingly, when we co-treated cells with IFNα and progestin (R5020), we found that progestin-treatment attenuated the IFNα-induced ISG transcriptional response, reducing it to near baseline levels (Fig 5). We concluded from these data that ligand-activated PR can repress ISG transcripts in response to IFNα.
Figure 5. Interferon-activated ISG expression is repressed by ligand-activated PR.
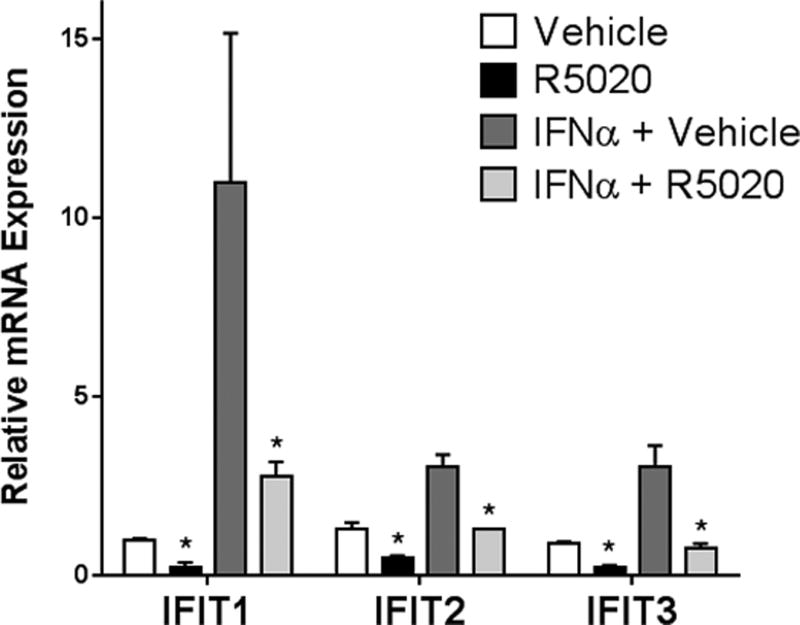
Following an 18hr starvation in serum-free media, cells were treated for 18hr with IFNα (20 IU/ml) or vehicle (water) for 18hr, followed by R5020 (10nM) or vehicle (EtOH) for 6hr. Isolated RNA was analyzed for expression of IFIT1, IFIT2, and IFIT3. Gene expression values were normalized to an internal control (β-actin). Error bars represent SD between biological triplicates. Asterisks represent statistical significance between the respective vehicle and R5020-treated groups; p < 0.05, as determined using an unpaired Student’s t-test. This experiment was performed in triplicate, and a representative experiment is shown here.
PR DNA-binding needed for ISG transcriptional repression
There are many potential mechanisms through which PR can mediate transcriptional repression of target genes (25). To determine if DNA-binding by PR is required to mediate ISG transcriptional repression, we employed a well characterized DNA-binding domain (DBD) mutant of PR (PR-mDBD). This mutant contains a single point mutation at Cys587, located within the first zinc finger of the PR DBD, which abolishes PR’s ability to bind DNA (26). Using T47D-PRB-mDBD cells, we measured the capacity of mDBD PR to repress ISG transcription. T47D cells stably expressing wt PR-B (T47D-YB, described above) robustly repress ISG transcription in response to ligand; this effect is lost in T47D-PRB-mDBD cells (Fig 6A). These data suggest PR binding to DNA is required for ISG transcriptional repression. IFNα-activated ISG transcription is regulated through ISREs, DNA sequences where the ISGF3 complex (STAT1, STAT2, and IRF9) binds and activates transcription. To test if PR repressed transcription via direct or indirect binding to ISRE sequences, we used an ISRE-luciferase reporter construct stably expressed in Hela or Hela-PR cells. ISRE-linked luciferase activity was unaffected by the presence of PR or its ligand in either cell line (Fig 6B); no changes were seen basally or in response to IFNα treatment. These data imply that PR does not repress ISG transcription by binding to or blocking the ISRE promoter element directly.
Figure 6. PR DNA-binding required for ISG transcriptional repression.
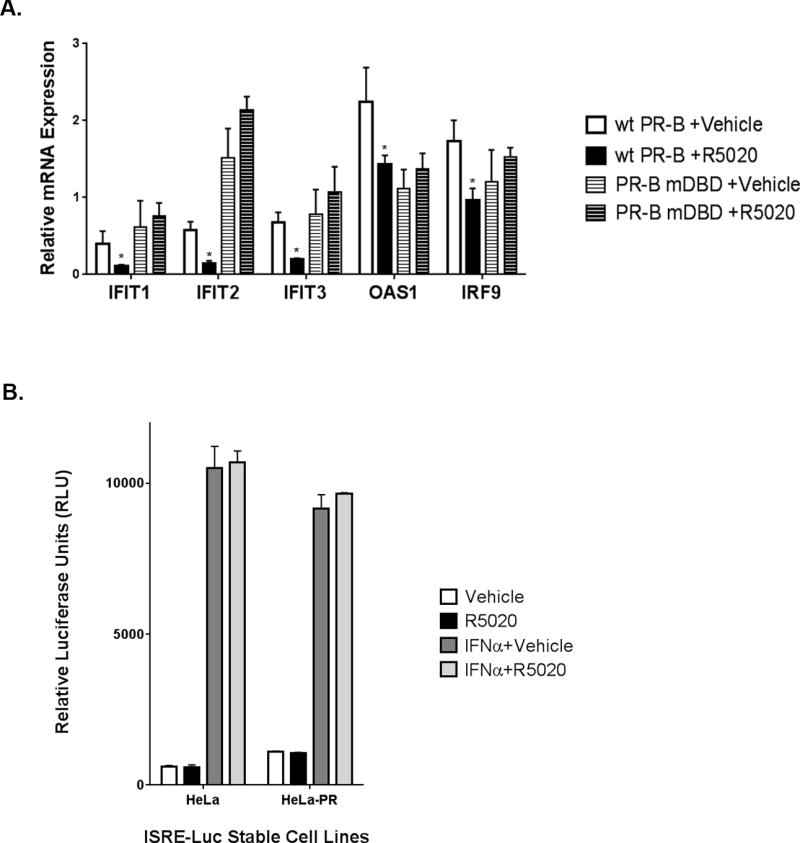
(A). Following an 18hr starvation in serum-free media, T47D-YB or T47D-PRB-mDBD cells were treated for 6hr with vehicle or 10nM R5020 for 6hr. Isolated RNA was analyzed for select genes. Gene values were normalized to an internal control (β-actin). Error bars represent SD between biological triplicates. Asterisks represent statistical significance; p < 0.05, as determined using an unpaired Student’s t-test. (B). HeLa and HeLa-PR cells that were stably transfected with an ISRE-luciferase were starved for 24hr, followed by an 18hr co-treatment with interferon-alpha (IFNα) (or water) or 10nM R5020 (or EtOH). Luciferase assays were performed as described in the ‘Materials and Methods’ section. Error bars represent SD of biological replicates. The experiments in (A) and (B) were performed in triplicate, and representative experiments are shown here.
PR is recruited to ISG enhancers
To determine if PR is directly recruited to promoters/enhancers that regulate ISG transcription, we used chromatin-immunoprecipitation (ChIP) to detect PR binding at ISG-regulatory regions. As mentioned above, ISGs are regulated by promoter proximal ISREs, typically within 100bp of ISG transcriptional start sites (TSS) (27). Using published ChIP-seq data for ligand-activated PR binding (6), we identified that all ISGs we assayed exhibited PR binding in their promoter/enhancer regions. It is well documented that nuclear receptors often bind in intragenic or distal (> 40kb away from the TSS) enhancer regions of the genes they regulate (28–32); PR binding to ISG enhancers falls well within this range (−18kb to +5kb). We identified individual PREs within these binding sites using in silico analysis. Figure 7A (left) highlights the position of the PREs and ISREs (ISREs defined in (27) or through ENCODE STAT1/2 ChIP-seq binding data (33–35)) in relation to the TSS for select genes assayed via ChIP-qPCR for PR and ISGF3 (STAT1/STAT2/IRF9) binding. Using PR ChIP-qPCR analysis following R5020 treatment, we showed robust PR recruitment to enhancer regions of multiple ISGs (Fig 7A and Fig S4); recruitment of PR to the IFIT3 promoter (Fig 7A) is shown. All subsequent ChIP-qPCR results will be shown for the IFIT3 promoter as a representative example of protein recruitment to all ISG enhancer regions assayed and summarized in Fig 7A.
Figure 7. PR diminishes recruitment of STAT2/IRF9 to ISG promoters.
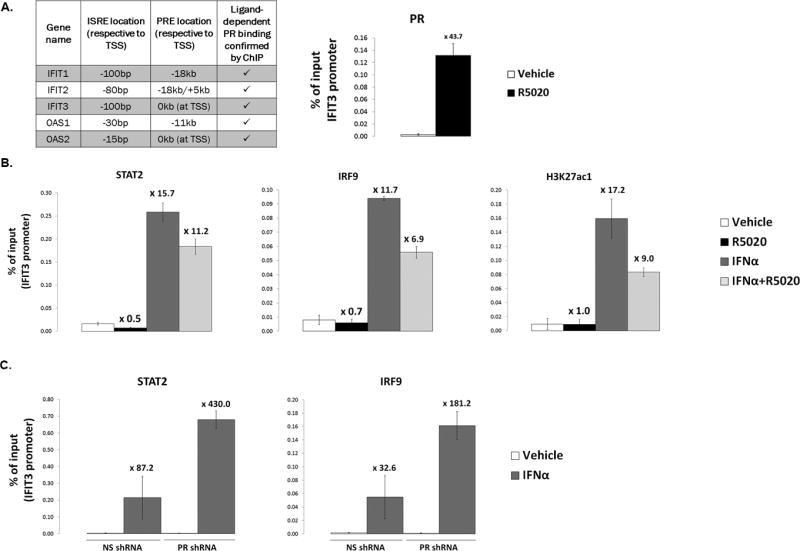
(A). Left: table summary of ISG promoters/enhancers assayed by ChIP-qPCR for PR, STAT2, and IRF9 recruitment. Location of interferon-stimulated response elements (ISREs) and progesterone receptor response elements (PREs) from the transcriptional start site (TSS) of each gene is shown in kilobases (kb). Right: T47D-co cells were serum-starved for 18hr. Cells were then treated with 10nM R5020 or vehicle for 30min. Fixed lysates were subjected to ChIP with antibodies against PR or a species-specific IgG (control; not shown), and qPCR was performed on the isolated DNA using primers designed to amplify the IFIT3 promoter. A percentage of ChIP’d DNA over input DNA is shown. (B). T47D-co cells were serum-starved for 18hr. Cells were then treated with 10nM R5020, 1000IU/ml IFNα, or a co-treatment of both (or appropriate vehicle controls). Fixed lysates were subjected to ChIP with antibodies against STAT2, IRF9, H3K27ac1 or a species-specific IgG (control; not shown), and qPCR was performed on the isolated DNA using primers designed to amplify the IFIT3 promoter. A percentage of ChIP’d DNA over input DNA is shown. (C). ChIP experiments were performed as in (B), but T47D-co NS and PR shRNA cells were used, with only IFNα treatment. All ChIP experiments were performed in triplicate; a representative experiment is shown here. Fold-recruitment in treated conditions (R5020, IFNα, or combo), as compared to vehicle treatment, is displayed above each bar. Error bars represent SD of technical replicates.
Although STAT1/STAT2/IRF9 historically comprise the ISGF3 transcription factor complex, IRF9 and STAT2 complexed together (in the absence of STAT1) can recapitulate ISRE-mediated transcription (36). We therefore focused on how PR activation alters STAT2/IRF9 recruitment to ISG promoter regions. We observed robust recruitment of STAT2 and IRF9 to ISREs following treatment with IFNα (Fig 7B). Interestingly, interferon-stimulated STAT2/IRF9 recruitment was decreased in the presence of PR ligand. Basal STAT2/IRF9 occupancy in the absence of interferon is too low (at/above IgG levels) to generate a consistently robust signal from ChIP-qPCR. However, a trend exists towards STAT2/IRF9 loss when comparing vehicle and R5020 in the absence of IFNα (Fig 7B – compare vehicle and R5020 bars). This observation (loss of STAT2/IRF9) is potentiated when IFNα treatment, in combination with PR ligand, is used. Moreover, we see a potent increase in H3K27ac1, a histone mark indicative of activated transcription and open chromatin, following treatment with interferon. Treatment with PR ligand decreases the levels of H3K27ac1, suggesting that recruitment of PR (and loss of STAT2/IRF9) leads to active repression of these enhancers. Finally, in T47D-co cells where PR expression was knocked-down using shRNA directed against PR (or a non-silencing control; NS), recruitment of STAT2/IRF9 was potently enhanced in response to IFNα, indicating that the presence of PR (ligand-independent) decreases ISG promoter occupancy of ISGF3 components (Fig 7C). Cumulatively, these data suggest that PR blocks/decreases efficient STAT2/IRF9 recruitment to ISRE regulatory regions, thereby decreasing transcription of ISGs. The ligand-dependent loss of STAT2/IFR9 occurs when the PRE and ISRE sequences are both located in the proximal promoter regions (as is the case with IFIT3), as well as when the PR binding sites occurs upstream of the ISRE (data not shown). The details regarding how PR recruitment destabilizes STAT2/IRF9 DNA binding/recruitment are the subject of our ongoing experiments.
Discussion
Herein, we demonstrate that PR transcriptionally represses ISGs in human breast cancer. This represents a novel class of genes previously unknown to be regulated by PR. Although regulation of ISGs has been well defined in response to interferons, PR-dependent repression contributes a novel layer of ISG regulation. We show PR-mediated ISG repression in breast cancer cell lines, as well as in tumor explants from breast cancer patients. Additionally, ISG repression exists in response to PR ligand, as well as in unliganded (basal) conditions. These data indicate a concerted PR-dependent effort aimed at maintaining low ISG levels in breast cancer.
The cancer immunoediting hypothesis highlights that the innate and adaptive immune responses work together to flag early neoplastic lesions for immune-mediated elimination (11). An early mediator of this elimination process is activation of type I interferon-signaling (10). Thus, suppression of type I interferon-signaling may help developing tumors evade the critical early steps of immune recognition and subsequent clearance, allowing nascent tumors to progress. In vivo studies support this notion. For example, female STAT1 knockout mice, lacking a key upstream activator of type I interferon-signaling, develop mammary gland adenocarcinomas (37). We show that PR represses the end product of interferon-signaling, ISG transcription, suggesting that progesterone, working through PR, may be a key player in tumor immune evasion. A number of previous observations support the importance of the mechanism we described, in the development of breast cancer. For example, 70% of breast cancers are ER/PR-positive at the time of diagnosis, and recent data has suggested a positive correlation between PR positivity in benign, normal breast tissue and risk for the development of breast cancer (38). Moreover, >90% of the mammary gland tumors that form in the STAT1-deficient mice are ER/PR-positive and have gene expression profiles that mimic the luminal ER/PR-positive molecular subtype of breast cancer (37). These data suggest there is pro-tumorigenic crosstalk between PR and interferon-signaling pathways.
Downregulation of multiple components of interferon-signaling have been correlated with increased tumor incidence and metastasis. Mice lacking IFNAR, the IFNα-binding receptor that initiates JAK/STAT-signaling which culminates in ISG transcription, have enhanced tumor development, impaired ability to reject syngeneic/allogenic tumors, and accelerated metastasis in a spontaneous mouse mammary gland tumor model (39, 40). A recent report shows that IFNAR inactivation in tumor-associated stroma leads to an immune-privileged niche for developing colon cancers (41). Type I interferons, in particular, and dendritic cells have been shown to be critical to tumor cell rejection, and therefore immune surveillance (42). Moreover, interferon regulatory factor 7 (IRF7) and its downstream targets (many of which are ISGs shown herein to be repressed by PR), are suppressed in the aforementioned mammary gland tumor metastasis model; restoration of IRF7-signaling suppressed bone metastasis (39). Finally, expression of an interferon-gene signature in a cohort of breast cancer patients was correlated to a lower frequency of breast cancer metastasis (43). Cumulatively, these data suggest mounting evidence that interferon signaling is correlated with tumor development and metastasis. Conversely, high/increased ISG expression has been linked to poor prognosis in more advanced tumors. An interferon-related DNA damage resistance signature (IRDS) has been defined by Weichselbaum and Minn that is associated with chemotherapy, radiation, and most recently, immunotherapy resistance in multiple tumor types, including breast (12, 44–46). Therefore, ISG expression, and subsequently the effect of ISGs on tumor growth, may vary by tumor stage (early vs late).
Other nuclear receptors, most notably the glucocorticoid receptor (GR), have been implicated in modulating interferon and inflammatory responses. Inhibitory interactions between GR and AP1/NF-kB have been shown to inhibit a broad range of inflammatory functions, largely through a mechanism termed transrepression. In this mechanism, GR tethers to DNA-bound transcription factors/regulatory proteins and affects co-factor recruitment and subsequent gene regulation (reviewed in (47)). Of note, GR has been shown to repress ISG transcription in a cell-type specific manner in macrophages by squelching away an ISGF3 co-factor, GRIP1 (glutamate receptor interacting protein 1), needed for interferon-dependent activation of ISGs in macrophages (48). In this instance, the repression was independent of GR binding to DNA, though ligand-activated GR effectively repressed ISRE-luciferase activity. This is in contrast to our findings (Fig 6B) where PR ligand treatment was not able to repress an ISRE-luciferase construct, implying that PR-dependent ISG repression occurs via a mechanism independent of co-factor squelching. Our data suggest that PR-mediated repression of ISGs in breast cancer cells occurs through a different mechanism than that proposed for GR-mediated ISG repression in macrophages, perhaps indicating cell type- and nuclear receptor-specific regulation. Recent work, however, suggests that select GR-dependent gene repression events may require direct GR binding to DNA through GR response elements (GREs). These GREs are in close proximity to AP1 and NF-kB binding sites. GRE-mediated binding of GR leads to recruitment of GRIP1, the aforementioned ISGF3 co-factor and known GR co-repressor, leading to repressive changes in the chromatin and subsequent gene repression (49). This mechanism has similarities to the mechanism we propose herein for PR-mediated, and PRE-dependent, ISG transcriptional repression. Because we observe decreased recruitment of ISGF3 components (STAT2 and IRF9) to ISRE promoter sequences following PR activation and recruitment of PR to PREs, we favor a model where protein displacement or steric competition occurs between PR and STAT2/IRF9. We recognize that transcriptional repression is complex, and a combination of mechanisms may contribute to PR-dependent ISG repression. Our future work is focused on detailing this mechanism(s).
Although PR gene activation has been well characterized, the mechanisms through which PR represses transcription remain poorly understood. Various mechanisms have been put forth for PR-mediated transcriptional repression, and are similar to what has been well characterized for GR-mediated repression (squelching of co-factors, recruitment of co-repressors, chromatin remodeling; reviewed in (25)). A recent study details ligand-dependent recruitment of PR, an HP1γ-LSD1 repressive complex, and BRG1 to repressed target gene promoters/enhancers. This repressive complex leads to ligand-dependent changes in chromatin architecture that result in transcriptional repression (50). Although we observed a decrease in activating histone marks at ISG enhancers following treatment with PR ligand (Fig 7B), preliminary data show that knockdown of BRG1, LSD1 or HP1γ had no effect on PR-dependent transcriptional repression of ISGs (not shown), suggesting an alternative (non- HP1γ/LSD1-dependent) mechanism for PR-mediated ISG transcriptional repression. Experiments to investigate PR-dependent recruitment of other chromatin modifiers to ISG enhancers are currently underway.
In summary, our results show novel PR-dependent repression of ISG transcription. These data have significant implications for the regulation of interferon-signaling in breast cancer, and provide a putative mechanism through which nascent breast cancers may avoid immune surveillance. Future directions will be aimed at understanding what effect PR-dependent downregulation of ISGs has on breast cancer development and progression.
Supplementary Material
Acknowledgments
The Center for Functional Cancer Epigenetics (CFCE) at the Dana-Farber Cancer Institute provided the computing infrastructure for RNA-seq analysis. We thank Drs. Joseph Fontes (KUMC), Chad Slawson (KUMC) and Todd Knutson (Minnesota) for critical scientific input.
This work was supported by NCI R00CA166643 (CRH), DOD BCRP W81XWH-16-1-0320 (CRH), Susan G Komen Foundation CCR16376147 (CRH), V Foundation V2015-025 (CRH), and the NCI Cancer Center Support Grant P30 CA168524 (CRH).
Footnotes
The authors declare no conflict of interest.
References
- 1.Doan TB, Graham J, Clarke C. Emerging functional roles of nuclear receptors in breast cancer. J Mol Endocrinol. 2017 doi: 10.1530/JME-16-0082. [DOI] [PubMed] [Google Scholar]
- 2.Brisken C. Progesterone signalling in breast cancer: a neglected hormone coming into the limelight. Nature reviews Cancer. 2013;13(6):385–96. doi: 10.1038/nrc3518. [DOI] [PubMed] [Google Scholar]
- 3.Hagan CR, Lange CA. Molecular determinants of context-dependent progesterone receptor action in breast cancer. BMC medicine. 2014;12:32. doi: 10.1186/1741-7015-12-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson GL, Limacher M, Assaf AR, Bassford T, Beresford SA, Black H, et al. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized controlled trial. JAMA. 2004;291(14):1701–12. doi: 10.1001/jama.291.14.1701. [DOI] [PubMed] [Google Scholar]
- 5.Carroll JS, Hickey TE, Tarulli GA, Williams M, Tilley WD. Deciphering the divergent roles of progestogens in breast cancer. Nature reviews Cancer. 2017;17(1):54–64. doi: 10.1038/nrc.2016.116. [DOI] [PubMed] [Google Scholar]
- 6.Mohammed H, Russell IA, Stark R, Rueda OM, Hickey TE, Tarulli GA, et al. Progesterone receptor modulates ERalpha action in breast cancer. Nature. 2015;523(7560):313–7. doi: 10.1038/nature14583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daniel AR, Gaviglio AL, Knutson TP, Ostrander JH, D’Assoro AB, Ravindranathan P, et al. Progesterone receptor-B enhances estrogen responsiveness of breast cancer cells via scaffolding PELP1- and estrogen receptor-containing transcription complexes. Oncogene. 2014 doi: 10.1038/onc.2013.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singhal H, Greene ME, Tarulli G, Zarnke AL, Bourgo RJ, Laine M, et al. Genomic agonism and phenotypic antagonism between estrogen and progesterone receptors in breast cancer. Sci Adv. 2016;2(6):e1501924. doi: 10.1126/sciadv.1501924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol. 2014;32:513–45. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheon H, Borden EC, Stark GR. Interferons and their stimulated genes in the tumor microenvironment. Semin Oncol. 2014;41(2):156–73. doi: 10.1053/j.seminoncol.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331(6024):1565–70. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 12.Minn AJ. Interferons and the Immunogenic Effects of Cancer Therapy. Trends Immunol. 2015;36(11):725–37. doi: 10.1016/j.it.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hagan CR, Knutson TP, Lange CA. A Common Docking Domain in Progesterone Receptor-B links DUSP6 and CK2 signaling to proliferative transcriptional programs in breast cancer cells. Nucleic Acids Res. 2013 doi: 10.1093/nar/gkt706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hagan CR, Regan TM, Dressing GE, Lange CA. ck2-Dependent Phosphorylation of Progesterone Receptors (PR) on Ser81 Regulates PR-B Isoform-Specific Target Gene Expression in Breast Cancer Cells. Mol Cell Biol. 2011;31(12):2439–52. doi: 10.1128/MCB.01246-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–73. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 16.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Faivre EJ, Lange CA. Progesterone receptors upregulate Wnt-1 to induce epidermal growth factor receptor transactivation and c-Src-dependent sustained activation of Erk1/2 mitogen-activated protein kinase in breast cancer cells. Mol Cell Biol. 2007;27(2):466–80. doi: 10.1128/MCB.01539-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Casella G, Berger RL. Statistical Inference. Duxbury Press; 2001. [Google Scholar]
- 19.Horwitz KB, Koseki Y, McGuire WL. Estrogen control of progesterone receptor in human breast cancer: role of estradiol and antiestrogen. Endocrinology. 1978;103(5):1742–51. doi: 10.1210/endo-103-5-1742. [DOI] [PubMed] [Google Scholar]
- 20.Horwitz KB, McGuire WL. Estrogen control of progesterone receptor induction in human breast cancer: role of nuclear estrogen receptor. Adv Exp Med Biol. 1979;117:95–110. doi: 10.1007/978-1-4757-6589-2_5. [DOI] [PubMed] [Google Scholar]
- 21.Sartorius CA, Groshong SD, Miller LA, Powell RL, Tung L, Takimoto GS, et al. New T47D breast cancer cell lines for the independent study of progesterone B- and A-receptors: only antiprogestin-occupied B-receptors are switched to transcriptional agonists by cAMP. Cancer Res. 1994;54(14):3868–77. [PubMed] [Google Scholar]
- 22.Dressing GE, Knutson TP, Schiewer MJ, Daniel AR, Hagan CR, Diep CH, et al. Progesterone receptor-cyclin d1 complexes induce cell cycle-dependent transcriptional programs in breast cancer cells. Mol Endocrinol. 2014;28(4):442–57. doi: 10.1210/me.2013-1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hagan CR, Daniel AR, Dressing GE, Lange CA. Role of phosphorylation in progesterone receptor signaling and specificity. Mol Cell Endocrinol. 2012;357(1–2):43–9. doi: 10.1016/j.mce.2011.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hagan CR, Faivre EJ, Lange CA. Scaffolding actions of membrane-associated progesterone receptors. Steroids. 2009;74(7):568–72. doi: 10.1016/j.steroids.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Santos GM, Fairall L, Schwabe JW. Negative regulation by nuclear receptors: a plethora of mechanisms. Trends Endocrinol Metab. 2011;22(3):87–93. doi: 10.1016/j.tem.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tung L, Mohamed MK, Hoeffler JP, Takimoto GS, Horwitz KB. Antagonist-occupied human progesterone B-receptors activate transcription without binding to progesterone response elements and are dominantly inhibited by A-receptors. Mol Endocrinol. 1993;7(10):1256–65. doi: 10.1210/mend.7.10.8123133. [DOI] [PubMed] [Google Scholar]
- 27.Testoni B, Vollenkle C, Guerrieri F, Gerbal-Chaloin S, Blandino G, Levrero M. Chromatin dynamics of gene activation and repression in response to interferon alpha (IFN(alpha)) reveal new roles for phosphorylated and unphosphorylated forms of the transcription factor STAT2. J Biol Chem. 2011;286(23):20217–27. doi: 10.1074/jbc.M111.231068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rao NA, McCalman MT, Moulos P, Francoijs KJ, Chatziioannou A, Kolisis FN, et al. Coactivation of GR and NFKB alters the repertoire of their binding sites and target genes. Genome Res. 2011;21(9):1404–16. doi: 10.1101/gr.118042.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reddy TE, Pauli F, Sprouse RO, Neff NF, Newberry KM, Garabedian MJ, et al. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res. 2009;19(12):2163–71. doi: 10.1101/gr.097022.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carroll JS, Brown M. Estrogen receptor target gene: an evolving concept. Mol Endocrinol. 2006;20(8):1707–14. doi: 10.1210/me.2005-0334. [DOI] [PubMed] [Google Scholar]
- 31.Welboren WJ, van Driel MA, Janssen-Megens EM, van Heeringen SJ, Sweep FC, Span PN, et al. ChIP-Seq of ERalpha and RNA polymerase II defines genes differentially responding to ligands. EMBO J. 2009;28(10):1418–28. doi: 10.1038/emboj.2009.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nielsen R, Pedersen TA, Hagenbeek D, Moulos P, Siersbaek R, Megens E, et al. Genome-wide profiling of PPARgamma:RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis. Genes Dev. 2008;22(21):2953–67. doi: 10.1101/gad.501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan KK, Cheng C, et al. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012;489(7414):91–100. doi: 10.1038/nature11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang J, Zhuang J, Iyer S, Lin XY, Greven MC, Kim BH, et al. Factorbook.org: a Wiki-based database for transcription factor-binding data generated by the ENCODE consortium. Nucleic Acids Res. 2013;41(Database issue):D171–6. doi: 10.1093/nar/gks1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, Greven MC, et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012;22(9):1798–812. doi: 10.1101/gr.139105.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kraus TA, Lau JF, Parisien JP, Horvath CM. A hybrid IRF9-STAT2 protein recapitulates interferon-stimulated gene expression and antiviral response. J Biol Chem. 2003;278(15):13033–8. doi: 10.1074/jbc.M212972200. [DOI] [PubMed] [Google Scholar]
- 37.Chan SR, Vermi W, Luo J, Lucini L, Rickert C, Fowler AM, et al. STAT1-deficient mice spontaneously develop estrogen receptor alpha-positive luminal mammary carcinomas. Breast Cancer Res. 2012;14(1):R16. doi: 10.1186/bcr3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oh H, Eliassen AH, Wang M, Smith-Warner SA, Beck AH, Schnitt SJ, et al. Expression of estrogen receptor, progesterone receptor, and Ki67 in normal breast tissue in relation to subsequent risk of breast cancer. NPJ Breast Cancer. 2016:2. doi: 10.1038/npjbcancer.2016.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bidwell BN, Slaney CY, Withana NP, Forster S, Cao Y, Loi S, et al. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nature medicine. 2012;18(8):1224–31. doi: 10.1038/nm.2830. [DOI] [PubMed] [Google Scholar]
- 40.Picaud S, Bardot B, De Maeyer E, Seif I. Enhanced tumor development in mice lacking a functional type I interferon receptor. J Interferon Cytokine Res. 2002;22(4):457–62. doi: 10.1089/10799900252952244. [DOI] [PubMed] [Google Scholar]
- 41.Katlinski KV, Gui J, Katlinskaya YV, Ortiz A, Chakraborty R, Bhattacharya S, et al. Inactivation of Interferon Receptor Promotes the Establishment of Immune Privileged Tumor Microenvironment. Cancer cell. 2017;31(2):194–207. doi: 10.1016/j.ccell.2017.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. The Journal of experimental medicine. 2011;208(10):1989–2003. doi: 10.1084/jem.20101158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Snijders AM, Langley S, Mao JH, Bhatnagar S, Bjornstad KA, Rosen CJ, et al. An interferon signature identified by RNA-sequencing of mammary tissues varies across the estrous cycle and is predictive of metastasis-free survival. Oncotarget. 2014;5(12):4011–25. doi: 10.18632/oncotarget.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Minn AJ, Wherry EJ. Combination Cancer Therapies with Immune Checkpoint Blockade: Convergence on Interferon Signaling. Cell. 2016;165(2):272–5. doi: 10.1016/j.cell.2016.03.031. [DOI] [PubMed] [Google Scholar]
- 45.Weichselbaum RR, Ishwaran H, Yoon T, Nuyten DS, Baker SW, Khodarev N, et al. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proc Natl Acad Sci U S A. 2008;105(47):18490–5. doi: 10.1073/pnas.0809242105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi HJ, Lui A, Ogony J, Jan R, Sims PJ, Lewis-Wambi J. Targeting interferon response genes sensitizes aromatase inhibitor resistant breast cancer cells to estrogen-induced cell death. Breast Cancer Res. 2015;17:6. doi: 10.1186/s13058-014-0506-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ratman D, Vanden Berghe W, Dejager L, Libert C, Tavernier J, Beck IM, et al. How glucocorticoid receptors modulate the activity of other transcription factors: a scope beyond tethering. Mol Cell Endocrinol. 2013;380(1–2):41–54. doi: 10.1016/j.mce.2012.12.014. [DOI] [PubMed] [Google Scholar]
- 48.Flammer JR, Dobrovolna J, Kennedy MA, Chinenov Y, Glass CK, Ivashkiv LB, et al. The type I interferon signaling pathway is a target for glucocorticoid inhibition. Mol Cell Biol. 2010;30(19):4564–74. doi: 10.1128/MCB.00146-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Uhlenhaut NH, Barish GD, Yu RT, Downes M, Karunasiri M, Liddle C, et al. Insights into negative regulation by the glucocorticoid receptor from genome-wide profiling of inflammatory cistromes. Mol Cell. 2013;49(1):158–71. doi: 10.1016/j.molcel.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nacht AS, Pohl A, Zaurin R, Soronellas D, Quilez J, Sharma P, et al. Hormone-induced repression of genes requires BRG1-mediated H1.2 deposition at target promoters. EMBO J. 2016;35(16):1822–43. doi: 10.15252/embj.201593260. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.