

Abstract
The three dimensional folding of mammalian genomes is cell-type specific and difficult to alter suggesting that it is an important component of gene regulation. However, given the multitude of chromatin associating factors, the mechanisms driving the co-localization of active chromosomal domains and the role of this organization in regulating the transcription program in adipocytes are not clear. Analysis of genome-wide chromosomal associations revealed cell type-specific spatial clustering of adipogenic genes in 3T3-L1 cells. Time course analysis demonstrated that the adipogenic "hub", sampled by PPARγ and Lpin1, undergoes orchestrated reorganization during adipogenesis. Coupling the dynamics of genome architecture with multiple chromatin datasets indicated that among all the transcription factors tested, RXR is central to genome reorganization at the beginning of adipogenesis. Interestingly, at the end of differentiation, the adipogenic hub was shifted to an H3K27me3 repressive environment in conjunction with attenuation of gene transcription.
We propose a stage-specific hierarchy for the activity of transcription factors contributing to the establishment of an adipogenic genome architecture that brings together the adipogenic genetic program. In addition, the repositioning of this network in a H3K27me3-rich environment at the end of differentiation may contribute to the stabilization of gene transcription levels and reduce the developmental plasticity of these specialized cells.
Keywords: Chromosome conformation, Adipogenesis, Transcription, Transcription Factor
Graphical abstract
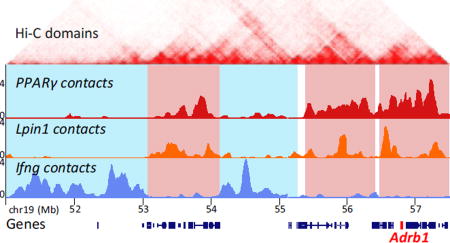
Inter-chromosomal associations shared by the adipogenic genesPPARγ and Lpin1, while distinct from the compartment of the non-adipogenic gene Ifng. At the beginning of adipogenesis the adipogenic compartment is shaped by spatial clustering of binding sites of the adipogenic transcription factor RXR. At the end of differentiation, the adipogenic hub is shifted to an H3K27me3 repressive environment in conjunction with attenuation of gene transcription.
Introduction
Mammalian genomes are folded in the 3D nuclear space in a non-random manner related to cell-type and function. The specific genomic spatial organization provides an important regulatory level of transcriptional activity [1–4]. High-resolution 3C chromatin interaction maps demonstrated that metazoan genomes are segmented into chromosomal domains of internal frequently associating loci, termed Topologically Associating Domains (TADs) [5–7]. TADs were shown to be evolutionarily conserved and give rise to chromosomal structures resembling a string of beads. In contrast to the mostly constant TAD boundaries, selective inter-TAD associations give rise to cell-type specific chromosomal compartments, which are developmentally regulated [3,5,6,8–10]. This cell-type specific high-order chromosomal organization brings together genes and regulatory chromatin loci, suggesting that it is a key determinant of cell identity.
Yet, it is not clear how the spatial ensemble of genes, histone marks and transcription factor binding sites at the associated loci contribute to the establishment of cell-type specific genome architecture and regulation of gene transcription. Recent studies in functional differentiation models indicate that chromatin binding of lineage-determining transcription factors (TFs) leads to spatial clustering of these binding sites, and thereby dictates high-order genome organization. For example, spatial association of chromosomal loci harbouring binding sites for the lineage determining transcription factors STAT4 or STAT6 in multi-potent naive cells, leads to cell-type specific genome architecture of Th1 or Th2 lymphocytes, respectively [3].
Similarly, in B cells, genome re-organization during developmental progression from the multipotent pre-pro-B-cells to the committed pro-B cells is associated with the histone acetyltransferase p300 and binding of the transcription factors E2A or PU.1 [8]. The importance of TFs as genome organizers was also demonstrated in pluripotent stem cell differentiation [11–13]. Interestingly, during stem cell differentiation the spatial position of genes can alternate from active, TF-enriched, to polycomb-repressed (H3K27me3 enriched) environments, to suppress pluripotency genes and to promote a specialized gene transcription program [11]. This further demonstrates a link between genome architecture and gene regulation. However the dichotomy of active versus repressed compartments, defined by the chromatin features, is often an oversimplification. Some domains may carry the H3K27me3 mark together with enhancer or active chromatin marks, both in undifferentiated and specialized cells [14–18]. Thus the complexity, the reciprocity, and the role of such compartments in gene regulation are far from understood.
Since differentiation is a dynamic process, it is impossible to identify the factors contributing to cell-type specific genome organization from only the differentiation end-point. Thus, to delineate the complex multifactorial process giving rise to this organization, we followed and integrated the dynamic shaping of genome 3D organization together with multiple chromatin features at high temporal resolution over the course of 3T3-L1 adipogenic differentiation, one of the most extensively characterized differentiation processes [19]. Moreover the spatial positioning of adipogenic genes during adipogenesis is dynamic, and is yet to be characterized at the genome-wide level [20–22]. Adipogenic differentiation of 3T3-L1 is initiated by a cocktail of inducers which activates a network of transcription factors in various signaling pathways. The major transcription factors in adipogenesis, including the key pro-adipogenic factors C/EBPs and PPARγ, orchestrate the dynamic progression of adipocyte differentiation transcriptional network in a sequential and combinatorial manner [19,23].
Here, we characterized the changes during the differentiation process in the spatial chromosomal environments of key adipogenic gene loci, PPARγ, an essential adipogenic determining transcription factor, and Lpin1, encoding a co-activator of PPARγ and enzyme in triglyceride synthesis [24–26]. These genes are highly significant for adipocyte differentiation and regulation of lipid metabolism. Notably, polymorphisms in PPARγ and Lpin1 genes are associated with metabolic syndromes and type 2 diabetes (T2D), and PPARγ activation by insulin-sensitizing drugs can be used in T2D treatment [27,28].
By combining determinations of genome architecture with dynamics of multiple chromatin datasets we demonstrate that the spatial organization of the adipogenic genes is highly cell-type specific and changes in a coordinated manner during differentiation. Among the adipogenic TFs investigated, RXR binding stands out as key in genome reorganization at the beginning of adipogenesis. While the adipogenic hub was progressively enriched with adipogenic TF binding, at the end of differentiation it was shifted to a H3K27me3 repressive environment in conjunction with attenuation of gene transcription. This repositioning may suggest an epigenetic mechanism to stabilize the transcription program of the specialized cell.
Results
Lineage-specific spatial organization of key adipogenic gene loci
Synchronous induction of 3T3-L1 pre-adipocyte differentiation initiates series of genome-wide events giving rise to a highly homogeneous population (~95%) of terminally differentiated cells, as assessed by lipid staining (not shown). Although the tightly orchestrated cascades of transcription factor binding and chromatin modifications along the chromatin fiber are relatively well characterized, it is not well understood how these events converge spatially to give rise to the genome architecture of adipocytes. We first used 4C-seq to determine the changes in chromatin interactions at different time points during 3T3-L1 adipocyte differentiation for two adipocyte specific genes PPARγ (Chr6), one of the key transcription factors in adipogenic differentiation, and Lpin1 (Chr12), an enzyme involved in triglyceride synthesis. Notably, PPARγ and Lpin1 undergo similar transcription dynamics during adipogenesis [29]. Lipin-1 also functions as a transcriptional coactivator of PPARγ and is required for adipocyte differentiation. For comprehensive capture of the entire chromosomal interactome, high complexity 4C libraries were sequenced to high depth. Similarly to previous 4C-seq datasets from our group and from other labs, the majority (>75%) of reads were derived from the cis chromosome. These reads encompassed approximately 35% of the total positive HindIII fragments in the 4C dataset, reflecting the high probability of intra-chromosomal associations (Table S1) [30–32]. The 4C libraries captured intra- and inter-chromosomal contacts that were defined based on a score assigned to every HindIII site. We retrieved the HindIII sites with the top 5% score (“positive HindIII sites”) for each sample, thereby providing equivalent coverage of contact regions throughout differentiation. Clusters of more than fifteen consecutive positive HindIII sites were defined as a contact domain (Table S2). The contact domains for all time points were then merged together to represent the PPARγ and Lpin1 interactome throughout differentiation. Overall, we identified 944 and 1109 contact domains encompassing 288 and 265 Mb for PPARγ and Lpin1, respectively. The inter-chromosomal contact domains ranged from 100kb to 2Mb, similar to the sizes of high resolution topologically associating domains (TADs) [33] (Fig. 1A, B). The capture of low-abundant inter-chromosomal contact domains at the resolution of TADs, which define chromosomal segments of local high contact frequency, indicates the high complexity of the 4C libraries. These data reveal a remarkable overlap between the spatial localization of the two adipogenic gene loci, which are located on different chromosomes (Fig. 1A, C highlighted in red, ~35% shared inter-chromosomal contacts, p=1.9×10−33 using a hypergeometric test for the probability of observing the overlap by chance). In contrast, the interactome of a non-adipogenic gene, Ifng, was strikingly disjoint with only ~2% shared inter-chromosomal contacts (Fig. 1A, C highlighted in blue, p~10−5 using a hypergeometric test for the probability of observing the separation by chance). In addition, the interactomes of these three gene loci showed low (3%–7%, p=0.08–0.53) overlap in B lymphocytes in which they are not expressed (Fig. 1C, D). Overall, the 3T3-L1-specific spatial compartmentalization of adipogenic genes indicates cell type-specific regulation of genome high-order organization.
Fig. 1. Adipocyte-specific genome architecture.

(A) Examples of 4C-seq inter-chromosomal association profiles in 3T3-L1 preadipocytes showing contact domains shared by PPARγ and Lpin1 (highlighted in red), while distinct from Ifng chromosomal contacts (highlighted in blue). Adrb1 adipogenic gene is highlighted in red on the x-axis. Y-axis indicates p-score. Local intra-chromosomal contact domains (TADs) from Hi-C in murine CH12-LX cells are shown on top [33]. (B) Histograms of domain sizes from high resolution mouse CH12-LX TADs [33], PPARγ, Lpin1 and Ifng 4C domains. (C) Venn diagram showing overlap (in base pairs) of the inter-chromosomal interactomes in 3T3-L1 and B lymphocytes. The lowest overlap in 3T3-L1 is between Ifng and the adipogenic genes PPARγ and Lpin1. (D) Examples of 4C-seq inter-chromosomal association profiles in 3T3-L1 and B lymphocytes at C/EBP-β, C/EBP-α and Scd1 adipogenic gene loci (highlighted in red). Y axis indicates p-score. Chromosomal coordinates in Mb of mouse mm9 genome build are indicated on top.
Spatial compartmentalization of the adipogenic genetic program
We noted that the PPARγ and Lpin1 loci associate with each other in the nuclear space, and that their chromosomal contacts contain additional adipogenic genes (Fig. 1A, 1D). For example, the C/EBP-β (Chr 2), C/EBP-α (Chr 7) and Scd1 (Chr19) gene loci associate with both the PPARγ and Lpin1 loci, specifically in 3T3-L1 cells (Fig. 1D). Indeed global analysis indicated that the chromosomal contacts in adipogenesis are highly enriched for adipogenic genes (GO terms). Notably the proportion of adipogenic GO terms (~30% of all adipogenic GO terms in PPARγ and ~35% in Lpin1 contact domains) was significantly higher than the overall enrichment for genes in their spatial compartments (p<0.05 for PPARγ and p<0.001 for Lpin1) (Fig. 2A). Remarkably, ~50% of all adipogenic genes were located in the contact regions of both genes together (Table S3). This enrichment of adipogenic genes was not found in the interactome of the negative control Ifng gene in the same cells nor in the interactome of PPARγ and Lpin1 in B lymphocytes (Fig. 2A).
Fig. 2. Spatial clustering of adipogenic genes and transcription factor binding sites.

(A) PPARγ and Lpin1 contact regions are enriched for adipogenic genes. The proportion of genes within 4C contact regions of PPARγ and Lpin1 (Ifng interactome serves as a negative control) relative to the total genes in the mouse genome (blue bars) and the proportion of adipogenic genes (from 134 adipogenic GO terms) within 4C contact regions (red bars) in adipocytes (left) and B cells (right). **p<0.001, *p<0.05, hypergeometric test. (B) The proportion of 200kb windows with expressed genes within 4C contact regions of Ifng, PPARγ, Lpin1 and genome-wide **p<0.001, proportional test. (C) Similar analysis for the proportion of 200kb windows with expressed adipogenic genes. (D) Spearman correlations of PPARy, Lpin1 and Ifng 4C-seq data with genomic features throughout adipogenesis. Color Key = Spearman values. (E) Example of PPARγ and Lpin1 chromosomal contact domains in adipocytes (day 6) (highlighted in red) enriched for PPARγ binding (black track). Y-axis indicates p score. Chromosomal coordinates in Mb of mouse mm9 genome build are indicated on top. (F) Log2 ratios of RNAPII occupancy on each day relative to day 0.
To determine whether the enrichment for adipogenic genes is related to the enrichment for active genes in the adipogenic hub, we partitioned the genome to non-overlapping 200kb windows with or without expressed genes. The spatial compartment of the adipogenic genes PPARγ and Lpin1 was active relative to the overall genome (Fig 2B). However, the enrichment of adipogenic genes in the active compartment within the PPARγ and Lpin1 interactome was indistinguishable from the rest of the active genome (Fig 2C), indicating that the adipogenic hub is enriched for active genes including, but not limited to, those with adipogenic functions. Thus the spatial clustering of the adipogenic genetic program represents the cell-type specific genome architecture of adipocytes.
To further define the features of the adipogenic nuclear architecture, we correlated our 4C-seq data with genome-wide profiles of chromatin accessibility as determined by DNase-seq, as well as transcription factor binding and histone modifications as determined by chromatin immunoprecipitation (ChIP)-seq in 3T3-L1 cells [14,29,34–37]. In agreement with previous studies from our group as well as those of others [3,10,38,39] high correlation was found between 4C-seq and regulatory elements, as determined by DNase I hypersensitivity (Fig. 2D). Similarly, a high correlation was found between regions identified by 4C-seq and transcription factors determining adipogenic cell fate such as PPARγ, as well as for histone marks of active enhancers such as H3K27ac, H3K4me1, H3K4me2, P300 and MED1 (Fig. 2D, E). A lower correlation was found for features of active genes such as H3K4me3, H3K36me3, and RNA polymerase II occupancy, and this correlation was not related to the transcriptional level of PPARγ and Lpin1 (Fig. 2F). No correlation was found between PPARγ and Lpin1 interacting regions and the inactive chromatin marks H3K27me3 and H3K9me3 (Fig. 2D). Similarly to the combined 4C datasets, PPARγ and Lpin1 interacting regions were predominantly correlated with TF binding and enhancer chromatin at each of the time points along adipogenesis (Fig. 3). The correlation between the adipogenic interactome and overall transcription level, which is lower than the correlation with transcription factor binding, is not surprising and supports the proposed role of TF binding in genome high-order organization by spatial clustering of their binding loci [3,8,11–13,38,40]. In order to depict the hierarchy and combinatorial interplay among factors in establishing genome architecture states we next characterized the dynamics of genome reorganization during adipogenesis.
Fig 3. Adipgenic genes spatial environment enriched for transcription binding sites throughout adipogenesis.

(A–D) Spearman correlations between each bait (indicated at the bottom of each lane) interactome on days 0, 1, 2 and 6, and genomic events. Color Key = Spearman values.
Orchestrated dynamics of genome architecture along adipogenesis
3T3-L1 adipogenesis progresses through the activity of two separate waves of adipogenic TF networks [19]. The adipogenic cocktail induces TFs that act in the first wave (e.g. RXR, C/EBP-β) which activate TFs of the second wave (e.g. PPARγ, C/EBP-α). The second wave of TFs drives final adipocyte differentiation by activating the adipogenic gene program [19]. Importantly, the first wave (day 1) is associated with substantial activation of a large number of regulatory sites, which become accessible for cooperative binding of TFs (3.5 fold induction in DHS sites) [36]. Thus, in addition to pre-adipocytes (day 0) and mature adipocytes (day 6 after induction) our measurements of nuclear architecture at days 1 and 2 represent key intermediate stages of differentiation [36].
Interestingly, the comparison between 4C datasets from different days revealed that more than half of the inter-chromosomal contact regions of PPARγ and Lpin1 in mature adipocytes are shared with the contacts detected at the previous intermediate time points.
To characterize the dynamics of genome spatial reorganization, we clustered contacts exhibiting similar dynamic patterns using K-means cluster analysis (see Methods). To avoid bias from the inherent variation in contact frequency between inter- and intra-chromosomal associations, we included only inter-chromosomal associations in the analysis. PPARγ and Lpin1 interactomes (inter-chromosomal contact regions) exhibited five distinct clusters of contacts with similar temporal behavior (Fig. 4A, C, D, E). Clusters 1–4 displayed transient contacts elevated on one of the days. Cluster 5, on the other hand, contained contacts that remained stable throughout the differentiation process. Strikingly, although the PPARγ and Lpin1 genes reside on different chromosomes, the proportion of contacts in each cluster of dynamic contacts was similar (Fig. 4B). In contrast, the trends were markedly different for the negative control, Ifng locus, which was measured from the same 4C libraries used for PPARγ and Lpin1 (Fig. 4F, G). In fact, the Ifng interactome remained stable overall during adipogenesis, and accordingly, the algorithm could not produce a good estimation for the number of clusters (Fig. 4C). These findings suggest that the dynamics of adipogenic genes during adipogenesis reflects a remarkable coordinated and regulated formation of an adipogenic hub(s). In line with the adipogenic predisposition of 3T3-L1 cells, the proportion of adipogenic genes was highest in the stable contacts (cluster 5, Table S4, Fig. 4H).
Fig. 4. Common chromosomal reorganization dynamics at adipogenic gene loci.

(A) Cluster analysis of PPARγ and Lpin1 contact regions identified by 4C-seq reveal distinct temporal profiles of chromosomal associations. Each cluster is presented in a plot where the X axis indicates the time points in the differentiation. Y axis represents relative 4C signal. Grey lines represent contact domains, light-blue and red lines represent the mean and the medoid, respectively. (B) Pie chart representing the proportion of contact regions in each cluster for each bait. (C) The number of clusters (1 to 9) and the statistical ratio (Gapk) calculated for each 4C dataset showing that for both PPARγ and Lpin1, the best K is 5 (indicated by a circle). For Ifng no such K is obtained. Red bars indicates SE, n=60. Representative examples of 4C regions in each cluster of PPARγ (D) and Lpin1 (E) are highlighted in screen shots from the UCSC Genome Browser. (F, G) Cluster analysis and example of contacts for Ifng 4C. Y-axis indicates p score. Chromosomal coordinates in Mb of mouse mm9 genome build are indicated. (H) The proportion of adipogenic genes in each dynamic cluster from the total of adipogenic genes in the chromosomal interactome of PPARγ (left) and Lpin1 (right). (I) Transcription dynamics (log2 ratios of RNAPII occupancy at each day relative to day 0) of adipogenic genes in cluster 5 (left) and cluster 4 (right), of both PPARγ and Lpin1.
However the dynamics of transcriptional activity were not mirrored in the dynamics of chromosomal reorganization. Although some of the adipogenic genes which were stably associated with both PPARγ and Lpin1 throughout adipogenesis (cluster5) were stably expressed (for example Socs7 and Axin1), others robustly increased at day1 and then gradually decreased (for example Lrp5 and Zfp385a), while Scd1 transcription continuously increased until day 4 and was then attenuated (Fig. 4I). The dynamics of Ero1l transcription were similar to Scd1; however Ero1l joins the spatial compartment of PPARγ and Lpin1 only at the last stage of adipogenesis (Fig. 4, cluster 4). These results are expected given that the correlation between chromosomal associations and transcription was not high in adipocytes (Fig. 2, Fig. 3) nor in other cell types [3,38,40]. These results reflect the complex relationship between gene regulation and genome architecture, as evident for other regulatory layers of gene transcription. For example, transcription factor binding may promote, inhibit, or potentiate gene transcription [41,42]. Thus, to define the molecular basis of genome architecture during adipogenesis, we searched for coordinated dynamics between genome reorganization and other chromatin processes (e.g. histone modifications and TF binding).
Hierarchy of transcription factors and epigenetic states in the establishment of the adipogenic genome in 3D
Differentiation is a complex process regulated by multiple transcription factors, chromatin modifiers and readers working in various sequential and combinatorial manners. The temporal dynamics of chromosomal contacts showed overall transient profiles characterized by increases and decreases at single time points. Thus, for understanding how the multifactorial processes at the local chromatin level are linked to changes in chromosomal associations, we decided to separately focus on each transition during adipocyte development. By assessing the relative enrichment of "genomic regulatory events" (i.e. TF binding, chromatin accessibility, histone modifications) in the dynamic chromosomal contacts, we expected to infer their contribution to 3D chromosomal reorganization. Since the chromosomal domains used for the cluster analysis were merged from overlapping contacts across adipogenesis, they tend to be broader than contact domains detected on single days. Thus, to improve the resolution of our analysis we focused on contacts which were detected on a given day relative to the previous one. To determine the relative enrichment of the ChIP signal from of a single day in dynamically associated chromatin, we calculated ratios between the ChIP signals per Kb in the day-specific contacts. Calculating the ratio of the ChIP signal of a particular day (e.g. day 1) in the "gained" (e.g. day 1-specific) vs "lost" (e.g. day 0-specific) contacts in each transition provides internal normalization of the ChIP data and a uniform scale for comparing different days and factors. To present enrichment (ratio >1) and depletion (0<ratio<1) on a similar scale, log2 ratios are shown.
In the first stage of differentiation (day 0 to day1), day1–specific contacts were enriched in chromatin accessible sites (as assessed by DNaseI hypersensitivity) relative to day0-specific contacts in both PPARγ and Lpin1 (higher log2 ratio, Fig. 5A, B). Chromatin accessible loci mainly reflect TF binding sites. Indeed, binding of RXR and C/EBP-β, key TFs of this stage, is enriched in the day1–specific contacts. Notably, the relative enrichment of RXR binding is higher than that of C/EBP-β (Fig. 5A, B). Thus, at the first stage (day 0 to day1) of differentiation, the changes in the spatial environment of adipogenic genes are associated with regulatory sites and binding of transcription factors, particularly binding of RXR. Unexpectedly, in contrast to the first stage, such enhancement of regulatory sites and binding of lineage determining TFs was not observed at the contacts acquired at the final stage of differentiation (day 2 to day 6) (Fig. 5A, B). In fact, we noted relative depletion of regulatory sites and binding of lineage determining TFs of the first and the second wave at the mature adipocyte-specific contacts. This indicates lower enrichment of TF binding in genomic regions that joined the spatial environments of PPARγ and Lpin1 relative to the regions that dissociated from the hub.
Fig. 5. Concordant dynamics of genome high-order reorganization and chromatin structure.

The relative enrichment of transcription factors, CTCF, RNA-PolII and histone marks at each transition point during adipogenesis. Enrichment was calculated as the ratio between ChIP-seq signal on the contacts joined relative to the contact domains that disjointed from PPARγ (A) and Lpin1 (B) in each transition. The ratios are presented on a log2 scale. (N.A - ChIP data for a particular day is not available). Normalized H3K27me3 peaks (peaks per kb per total peaks *1000) through the differentiation at the chromosomal contacts of PPARγ (C) and Lpin1 (D) in mature adipocytes (day 6).
On the other hand, the specific contacts of day 6 were enriched for H3K27me3 transcriptional repressive mark relative to day 2-specific contacts. Interestingly, overall H3K27me3 levels (ChIP signal) at the day 6- specific associating loci remained stable relative to previous days (Fig. 5C, D), suggesting that the enrichment in repressive chromatin at the adipogenic compartment is due to recruitment of chromosomal loci decorated with H3K27me3 rather than an increase in H3K27 tri methylation at the interacting loci.
Similarly to PPARγ contacts in the first stage of adipogenesis, in the intermediate transition, day 2 -specific contacts of Lpin1 are enriched in regulatory sites (DHS) and in RXR and C/EBP-β binding relative to day 1-specific contacts (Fig. 5 A, B). Interestingly, in the PPARγ locus, day 2-specific contacts were not enriched in regulatory sites or in binding of lineage-determining TFs relative to the contacts that were lost in this transition (Fig. 5A), similarly to the pattern observed in the final stage of adipogenesis. Thus, despite the similarity in the interactomes of PPARγ and Lpin1 and their dynamics along adipogenesis, there are some differences in the timeline of the reshaping of the interactome between these two adipogenic genes along the differentiation process.
Discussion
In this study, we documented the reorganization of the adipogenic gene loci, PPARγ and Lpin1, along the progression of 3T3-L1 adipogenesis. Despite being on different chromosomes, the two genes share many of their genome-wide long-range chromosomal associations along adipogenesis and create an adipogenic –specific sub nuclear compartment, or "hub". This hub is very robust, as chromosomal associations of the two gene loci comprise 50% of all adipogenic genes from different chromosomes. This exquisite spatial clustering of the adipogenic genetic program during adipogenesis reflects the functional aspects of nuclear architecture and suggests that it is a hallmark of the cellular adipogenic identity. Integration of 4C data with chromatin-immunoprecipitation (ChIP) and DNaseI hypersensitivity (DHS) data on these cells demonstrated a positive correlation with active regulatory sites, but lower correlation with transcriptional activity. Thus, although these environments are transcriptionally active, the interplay between gene positioning and transcription is more complex than simple induction of gene transcription within the adipogenic hub. We have previously shown that the hormone-activated glucocorticoid receptor (GR) transcription factor, induces and represses genes which are spatially associated in cell type-specific active compartments [38]. These results suggest that active hubs are organized to support regulated processes including maintenance of cell-type specific transcriptional programs as well as responses to signals.
The significant correlation of the adipogenic interactome with regulatory elements and TF binding loci is consistent with the role of TFs in organizing the genome by spatial clustering of their genomic binding loci, as was shown in several differentiation paradigms [3,8,11–13]. However defining cooperative and hierarchical roles of different fate determining transcription factors and other chromatin factors in reconstructing the nuclear space of the mature cell requires these links to be captured along different steps of differentiation.
The analysis of 3D reorganization of the genome along adipogenesis revealed an overall shared dynamics of the two adipogenic genes, suggesting common dynamics of the adipogenic hub. Importantly, approximately half of the contact regions of PPARγ and Lpin1 in mature adipocytes were already established in 3T3L1 pre-adipocytes, reflecting the adipogenic potential (primed state) of the 3T3-L1 pre-adipocyte line. The commitment of 3T3-L1 cells to the adipogenic fate provides a narrow developmental window that allows fine dissection of the dynamics of 3D genome organization and its associated molecular events. Since adipogenesis requires the sequential participation of several TFs, analysis at several intermediate time points allowed us to dissect the factors contributing to the reshaping of the nuclear architecture along differentiation. C/EBP-β, RXR and PPARγ are key adipogenic TFs. C/EBP-β binds to chromatin at the pre-adipocyte stage. Induction of differentiation leads to extensive chromatin remodeling and RXR recruitment to these sites at day 1 of adipogenesis. PPARγ expression is induced at day 2, and the protein is recruited to these open regulatory sites where it heterodimerizes with RXR and promotes the adipogenic transcription program [36].
The changes in the spatial environment of adipogenic genes at the first stage (day 0 to day1) of differentiation are predominantly associated with spatial focusing of RXR binding sites, suggesting that RXR is the key factor mediating genome organization at first stage. This is consistent with the activation of RXR on day1, and its binding to a subset of pre-adipogenic C/EBP-β binding loci [29]. Thus genome reorganization may lead to selective enrichment of active C/EBP-β binding sites at the spatial environment of adipogenic genes. This interplay between C/EBP-β and RXR may shed new light on a recent study showing that the spatial organization of the PPARγ locus in the first hours of differentiation requires C/EBP-β [43]. Since prior to induction of adipogenesis in 3T3-L1 cells, C/EBP-β binding marks a subset of hotspots to which other adipogenic TFs including RXR subsequently bind [36], it is possible that C/EBP-β binding affects nuclear organization indirectly by establishing sites for RXR binding.
Notably, although PPARγ expression and chromatin binding is initially observed at day 2 and then extends to the entire adipogenic genome by day 6 [29,44], PPARγ binding is negatively associated with changes in 3D genome organization. A possible explanation is that PPARγ binds to regulatory sites that were already primed and occupied by TF, acting upstream to PPARγ, which are more dominant in the establishment of the cell-type specific chromosomal organization. Hence, the relative enrichment for MED1 and DHS in the first transition suggests that the dominance of RXR binding in this transition is due to coupling of enhancer activation. Surprisingly, in contrast to the relative reduction in PPARγ binding in mature adipocytes (day 6), the spatial environment of both PPARγ and Lpin1, is relatively enriched for the H3K27me3 transcriptional repressive signature. Since the levels (ChIP signal) of H3K27me3 at the associating loci remain stable, the enrichment in repressive chromatin at the adipogenic compartment is likely a consequence of spatial joining of loci decorated with H3K27me3 at this stage of differentiation. This enrichment of repressive chromatin through long-range chromatin association may be linked to numerous previous observations of overall attenuation of the transcription rate at the conclusion of terminal differentiation in adipogenesis and other differentiation paradigms (Fig. 5E) [45–49]. This ensuing decrease in gene transcription is subtle, relative to the robust induction of cell type-specific genes during differentiation.
Our analyses indicate that the transition in H3K27me3 enrichment in the spatial environment of PPARγ precedes that of Lpin1, although the transcriptional dynamics of both genes are similar. This is not surprising given that such variation in the dynamics of local chromatin decoration was reported for PPARγ and the adipogenic transcription factor C/EBPa, although the two genes are induced with similar kinetics. The H3K27me3 signal is high at C/EBPα in pre-adipocytes and decreases dramatically upon gene activation, while this repressive modification remains low at PPARγ throughout 3T3-L1 adipogenesis. On the other hand, PPARγ induction is coupled with an elevated H3K4me3 signal, while this active mark is high at C/EBPα prior to its activation [50]. Thus although these genes are marked by high H3K4me3 and low H3K27me3 signals in mature adipocytes, the dynamics of their loading suggests that it is the combination of these and other features that determine the transcriptional activity. Analogously, at the intermediate stage (day 1 to day 2), H3K27me3 is increased in the PPARγ environment, while TFs and active chromatin marks decrease at the later (day 2 to day 6) transition. This suggests that the attenuation of transcription requires both an increase in the repressive signal, and a decrease in active chromatin. In addition, it is possible that high-order genome organization provides a regulatory function that is downstream to more direct mechanisms acting in cis. Further studies of terminal and functional differentiation systems are required to understand the link between genome reorganization and transcriptional attenuation at the end of differentiation, which may serve to stabilize the proper transcription program.
On the basis of our findings, we propose the existence of an adipogenic hub, rich in adipogenic genes, and specific to cells undergoing adipogenesis, indicating that the 3D genome organization supports the regulatory networks that establish and maintain cell identity. This hub is found to some extent in pre-adipocytes, changes along adipogenesis, and is strongly related to regulatory elements and lineage-determining TFs. The high-temporal resolution analysis suggests that RXR is a major player in the establishment of adipogenic specific 3D genome organization at the early stages of differentiation, whereas the spatial adipogenic organization is more related to the repressive environment at later stages of adipogenesis. These data support an essential role for transcription factors in shaping the nuclear environment that supports cell identity along with their role in gene transcription regulation.
Materials and Methods
3T3-L1 cell culture
3T3-L1 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) containing 4500 mg/liter glucose supplemented with 10% calf serum (Sigma), 100 µg/ml streptomycin, 62.5 µg/ml penicillin, 8 µg/ml biotin, and 8 µg/ml pantothenic acid.
Differentiation of 3T3-L1 fibroblasts to adipocytes was achieved as described previously (Helledie et al, 2002). Namely, 2-day post-confluent 3T3-L1 cells (designated day 0 cells) were exposed to DMEM containing 10% FCS (Invitrogen) supplemented with 1 µM dexamethasone (Sigma), 0.5 mM 3-isobutyl-1-methylxanthine (Aldrich), and 1 µg/ml insulin (Roche Molecular Biochemicals). At day 2, cells were fed DMEM containing FCS and 1 µg/ml insulin, thereafter cells were maintained in DMEM and FCS.
4C-seq
The 4C assay was performed according to the method previously described (Simonis et al. 2006; Hakim et al. 2011). Cells were fixed with 2% formaldehyde for 10 min, cross-linked chromatin was digested overnight with excess of HindIII enzyme (New England Biolabs) and then DNA ends were ligated under dilute conditions that favor junctions between cross-linked DNA fragments. The ligation junctions were then circularized by digestion with the DpnII four-base restriction enzyme (New England Biolabs) followed by ligation. Baits were amplified with the following inverse PCR primers: PPARg- m_Pparg_DpnII_F: TGTTGAACAAATGAATGAAATAGAATG; m_Pparg_HindIII_R: GAGATGGCAAGCCTTAAGCTT, Lpin1- m_Lpin1_H4_R: GTGTTGTGACACAGGCCTTC; m_Lpin1_D4_F: CTGGGTGATAAGTTGGGGTT, Ifng-m_Ifng_DpnII_F: CTCTATCTCCAGAGGAGCCC; m_Ifng_Hind_R: GACTCTCCGGTGAACAAAGC. In order to multiplex the samples, 5-base barcodes were added to the primers on the HindIII cut side: day 0- replica 1 ATCGA/replica 2 GCATG; day 1- replica 1 TAGCA/replica 2 CGTAG; day 2- replica 1 ATGCG/ replica 2 CGATA; day 6 – replica 1 GCTAC/ replica 2 ACGTC. B cells samples were without indexes. Libraries were sequenced on an Illumina HiSeq 2500 platform.
4C-seq analysis
Reads were sorted into different fastq files for each bait/viewpoint and time point according to the bait and indexes sequences and aligned to the mouse (mm9) genome using BOWTIE. Reads were then counted for each HindIII site. To avoid possible quantitative PCR or sequencing bias [51] the data were transformed to represent unique coverage (>1 reads per HindIII site was set to 1). The 10Mb centered at the viewpoint were excluded from the analysis, as contact frequency at this range is significantly higher than at the rest of the genome. To identify contact loci, i.e. genomic loci with higher sequence capture frequency than expected, a p score (= −log10 p-value) was calculated for each HindIII site using a one tailed binomial test with 100Kb running window centered at the HindIII site and the entire chromosome as background. Since intra- and inter-chromosomal associations are typically different in intensity and size, the top 10% intra-chromosomal and the top 5% inter-chromosomal HindIII sites were considered positive (FDR ~0.04). Contact regions were determined as clusters of consecutive positive HindIII sites of 100Kb or greater. To avoid loci with high p score that may result from low HindIII content, only clusters containing at least15 HindIII sites were considered.
Overlap between 4C contact regions was calculated for the merged 4C contact regions detected for each bait. A hypergeometric test was used to determine significantly high overlap between baits (calculating P[X > x]) or significantly low overlap between baits (calculating P[X <= x])
Enrichment of genes that are related to adipogenesis within contact regions was calculated for the 134 adipogenic GO terms (from GO database) relatively to all mouse genes (24,783 genes from the RefSeq database). A hypergeometric test was used to determine significance. Active genes were determined as genes with at least 1 cpm at all time points during differentiation. RNA-seq from GSE95533 was used for this analysis. The genome was divided into 200kb windows and the number of windows overlapping at least 50% of the contact regions was determined. A similar analysis was performed for the overlaps of windows with active genes with contact regions. A proportional test was used to determine significant enrichment of the windows.
To identify contacts with similar temporal profiles, we first merged overlapping contact regions from all the time points. To normalize for the amount of total HindIII sites and reads at all time points, quantile-normalization was applied for the ratio of HindIII sites covered with reads / total HindIII sites at a given region at a given time point. K-means cluster analysis for the merged dataset was performed by the PAM algorithm in R programming using the squared Euclidean distance. The number of clusters, K, was determined by the function 'clusGap' in R that calculates a goodness of clustering measure by testing various values of K. Both functions are implemented in the 'cluster' package (available at http://cran.r-project.org/web/packages/cluster/index.html).
DHS-seq and ChIP-seq
DHS-seq data and ChIP seq data were taken from GSE13511, GSE27826, GSE21898, GSE73434, GSE95533, GSE27450 and GSE20752 [14,29,34–37,52]. For detecting regulatory loci and transcription factor binding sites, hotspot algorithm was applied as described [36,53]. This algorithm identifies regions of local enrichment which are calculated based on the binomial distribution of a 250bp target window relative to 200kb surrounding window (local background). The hotspots were filtered by selecting data specific tag density thresholds based on visual inspection of a large number of low signal peaks.
Genome-wide correlation
To maintain the quantitative aspect of each genomic data set despite their different genomic scales, a fixed resolution profile of each genomic data was obtained by computing the total number of reads within contact regions (for 4C), genes (for transcription) or peaks (for ChIP peaks or DHS hotspots) in a fixed 100 kb window, sliding along the genome at 50-kb increments. The resulting profiles were used for calculating the Spearman correlation coefficient over the genome between 4C and each of the other profiles.
Enrichment analysis between consecutive days
The chromosomal contacts that were gained or lost between consecutive 4C measurements were determined using bedtools multiIntersectBed function, which allowed separation of the genome covered by the domains of two consecutive 4C measurements into overlapping or non-overlapping intervals. Non-overlapping intervals between 4C datasets from two timepoints were defined as day-specific. The signal of each regulatory event (DHS hotspots, ChIP peaks) within the day-specific contact was calculated as reads per Kb, and the log2 ratios between the signals of regulatory events on consecutive days were determined.
Supplementary Material
Table S1: 4C reads distribution in each sample and library complexity
Table S2: Contact domains analysis for each bait in adipogenic cells throughout the differentiation and in B cells
Table S3: Adipogenic genes, termed by GO, within PPARγ and Lpin1 contact domains
Table S4: Genes within PPARγ and Lpin1 clusters
Acknowledgments
This work is supported by the Israel Science Foundation (grant 748/14), Marie Curie Integration grant (CIG)- FP7-PEOPLE-20013-CIG-618763 and I-CORE Program of the Planning and Budgeting Committee and The Israel Science Foundation grant no. 41/11. A.S.P is supported by the Nehemia Levtzion Fellowship. This research is supported in part by the Intramural Research Program of the National Institutes of Health at the National Institute on Aging.
Abbreviations
- TF
transcription factor
- RXR
retinoid X receptor
- PPARγ
Peroxisome proliferator-activated receptor gamma
- Lpin1
Lipin 1
- TAD
topologically associating domain
- 4C
circular chromosome conformation structure
Footnotes
Database
All sequence data reported in this paper have been deposited at GEO (http://www.ncbi.nlm.nih.gov/geo/) (GSE92475)
Data Accessibility
Research data pertaining to this article is located at figshare.com
Table 1: 4C reads distribution in each sample and library complexity
Table 2: Contact domains analysis for each bait in adipogenic cells throughout the differentiation and in B cells
Table 3: Adipogenic genes, termed by GO, within PPARγ and Lpin1 contact domains
Table 4: Genes within PPARγ and Lpin1 clusters
Author Contributions
O.H. designed the experiment together with S.M, M.-H.S. and R.S. O.H and T.K. supervised the project. M.S. and R.S. performed the experiments with assistance from R.N. A.S.P conducted all bioinformatics analyses. O.H., M.S. and A.S.P wrote the manuscript. All authors discussed the results and implications and commented on the manuscript at all stages. M.-H.S. performed preliminary bioinformatics analysis and advised on statistical methods.
References
- 1.Schwartz M, Hakim O. 3D view of chromosomes, DNA damage, and translocations. Curr. Opin. Genet. Dev. 2014;25:118–25. doi: 10.1016/j.gde.2013.12.008. [DOI] [PubMed] [Google Scholar]
- 2.Joffe B, Leonhardt H, Solovei I. Differentiation and large scale spatial organization of the genome. Curr. Opin. Genet. Dev. 2010;20:562–9. doi: 10.1016/j.gde.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 3.Hakim O, Sung M-H, Nakayamada S, Voss TC, Baek S, Hager GL. Spatial congregation of STAT binding directs selective nuclear architecture during T-cell functional differentiation. Genome Res. 2013;23:462–72. doi: 10.1101/gr.147652.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bridger JM, Arican-Gotkas HD, Foster HA, Godwin LS, Harvey A, Kill IR, Knight M, Mehta IS, Hassan M, Bridger AJM, Arican-Gotkas HD, Ahmed MH, Foster HA, Godwin LS, Harvey A, Knight M, Mehta IS. The Non-random Repositioning of Whole Chromosomes and Individual Gene Loci in Interphase Nuclei and Its Relevance in Disease, Infection, Aging, and Cancer. Adv. Exp. Med. Biol. 2007;773:978–1. doi: 10.1007/978-1-4899-8032-8_12. [DOI] [PubMed] [Google Scholar]
- 5.Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–80. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, Gribnau J, Barillot E, Bluthgen N, Dekker J, Heard E. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485:381–5. doi: 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, Parrinello H, Tanay A, Cavalli G. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell. 2012;148:458–72. doi: 10.1016/j.cell.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 8.Lin YC, Benner C, Mansson R, Heinz S, Miyazaki K, Miyazaki M, Chandra V, Bossen C, Glass CK, Murre C. Global changes in the nuclear positioning of genes and intra- and interdomain genomic interactions that orchestrate B cell fate. Nat. Immunol. 2012;13:1196–204. doi: 10.1038/ni.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Phillips-Cremins JE, Sauria MEG, Sanyal A, Gerasimova TI, Lajoie BR, Bell JSK, Ong C-T, Hookway TA, Guo C, Sun Y, Bland MJ, Wagstaff W, Dalton S, McDevitt TC, Sen R, Dekker J, Taylor J, Corces VG. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell. 2013;153:1281–95. doi: 10.1016/j.cell.2013.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W, Diao Y, Liang J, Zhao H, Lobanenkov VV, Ecker JR, Thomson JA, Ren B. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015;518:331–336. doi: 10.1038/nature14222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Denholtz M, Bonora G, Chronis C, Splinter E, de Laat W, Ernst J, Pellegrini M, Plath K. Long-Range Chromatin Contacts in Embryonic Stem Cells Reveal a Role for Pluripotency Factors and Polycomb Proteins in Genome Organization. Cell Stem Cell. 2013;13:602–16. doi: 10.1016/j.stem.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wei Z, Gao F, Kim S, Yang H, Lyu J, An W, Wang K, Lu W. Klf4 organizes long-range chromosomal interactions with the oct4 locus in reprogramming and pluripotency. Cell Stem Cell. 2013;13:36–47. doi: 10.1016/j.stem.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 13.de Wit E, Bouwman BAM, Zhu Y, Klous P, Splinter E, Verstegen MJAM, Krijger PHL, Festuccia N, Nora EP, Welling M, Heard E, Geijsen N, Poot RA, Chambers I, de Laat W. The pluripotent genome in three dimensions is shaped around pluripotency factors. Nature. 2013;501:227–31. doi: 10.1038/nature12420. [DOI] [PubMed] [Google Scholar]
- 14.Matsumura Y, Nakaki R, Inagaki T, Yoshida A, Kano Y, Kimura H, Tanaka T, Tsutsumi S, Nakao M, Doi T, Fukami K, Osborne TF, Kodama T, Aburatani H, Sakai J. H3K4/H3K9me3 Bivalent Chromatin Domains Targeted by Lineage-Specific DNA Methylation Pauses Adipocyte Differentiation. Mol. Cell. 2015;60:584–596. doi: 10.1016/j.molcel.2015.10.025. [DOI] [PubMed] [Google Scholar]
- 15.Wongtrakoongate P, Riddick G, Fucharoen S, Felsenfeld G. Association of the Long Non-coding RNA Steroid Receptor RNA Activator (SRA) with TrxG and PRC2 Complexes. PLOS Genet. 2015;11:e1005615. doi: 10.1371/journal.pgen.1005615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Noordermeer D, Leleu M, Schorderet P, Joye E, Chabaud F, Duboule D. Temporal dynamics and developmental memory of 3D chromatin architecture at Hox gene loci. Elife. 2014;3 doi: 10.7554/eLife.02557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eng D, Vogel WK, Flann NS, Gross MK, Kioussi C. Genome-wide mapping of chromatin state of mouse forelimbs. Open Access Bioinformatics. 2014;6:1–11. doi: 10.2147/OAB.S59043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim T-KK, Koche RP, Lee W, Mendenhall E, O’donovan A, Presser A, Russ C, Xie X, Meissner A, Wernig M, Jaenisch R, Nusbaum C, Lander ES, Bernstein BE. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Siersbak R, Mandrup S. Transcriptional networks controlling adipocyte differentiation. Cold Spring Harb. Symp. Quant. Biol. 2011;76:247–55. doi: 10.1101/sqb.2011.76.010512. [DOI] [PubMed] [Google Scholar]
- 20.Charó NL, Rodriguez Ceschan MI, Galigniana NM, Toneatto J, Piwien-Pilipuk G. Organization of nuclear architecture during adipocyte differentiation. Nucleus. 2016;7:249–269. doi: 10.1080/19491034.2016.1197442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuroda M, Tanabe H, Yoshida K, Oikawa K, Saito A, Kiyuna T, Mizusawa H, Mukai K. Alteration of chromosome positioning during adipocyte differentiation. J Cell Sci. 2004;117:5897–5903. doi: 10.1242/jcs.01508. [DOI] [PubMed] [Google Scholar]
- 22.Szczerbal I, Foster Ha, Bridger JM. The spatial repositioning of adipogenesis genes is correlated with their expression status in a porcine mesenchymal stem cell adipogenesis model system. Chromosoma. 2009;118:647–63. doi: 10.1007/s00412-009-0225-5. [DOI] [PubMed] [Google Scholar]
- 23.Zhou H, Kaplan T, Li Y, Grubisic I, Zhang Z, Wang PJ, Eisen MB, Tjian R. Dual functions of TAF7L in adipocyte differentiation. Elife. 2013;2:e00170. doi: 10.7554/eLife.00170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim HE, Bae E, Jeong D-Y, Kim M-J, Jin W-J, Park S-W, Han G-S, Carman GM, Koh E, Kim K-S. Lipin1 regulates PPARγ transcriptional activity. Biochem. J. 2013;453:49–60. doi: 10.1042/BJ20121598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koh Y-K, Lee M-Y, Kim J-W, Kim M, Moon J-S, Lee Y-J, Ahn Y-H, Kim K-S. Lipin1 is a key factor for the maturation and maintenance of adipocytes in the regulatory network with CCAAT/enhancer-binding protein alpha and peroxisome proliferator-activated receptor gamma 2. J. Biol. Chem. 2008;283:34896–906. doi: 10.1074/jbc.M804007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reue K, Zhang P. The lipin protein family: dual roles in lipid biosynthesis and gene expression. FEBS Lett. 2008;582:90–6. doi: 10.1016/j.febslet.2007.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bego T, Dujic T, Mlinar B, Semiz S, Malenica M, Prnjavorac B, Ostanek B, Marc J, Causević A. Association of PPARG and LPIN1 gene polymorphisms with metabolic syndrome and type 2 diabetes. Med. Glas. (Zenica) 2011;8:76–83. [PubMed] [Google Scholar]
- 28.Bermúdez V, Finol F, Parra N, Parra M, Pérez A, Peñaranda L, Vilchez D, Rojas J, Arráiz N, Velasco M. PPAR-γ Agonists and Their Role in Type 2 Diabetes Mellitus Management. Am. J. Ther. 2010;17:274–283. doi: 10.1097/MJT.0b013e3181c08081. [DOI] [PubMed] [Google Scholar]
- 29.Nielsen R, Pedersen TA, Hagenbeek D, Moulos P, Siersbaek R, Megens E, Denissov S, Børgesen M, Francoijs K-J, Mandrup S, Stunnenberg HG. Genome-wide profiling of PPARgamma:RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis. Genes Dev. 2008;22:2953–67. doi: 10.1101/gad.501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hakim O, Resch W, Yamane A, Klein I, Kieffer-Kwon K-R, Jankovic M, Oliveira T, Bothmer A, Voss TC, Ansarah-Sobrinho C, Mathe E, Liang G, Cobell J, Nakahashi H, Robbiani DF, Nussenzweig A, Hager GL, Nussenzweig MC, Casellas R, Keiffer-Kwon K-R, Jankovic M, Oliveira T, Bothmer A, Voss TC, Ansarah-Sobrinho C, Liang G, Mathe E, Cobell J, Nakahashi H, Robbiani DF, Nussenzweig A, Hager GL, Nussenzweig MC, Casellas R. DNA damage defines sites of recurrent chromosomal translocations in B lymphocytes. Nature. 2012;484:69–74. doi: 10.1038/nature10909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Medvedovic J, Ebert A, Tagoh H, Tamir IM, Schwickert TA, Novatchkova M, Sun Q, Huis In ’t Veld PJ, Guo C, Yoon HS, Denizot Y, Holwerda SJB, de Laat W, Cogné M, Shi Y, Alt FW, Busslinger M. Flexible long-range loops in the VH gene region of the Igh locus facilitate the generation of a diverse antibody repertoire. Immunity. 2013;39:229–44. doi: 10.1016/j.immuni.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wijchers PJ, Geeven G, Eyres M, Bergsma AJ, Janssen M, Verstegen M, Zhu Y, Schell Y, Vermeulen C, de Wit E, de Laat W. Characterization and dynamics of pericentromere-associated domains in mice. Genome Res. 2015;25:958–969. doi: 10.1101/gr.186643.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Siersbak R, Madsen JGS, Javierre BM, Nielsen R, Bagge EK, Cairns J, Wingett SW, Traynor S, Spivakov M, Fraser P, Mandrup S. Dynamic Rewiring of Promoter-Anchored Chromatin Loops during Adipocyte Differentiation. Mol. Cell. 2017;66:420–435. e5. doi: 10.1016/j.molcel.2017.04.010. [DOI] [PubMed] [Google Scholar]
- 35.Schmidt SF, Jorgensen M, Chen Y, Nielsen R, Sandelin A, Mandrup S. Cross species comparison of C/EBPα and PPARγ profiles in mouse and human adipocytes reveals interdependent retention of binding sites. BMC Genomics. 2011;12:152. doi: 10.1186/1471-2164-12-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Siersbak R, Nielsen R, John S, Sung M-H, Baek S, Loft A, Hager GL, Mandrup S. Extensive chromatin remodelling and establishment of transcription factor “hotspots” during early adipogenesis. EMBO J. 2011;30:1459–72. doi: 10.1038/emboj.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steger DJ, Grant GR, Schupp M, Tomaru T, Lefterova MI, Schug J, Manduchi E, Stoeckert CJ, Lazar MA. Propagation of adipogenic signals through an epigenomic transition state. Genes Dev. 2010;24:1035–44. doi: 10.1101/gad.1907110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hakim O, Sung M-H, Voss TC, Splinter E, John S, Sabo PJ, Thurman RE, Stamatoyannopoulos JA, de Laat W, Hager GL. Diverse gene reprogramming events occur in the same spatial clusters of distal regulatory elements. Genome Res. 2011;21:697–706. doi: 10.1101/gr.111153.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–93. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hakim O, Sung M-HH, Hager GL. 3D Shortcuts to Gene Regulation. Curr. Opin. Cell Biol. 2010;22:305–313. doi: 10.1016/j.ceb.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grbesa I, Hakim O. Genomic effects of glucocorticoids. Protoplasma. 2016:1–11. doi: 10.1007/s00709-016-1063-y. [DOI] [PubMed] [Google Scholar]
- 42.Siersbak R, Rabiee A, Nielsen R, Sidoli S, Traynor S, Loft A, Poulsen LLC, Rogowska-Wrzesinska A, Jensen ON, Mandrup S. Transcription factor cooperativity in early adipogenic hotspots and super-enhancers. Cell Rep. 2014;7:1443–55. doi: 10.1016/j.celrep.2014.04.042. [DOI] [PubMed] [Google Scholar]
- 43.LeBlanc SE, Wu Q, Barutcu AR, Xiao H, Ohkawa Y, Imbalzano AN. The PPARγ locus makes long-range chromatin interactions with selected tissue-specific gene loci during adipocyte differentiation in a protein kinase A dependent manner. PLoS One. 2014;9:e86140. doi: 10.1371/journal.pone.0086140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Siersbaek R, Nielsen R, Mandrup S. PPARgamma in adipocyte differentiation and metabolism--novel insights from genome-wide studies. FEBS Lett. 2010;584:3242–9. doi: 10.1016/j.febslet.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 45.Burton GR, Nagarajan R, Peterson CA, McGehee RE. Microarray analysis of differentiation-specific gene expression during 3T3-L1 adipogenesis. Gene. 2004;329:167–85. doi: 10.1016/j.gene.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 46.Jessen BA, Stevens GJ. Expression profiling during adipocyte differentiation of 3T3-L1 fibroblasts. Gene. 2002;299:95–100. doi: 10.1016/s0378-1119(02)01017-x. [DOI] [PubMed] [Google Scholar]
- 47.Keller MA, Addya S, Vadigepalli R, Banini B, Delgrosso K, Huang H, Surrey S. Transcriptional regulatory network analysis of developing human erythroid progenitors reveals patterns of coregulation and potential transcriptional regulators. Physiol. Genomics. 2006;28 doi: 10.1152/physiolgenomics.00055.2006. [DOI] [PubMed] [Google Scholar]
- 48.Koenig KM, Sun P, Meyer E, Gross JM. Eye development and photoreceptor differentiation in the cephalopod Doryteuthis pealeii. Development. 2016;143 doi: 10.1242/dev.134254. [DOI] [PubMed] [Google Scholar]
- 49.Stewart R, Rascón CA, Tian S, Nie J, Barry C, Chu L-F, Ardalani H, Wagner RJ, Probasco MD, Bolin JM, Leng N, Sengupta S, Volkmer M, Habermann B, Tanaka EM, Thomson JA, Dewey CN. Comparative RNA-seq Analysis in the Unsequenced Axolotl: The Oncogene Burst Highlights Early Gene Expression in the Blastema. PLoS Comput. Biol. 2013;9:e1002936. doi: 10.1371/journal.pcbi.1002936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Q, Ramlee MK, Brunmeir R, Villanueva CJ, Halperin D, Xu F. Dynamic and distinct histone modifications modulate the expression of key adipogenesis regulatory genes. Cell Cycle. 2012;11:4310–22. doi: 10.4161/cc.22224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Splinter E, de Wit E, van de Werken HJG, Klous P, de Laat W. Determining long-range chromatin interactions for selected genomic sites using 4C-seq technology: From fixation to computation. Methods. 2012 doi: 10.1016/j.ymeth.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 52.Mikkelsen TS, Xu Z, Zhang X, Wang L, Gimble JM, Lander ES, Rosen ED. Comparative epigenomic analysis of murine and human adipogenesis. Cell. 2010;143:156–69. doi: 10.1016/j.cell.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baek S, Sung M, Hager GL. Quantitative Analysis of Genome-Wide Chromatin Remodeling. 2012;833:433–441. doi: 10.1007/978-1-61779-477-3_26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: 4C reads distribution in each sample and library complexity
Table S2: Contact domains analysis for each bait in adipogenic cells throughout the differentiation and in B cells
Table S3: Adipogenic genes, termed by GO, within PPARγ and Lpin1 contact domains
Table S4: Genes within PPARγ and Lpin1 clusters