

Abstract
Vaccine elicitation of protective antibody responses has proved difficult for a number of important human pathogens, including HIV-1. The amount of somatic hypermutation associated with the development of broadly neutralizing antibodies against HIV has not been achieved using conventional immunization strategies. An underexplored aspect of vaccine design is modulation of antigen kinetics. Immunization strategies with extended antigen availability have recently been shown to enhance humoral responses. In this review, we explore the mechanisms through which sustained antigen availability can enhance germinal center responses and the potency of antibody responses. These potential mechanisms include shifting B cell recognition away from non-neutralizing immunodominant epitopes, altered kinetics of immune complex deposition, improved T follicular helper (Tfh) cell responses, enhanced affinity maturation, and enhanced development of B cell memory. Finally, we discuss immunization strategies that result in extended antigen availability.
Introduction
Most licensed human vaccines rely on antibody-mediated responses for protection. Those responses are primarily dependent on CD4+ T cells and germinal centers (GCs). GCs are sites within lymphoid organs where B cells undergo B cell receptor (BCR) somatic hypermutation (SHM) to enhance BCR affinity for antigen. Knowledge of the GC processes can almost certainly improve rational vaccine design, if parameters that modulate those processes can be understood. The use of model protein antigens has provided considerable insight into the mechanisms underlying GC and antibody responses. However, the use of simple model antigens most likely does not reflect the immunological challenges that more complex pathogen antigens present, which have potently been driven by eons of evolution to be difficult for host B cells to recognize and neutralize. Few mutations are required for development of high-affinity antibodies against most simple model antigens, including the most commonly studied model antigen 4-hydroxy-3-nitrophenyl acetyl (NP), which only requires a single BCR amino acid mutation to develop high affinity antibodies [1]. Protective antibodies against some pathogens, including HIV-1, contain high numbers of amino acid mutations (> 10) and develop over extended periods of time during infection [2,3]. Lastly, the lifespan of GCs elicited by model antigens can also be short compared to even acute natural infections, where there is frequently a prolonged supply of antigen and GC reactions can last many weeks [4]. Thus, experimental studies of more complex antigens are necessary to study the importance of GC parameters involved in the development of potent antibodies against difficult epitopes on pathogens [5].
One example of a difficult pathogen antigen for B cell recognition and neutralization is HIV envelope (Env). Approximately 10% of HIV+ individuals develop potent broadly neutralizing antibodies (bnAbs) targeting HIV Env [3]. These bnAbs take multiple years to develop and accumulate more amino acid mutations than antibodies generated during conventional immunizations. Many HIV bnAbs require rare SHM events, including deletions or combinatorial mutations (e.g., addition of a new disulfide bond across a CDR loop). Longitudinal analyses of BCR and viral lineages throughout HIV infection has provided clear evidence that bnAbs undergo high amounts of affinity maturation before obtaining their broadly neutralizing activity [6,7]. The development of bnAbs via immunization is a major challenge and it is likely that certain conditions that resemble natural infection, including persistent antigen presence, are required for HIV bnAb development [8].
A promising avenue in rational vaccine design for modulating GCs is the sustained delivery of antigen, which can more mimic natural infection. Sporadic studies more than a decade ago found that controlled release of antigen over a longer period of time could result in stronger immune responses than conventional bolus injections [9–11]. More recent studies have revisited this concept with substantial success [12–14]. Here we describe several mechanisms through which sustained antigen availability may modulate the GC response to enhance the humoral response. These mechanisms include 1) increased availability of native antigen, 2) increased immune complex deposition, 3) modulation of Tfh help and affinity maturation, and 4) modulation of memory B cell formation. Lastly, we discuss the implications of these immunological processes and extended antigen strategies for vaccine design.
Availability of Intact Protein Antigen
GC B cells with the highest affinity for antigen are selected to survive and proliferate based on the ability of the B cell to strip antigen from follicular dendritic cells (FDCs) and subsequently receive help from Tfh cells. One should consider how that process aligns with conventional immunizations. Conventional protein immunizations deliver antigen and adjuvant in a single bolus injection. A potential shortcoming of that strategy is that it is not synchronized with the GC response. The GC response peaks weeks after initial antigen exposure [15]. It is likely that the highest number of B cells are undergoing affinity maturation weeks after initial antigen exposure. It is important to consider that all proteins have a half-life and are susceptible to degradative processes over time. Thus, for proteins that don’t exhibit exceptional stability, it is likely that at the peak of the GC response after a conventional immunization much of the antigen presented by FDCs to GC B cells is nonnative protein and protein degradation products, which expose epitopes that are normally hidden or nonexistent on the native form of the protein (Figure 1). That is a potentially problematic and counterproductive situation. There are data suggesting that in some cases, nonnative epitopes can be immunodominant and distract the GC response from relevant targets.
Figure 1. Slow immunogen release improves the availability of intact antigen.
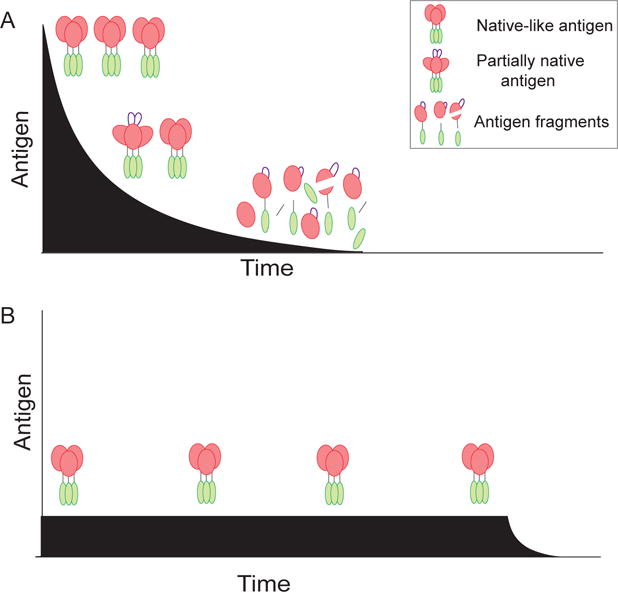
(A) Soluble immunogen can lose native structure due to lability or proteolysis over time. This can lead to exposure of irrelevant and potentially distracting epitopes. (B) Sustained release of immunogen limits the amount of soluble antigen available to proteases, and ensures availability of intact immunogen at later times during the immune response.
HIV Env is one such example. Env is the only target of HIV-neutralizing antibodies. The protein is made up of three gp120 and three gp41 molecules. Immunization with gp120 or labile Env fails to elicit neutralizing antibodies against most circulating HIV strains (frequently called Tier 2 and 3 viruses) [16,17]. Most antibodies from these immunizations are directed against the V3 loop, which is normally hidden within the intact trimer, and thus such antibodies are largely irrelevant. Disorganization or degradation of the Env trimer can result in the exposure of the V3 loop and other non-neutralizing epitopes, which could potentially be immunodominant over neutralizing epitopes. We recently demonstrated proof of concept of antigen integrity impacting GC responses and B cell epitope dominance. The response against the V3 loop can be drastically reduced in mice by sequestering the Env trimer and adjuvant in osmotic minipumps [13]. Osmotic minipumps are nonmechanical delivery systems implanted subcutaneously to continuously supply material at a fixed rate over several days or weeks. Each immunization of immunogen plus adjuvant was slowly delivered over one or two weeks. After three immunizations, the antibody response of conventional bolus-immunized mice was frequently dominated by the V3-response. In contrast, the V3-response was no longer dominant in osmotic pump immunized mice. Sustained release of native trimers was sufficient to shift B cell recognition towards more native epitopes. Naïve B cells that target neutralizing epitopes are likely immunorecessive for difficult to neutralize pathogens (i.e., rare and/or of very low affinity compared to immunodominant epitope targets). The likelihood that rare B cells with neutralizing specificity will be successfully recruited increases when most, if not all, antigen in the immunization maintains its native form. Sustained antigen delivery is therefore an attractive strategy in vaccine design for labile antigens, or antigens for which the B cells specific to critical epitopes are known or suspected of being immunorecessive.
Immune Complex Deposition
FDCs concentrate and present intact antigen in the form of immune complexes to B cells in the light zone (LZ) of the GC [18,19]. B cells test their evolving affinity on FDC immune complexes. B cells with BCRs with higher affinity to the antigen are selected to survive and undergo an additional round of affinity maturation or develop into plasma cells. The resulting higher-affinity IgG from newly generated plasma cells can then form new immune complexes, replacing the lower affinity immune complexes in a recursive process. This results in longer retention of antigen on FDCs (Figure 2). If concentrations of high affinity antibody become sufficiently high, all antigen becomes complexed with antibody. This can lead to GC termination [20,21]. The effects of extended antigen availability immunization strategies on antigen retention by FDCs and the antibody response were recently explored [12]. Mice that were given repeated, exponentially-increasing immunization doses retained antigen in their draining LNs significantly longer than mice given a single bolus. This was correlated with improved immune complex formation on FDCs. Immunogen was provided during a window of time when antigen-specific antibody was available for immune complex formation (> d6 post-immunization). This shift in immune complex kinetics is significant, as the peak of the GC response can frequently occur multiple weeks after an immunization. More antigen on FDCs during later stages of a GC likely increases GC B cell clonotypic diversity (Figure 2), allowing for significantly more BCR sequence space to be explored for high affinity BCR mutations. During a conventional immunization, most antigen presentation, and thus B cell selection, likely occurs prior to the peak GC response. Interestingly, administration of the same dosing profile over different time frames resulted in differences in the humoral response. Immunization over one or three weeks resulted in significantly lower antibody titers compared to immunization over two weeks [12,13]. Altogether, these data indicate that extended antigen availability improves immune complex formation and thus improves the magnitude and quality of GCs.
Figure 2. Sustained immunogen release enhances immune complex deposition and GCs.
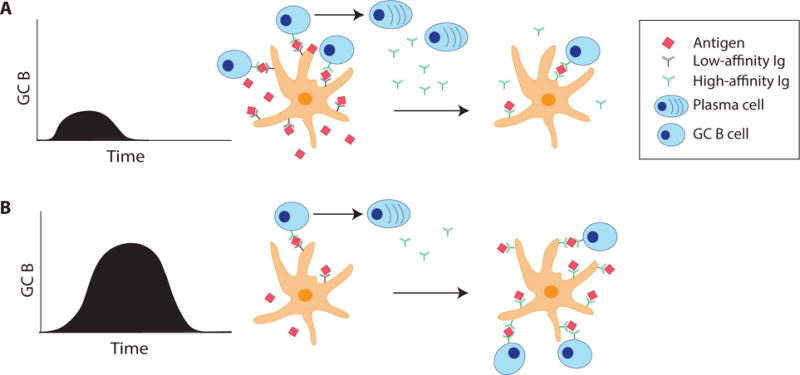
Low-affinity antibodies bind antigen as immune complexes and are deposited on follicular dendritic cells (FDCs). Some B cells that have sufficient affinity to antigen will differentiate into plasma cells early in the immune response and secrete higher affinity antibodies, which then bind remaining free antigen. (A) During a conventional protein immunization, most antigen is presented to B cells early relative to the peak of the GC response. Thus, B cell affinity maturation rates may peak relatively early under those conventional immunization conditions. (B) Sustained antigen release immunization introduces new antigen during a time window when higher affinity IgG is produced, allowing for increased immune complex formation and deposition onto FDCs. This antigen is retained at higher concentrations and for longer durations on FDCs. Thus, the magnitude of the GC response is larger, and B cell affinity maturation rates may be sustained better than under conventional immunization conditions.
Tfh Cell Help and Affinity Maturation
T follicular helper (Tfh) cells are required for GC development and function [22]. GC B cells with the highest affinity capture and present the most antigen, in the form of peptide-MHCII complexes to GC Tfh cells in the LZ. GC Tfh cells selectively help B cells that present the most antigen by providing signals essential for GC B cell survival, proliferation, and mutation [23,24]. Surviving GC B cells migrate to the dark zone (DZ), where they proliferate and undergo SHM. Mutated GC B cells migrate back to the LZ, where the highest affinity B cells are selected again by GC Tfh cells. The amount of help provided by GC Tfh to GC B cells determines the rate at which B cells proliferate and the number of mutations the B cell acquires within a given selection cycle [24,25]. GC Tfh cells therefore control the amount of clonotypic BCR diversification within a GC. High affinity antibodies against simple antigens require only a few mutations. Thus, the BCR diversity pool (i.e. the number of GC B cells) necessary to develop these antibodies is relatively small. Hence, stringent Tfh help, in the form of limited numbers of GC Tfh cells, is likely optimal for affinity maturation to simple antigens. In contrast, to develop high affinity antibodies against more complex antigens, high rates of SHM are likely required. Therefore, higher numbers of GC Tfh cells or greater GC Tfh help is needed in GC responses to complex antigens to maintain a larger pool of diverse BCRs and increase the possibility of developing high affinity nAbs (Figure 3). Lastly, strong GC Tfh help is likely essential for the development of some bnAbs that specifically require rare mutation events like the addition of disulfide bonds [26]. Generation of two cysteine mutations within a given GC cycle may be necessary as a single cysteine mutation is likely detrimental to BCR affinity. With strong GC Tfh help, GC B cells can accumulate multiple mutations in a single round of SHM. Thus, enhancement of the Tfh cell response is likely of value in the context of immunization against antigens that are difficult for B cell to recognize.
Figure 3. A model of Tfh help modulation by extended immunogen release.
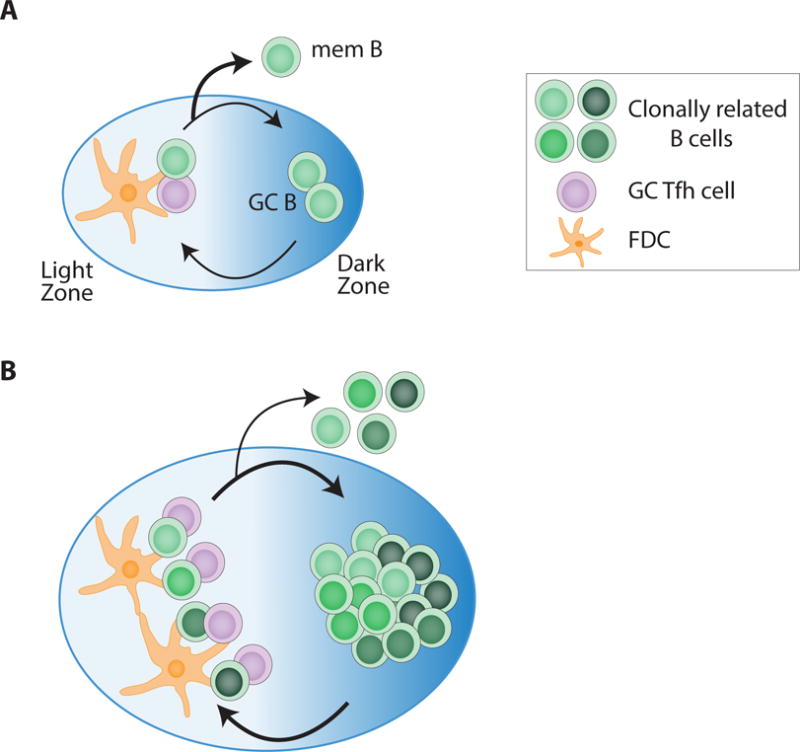
(A) During a conventional immunization, the presence of low numbers of GC Tfh cells usually restricts the number of GC B cells, limiting BCR diversity. (B) Extended immunogen release can support increased GC Tfh cell numbers, or greater GC Tfh help quality. More GC B cells are able to proliferate and undergo SHM in the presence of increased GC Tfh cell help. More GC Tfh help both generates a larger pool of clonotypic BCR diversity and lowers competition between B cells, allowing for the generation of GC B cells with more complex BCR antigen recognition features.
Based on the basic biology of CD4 T cells, and Tfh cells in particular [27,28], it was likely that immunization strategies utilizing sustained antigen delivery would increase the abundance of Tfh cells. Indeed, continuous antigen release over 1–2 weeks using osmotic pumps in mice resulted in higher frequencies of GC Tfh, Tfh and GC B cells [12]. Strikingly, non-human primates (NHPs) immunized with HIV Env via two week osmotic pumps had dramatically increased autologous HIV nAbs compared to animals given a conventional bolus immunization [14]. Furthermore, HIV nAbs also developed in osmotic pump immunized animals more rapidly than conventional animals, suggesting that extended antigen release can accelerate the generation of GC B cells capable of neutralizing HIV. Osmotic pump immunized NHPs had significantly higher frequencies of GC Tfh cells. Interestingly, higher frequencies of Ki67+ GC Tfh were observed in pump animals compared to the conventionally immunized animals. Altogether, these data demonstrate that sustained antigen administration alters the GC response and can have an impressive impact on the resulting antibody response. Whether an increase in GC Tfh cell frequency results in increased rates of GC B cell SHM remains to be determined. Overall, it is now of great interest to directly investigate the effect of extended antigen availability on Tfh cell biology and the Tfh-dependent processes of SHM and affinity maturation against difficult antigens.
Development of Memory
Vaccination strategies to elicit memory B cells (MBCs) that can reenter GCs are attractive, as it is highly unlikely a single conventional immunization will allow for the extensive mutations required for neutralization of some pathogens. Few, if any, studies have explored the effect of vaccination with persistent antigen on the development of memory. Antigen kinetics may affect the pathways that control the decisions to differentiate into MBCs or PCs. B cells with lower affinity preferentially become MBCs early relative to the peak of the GC response [29,30]. Immunizations with sustained antigen may delay MBC development relative to a conventional bolus. This may allow GC B cells to accumulate more mutations, and thus higher affinity, before exiting GCs as MBCs. There is currently no evidence to support or refute this hypothesis, but insights into these processes may provide considerable guidance in vaccine design.
Delivery systems and adjuvants
Osmotic pumps have been used for sustained immunization delivery in several proof-of-principle animal studies. While surgical implantation of osmotic pumps may be feasible for a Phase I human vaccine trial, osmotic pumps are clearly impractical for large scale human vaccine trials. Near daily repeated injections are also impractical for large scale human vaccine trials. More pragmatic delivery systems include slow release microneedles implanted through the skin with a patch and injectable microparticles [31–33]. Additionally, it has long been believed that some adjuvants, most notably alum, modulate humoral immune responses via a ‘depot effect’, in which antigen is retained at the site of injection and slowly released [34,35]. However, the adjuvanticity of alum is not dependent on depot formation. Surgical removal of alum depots two hours after injection did not affect the magnitude of the antibody response in mice [36]. Nevertheless, it remains unknown how much of the effect of other adjuvants on GCs is due to extended antigen release. The formation of an adjuvant antigen depot in and of itself is not informative, as it is not clear that the antigen trapped in the depot becomes available to lymphocytes at later times. The duration and kinetics of antigen release are unknown for many adjuvants. Thus, these recent advances demonstrating the value of extended antigen release highlight that a mechanistic understanding of the depot effects of adjuvants is needed. Such experiments may need to be done with difficult or immunorecessive antigens to accurately assess their impact on Tfh cells, GCs, and humoral immunity, for the reasons described above.
Summary
Protein vaccines safely elicit protective responses in humans and have greatly improved public health globally. For some pathogens with difficult neutralizing epitopes, nontraditional vaccination strategies may be required for long-lived immunity. It will be important to further understand how modulating immunogen kinetics impacts Tfh cell help, SHM, and MBC formation. Insights into the kinetics of the GC response, particularly in humans and NHPs, will benefit the efforts to optimize vaccination against difficult targets.
Highlights.
Extended antigen delivery immunization results in stronger humoral immune responses
Germinal centers are enhanced by extended antigen delivery
Slow antigen release can limit responses targeting non-native epitopes
Acknowledgments
We thank Crotty lab members for helpful discussions. This work was supported by UM1 AI100663 (CHAVI-ID) and R01 AI134796 (S.C.) from the NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
We wish to confirm that there are no known conflicts of interest associated with this publication.
References
- 1.Weiss U, Rajewsky K. The repertoire of somatic antibody mutants accumulating in the memory compartment after primary immunization is restricted through affinity maturation and mirrors that expressed in the secondary response. Journal of Experimental Medicine. 1990;172:1681–1689. doi: 10.1084/jem.172.6.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haynes BF, Moody MA, Alam M, Bonsignori M, Verkoczy L, Ferrari G, Gao F, Tomaras GD, Liao H-X, Kelsoe G. Progress in HIV-1 vaccine development. J Allergy Clin Immunol. 2014;134:3–10. doi: 10.1016/j.jaci.2014.04.025. quiz 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Landais E, Huang X, Havenar-Daughton C, Murrell B, Price MA, Wickramasinghe L, Ramos A, Bian CB, Simek M, Allen S, et al. Broadly Neutralizing Antibody Responses in a Large Longitudinal Sub-Saharan HIV Primary Infection Cohort. PLoS Pathog. 2016;12:e1005369. doi: 10.1371/journal.ppat.1005369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adachi Y, Onodera T, Yamada Y, Daio R, Tsuiji M, Inoue T, Kobayashi K, Kurosaki T, Ato M, Takahashi Y. Distinct germinal center selection at local sites shapes memory B cell response to viral escape. J Exp Med. 2015;212:1709–1723. doi: 10.1084/jem.20142284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Havenar-Daughton C, Lee JH, Crotty S. Tfh cells and HIV bnAbs, an immunodominance model of the HIV neutralizing antibody generation problem. Immunological Reviews. 2017;275:49–61. doi: 10.1111/imr.12512. [DOI] [PubMed] [Google Scholar]
- 6.Wu X, Zhou T, Zhu J, Zhang B, Georgiev I, Wang C, Chen X, Longo NS, Louder M, McKee K, et al. Focused evolution of HIV-1 neutralizing antibodies revealed by structures and deep sequencing. Science. 2011;333:1593–1602. doi: 10.1126/science.1207532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7′.Liao H-X, Lynch R, Zhou T, Gao F, Alam SM, Boyd SD, Fire AZ, Roskin KM, Schramm CA, Zhang Z, et al. Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature. 2013;496:469–476. doi: 10.1038/nature12053. This study demonstrates extensive somatic hypermutation of a B cell lineage that became an HIV broadly neutralizing antibody over several years. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borrow P, Moody MA. Immunologic characteristics of HIV-infected individuals who make broadly neutralizing antibodies. Immunological Reviews. 2017;275:62–78. doi: 10.1111/imr.12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kemp JM, Kajihara M, Nagahara S, Sano A, Brandon M, Lofthouse S. Continuous antigen delivery from controlled release implants induces significant and anamnestic immune responses. Vaccine. 2002;20:1089–1098. doi: 10.1016/s0264-410x(01)00444-3. [DOI] [PubMed] [Google Scholar]
- 10.Higaki M, Azechi Y, Takase T, Igarashi R, Nagahara S, Sano A, Fujioka K, Nakagawa N, Aizawa C, Mizushima Y. Collagen minipellet as a controlled release delivery system for tetanus and diphtheria toxoid. Vaccine. 2001;19:3091–3096. doi: 10.1016/s0264-410x(01)00039-1. [DOI] [PubMed] [Google Scholar]
- 11.Ehrenhofer C, Opdebeeck JP. The effects of continuous and intermittent delivery of antigens of boophilus microplus on the development of murine antibodies. Parasitology Today. 1995;11:445. doi: 10.1016/0304-4017(94)00747-z. [DOI] [PubMed] [Google Scholar]
- 12••.Tam HH, Melo MB, Kang M, Pelet JM, Ruda VM, Foley MH, Hu JK, Kumari S, Crampton J, Baldeon AD, et al. Sustained antigen availability during germinal center initiation enhances antibody responses to vaccination. Proc Natl Acad Sci USA. 2016 doi: 10.1073/pnas.1606050113. This study demonstrates that extended immunization delivery results in longer retention of antigen, larger GCs, more GC Tfh cells, and stronger antibody responses. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13•.Hu JK, Crampton JC, Cupo A, Ketas T, van Gils MJ, Sliepen K, de Taeye SW, Sok D, Ozorowski G, Deresa I, et al. Murine Antibody Responses to Cleaved Soluble HIV-1 Envelope Trimers Are Highly Restricted in Specificity. J Virol. 2015;89:10383–10398. doi: 10.1128/JVI.01653-15. This study demonstrated extended immunogen release can shift B cell recognition away from nonnative epitopes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14••.Pauthner M, Havenar-Daughton C, Sok D, Nkolola JP, Bastidas R, Boopathy AV, Carnathan DG, Chandrashekar A, Cirelli KM, Cotrell CA, et al. Elicitation of Robust Tier 2 Neutralizing Antibody Responses in Nonhuman Primates by HIV Envelope Trimer Immunization Using Optimized Approaches. Immunity. 2017;46:1073–1088.e6. doi: 10.1016/j.immuni.2017.05.007. This large study shows slow continuous delivery of antigen via osmotic pump immunization results in earlier and stronger autologous neutralizing antibody responses in non-human primates. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Havenar-Daughton C, Carnathan DG, Torrents de la Peña A, Pauthner M, Briney B, Reiss SM, Wood JS, Kaushik K, van Gils MJ, Rosales SL, et al. Direct Probing of Germinal Center Responses Reveals Immunological Features and Bottlenecks for Neutralizing Antibody Responses to HIV Env Trimer. Cell Rep. 2016;17:2195–2209. doi: 10.1016/j.celrep.2016.10.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mascola JR, Montefiori DC. The role of antibodies in HIV vaccines. Annu Rev Immunol. 2010;28:413–444. doi: 10.1146/annurev-immunol-030409-101256. [DOI] [PubMed] [Google Scholar]
- 17.Sliepen K, Sanders RW. HIV-1 envelope glycoprotein immunogens to induce broadly neutralizing antibodies. Expert Rev Vaccines. 2016;15:349–365. doi: 10.1586/14760584.2016.1129905. [DOI] [PubMed] [Google Scholar]
- 18.Heesters BA, Myers RC, Carroll MC. Follicular dendritic cells: dynamic antigen libraries. Nature Publishing Group. 2014;14:495–504. doi: 10.1038/nri3689. [DOI] [PubMed] [Google Scholar]
- 19.Heesters BA, van der Poel CE, Das A, Carroll MC. Antigen Presentation to B Cells. Trends Immunol. 2016;37:844–854. doi: 10.1016/j.it.2016.10.003. [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Meyer-Hermann M, George LA, Figge MT, Khan M, Goodall M, Young SP, Reynolds A, Falciani F, Waisman A, et al. Germinal center B cells govern their own fate via antibody feedback. Journal of Experimental Medicine. 2013;210:457–464. doi: 10.1084/jem.20120150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pape KA, Taylor JJ, Maul RW, Gearhart PJ, Jenkins MK. Different B cell populations mediate early and late memory during an endogenous immune response. Science. 2011;331:1203–1207. doi: 10.1126/science.1201730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. 2014;41:529–542. doi: 10.1016/j.immuni.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Victora GD, Schwickert TA, Fooksman DR, Kamphorst AO, Meyer-Hermann M, Dustin ML, Nussenzweig MC. Germinal center dynamics revealed by multiphoton microscopy with a photoactivatable fluorescent reporter. Cell. 2010;143:592–605. doi: 10.1016/j.cell.2010.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gitlin AD, Shulman Z, Nussenzweig MC. Clonal selection in the germinal centre by regulated proliferation and hypermutation. Nature. 2014;509:637–640. doi: 10.1038/nature13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25••.Gitlin AD, Mayer CT, Oliveira TY, Shulman Z, Jones MJK, Koren A, Nussenzweig MC. T cell help controls the speed of the cell cycle in germinal center B cells. Science. 2015;349:643–646. doi: 10.1126/science.aac4919. This study showed that rates of B cell proliferation and somatic hypermutation are directly proportional to the amount of antigen presented by GC B cells to GC Tfh cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doria-Rose NA, Schramm CA, Gorman J, Moore PL, Bhiman JN, DeKosky BJ, Ernandes MJ, Georgiev IS, Kim HJ, Pancera M, et al. Developmental pathway for potent V1V2-directed HIV-neutralizing antibodies. Nature. 2014;509:55–62. doi: 10.1038/nature13036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Obst R, van Santen H-M, Mathis D, Benoist C. Antigen persistence is required throughout the expansion phase of a CD4(+) T cell response. J Exp Med. 2005;201:1555–1565. doi: 10.1084/jem.20042521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baumjohann D, Preite S, Reboldi A, Ronchi F, Ansel KM, Lanzavecchia A, Sallusto F. Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity. 2013;38:596–605. doi: 10.1016/j.immuni.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 29•.Shinnakasu R, Inoue T, Kometani K, Moriyama S, Adachi Y, Nakayama M, Takahashi Y, Fukuyama H, Okada T, Kurosaki T. Regulated selection of germinal-center cells into the memory B cell compartment. Nat Immunol. 2016;17:861–869. doi: 10.1038/ni.3460. This study demonstrates memory B cells form from GC B cells of relatively low affinity that receive less Tfh cell help. This process of memory B cell development is regulated by Bach2 expression in the GC B cells. [DOI] [PubMed] [Google Scholar]
- 30.Weisel FJ, Zuccarino-Catania GV, Chikina M, Shlomchik MJ. A Temporal Switch in the Germinal Center Determines Differential Output of Memory B and Plasma Cells. Immunity. 2016;44:116–130. doi: 10.1016/j.immuni.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31•.DeMuth PC, Li AV, Abbink P, Liu J, Li H, Stanley KA, Smith KM, Lavine CL, Seaman MS, Kramer JA, et al. Vaccine delivery with microneedle skin patches in nonhuman primates. Nat Biotechnol. 2013;31:1082–1085. doi: 10.1038/nbt.2759. This was the first study to immunize NHPs using transdermal patches. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeMuth PC, Min Y, Irvine DJ, Hammond PT. Implantable silk composite microneedles for programmable vaccine release kinetics and enhanced immunogenicity in transcutaneous immunization. Adv Healthc Mater. 2014;3:47–58. doi: 10.1002/adhm.201300139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Slobbe L, Medlicott N, Lockhart E, Davies N, Tucker I, Razzak M, Buchan G. A prolonged immune response to antigen delivered in poly (epsilon-caprolactone) microparticles. Immunol Cell Biol. 2003;81:185–191. doi: 10.1046/j.1440-1711.2003.01155.x. [DOI] [PubMed] [Google Scholar]
- 34.Tritto E, Mosca F, De Gregorio E. Mechanism of action of licensed vaccine adjuvants. Vaccine. 2009;27:3331–3334. doi: 10.1016/j.vaccine.2009.01.084. [DOI] [PubMed] [Google Scholar]
- 35.Oleszycka E, Lavelle EC. Immunomodulatory properties of the vaccine adjuvant alum. Curr Opin Immunol. 2014;28:1–5. doi: 10.1016/j.coi.2013.12.007. [DOI] [PubMed] [Google Scholar]
- 36.Hutchison S, Benson RA, Gibson VB, Pollock AH, Garside P, Brewer JM. Antigen depot is not required for alum adjuvanticity. FASEB J. 2012;26:1272–1279. doi: 10.1096/fj.11-184556. [DOI] [PMC free article] [PubMed] [Google Scholar]