

Abstract
Distinguishing between indolent and aggressive prostate adenocarcinoma (PCa) remains a priority to accurately identify patients who need therapeutic intervention. SIN3B has been implicated in the initiation of senescence in vitro. Here we show that in a mouse model of prostate cancer, SIN3B provides a barrier to malignant progression. SIN3B was required for PTEN-induced cellular senescence and prevented progression to invasive PCa. Furthermore, SIN3B was downregulated in human PCa correlating with upregulation of its target genes. Our results suggest a tumor suppressor function for SIN3B that limits PCa progression, with potential implications for the use of SIN3B and its target genes as candidate diagnostic markers to distinguish indolent from aggressive disease.
Keywords: Sin3, Prostate, Mouse Model, Senescence, Invasion
Introduction
Prostate adenocarcinoma (PCa) is the 2nd most common cancer type in American men with approximately 230,000 new patients diagnosed each year, equating to about 1 in 7 men being diagnosed with PCa in his lifetime (1). Despite the prevalence of PCa, the 5-year survival rate for local or regional disease (Stages I – early IV) is nearly 100%. However, men with invasive or metastatic disease (Stage IV) have a significantly reduced survival rate of about 28% (2). Therefore, defining the molecular mechanisms that restrict prostate cancer progression remains a priority to improve the identification of patients in need of therapeutic intervention. Clinical monitoring of prostate cancer currently relies on histo-pathological and chemical indicators, such as Gleason scoring and PSA levels. Unfortunately, the use of these parameters to manage prostate cancer treatment has proven largely inadequate (3,4).
As a potential mechanism that restricts cancer progression, cellular senescence has generated much interest. Cellular senescence is a stable form of cell cycle arrest that can be triggered by different stimuli including serial passaging, DNA damage, activation of oncogenes or loss of a tumor suppressor (5,6). Senescent cells have been identified in preneoplastic lesions of several solid tumor types, including prostatic intraepithelial neoplasias (PINs), but are rarely found in normal prostate or PCa (7). Based on these findings, cellular senescence has been hypothesized to prevent cancer progression, through its ability to trigger stable cell cycle exit and prevent the proliferation of potentially deleterious cells.
Mouse models of cancer have enabled a greater understanding of the biology of tumor initiation and progression, leading to improved diagnostic and therapeutic strategies (8,9). Various attempts have been made to recapitulate PCa in the mouse. Among these, somatic inactivation of Phosphatase and TENsin homolog (PTEN) in prostate epithelium most accurately models the initiation of prostate cancer. Importantly, approximately 70% of human primary prostate tumors harbor loss or alteration of at least one PTEN allele (10). In mice, prostate specific inactivation of Pten causes the generation of hyperplasia early in life, followed by the generation of PINs that rarely progress to PCa. More recently, it was demonstrated that Pten-deletion in the prostate leads to the activation of a TRP53-dependent senescence response, termed PTEN-loss-induced cellular senescence or PICS (11). Importantly, combined inactivation of Pten and Trp53 in the prostate alleviates PICS and accelerates the progression to invasive PCa (7). However, due to the wide range of TRP53 targets in vivo, it has been difficult to precisely delineate the specific effector programs engaged upon loss of PTEN that trigger senescence and restrict cancer progression in early prostate lesions.
In mammals, the SIN3 family consists of two proteins, SIN3A and SIN3B, which are ubiquitously expressed. SIN3 proteins are evolutionary conserved noncatalytic scaffold components of large transcriptional repressive complexes that are recruited to genomic loci through their interaction with sequence specific transcription factors (12,13). Repression of gene transcription is achieved at least in part through the association of SIN3 proteins with the histone deacetylases HDAC1/2. While genetic inactivation of Sin3a leads to cell death in a wide range of cell types (14), SIN3B is dispensable for cellular viability and proliferation. However, Sin3b null mice die during late embryogenesis, likely due to the inability of different cellular compartments to differentiate appropriately (14,15). Recently, we have demonstrated that the genetic inactivation of Sin3b renders mouse embryonic fibroblasts refractory to senescence triggered by ectopic expression of oncogenic RAS (15). Additionally, using a mouse model for pancreatic cancer, we showed that SIN3B expression is upregulated in PanINs (Pancreatic Intraepithelial Neoplasia) and that the genetic deletion of Sin3b in the pancreas prevents KRAS-induced cellular senescence (12,16). Surprisingly, the inactivation of a SIN3B-dependent senescence program in a mouse model of pancreatic cancer protected against cancer progression, likely due to the decrease of senescence-associated inflammation. The paradoxical outcome resulting from inactivation of senescence in this model warrants further investigation of the relationship between senescence and cancer progression in specific contexts in vivo. Based on the requirement of SIN3B for the induction of OIS both in vitro and in vivo and since PICS is believed to prevent the progression of PIN lesions, we sought to investigate the necessity of SIN3B for PICS and elucidate its contribution to prostate cancer initiation and progression. Using genetic inactivation of Pten to model prostate cancer in the mouse, we demonstrate here that SIN3B protects against prostate cancer progression by promoting PICS and limiting cellular proliferation in pre-neoplastic lesions. Furthermore, we identify that downregulation of SIN3B expression is a common event in human PCa. This study identifies the chromatin-associated SIN3B as a critical suppressor of prostate cancer progression through the induction of the senescence process.
Materials and Methods
Animal models
The Pten L/L mice were a gift from Pier Paolo Pandolfi. Pb-Cre4 mice were purchased from The Jackson Laboratory. The Sin3b+/− and Sin3bL/L strains have been described previously (15). The strains were mated to obtain mice with the correct genotypes. All animals were maintained in a mixed C57BL/6-FVB background.
Cell lines
PC3 (CRL-1435) human prostate cancer cell lines were obtained from ATCC (2016). LNCaP C4-2 human prostate cancer cell lines were obtained from the Characterized Cell Line Core Facility at MD Anderson Cancer Center (Houston, TX; 2016). All experiments were performed less than one year after acquisition, and at low passage number (<10). Both cell lines have been authenticated and mycoplasma tested by Genetica (Burlington, NC), as well as through the use of the Venor GeM Mycoplasma Detection Kit ( Sigma, MP0025).
Cell culture
PC3 cells were cultured in Ham’s F-12 (Cellgro), 10% fetal bovine serum, and 1% penicillin/streptomycin (Cellgro). C4-2 cells were cultured in RPMI (Cellgro), 10% fetal bovine serum, and 1% penicillin/streptomycin (Cellgro). All cultures were maintained in 5% CO2 at 37°C.
Histology and IHC
Mouse prostates were fixed in 10% formalin (Fisher) and processed for paraffin embedding. Histology was performed at the NYU School of Medicine Histopathology Core Facility. Five-micron sections were deparaffinized, stained with Gill’s hematoxylin and eosin Y, followed by an alcohol dehydration series and mounting (Permount; Fisher). For IHC, deparaffinized five-micron sections were rehydrated and quenched in 3% hydrogen peroxide for 15 minutes. Antigen retrieval was performed in 10 mM sodium citrate and 0.1% Tween-20 (pH 6.0) for 15 minutes in a microwave oven. Blocking was done in 5% serum, 1% BSA, and 0.1% Tween-20 for 1 hour at room temperature, followed by incubation with primary antibodies diluted in 1% BSA overnight at 4°C. The following primary antibodies were used: rabbit anti-SIN3B (Novus Biologicals); rabbit anti-HP1γ (phospho S93) (Abcam); rabbit anti-PTEN (Cell Signaling D4.3 XP), rabbit anti-pAKT (phospho S473) (Cell Signaling D9E), rabbit anti-KI67 (Lab Vision SP6), rabbit anti-P21 (Santa Cruz C-19), rabbit anti-alpha smooth muscle actin (Abcam ab5694), mouse anti-CK18 (Abcam C-04), rabbit anti-AR (Santa Cruz N-20), rabbit anti-CK5 (BioLegend Poly19055), and rabbit anti-IL6 (Santa Cruz M-19). After incubating with secondary biotinylated antibodies and solution T.U. horseradish peroxidase streptavidin (both from Vector Laboratories), sections were developed with DAB Peroxidase Substrate Kit (Vector Laboratories). After counterstaining with Gill’s hematoxylin (Sigma-Aldrich), slides were subjected to an alcohol dehydration series and mounted with Permount (Fisher). Slides were examined on a Zeiss AxioImager A2 microscope.
Infection of prostate cancer cells
A PTRIPZ lentiviral doxycycline-inducible short hairpin against human SIN3B was purchased from Dharmacon, V3THS_315587 (AGGCTGTAGACATCGTCCA). PC3 and LNCaP C4-2 cells were infected with either shSIN3B or a shScramble for 3 days and selected via 1μg/mL puromycin for 5 days. Doxycycline was added for 3 days and knockdown was assessed via western blot.
Immunoblot analysis
Cells were lysed in 1× RIPA buffer (1% NP-40, 0.1% SDS, 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.5% sodium deoxycholate, 1 mM EDTA), 0.5 μM DTT, 25 mM NaF, 1 mM sodium vanadate, 1 mM PMSF, and protease inhibitors. The following primary antibodies were used: rabbit anti-SIN3B (Santa Cruz AK12) and mouse anti-α-TUBULIN (Sigma-Aldrich T9026). After incubation with either the secondary IRDye Alexa Fluor 680 goat anti-mouse antibody or 800 goat anti-rabbit antibodies (Odyssey), the membranes were visualized with the Odyssey Infrared Imaging System (Li-Cor).
SA-β-gal assay
Prostate tissue was flash-frozen and embedded in OTC compound. Six-micron sections were fixed with 2% formaldehyde/0.2% glutaraldehyde in PBS for 3 to 5 minutes, washed with PBS, and stained at 37°C for 12 to 16 hours in X-Gal solution (1 mg/ml X-Gal, 5 mM potassium ferrocyanide, 5 mM potassium ferricyanide, and 1 mM MgCl2 in PBS at pH 6.0). After counterstaining with Nuclear Fast Red Solution (Ricca), slides were subjected to an alcohol dehydration series and mounted with Permount (Fisher). Slides were examined on Zeiss AxioImager A2 microscope.
Transwell Migration Assay
30,000 PC3 or LNCaP C4-2 cells stably expressing a short hairpin directed against either Scramble or human SIN3B were suspended in serum-free Ham’s F-12 for PC3 cells (Cellgro) or RPMI for LNCaP C4-2 cells (Cellgro) supplemented with 1% penicillin/streptomycin (Cellgro) and plated on top of an eight-micron pore size transparent PET membrane 24-well transwell cell culture insert (Falcon). Ham’s F-12 medium (Cellgro) or RPMI medium (Cellgro) supplemented with 10% FBS and 1% penicillin/streptomycin (Cellgro) was added to each well creating a serum gradient. After 24 hrs., transwells were removed, fixed in 1% glutaraldehyde, and stained with crystal violet. Cells traversing the membrane were counted and reported as a percentage of the total cells plated.
Scratch Assay
PC3 and LNCaP C4-2 cells were grown in 6-well plates until confluent. A p200 tip was used to create vertical scratches. Pictures were taken each day until the scratch was completely closed. Using ImageJ, the average size of the scratch was measured using the average pixel length of ten drawn lines. The data are presented as a percent distance of the original scratch distance.
Isolation and analysis of prostate immune infiltration
To obtain single cell suspensions, prostates were processed with slight modification as described by Lukacs et al (17). Briefly, prostates were harvested, washed with PBS, and placed in 1 mL dissecting medium (DMEM, 10% FBS, 1% penicillin/streptomycin). Prostates were briefly minced and collagenase I (Sigma, 1 mg/mL final concentration) was added followed by incubation at 37°C for 2 h while shaking. Tissue suspensions were pelleted, supernatant removed, and 1 mL of warm Trypsin/0.05% EDTA (Gibco) was added followed by incubation at 37 °C for 5 min. Tissue suspensions were mixed and 0.5 mL of dissecting medium containing 500 U DNase I (Roche) was added. Tissue suspensions were then passed through a 20-G syringe five times and subsequently filtered through 70 μm nylon mesh. Cells were pelleted and resuspended in 1 mL FACS buffer (PBS, 2% FBS). Cells were blocked with purified rat IgG (Sigma, 20 μg/ml) for 15 min on ice and stained with antibodies for 30 min on ice. The following antibodies were used for analysis (from Biolegend or BD Pharmingen): anti-CD45.2 (104), anti-B220 (RA3-6B2), anti-CD4 (RM4-5), anti-CD8 (53-6.7), anti-GR1 (RB6-8C5), anti-CD11B (M1-70), and anti-F4/80 (BM-8). 4′,6-diamidino-2-phenylindole (DAPI, 500 ng ml−1) or 7-amino-actinomycin D (7AAD, 1 ug ml−1) was added following staining for the exclusion of dead cells. Cells were analyzed on a LSRII (BD). Data were analyzed with FlowJo software (Treestar).
Blinded pathological analysis
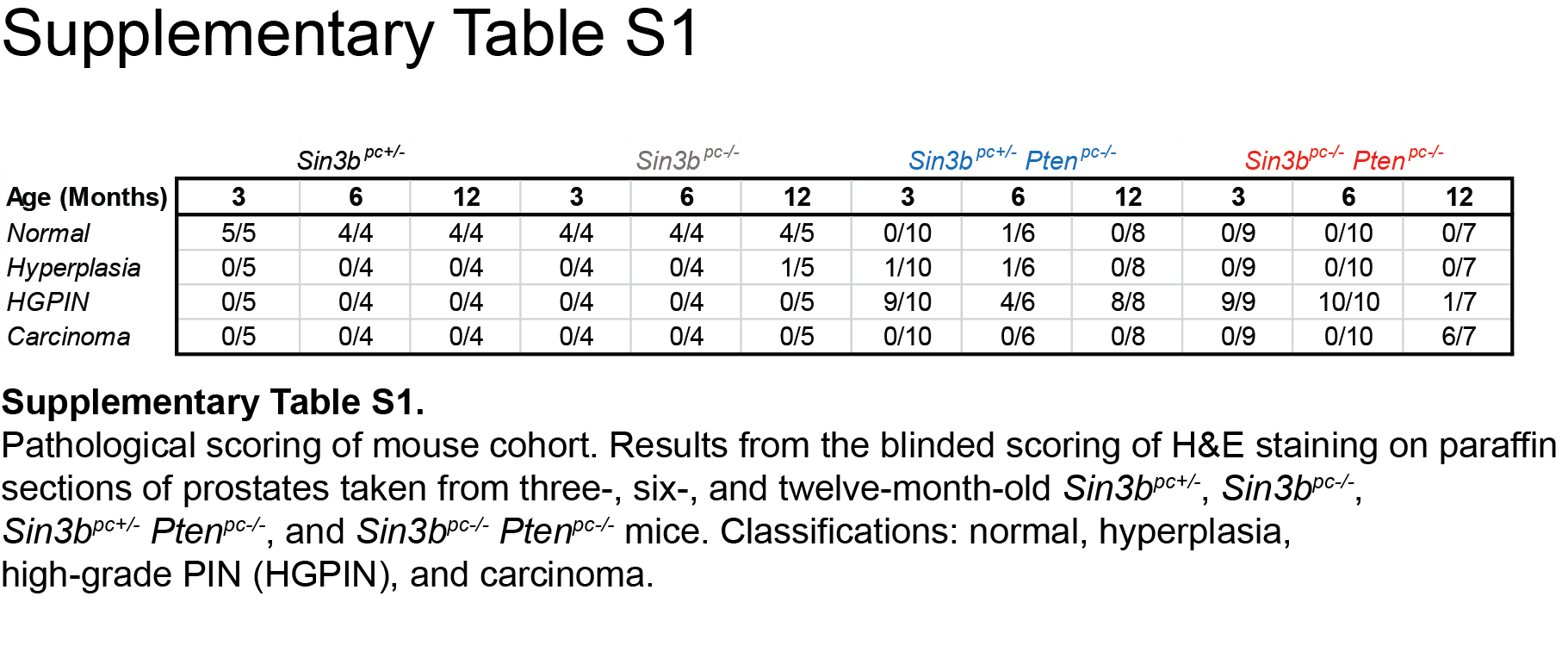
H&E samples for each prostate were numbered, randomized and given to F.M.D to score the pathology. Samples were scored as: normal, hyperplasia, HGPIN, and Carcinoma. Scores were tabulated and reported as a percentage of total number of prostates of the respective age and genotype.
Statistics
All data were analyzed by Student’s t test (unpaired, 2-tailed) and results were considered significant at P < 0.05. Results are presented as mean ± SEM. Survival curves were plotted by the Kaplan-Meier method and are compared by the log-rank test for significance.
Study approval
All animal procedures were approved by the NYU School of Medicine Institutional Animal Care and Use Committee.
Results
Generation of a mouse cohort with prostate-specific Pten and/or Sin3b deletion
We have previously demonstrated that SIN3B is required for cellular senescence upon exposure to stress (12,16). Therefore, we hypothesized that SIN3B may prevent prostate cancer progression by promoting PICS in PINs. To directly assess the contribution of SIN3B in prostate cancer progression, we generated a genetically modified mouse cohort harboring a probasin-driven Cre transgene (Pb-Cre4) in combination with conditional knockout alleles for both Pten and Sin3b. Expression of the Pb-Cre4 transgene results in Cre recombinase activity in all lobes of the mouse prostate, albeit to various degrees depending on the location. Specifically, 5% of the cells were shown to exhibit Cre-mediated recombination of a reporter allele in the anterior lobe, 10% in the dorsal lobe, 50% in the ventral lobe and 95% in the lateral prostate lobes (18). To confirm the efficient genetic inactivation of Sin3b and Pten (hereafter referred to as Ptenpc−/− and Sin3bpc−/− alleles) in the corresponding mouse prostates, we performed immunohistochemistry (IHC) on the dorsolateral and anterior prostate lobes of three-month-old mice of different genotypes. SIN3B is expressed at low levels in normal, Sin3b+/− prostate epithelium, but is strongly upregulated in PINs resulting from Pten deletion (Fig. 1A and Supplementary Fig. S1A). As expected, SIN3B expression was undetectable in Sin3bpc−/− prostates and greatly diminished in Sin3bpc−/− Ptenpc−/− compared to their Sin3bpc+/− Ptenpc−/− littermates. In addition, PTEN was expressed at high levels in the prostate regardless of the Sin3b status (Fig. 1B). Finally, in Sin3bpc+/− Ptenpc−/− prostates, no expression of PTEN was detected, correlating with an increase in phosphorylated-AKT (pAKT) (Fig. 1B, C, and Supplementary Fig. S1B). Importantly, this pathway appeared unaffected by the genetic inactivation of Sin3b (Fig. 1C and Supplementary Fig. S1C).
Figure 1.

Deletion of Sin3b does not impair loss of PTEN-driven AKT activation. A, Representative images of immunohistochemistry for SIN3B on paraffin sections of the dorsolateral lobe of prostates taken from three-month-old Sin3bpc+/−, Sin3bpc−/−, Sin3bpc+/− Ptenpc−/−, and Sin3bpc−/− Ptenpc−/− mice. B, PTEN. C, pAKT. Top Panel = 10× magnification. Bottom Panel = 40× magnification. Scale bar = 200 μm. n = 3 for each genotype.
Combined inactivation of Sin3b and Pten leads to accelerated PCa progression
To determine the role of SIN3B in prostate cancer initiation and progression, mice of all four genotypes (Sin3bpc+/−, Sin3bpc−/−, Sin3bpc+/− Ptenpc−/−, and Sin3bpc−/− Ptenpc−/−) were sacrificed at three, six, and twelve months of age, and their prostates were analyzed for morphological differences. Sin3bpc−/− prostates developed normally and did not exhibit any pathology (Fig. 2). Sin3bpc+/− Ptenpc−/− prostates exhibited increased size and weight as early as three months (Fig. 2A, B), consistent with previous reports (7). Strikingly, combined deletion of Pten and Sin3b in the prostate resulted in a dramatic change in the prostate morphology and a significant increase in prostate weight at twelve months (Fig. 2A, B, Supplementary Fig. S2A). These phenotypes suggest accelerated prostate cancer progression upon combined genetic inactivation of Sin3b and Pten. To confirm these observations at the histological level, we analyzed prostate sections from each of the corresponding genotypes at each time point. Sin3bpc−/− prostates displayed normal histology, comparable to that of age-matched Sin3bpc+/− mice, up to one-year of age, indicating that Sin3b loss is not sufficient to promote prostate cancer initiation (Fig. 2C–E). Prostate tissue from Sin3bpc+/− Ptenpc−/− and Ptenpc−/− Sin3bpc−/− mice were virtually indistinguishable at the histological level up to six-months of age. Both genotypes presented with a comparable incidence of hyperplasia and High-Grade (HG) PIN (Fig. 2C–E, Fig. 3A). At twelve months, all Sin3bpc+/− Ptenpc−/− prostates evaluated exhibited HGPIN (8/8 mice) (Fig. 2E, Fig. 3A, Supplementary Table S1). By contrast, twelve-month-old Sin3bpc−/− Ptenpc−/− mice had developed aggressive PCa with high penetrance (6/7 mice) as evidenced by histological analysis and staining for luminal cell makers (Fig. 2E, Fig. 3A, Supplementary Fig. S2B, Supplementary Table S1). Consistently, Sin3bpc−/− Ptenpc−/− mice had worse overall survival compared to Sin3bpc+/− Ptenpc−/− mice correlating with their increased tumor burden (p=0.0065, Fig. 3B). We suspect their accelerated death was due to bladder obstruction and subsequent renal failure (Supplementary Fig. S2A). Despite the lethal nature of these tumors, no macroscopic metastases were detected in the lung or lumbar lymph nodes. Taken together, these data indicate that SIN3B provides a barrier against PCa progression driven by Pten deletion.
Figure 2.

SIN3B prevents Pten deletion-induced prostate cancer. A, Representative images of prostates taken from twelve-month-old Sin3bpc+/−, Sin3bpc−/−, Sin3bpc+/− Ptenpc−/−, and Sin3bpc−/− Ptenpc−/− mice. n = 4, 5, 8, 7 respectively. B, Weight in grams of whole prostates taken from three, six and twelve month-old Sin3bpc+/− (n = 5, 4, 5), Sin3bpc−/− (n = 4, 4, 5), Sin3bpc+/− Ptenpc−/− (n = 10, 6, 8), and Sin3bpc−/− Ptenpc−/− (n = 9, 10, 7) mice, respectively. * p<0.05 via student’s t-test. C–E, Representative images of H&E staining on paraffin sections of the dorsolateral lobe of prostates taken from three-, six-, and twelve-month-old Sin3bpc+/− (n = 5, 4, 5), Sin3bpc−/− (n = 4, 4, 5), Sin3bpc+/− Ptenpc−/− (n = 10, 6, 8), and Sin3bpc−/− Ptenpc−/− (n = 9, 10, 7) mice, respectively. Top Panel = 10× magnification. Bottom Panel = 40× magnification. Scale bar = 200 μm.
Figure 3.

Loss of Sin3b-induced PCa is lethal and occurs late in life. A, Kaplan-Meier survival curve of Sin3bpc+/− Ptenpc−/− (black, n = 19) and Sin3bpc−/− Ptenpc−/− (red, n = 15) mice. ** p=0.0065. B, Quantification of pathological scoring conducted using H&E stained sections taken from three, six, and twelve-month-old Sin3bpc+/− (n = 5, 4, 4), Sin3bpc−/− (n = 4, 4, 5), Sin3bpc+/− Ptenpc−/− (n = 10, 6, 8), and Sin3bpc−/− Ptenpc−/− (n = 9, 10, 7) mice, respectively.
SIN3B protects against locally invasive PCa
The ability of tumor cells to invade the basement membrane is the first step towards metastatic disease and is indicative of a poor prognosis. Based on our observation that combined inactivation of Sin3b and Pten accelerates PCa progression, we sought to determine whether the tumors derived from Sin3bpc−/− Ptenpc−/− mice exhibited increased invasive potential. Histological analysis and staining for AR and CK5, markers for prostate luminal and basal cells, respectively, of the dorsolateral lobe from twelve-month-old Sin3bpc+/− Ptenpc−/− and Sin3bpc−/− Ptenpc−/− mice revealed that while Sin3bpc+/− Ptenpc−/− prostate glands containing HGPIN retained basement membrane integrity and displayed little to no invasion of luminal cells into the stroma, Sin3bpc−/− Ptenpc−/− prostates exhibited local invasion of luminal cells into the stroma and loss of basement membrane integrity (Fig. 4A–C). Another indication of advanced carcinoma is the presence of activated fibroblasts expressing the marker alpha smooth muscle actin (αSMA). We performed staining against αSMA to access the presence of activated fibroblasts in both Sin3bpc+/− Ptenpc−/− and Sin3bpc−/− Ptenpc−/− prostates. While Sin3bpc+/− Ptenpc−/− prostates contained negligible staining for αSMA, Sin3bpc−/− Ptenpc−/− prostates exhibited abundant positive staining (Fig. 4D). Taken together, these data suggest that the expression of SIN3B is protecting against an advanced, locally invasive disease.
Figure 4.

SIN3B protects against aggressive PCa. A, Representative images of H&E staining on paraffin sections of the dorsolateral lobe of prostates taken from 12-month-old Sin3bpc+/− Ptenpc−/− (n = 8) and Sin3bpc−/− Ptenpc−/− (n = 7) mice. B, Immunohistochemistry for AR on serial paraffin sections of the dorsolateral lobe of prostates taken from twelve-month-old Sin3bpc+/− Ptenpc−/− mice (n = 3) and Sin3bpc−/− Ptenpc−/− (n = 3) mice. C, Immunohistochemistry for CK5 on serial paraffin sections of the dorsolateral lobe of prostates taken from twelve-month-old Sin3bpc+/− Ptenpc−/− (n = 3) and Sin3bpc−/− Ptenpc−/− (n = 3) mice. Top Panel = 10× magnification. Bottom Panel = 40× magnification. Scale bar = 200 μm. D, Immunohistochemistry for αSMA on paraffin sections of the dorsolateral lobe of prostates taken from twelve-month-old Sin3bpc+/− Ptenpc−/− (n = 3) and Sin3bpc−/− Ptenpc−/− (n = 3) mice. Top Panel = 10× magnification. Bottom Panel = 40× magnification. Scale bar = 200 μm. E, Immunoblot analysis for SIN3B and TUBULIN using 40 μg of whole cell extracts from PC3 cells stably transduced with either shRNA against Scramble or SIN3B. F, Immunoblot analysis for SIN3B and TUBULIN using 40 μg of whole cell extracts from LNCaP C4-2 cells stably transduced with either shRNA against Scramble or SIN3B. G, Scratch assay using cells lines in E. D0–3 represent days post scratch. H, Quantification of G. n=3, student’s t-test was used to assess significance at each time point. * p<0.05. I, Scratch assay using cells lines in F. D0–5 represent days post scratch. J, Quantification of I. n=3, student’s t-test was used to assess significance at each time point. * p<0.05. K, Transwell migration assay using cell lines in E. Average±SEM of n = 3 experiments. Student’s t-test was used to assess significance. * p<0.05. L, Transwell migration assay using cell lines in F. Average±SEM of n = 3 experiments. Student’s t-test was used to assess significance. P=0.08, NS.
To further assess the direct contribution of SIN3B in restricting prostate cancer cell invasion, we engineered human prostate cancer cells (PC3 and LNCaP C4-2) with reduced SIN3B expression via RNAi (Fig. 4E, 4F). To document the ability of these cells to migrate, we first performed scratch assays. SIN3B knockdown PC3 and C4-2 cells were able to migrate significantly faster after 24, and 48 to 72 hrs. than their scramble controls, respectively (Fig. 4G–J). The migratory behavior of SIN3B-depleted prostate cancer cells was also confirmed through the use of a transwell membrane assay. PC3 cells depleted for SIN3B exhibited a modest, yet significant increase in migration potential compared to scramble control, further confirming that SIN3B may function to restrict migration of prostate cancer cells (Fig. 4K). Of note, C4-2 cells depleted for SIN3B exhibited a slight, albeit not significant, increase in migration potential as well (Fig. 4L). Together, these results indicate that SIN3B restricts PCa cell invasion and prevents the development of aggressive PCa.
SIN3B is necessary for PICS in PINs
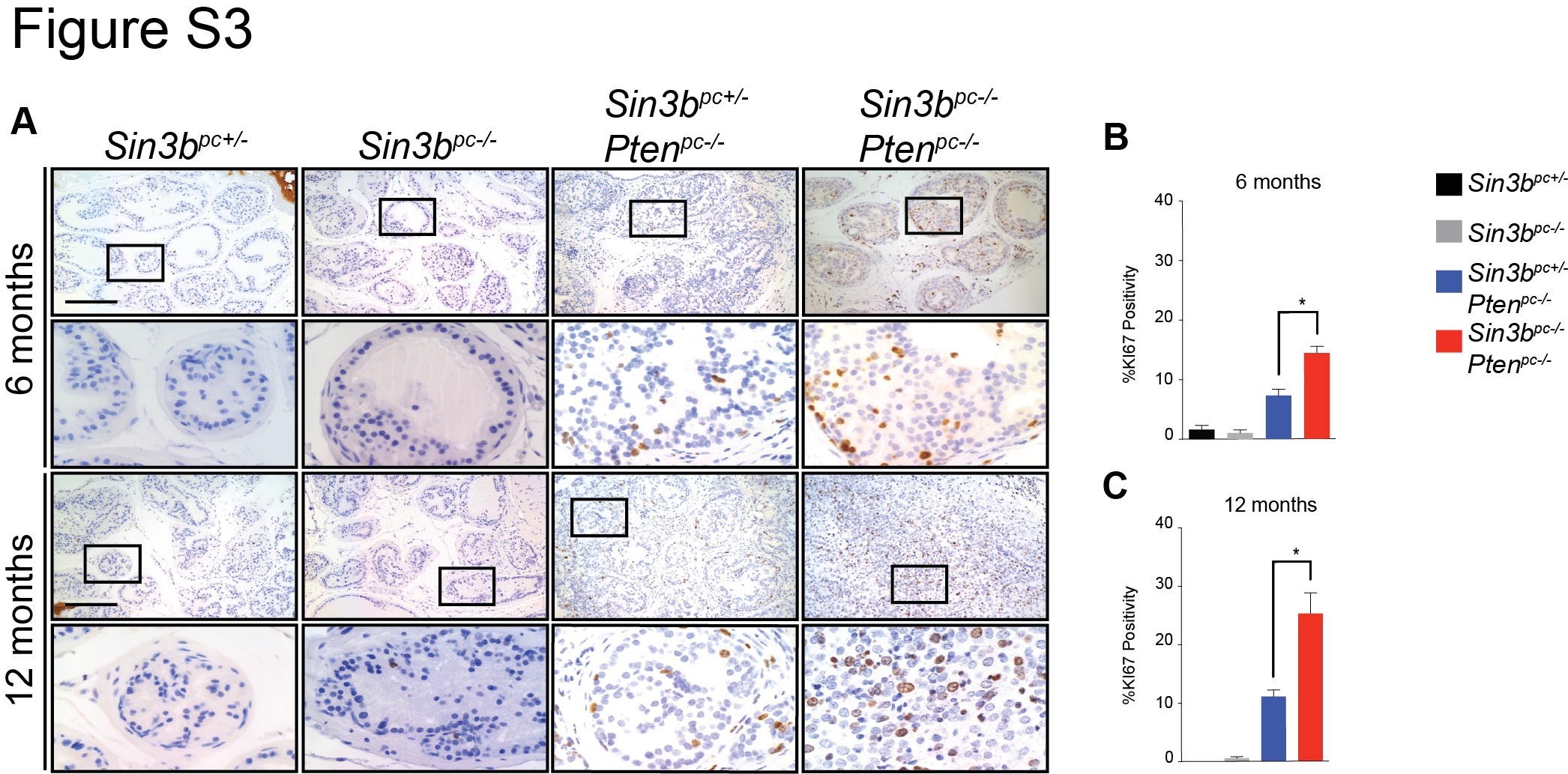
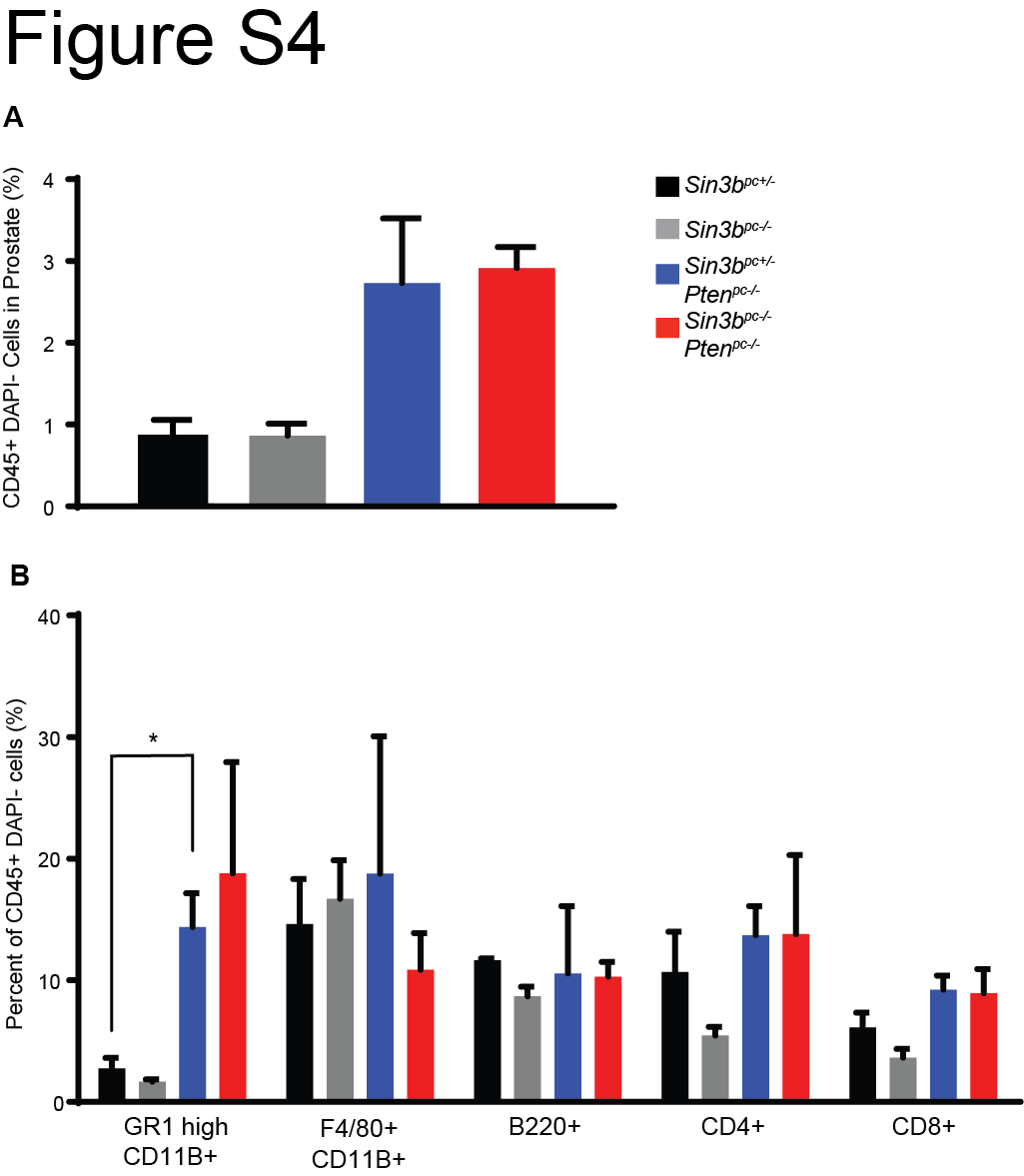
Senescence has been reported to function as a barrier against prostate cancer progression (7,11). Based on the requirement of SIN3B for oncogene-induced senescence (OIS) both in vitro and in vivo (12,16) and due to the significant increase of SIN3B protein levels in PIN lesions (Fig. 1A), we hypothesized that Sin3b loss promotes cancer progression by preventing PICS-associated cell cycle exit in the prostate. Prostate tissue from three-month-old Sin3bpc+/− Ptenpc−/− mice exhibited robust positive SA-β-Gal staining throughout the prostate epithelium, indicating a large number of senescent cells. By contrast, Sin3bpc−/− Ptenpc−/− prostates contained few SA-β-Gal positive epithelial cells, consistent with the requirement of SIN3B for the induction of senescence (Fig. 5A). To confirm the impact of Sin3b inactivation on PICS in the prostate, the expression of additional senescence markers was evaluated. Specifically, immunostaining revealed a significant decrease in positivity for CDKN1A (P21CIP1, P21) (19), phosphorylated HP1γ (pHP1γ) (20), and IL6 (21) on prostate sections of three-month-old Sin3bpc−/− Ptenpc−/− mice compared to age-matched Sin3bpc+/− Ptenpc−/− prostates (Fig. 5B, C, D). The decreased expression of senescence markers correlated with greater cellular proliferation in Sin3bpc−/− Ptenpc−/− prostate tissue compared to Sin3bpc+/− Ptenpc−/− prostates, as demonstrated by an increase in KI67 positive epithelium at three, six, and twelve months (Fig. 5B, E, Supplementary Fig. S3). Despite the increase in proliferation, no gross differences in tissue morphology were observed at either three or six months between Sin3bpc+/− Ptenpc−/− and Sin3bpc−/− Ptenpc−/− prostates (Fig. 2A, C), indicating that the increase in proliferation is unlikely a mere consequence of cancer progression, but rather reflects inherent differences in the proliferation properties of Sin3bpc−/− Ptenpc−/− epithelial cells. Recent studies have implicated the secretome of PTEN-induced senescent prostate epithelium in the recruitment of immune cells (22). However, while we observed a modest, but non-significant increase in total prostate infiltrating CD45+ immune cells, as well as a significant increase in GR1-high, CD11B+ cells in Sin3bpc+/− Ptenpc−/− compared to Sin3bpc+/− prostates consistent with previous reports (22), we did not observe differences in immune cell infiltration between Sin3bpc+/− Ptenpc−/− and Sin3bpc−/− Ptenpc−/− prostates (Supplementary Fig. S4). Taken together, these observations indicate that SIN3B cell autonomously promotes PICS in order to restrict cellular proliferation in the prostate epithelium and prevent PCa progression.
Figure 5.

SIN3B is required for PICS. A, Representative images of SA-β-Gal on fresh-frozen sections of the dorsolateral lobe of prostates taken from three-month-old Sin3bpc+/− Ptenpc−/−, and Sin3bpc−/− Ptenpc−/− mice. n = 2, 3 respectively. B, Representative images of immunohistochemistry for KI67, P21, pHP1γ, and IL6 on paraffin sections of the dorsolateral lobe of prostates taken from three-month-old Sin3bpc+/−, Sin3bpc−/−, Sin3bpc+/− Ptenpc−/−, and Sin3bpc−/− Ptenpc−/− mice. n = 3 each. Top Panel = 10× magnification. Bottom Panel = 40× magnification. Scale bar = 200 μm. C, Quantification of KI67 positive nuclei. n = 3, 3, 5, 5 respectively. Student’s t-test was used to assess significance. * p<0.05. D, P21. n = 3, 3, 4, 5 respectively. E, pHP1γ. n = 3, 3, 5, 6 respectively.
SIN3B levels and the SIN3B repression program are attenuated in prostate cancer in humans
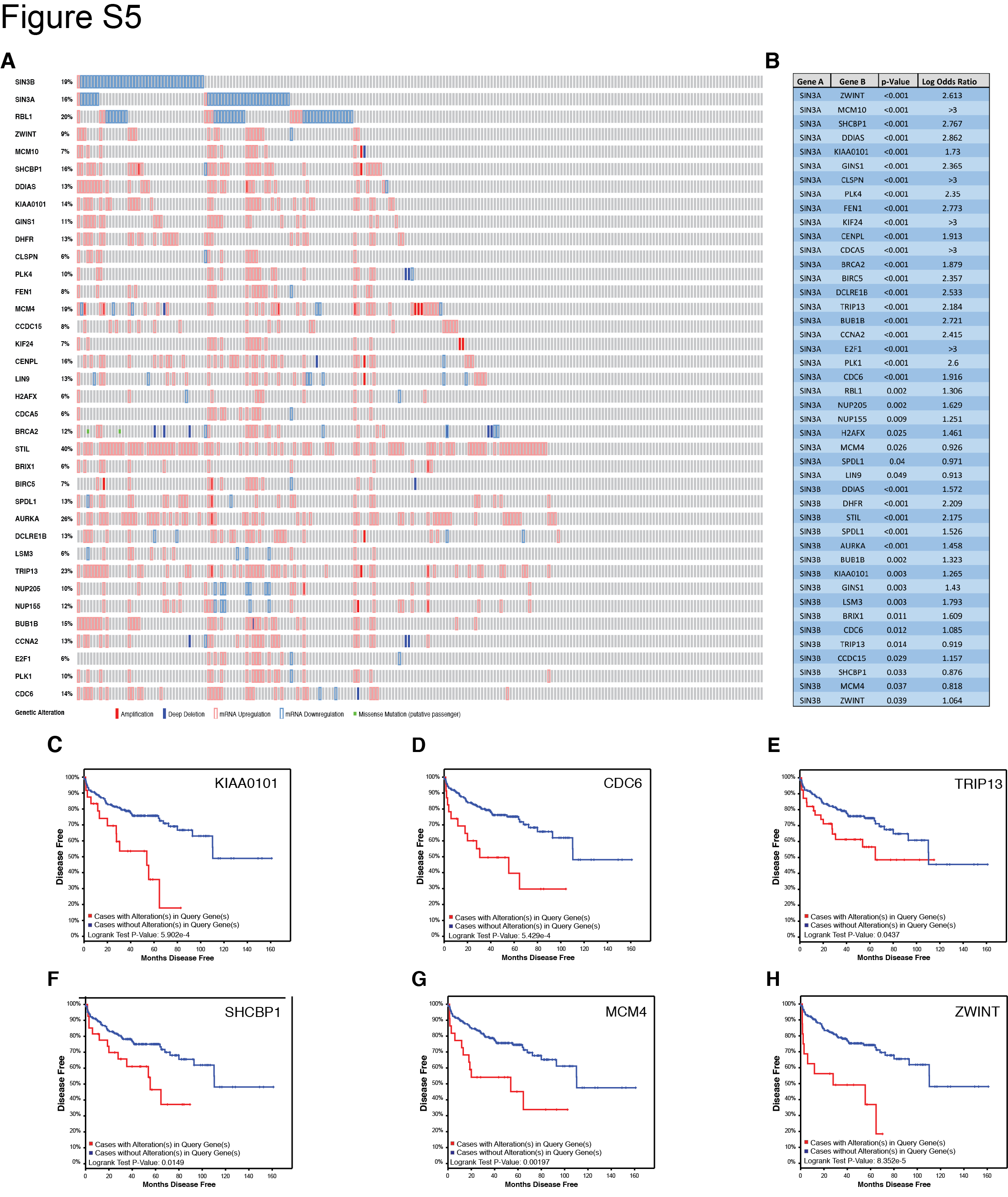
Our results suggest that SIN3B levels correlate with PCa progression in the mouse. To examine the clinical relevance of this finding, we analyzed available genome-wide expression data of human PCa tumors (23,24). Oncomine expression analysis using two independent studies showed that SIN3B is significantly downregulated in carcinoma compared to normal prostate tissue, and that SIN3B lies among the top 10% most downregulated transcripts in prostate cancer (ranked 1,384 out of 22,238 genes and 83 out of 8,603 genes, respectively) (Fig. 6A, B). We also found that SIN3B transcript levels are significantly downregulated in approximately 18% of sequenced tumors compared to the adjacent normal prostate tissue (Fig. 6C) (23,25,26). Importantly, expression of the SIN3B-related transcript SIN3A, a related essential component of the SIN3-HDAC complex, is also decreased in 15% of sequenced tumors (Fig. 6C) (23,25,26). There is, however, little overlap between the tumors exhibiting reduced SIN3B levels and those exhibiting reduced SIN3A levels, suggesting that loss of SIN3B and loss of SIN3A expression are mostly mutually exclusive in prostate cancer (Fig. 6C). We examined the impact of the downregulation of either SIN3B or SIN3A on disease-free survival, and found that neither had any significant effect. However, it is important to note that the functional disruption of the SIN3B complex can be achieved through multiple processes independently of SIN3B downregulation, including mutations or downregulation of any SIN3B-associated proteins. Therefore, rather than examining each individual component of the SIN3B complex, we investigated the correlation between SIN3B target gene levels, a read-out of the SIN3B complex function, and disease-free survival. We generated a list of potential direct SIN3B target genes based on ChIP-seq data in myoblasts (27) and RNA-seq data in T98G cells (unpublished observations), which we input into cBioPortal in tandem with SIN3B (and the related SIN3A). The comprehensive oncoprint data (Fig. S5A) shows a significant overlap between the downregulation of either SIN3B or SIN3A and the subsequent upregulation of their target genes. In total, we identified 16 genes and 28 genes that exhibit significant inverse correlation in expression levels with SIN3B or SIN3A, respectively (Fig. 6D, Fig. S5B). The expression levels of eight of these genes, including DDIAS and BUB1B, correlated with a significant decrease in disease-free survival after radical prostatectomy (Fig. 6E, 6F, Fig. S5C–H, Supplementary Table S2). Together, these data suggest that the downregulation of essential components of the SIN3-HDAC complex is a common event in human PCa, which correlates with the increased expression of its target genes resulting in a worse overall prognosis.
Figure 6.

The SIN3B complex is differentially regulated in human PCa. A and B, Oncomine box and whisker plots of SIN3B expression in normal and PCa human samples. A was derived from the study in (23) B was derived from the study in (24). Both plots have p-values < 0.05. C, Oncoprint plot of SIN3B and SIN3A, and their target genes DDIAS and BUB1B derived from cBioPortal using the study in reference (23,25,26). Amplification = filled red box, Deep Deletion = filled blue box, mRNA Upregulation = red outline, mRNA Downregulation = blue outline. D, Statistical analysis of the co-occurrence between the genes listed in C. E, Disease-free survival Kaplan-Meier curve of patients harboring DDIAS upregulation > 2 standard deviations derived using cBioPortal and the study in reference (23,25,26). F, Disease-free survival Kaplan-Meier curve of patients harboring BUB1B upregulation > 2 standard deviations derived using cBioPortal and the study in reference (23,25,26).
Discussion
Senescence, a permanent withdrawal from cell cycle, is hypothesized to protect against cancer initiation and progression, and is engaged in preneoplastic lesions. In the prostate, senescent cells are found in early PINs; however, their function and regulation in PCa progression have not been comprehensively elucidated. In this study, we demonstrate that SIN3B is required for senescence in PINs. Loss of SIN3B-dependent senescence resulted in accelerated disease progression to invasive PCa and worse survival in a prostate cancer-prone mouse model. Interestingly, while we observed an attenuated senescence program upon deletion of Sin3b in three-month-old animals, macroscopic and histological differences only became evident at twelve months of age. In vitro, deletion of Sin3b prevents entry into senescence in cells expressing oncogenic HRAS, but is not sufficient to sensitize cells to oncogenic transformation (12). This observation is similar to what is detected upon loss of RB1 and the RB1-related pathways (28,29). It is likely that in vivo, loss of senescence increases the likelihood for other molecular pathways to be activated and/or inactivated, driving cancer progression further. We believe the model described here may serve as a useful tool to identify these pathways.
The establishment of PICS involves multiple molecular pathways; yet, how these pathways interact has not been fully elucidated. Several mouse models of prostate cancer have been generated, relying on the inactivation of PICS for tumor progression. The deletion of Trp53, which like Sin3b deletion, attenuates PICS, leads to a phenotype reminiscent to the one we described here, albeit with decreased latency (7). At the molecular level, the ablation of Pten results in the stabilization of TRP53 through the inhibition of the TRP53/MDM2 interaction, which in turn results in the upregulation of CDKN1A, and ultimately drives cells into senescence (11,30,31). We have previously demonstrated that Sin3b-inactivated cells are refractory to the cell cycle exit elicited upon CDKN2A (P19ARF) overexpression, a well-established inhibitor of the MDM2-driven degradation of TRP53. In addition, we found that SIN3B levels are elevated in PINs in the mouse, consistent with our previous results demonstrating an increase in SIN3B levels upon entry into senescence (12,16,32). We and other groups have demonstrated the requirement for SIN3B to repress E2F target transcription in specific cell cycle exit settings (12,33). We suspect that the derepression of selected E2F targets in the absence of SIN3B may also directly contribute to the inability of cells to undergo PICS through the activation of cell cycle related genes. As such, the genetic inactivation of Sin3b would be expected to prevent senescence driven by Pten-deletion. Furthermore, the combined deletion of Cdkn1b and Pten in the mouse resulted in the development of epithelial tumors including PCa (34). Similarly, the introduction of an active form of AKT1 and the inactivation Cdkn1b specifically in the prostate abrogated the senescence process and increased proliferation which led to an aggressive and invasive pathology at a similar rate as demonstrated in this study (35). The similarities between the phenotypes detected upon deletion of these proteins and deletion of Sin3b, warrant a deeper investigation of the relationship between SIN3B and CDKN1A/B.
The molecular pathways responsible for the modulation of SIN3B expression throughout prostate cancer progression are unknown. Interestingly, we have recently determined that the polycomb group protein BMI1 directly represses the transcription of the SIN3B locus, similar to what has been observed for the INK4A/ARF locus (32,36,37). Consistently, ectopic expression of BMI1 in tandem with Pten inactivation in the mouse prostate leads to invasive PCa (38). Thus, it is tempting to speculate that the phenotype observed in our genetic model phenocopies the effects of increased BMI1 expression.
Currently, there is a significant need to identify prostate tumors that are at risk of progressing from indolent lesions to aggressive adenocarcinoma as the overtreatment of PCa remains a pervasive issue (3,39,40). As such, the necessity for prognostic markers that accurately predict aggressive disease remains paramount. We found that the deletion of Sin3b accelerated PCa progression, enhanced local invasion, and resulted in worse overall survival. Our data suggests that SIN3B levels are increased in PIN lesions, and subsequently downregulated in human PCa, correlating with the transcriptional increase of direct SIN3B targets. Thus, the expression of SIN3B and its known targets, as well as the abrogation of senescence, could potentially be used as a set of diagnostic markers, in tandem with current approaches, to better predict aggressive PCa. In addition, we feel that our model accurately replicates the human disease prior to metastatic progression, and can be exploited to study an aggressive carcinoma in situ to identify additional drivers of the metastatic disease.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
Financial Support: This work was funded by the NIH/NCI (R01CA148639 and R21CA155736) (G. David), the Samuel Waxman Cancer Research Foundation (G. David) and a pilot grant from the NYU Department of Urology (G. David). A.J. Bainor was supported by a predoctoral NIH training grant T32GM066704. D.J. Cantor was supported by a predoctoral NIH/NCI training grant (T32CA009161) and a predoctoral NIH/NCI NRSA (F30CA203047).
We would like to thank all the members of the David laboratory for helpful discussions during the preparation of this manuscript. We thank the NYU Cytometry and Cell Sorting Core for help with analysis and cell sorting. We thank the NYU Histopathology Core and Experimental Pathology Immunohistochemistry Core for help with sectioning and staining of mouse prostate tissues. We thank Dr. Elaine Wilson, PhD (NYU School of Medicine) for her gracious gift of the CK5 antibody, and Dr. Dafna Bar-Sagi, PhD for her gracious gift of the alpha smooth muscle actin antibody. We thank Dr. Jonathan Melamed, MD (NYU School of Medicine) for helpful discussions and analysis. Finally, we thank the NYU Department of Urology for their support.
Footnotes
Conflict of Interest: The authors declare no potential conflicts of interest.
References
- 1.Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2011;61:212–36. doi: 10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- 2.Hoffman RM. Clinical practice. Screening for prostate cancer. N Engl J Med. 2011;365:2013–9. doi: 10.1056/NEJMcp1103642. [DOI] [PubMed] [Google Scholar]
- 3.Schroder FH, Hugosson J, Roobol MJ, Tammela TL, Ciatto S, Nelen V, et al. Screening and prostate-cancer mortality in a randomized European study. N Engl J Med. 2009;360:1320–8. doi: 10.1056/NEJMoa0810084. [DOI] [PubMed] [Google Scholar]
- 4.Sartor AO, Hricak H, Wheeler TM, Coleman J, Penson DF, Carroll PR, et al. Evaluating localized prostate cancer and identifying candidates for focal therapy. Urology. 2008;72:S12–24. doi: 10.1016/j.urology.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 5.DiMauro T, David G. Ras-induced Senescence and its Physiological Relevance in Cancer. Current Cancer Drug Targets. 2010 doi: 10.2174/156800910793357998. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campisi J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 2001;11:S27–31. doi: 10.1016/s0962-8924(01)02151-1. [DOI] [PubMed] [Google Scholar]
- 7.Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–30. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Irshad S, Abate-Shen C. Modeling prostate cancer in mice: something old, something new, something premalignant, something metastatic. Cancer Metastasis Rev. 2013;32:109–22. doi: 10.1007/s10555-012-9409-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shen MM, Abate-Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev. 2010;24:1967–2000. doi: 10.1101/gad.1965810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cairns P, Okami K, Halachmi S, Halachmi N, Esteller M, Herman JG, et al. Frequent inactivation of PTEN/MMAC1 in primary prostate cancer. Cancer Res. 1997;57:4997–5000. [PubMed] [Google Scholar]
- 11.Alimonti A, Nardella C, Chen Z, Clohessy JG, Carracedo A, Trotman LC, et al. A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J Clin Invest. 2010;120:681–93. doi: 10.1172/JCI40535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grandinetti KB, Jelinic P, DiMauro T, Pellegrino J, Fernandez Rodriguez R, Finnerty PM, et al. Sin3B expression is required for cellular senescence and is up-regulated upon oncogenic stress. Cancer Res. 2009;69:6430–7. doi: 10.1158/0008-5472.CAN-09-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Silverstein RA, Ekwall K. Sin3: a flexible regulator of global gene expression and genome stability. Curr Genet. 2004 doi: 10.1007/s00294-004-0541-5. [DOI] [PubMed] [Google Scholar]
- 14.Dannenberg JH, David G, Zhong S, van der Torre J, Wong WH, Depinho RA. mSin3A corepressor regulates diverse transcriptional networks governing normal and neoplastic growth and survival. Genes Dev. 2005;19:1581–95. doi: 10.1101/gad.1286905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.David G, Grandinetti KB, Finnerty PM, Simpson N, Chu GC, Depinho RA. Specific requirement of the chromatin modifier mSin3B in cell cycle exit and cellular differentiation. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:4168–72. doi: 10.1073/pnas.0710285105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rielland M, Cantor DJ, Graveline R, Hajdu C, Mara L, Diaz Bde D, et al. Senescence-associated SIN3B promotes inflammation and pancreatic cancer progression. J Clin Invest. 2014;124:2125–35. doi: 10.1172/JCI72619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lukacs RU, Goldstein AS, Lawson DA, Cheng D, Witte ON. Isolation, cultivation and characterization of adult murine prostate stem cells. Nat Protoc. 2010;5:702–13. doi: 10.1038/nprot.2010.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu X, Wu J, Huang J, Powell WC, Zhang J, Matusik RJ, et al. Generation of a prostate epithelial cell-specific Cre transgenic mouse model for tissue-specific gene ablation. Mech Dev. 2001;101:61–9. doi: 10.1016/s0925-4773(00)00551-7. [DOI] [PubMed] [Google Scholar]
- 19.Stein GH, Drullinger LF, Soulard A, Dulic V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol Cell Biol. 1999;19:2109–17. doi: 10.1128/mcb.19.3.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang R, Adams PD. Heterochromatin and its relationship to cell senescence and cancer therapy. Cell Cycle. 2007;6:784–9. doi: 10.4161/cc.6.7.4079. [DOI] [PubMed] [Google Scholar]
- 21.Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, et al. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008;133:1019–31. doi: 10.1016/j.cell.2008.03.039. [DOI] [PubMed] [Google Scholar]
- 22.Di Mitri D, Toso A, Chen JJ, Sarti M, Pinton S, Jost TR, et al. Tumour-infiltrating Gr-1+ myeloid cells antagonize senescence in cancer. Nature. 2014;515:134–7. doi: 10.1038/nature13638. [DOI] [PubMed] [Google Scholar]
- 23.Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Singh D, Febbo PG, Ross K, Jackson DG, Manola J, Ladd C, et al. Gene expression correlates of clinical prostate cancer behavior. Cancer Cell. 2002;1:203–9. doi: 10.1016/s1535-6108(02)00030-2. [DOI] [PubMed] [Google Scholar]
- 25.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Oevelen C, Bowman C, Pellegrino J, Asp P, Cheng J, Parisi F, et al. The mammalian Sin3 proteins are required for muscle development and sarcomere specification. Mol Cell Biol. 2010;30:5686–97. doi: 10.1128/MCB.00975-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chicas A, Wang X, Zhang C, McCurrach M, Zhao Z, Mert O, et al. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell. 2010;17:376–87. doi: 10.1016/j.ccr.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peeper DS, Dannenberg JH, Douma S, te Riele H, Bernards R. Escape from premature senescence is not sufficient for oncogenic transformation by Ras. Nature cell biology. 2001;3:198–203. doi: 10.1038/35055110. [DOI] [PubMed] [Google Scholar]
- 30.Polager S, Ginsberg D. p53 and E2f: partners in life and death. Nat Rev Cancer. 2009;9:738–48. doi: 10.1038/nrc2718. [DOI] [PubMed] [Google Scholar]
- 31.Munoz-Espin D, Serrano M. Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol. 2014;15:482–96. doi: 10.1038/nrm3823. [DOI] [PubMed] [Google Scholar]
- 32.DiMauro T, Cantor DJ, Bainor AJ, David G. Transcriptional repression of Sin3B by Bmi-1 prevents cellular senescence and is relieved by oncogene activation. Oncogene. 2015;34:4011–7. doi: 10.1038/onc.2014.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rayman JB, Takahashi Y, Indjeian VB, Dannenberg JH, Catchpole S, Watson RJ, et al. E2F mediates cell cycle-dependent transcriptional repression in vivo by recruitment of an HDAC1/mSin3B corepressor complex. Genes & development. 2002;16:933–47. doi: 10.1101/gad.969202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Di Cristofano A, De Acetis M, Koff A, Cordon-Cardo C, Pandolfi PP. Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse. Nat Genet. 2001;27:222–4. doi: 10.1038/84879. [DOI] [PubMed] [Google Scholar]
- 35.Majumder PK, Grisanzio C, O’Connell F, Barry M, Brito JM, Xu Q, et al. A prostatic intraepithelial neoplasia-dependent p27 Kip1 checkpoint induces senescence and inhibits cell proliferation and cancer progression. Cancer Cell. 2008;14:146–55. doi: 10.1016/j.ccr.2008.06.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bracken AP, Kleine-Kohlbrecher D, Dietrich N, Pasini D, Gargiulo G, Beekman C, et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007;21:525–30. doi: 10.1101/gad.415507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 1999;397:164–8. doi: 10.1038/16476. [DOI] [PubMed] [Google Scholar]
- 38.Nacerddine K, Beaudry JB, Ginjala V, Westerman B, Mattiroli F, Song JY, et al. Akt-mediated phosphorylation of Bmi1 modulates its oncogenic potential, E3 ligase activity, and DNA damage repair activity in mouse prostate cancer. J Clin Invest. 2012;122:1920–32. doi: 10.1172/JCI57477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loeb S, Bjurlin MA, Nicholson J, Tammela TL, Penson DF, Carter HB, et al. Overdiagnosis and overtreatment of prostate cancer. Eur Urol. 2014;65:1046–55. doi: 10.1016/j.eururo.2013.12.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tosoian JJ, Carter HB, Lepor A, Loeb S. Active surveillance for prostate cancer: current evidence and contemporary state of practice. Nat Rev Urol. 2016;13:205–15. doi: 10.1038/nrurol.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.