

Abstract
T cells expressing CD19-specific chimeric antigen receptors (CARs) with endodomains that encode a signaling domain derived from CD3ζ and CD28 or 41BB have potent antitumor activity in early phase clinical studies for B-cell malignancies. Besides CD19-specific CARs, other approaches are actively being pursued to redirect T cells to CD19, including recombinant bispecific T-cell engager (BiTE) proteins or T cells genetically modified to express BiTEs (engager [ENG] T cells). Since BiTEs provide no costimulation, we investigated here if provision of costimulation through CD28 and 41BB enhances the effector function of CD19-ENG T cells. CD19-ENG T cells expressing CD80 and 41BBL on their cell surface (CD19-ENG.41BBL/CD80 T cells) were generated by retroviral transduction. CD19-ENG.41BBL/CD80 T cells retained their antigen specificity and had superior effector function compared to both unmodified T cells and CD19-ENG T cells expressing either CD80, 41BBL or no costimulatory molecule, as judged by cytokine (IFNγ and IL2) production, T-cell proliferation, and their ability to sequentially kill target cells. In vivo, CD19-ENG.41BBL/CD80 T cells had superior antileukemia activity in the BV173 xenograft model resulting in a survival advantage in comparison to CD19-ENG T cells. Thus, provision of costimulation is critical for the effector function of ENG T cells.
Keywords: Immunotherapy, Costimulation, CD19, bispecific T-cell engager, leukemia, lymphoma
Introduction
Hematological malignancies of B-cell origin are an important cause of cancer-related mortality since the prognosis of relapsed or refractory disease remains poor (1–4). In recent years, immunotherapeutic approaches have shown promise in the treatment of CD19+ hematological malignancies, including the adoptive transfer of T cells expressing CD19-specific chimeric antigen receptors (CARs) or the infusion of bispecific antibodies (BiTEs) to redirect T cells to CD19+ tumor cells (5–16).
Although CD19-targeting CAR T cells and BiTEs have been successful in clinical studies, however, both have been associated with toxicities including cytokine release syndrome (CRS) and neurotoxicity (7,10,11,14,17,18). Thus, exploration of alternative strategies to redirect the immune system towards CD19+ malignancies are needed. For example, T cells, genetically modified to secrete CD19-specific BiTEs (CD19-ENG T cells), kill not only CD19+ cells, but also recruit bystander T cells to tumor cells in an antigen specific manner (19,20). Although CD19-ENG T cells had antitumor activity in preclinical animal models, consistent IL2 production and T-cell expansion in vivo was dependent on the presence of costimulatory molecules on the cell surface of tumor cells (19).
Because most CD19+ malignancies do not express costimulatory molecules on their cell surface (19), we explored here if expressing the costimulatory molecules CD80 and/or 41BBL on the cell surface of CD19-ENG T cells enhanced their effector function. Our results indicate that costimulation with CD80 and 41BBL is required for optimal antigen-dependent CD19-ENG T-cell activation.
Materials and Methods
Cell lines and culture conditions
The Ph-positive chronic B lymphoblastic leukemia (ALL) cell line BV173 (German Collection of Microoganisms and Cell Cultures (DSMZ), Braunschweig, Germany), and the ALL cell line Nalm 6 (DSMZ) were used as CD19+ targets. The generation of firefly luciferase (ffLuc)-expressing BV173 (BV173.ffLuc) were described previously (21,22). K562 (chronic myelogenous leukemia; ATCC, Manassas, VA), and KG1a (acute myelogenous leukemia; ATCC) were used as negative controls. All cell lines were grown in RPMI 1640 (Thermo Scientific, Waltham, MA) except for KG1a (IMDM; Thermo Scientific). 293T cells (ATCC) were used for packaging retroviral vectors and grown in DMEM. All media was supplemented with 10–20% FBS (Thermo Scientific) and GlutaMAX-I (2 mmol/L; Invitrogen, Carlsbad, CA), and all cell lines were grown in standard (37°C, 5% CO2) tissue culture incubators.
Cell lines were purchased between 2012 and 2016. The Characterized Cell Line Core Facility at MD Anderson Cancer Center, Houston, Texas, performed cell line validation. Once thawed, cell lines were kept in culture for a maximum of three months before a new reference vial was thawed. All cell lines were tested on a regular basis for mycoplasma and were negative.
Generation of retroviral vectors
The generation of SFG retroviral vectors encoding the CD19- or EphA2-ENG molecule and mOrange were previously described (19,23). MSCV retroviral vectors encoding CD80, 41BBL, or 41BBL and CD80 were generated by subcloning CD80 from pORF.CD80 (Invivogen, San Diego, CA, USA), and/or 41BBL from pORF.41BBL (Invivogen) into MSCV-I-GFP(M) (provided by the late Elio Vanin, Northwestern University Feinberg School of Medicine, Chicago, IL). RD114-pseudotyped retroviral particles were generated as previously described (24).
Generation of engager T cells
All methods involving human subjects were carried out in accordance to the Declaration of Helsinki. Human peripheral blood mononuclear cells (PBMCs) from healthy donors were obtained under a Baylor College of Medicine IRB approved protocol, after acquiring informed consent. Retroviral transduction was done as previously described (25,26). PBMCs were stimulated on OKT3 (1μg/mL; CRL-8001, ATCC) and CD28 (1μg/mL; BD Bioscience) antibody-coated, non-tissue culture treated 24-well plates. Human interleukin 7 (IL7; 10ng/mL; Peprotech, Rocky Hill, NJ) and human interleukin 15 (IL15; 5ng/mL; Peprotech, Rocky Hill, NJ) were added to cultures on day 2. On day 3, T cells were transduced with retroviral particles on RetroNectin-coated plates (Takara Bio USA, Mountainview CA) in the presence IL7 (10ng/mL) and IL15 (5 ng/mL). T cells were subsequently expanded with IL7 and IL15. Non-transduced (NT) T cells were activated with OKT3/CD28 and expanded in parallel with IL7 and IL15. Cells were cultured for 7–10 days prior to being used for in vitro or in vivo experiments.
Flow cytometric analysis
Monoclonal antibodies (mAb) for the following markers were used for fluorescence activated cell sorting (FACS) analysis as described elsewhere (26): 41BBL (Clone C65–485; BD Biosciences, San Jose, CA) conjugated with GAM-APC antibody (BD Biosciences; cat. 550826), CD80-PerCP (eBioscience, San Diego, CA; cat. 46080942); CD3-APC (clone HIT3a; cat. 555342), CD4-PECy7 (clone SK3; cat. 560909), CD8-APCH7 (clone SK1; cat. 560179), CCR7-FITC (clone 150503; cat. 561271), CD62L-APC (clone DREG-56; cat. 559772), CD95-Pacific Blue (clone DX2; cat. 562616), and CD45RO-PercP (clone UCHL1; cat. 560607) (all BD Biosciences, Mountain View, CA). Isotype controls used were IgG1-FITC, IgG1 APC, IgG1Pe.Cy7, IgG1APC H7, IgG1 Pac Blue and IgG1 PercP.Cy7 (all from BD Biosciences). 10,000 cells (non-transduced [NT] or genetically modified) per sample were analyzed by a FACSCalibur instrument (BD Biosciences) using Cell Quest Software (BD Biosciences) and a BD Canto II instrument (BD Biosciences) using FACSDiva software (BD Biosciences) and analyzed using Kaluza Analysis 1.3 (Beckman Coulter) and FlowJo v10 (FlowJo LLC).
Coculture assays and ELISA
NT or genetically modified effector T cells were plated at a 2:1 effector to target (E:T) ratio with CD19+ (BV173) or CD19− (K562) target cells. Coculture supernatants were collected after 48 hours, snap frozen, and stored for cytokine analysis at a later time. IFNγ and IL2 concentrations were determined using ELISA kits (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions.
Cytotoxicity assay
Cytotoxic activity of ENG T cells against targets was determined by standard 51Chromium (Cr) release assay. 1×106 target cells were labeled with 50μCi 51Cr and incubated for 1 hour. Targets were then washed and 5×103 cells were cocultured with effector T cells at different effector to target (E:T) ratios. Supernatants were analyzed for radioactivity using a Wizard gamma counter Model 2470 (Perkin Elmer, Shelton CT) reader after 4 hour incubation. Percent lysis was calculated as previously described (21).
Sequential killing assay
To determine the cytolytic activity of T cells after repeat stimulations, we performed a sequential killing assay with GFP-positive tumor cell lines (BV173 and Nalm 6) as outlined in Supplementary Fig. S4. 1.5×105 GFP-positive tumor cells were plated at different E:T ratios (1:4 and 1:8) with T cells in a 48-well plate. Every 3 to 4 days, cells were mixed in the well, and a small aliquot of cells was removed for analysis. The remaining cells were washed once, resuspended in fresh, cytokine-free RPMI and 1.5×105 fresh tumor cells were added. Cells were stained with 7-AAD (BD Biosciences; cat. 559925) and anti-CD3-APC (BD Biosciences) before enumerating viable GFP+ (tumor) and CD3+ T cells by FACS analysis using Countbright Absolute Counting Beads (Life technologies, Eugene, OR). Coculture assays were also performed in a 24-well plate format in which tumor cells and T cells were plated at a 1:1 E:T ratio (5×105 cells each per well). After 5 days, cells were mixed in the well, and a small aliquot of cells was removed for analysis. The remaining cells were washed once, resuspended in fresh, cytokine-free RPMI and 5×105 fresh tumor cells were added.
In vivo experiments
Animal experiments were performed on a protocol approved by the Baylor College of Medicine Institutional Animal Care and Use Committee in accordance to the American Association for Laboratory Animal Science. Eight to 10-week old NSG mice (NOD.Cg-Prkdcscid/Il2rgtm1Wjl/SzJ; JAX Mice, Bar Harbor, ME) were sublethally irradiated with 120 cGy 24 hours before the intravenous (i.v.) injection of 3×106 BV173.ffLuc cells. Mice were treated with 1×106 CD19-ENG, CD19-ENG.CD80.41BBL or EphA2-ENG.CD80/41BBL T cells given i.v. on day 7 after tumor cell injection. The same model was used to track T cells in vivo, but in this case, unmodified BV173 cells and T cells genetically modified to express an enhanced green fluorescent protein ffLuc fusion gene (eGFP.ffLuc) were injected. Mice were imaged using the IVIS® system (IVIS, Xenogen Corp., Alameda, CA) as previously described (21), and euthanized at predefined endpoints (antitumor activity: day 80; T-cell persistence: day 100) or when they met euthanasia criteria in accordance with the Center for Comparative Medicine at Baylor College of Medicine.
Statistical analysis
Data were summarized using descriptive statistics. Comparisons of continuous variables among three or more groups were made by one-way ANOVA, while comparisons between two groups were made by t-test or Wilcoxon rank-sum test when appropriate. Multiple comparisons were adjusted by the Holm’s method. Survival times from tumor cell injection in the mouse experiments were analyzed by the Kaplan–Meier method and the Gehan-Wilcoxon test. GraphPad Prism 5 software (GraphPad software, Inc., La Jolla, CA), SAS 9.4, and R 3.3.2 were used for statistical analysis. P values < 0.05 were considered statistically significant.
RESULTS
Generation and characterization of CD19-ENG T cells
We generated T cells expressing CD19-ENG, CD19-ENG and CD80 (CD19-ENG.CD80), CD19-ENG and 41BBL (CD19-ENG.41BBL), and CD19-ENG with both 41BBL and CD80 (CD19-ENG.41BBL/CD80) by transducing CD3/CD28-activated T cells with one or two retroviral vectors encoding the respective transgenes (Fig. 1A). Five to seven days post transduction, genetically modified T cells were enumerated by FACS analysis for mOrange (CD19-ENG), CD80, and 41BBL expression (Fig. 1B,C). Mean mOrange expression ranged from 53.9% (±6.2%) to 79% (±3%) with no significant differences between transduced T-cell populations. CD19-ENG.CD80 (mean 55.9% ±4.4%) and CD19-ENG.41BBL/CD80 (mean 53.6% ±13%) T cells showed significantly higher expression of CD80 molecules when compared to CD19-ENG (p=0.0002 and p=0.004) and CD19-ENG.41BBL T cells (p=0.0002 and p=0.003). CD19-ENG.41BBL (mean 62.8% ±14%) and CD19-ENG.41BBL/CD80 T cells (mean 86.7% ±8%) expressed significantly more 41BBL than CD19-ENG and CD19-ENG.CD80 T cells (p=0.004 and p=0.0002, respectively). These results indicate the successful generation of CD19-ENG T cells coexpressing CD80 and/or 41BBL.
Figure 1. Generation of CD19-ENG T cells expressing CD80 and/or 41BB.
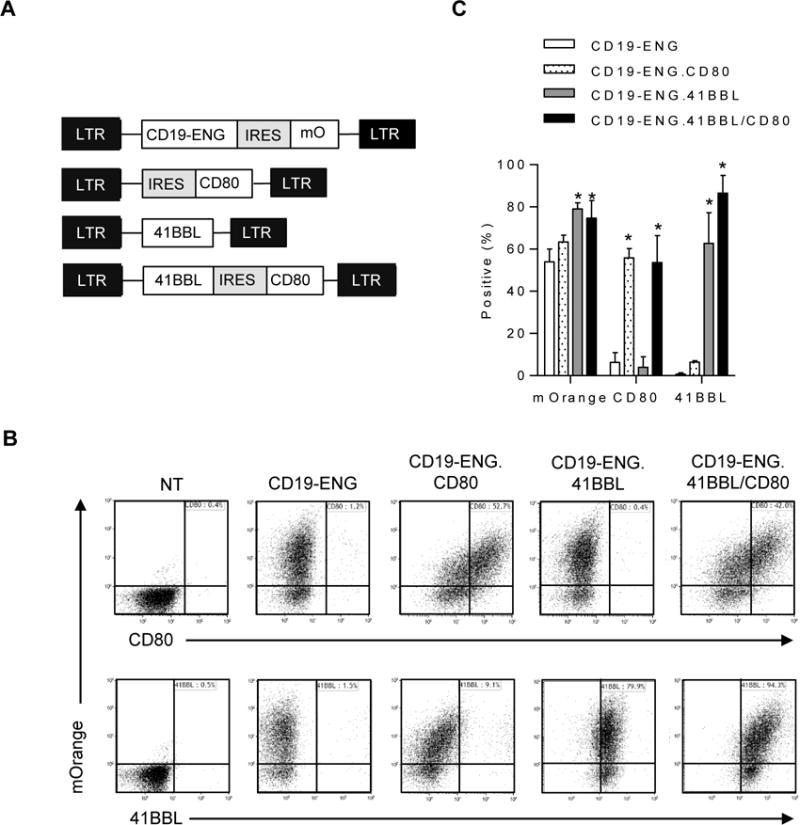
(A) Scheme of retroviral constructs. (B) Representative dot plots for CD19-ENG (mOrange), CD80 (PercP) and 41BBL (APC) 5 to 7 days post transduction of CD3/CD28-activated T cells with retroviral vectors. (C) Mean and SD of mOrange, CD80, and 41BBL expression (n=3; * p<0.05 compared to CD19-ENG T cells, two-tailed t-test).
Ten to 14 days post transduction, CD19-ENG T-cell populations were stained for CD3, CD4, CD8, CD45RO, CCR7, TIM3, LAG3, and PD-1 to determine if expression of costimulatory molecules changes their phenotype. The ratio of CD8+ to CD4+ T cells was 3 to 1 for T cells expressing CD19-ENG, and T cells had predominantly an effector memory RA (EMRA; CD45RO− CCR7−) phenotype (Fig. 2A,B; gating strategy in Supplementary Fig. S1B). Expressing CD80 and/or 41BBL on CD19-ENG T cells changed neither CD8:CD4 T-cell ratio nor phenotype. Non-transduced (NT) T cells had a similar CD8:CD4 ratio, but had a predominately naïve (N; CD45RO− CCR7+) and central memory (CM; CD45RO+ CCR7+) phenotype (Fig. 2A,B). PD-1 and LAG3 expression in T cells transduced with CD19-ENG, CD19-ENG.CD80, CD19-ENG.41BBL, or CD19-ENG.41BBL.CD80 was not significantly different from NT T cells, and no T-cell population expressed TIM3 (Fig. 2C,D; gating strategy in Supplementary Fig. S1C). Thus, expression of CD80 and/or 41BBL on the cell surface of CD19-ENG T cells does not change their phenotype. To further investigate the naïve cell population of NT T cells, we performed staining for CD62L and CD95, demonstrating a high percentage of T cells with a stem cell memory-like phenotype (CD45RO− CCR7+ CD62L+ CD95+ cells; Supplementary Fig. S2), which is consistent with previous findings by others (27–29).
Figure 2. Immunophenotype and exhaustion marker expression in CD19 ENG T cells.
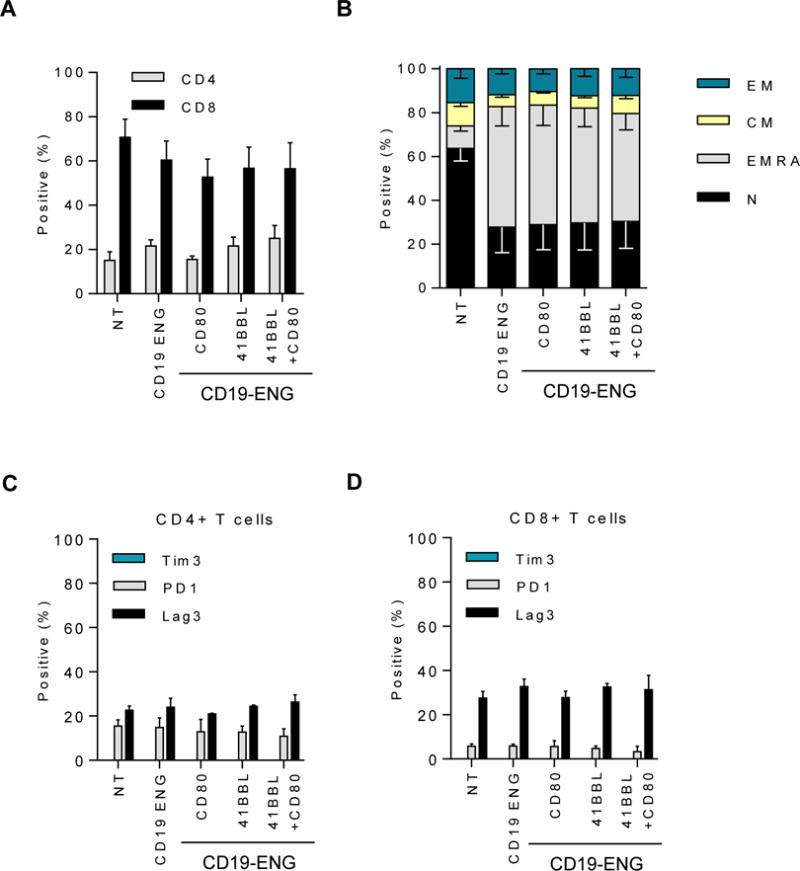
10 to 14 days post transduction, flow cytometric analysis was used to determine (A) Frequency of CD8+ and CD4+ T cells subsets (n=3, non-significant (NS), one-way ANOVA); (B) Frequency of naïve (CD45RO− CCR7+), effector memory (CD45RO+ CCR7−), central memory (CD45RO+ CCR7+), and EMRA (CD45RO− CCR7−) T cells (n=6; naïve and EMRA T cells: NT vs CD19-ENG +/− costimulatory molecules: p≤0.001, one-way ANOVA); Frequency of Tim3, PD-1, and LAG3 expression on (C) CD4+ cells and (D) CD8+ cells (n=3, NS, one-way ANOVA).
CD80 and 41BBL enhance antigen-dependent IFNγ and IL2 production
To evaluate if expressing CD80 and/or 41BBL on the cell surface of CD19-ENG T cells enhanced their effector function, we performed cytotoxicity assays and determined the production of IFNγ and IL2 after exposure to CD19+ target cells. CD19-ENG T cells killed CD19+ (BV173) but not CD19− (KG1a) tumor cells, and expression of CD80 and/or 41BBL did not change their specificity or cytolytic activity (Fig. 3A). This was confirmed for a second CD19+ target cell (Nalm 6; Supplementary Fig. S3). To confirm that expression of CD80 and 41BBL did not result in nonspecific target cell killing, we expressed both molecules in T cells transduced with an ENG molecule specific for an irrelevant antigen (EphA2-ENG). EphA2-ENG.41BBL/CD80 T cells did not kill BV173 or KG1a (Fig. 3A).
Figure 3. Antigen specificity and cytokine secretion by CD19-ENG T cells.
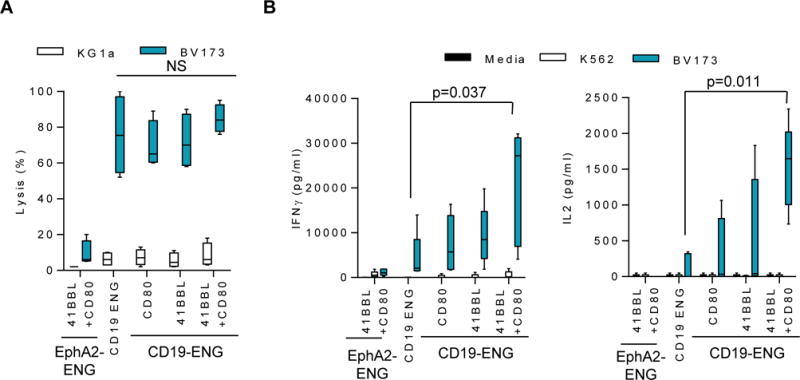
(A) Cytotoxicity assays were performed using CD19-ENG, CD19-ENG.CD80, CD19-ENG.41BBL, CD19-ENG.41BBL/CD80 and EphA2-ENG, 41BBL/CD80 T cells as effectors and CD19+ (BV173) and CD19− (KG1a) tumor cells as targets at a E:T ratio of 10:1. Percent lysis is plotted using Tukey box and whisker plots. (n=4; assay performed in triplicates). (B) Effector T cells were cocultured with CD19+ (BV173), CD19− (KG1a) tumor cells, or media only at a ratio of 2:1. After 48 hours, IFNγ and IL2 production was determined by ELISA and plotted using Tukey box and whisker plots (n=5; assay performed in duplicates, significance at p<0.05; one-way ANOVA).
To determine IFNγ and IL2 production after antigen exposure, CD19-ENG, CD19-ENG.CD80, CD19-ENG.41BBL, CD19-ENG.41BBL/CD80, or EphA2-ENG.41BBL/CD80 T cells were cultured with CD19+ (BV173) or CD19− (K562) cell lines, two cell lines that do not express the costimulatory molecules CD80, CD86, or 41BBL (19). After 24 hours, the concentration of IFNγ and IL2 in culture media was determined by ELISA. CD19-ENG.41BBL/CD80 T cells secreted significantly more IFNγ (p=0.037) and IL2 (p=0.011) in comparison to CD19-ENG T cells (Fig. 3B). Although CD19-ENG.CD80 and CD19-ENG.41BBL T cells also secreted more IFNγ and IL2 than CD19-ENG T cells, this difference did not reach significance. EphA2-ENG.41BBL/CD80 T cells did not produce significant levels of IFNγ and IL2 in the presence of CD19+ or CD19− targets, confirming antigen specificity (Fig. 3B). Thus, expression of CD80 and 41BBL on the cell surface of CD19-ENG T cells is required to significantly enhance their ability to secrete IFNγ and IL2 after antigen-specific T-cell activation.
CD80 and 41BBL enhance sequential antitumor activity
To evaluate if provision of costimulation enhanced the sequential killing capability of CD19-ENG T cells, we performed a sequential killing assay (outlined in Supplementary Fig. S4). BV173 or Nalm 6 cells were cocultured with different CD19-ENG T cell populations at an initial E:T ratio of 1:4 or 1:8. Every 3 to 4 days, T cells and tumor cells were enumerated before washing cells, resuspending in cytokine-free media, and adding fresh tumor cells. For BV173 at both E:T ratios and for Nalm 6 at an E:T ratio of 1:4, CD19-ENG.41BBL/CD80 T cells killed target cells up to the 7th time fresh tumor cells were added. CD19-ENG.CD80 T cells killed target cells up to the 5th, CD19-ENG.41BBL T cells up to the 4th, and CD19-ENG T cells up to the 2nd time, respectively (Fig. 4A–C; Supplementary Fig. S5A). For Nalm 6, at an initial E:T ratio of 1:8, CD19-ENG.41BBL.CD80 T cells could kill targets for up to the 4th time fresh tumor cells were added, with other CD19-ENG T cell populations being able to kill up to the 3rd time (Supplementary Fig. S5B). CD19-ENG.41BBL/CD80 T cells had the most consistent antitumor activity against both targets at both E:T ratios in comparison to CD19-ENG T cells (Fig. 5B). CD19-ENG.41BBL/CD80 T cells were also the only population that expanded significantly more than CD19-ENG in the presence of both targets and E:T ratios (Fig. 5A,B). Thus, our analysis demonstrated the following T-cell effector rank order: CD19-ENG.41BBL/CD80 > CD19-ENG.CD80 = CD19-ENG.41BBL > CD19-ENG T cells (Fig. 5). Due to the superior effector function of CD19-ENG.41BBL.CD80 T cells in all performed in vitro assays, we selected these cells for our in vivo experiments. Improved cytolytic activity of CD19-ENG.41BBL/CD80 and CD19-ENG.CD80 T cells was also confirmed in a coculture assay in which T cells were re-exposed to tumor cells every 5 days (Supplementary Fig. S6). Although CD19-ENG.41BBL/CD80 T cells were effective against both targets, T-cell expansion was limited in the presence of Nalm 6 cells. We, therefore, investigated whether a difference in PD-L1 expression existed between BV173 and Nalm 6 cells. At baseline, neither cell line expressed PD-L1, whereas IFNγ exposure induced PD-L1 expression in Nalm 6 cells but not in BV173 cells (Supplementary Fig. S7).
Figure 4. CD19-ENG.41BBL/CD80 T cells have improved ability to sequentially kill CD19+ target cells.
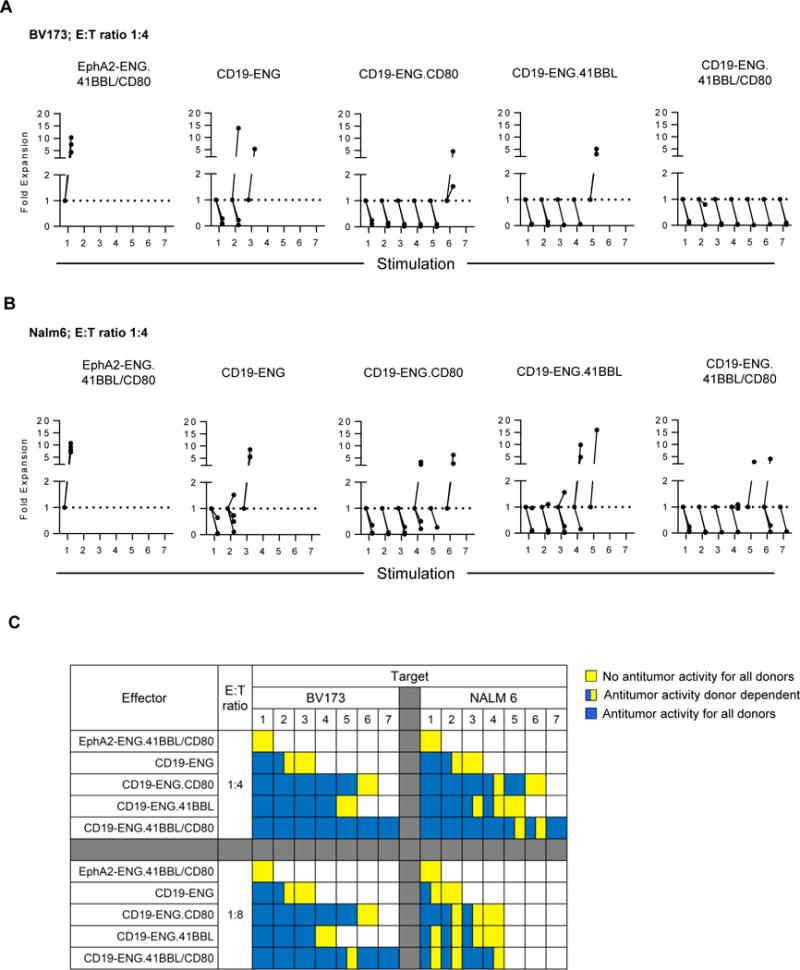
Sequential killing assays were performed as outlined in the material and methods section and Supplementary Fig S2. Absolute cell count of live tumor cells was obtained by flow cytometry using Countbright counting beads. (A,B) Live tumor cell number was plotted relative to tumor cell number at the start of each stimulation; (A) BV173 (n=3), (B) Nalm6 (n=4). (C) Summary data presented as a heatmap denoting no antitumor activity for all donors (yellow), donor-dependent antitumor activity (blue/yellow), or antitumor activity in all donors (blue). Numbers 1–7 under cell line names denote stimulation number. White boxes represent values not determined. The Wilcoxon rank sum test was used to determine significance (p<0.05).
Figure 5. CD19-ENG.41BBL/CD80 T cells have improved antigen-dependent proliferative capacity.
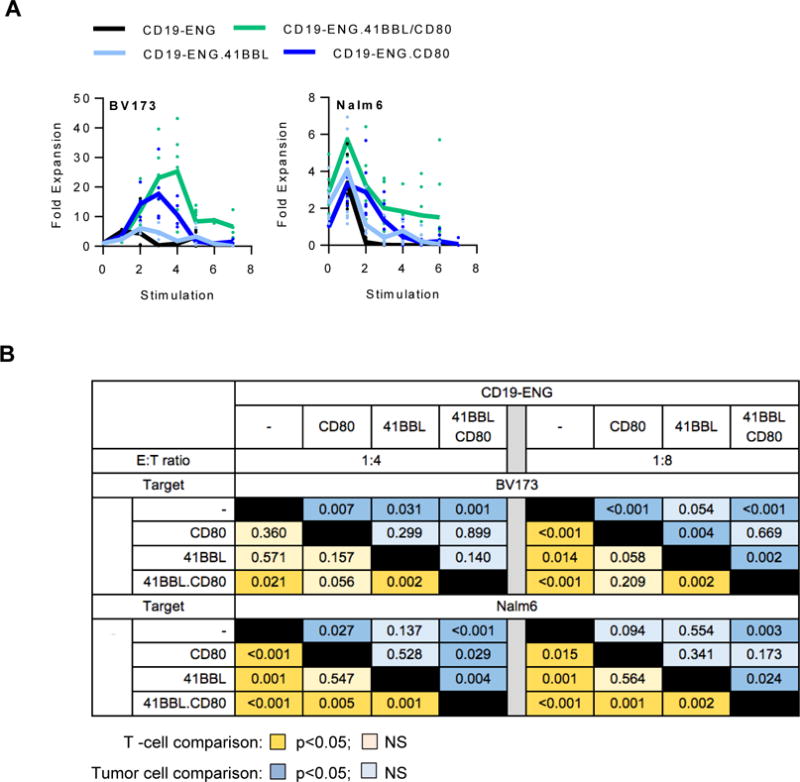
(A) Sequential killing assays were performed as outlined in the material and methods section and Supplementary Fig S2. Absolute cell count of live T cells was obtained by flow cytometry using Countbright counting beads. Fold T-cell expansion at each sequential stimulation is plotted. (B) Proliferation comparison table. Statistical significance was determined using the Wilcoxon rank sum test. Orange boxes denote comparison of T-cell numbers (dark orange: p <0.05, light orange p=NS). Blue boxes denote comparison of tumor cell numbers between different conditions (dark blue: p<0.05, light blue p=NS).
CD19-ENG.41BBL/CD80 T cells have superior antitumor activity in vivo
To evaluate the antitumor activity of CD19-ENG.41BBL/CD80 T cells, NSG mice were injected with 3×106 BV173.ffLuc cells, and on day 7 received a single i.v. dose of 1×106 CD19-ENG, CD19-ENG.41BBL/CD80, or EphA2-ENG.41BBL/CD80 T cells. Whereas CD19-ENG T cells had no antitumor activity at a cell dose of 1×106 as previously reported (19), 1×106 CD19-ENG.41BBL/CD80 T cells had potent antitumor activity in 9/10 mice as judged by bioluminescence imaging (Fig. 6A,B). Long-term follow up to 80 days revealed no weight loss or leukemia recurrence, resulting in a significant survival advantage of CD19-ENG.41BBL/CD80 T cell treated mice (p<0.0001; Fig. 6C,D). EphA2-ENG.41BBL/CD80 T cells had no antitumor activity.
Figure 6. CD19-ENG.41BBL/CD80 T cells have improved antitumor activity when compared to CD19 ENG T cells in vivo.
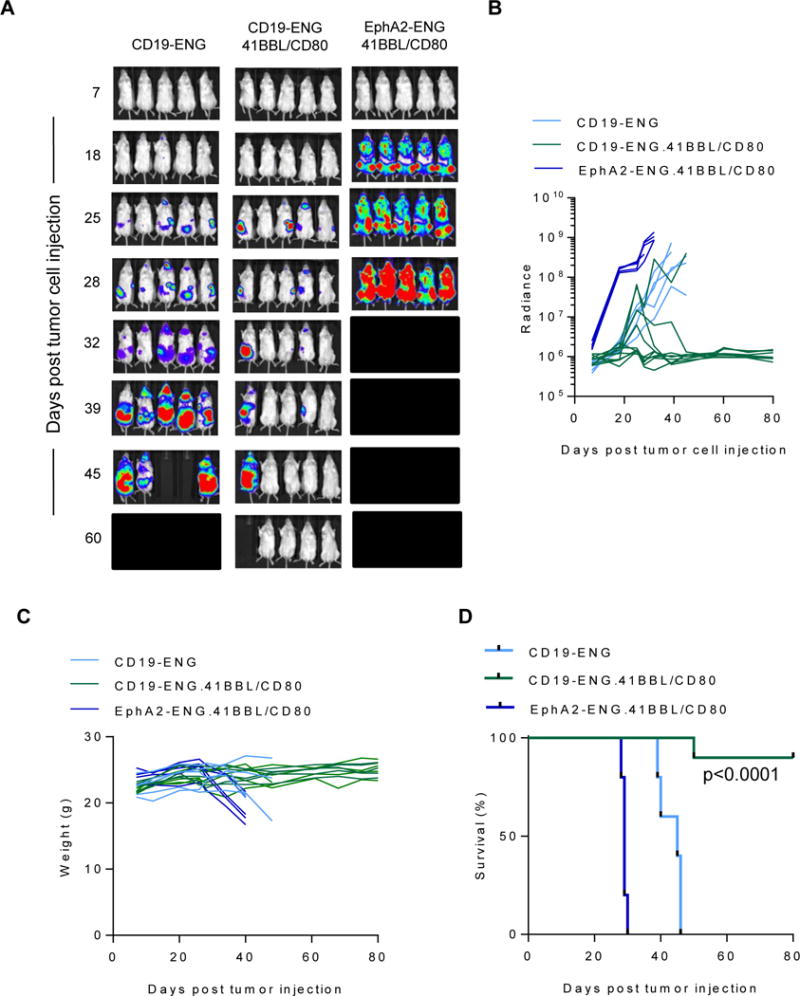
NSG mice were sublethally irradiated and injected i.v. with BV173.ffLuc. On day 7, mice received one i.v. dose of 1×106 CD19-ENG (n=5), CD19-ENG.41BBL/CD80 (n=9), or EphA2-ENG.41BBL/CD80 (n=5) T cells. Tumor growth was monitored by bioluminescence imaging. (A) Images of representative animals. (B) Bioluminescence signal (radiance = photons/sec/cm2/sr) over time is plotted. (C) Shown are weights of animals over the course of the experiment. (D) Kaplan-Meier survival curves for injected mice. Statistical significance was determined using the Wilcoxon rank sum test (p<0.0001).
CD19-ENG T cells do not expand in the BV173 leukemia model in vivo.(19) To evaluate if CD19-ENG.41BBL/CD80 T cells expand in vivo in an antigen-dependent manner, NSG mice were injected with 1×106 BV173 cells, and on day 7 received a single i.v. dose of 1×106 CD19-ENG.41BBL/CD80 or EphA2-ENG.41BBL/CD80 T cells, that were also genetically modified to express eGFP.ffLuc. Control mice that did not receive any tumor were injected with eGFP.ffLuc-expressing CD19-ENG.41BBL/CD80 T cells. Within the first 5 days post T-cell injection, CD19-ENG.41BBL/CD80 T cells expanded significantly in the femurs of tumor-bearing mice, as judged by bioluminescence imaging, in contrast to mice without tumors (Fig. 7A, B). EphA2-ENG.41BBL/CD80 T cells expanded much more slowly than CD19-ENG.41BBL/CD80 in tumor-bearing mice, demonstrating that the early expansion of CD19-ENG.41BBL/CD80 T cells post injection is antigen-specific. Starting 7 days post injection, no statistical significant difference in the bioluminescence signal among all three groups was seen. Long-term follow-up revealed that all three T-cell populations expanded until day 13. Afterwards, T cells in the CD19-ENG.41BBL/CD80 T cells +/− tumor groups contracted and persisted in low numbers until the end of the end of the experiment (day 93 post T-cell injection) with no evident weight loss (Supplementary Fig. S8A–C). Area under the curve analysis revealed a significantly greater (p<0.01) expansion and persistence of CD19-ENG.41BBL/CD80 T cells in the presence of tumor cells (Supplementary Fig. S8B). Tumor-bearing mice that had received EphA2-ENG.41BBL/CD80 T cells needed to be euthanized on day 13, which is consistent with tumor progression (Fig. 6).
Figure 7. CD19 ENG.41BBL/CD80 T cells expand in vivo.
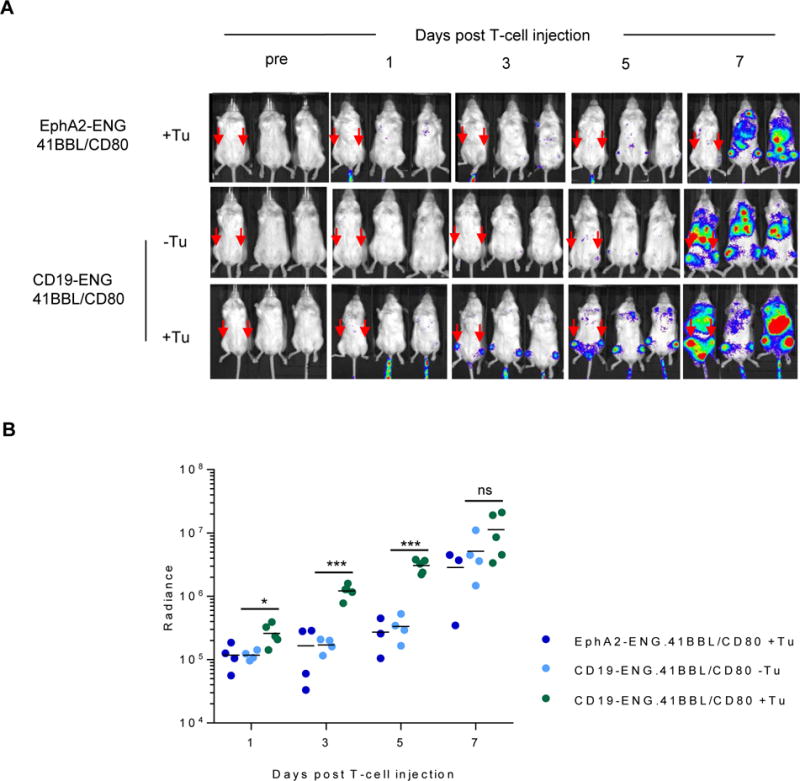
NSG mice were sublethally irradiated and injected i.v. with BV173. On day 7, mice received one i.v. dose of 1×106 CD19-ENG.41BBL/CD80 (n=5) or EphA2-ENG.41BBL/CD80 (n=4) T cells that were genetically modified with eGFP.ffLuc. Mice without tumors received one i.v. dose of 1×106 CD19-ENG.41BBL/CD80 (n=5). (A) Images of representative animals over time are shown. (B) Plots of bioluminescence signal (radiance = photons/sec/cm2/sr) of femurs (*p<0.05 for day 1, ***p<0.001 for days 3 and 5, p=ns for day 7, by Gehan-Wilcoxon test).
DISCUSSION
In this manuscript, we demonstrate that the expression of CD80 and 41BBL on the cell surface of CD19-ENG T cells enhances their effector function. Expression of CD80 and 41BBL had no impact on antigen specificity, but improved antigen-dependent cytokine secretion (IFNγ and IL2), T-cell expansion, and antitumor activity of CD19-ENG T cells.
Optimal T-cell activation requires antigen-specific CD3ζ stimulation (signal 1) and costimulation (signal 2). Upon proper costimulation, T cells produce cytokines or induce cytokine production by neighboring cells (signal 3), which is critical for their expansion (30–33). Comparisons of the effector function of T cells that express 1st generation CARs, containing only a CD3ζ endodomain, to 2nd generation CARs, whose endodomains contain the CD3ζ signaling domain plus those from costimulatory molecules such as CD28, 41BB, OX40, or CD27, has highlighted the need of costimulation for proper T cell activation (34–36). Whether T cells expressing a CAR that encodes two costimulatory endodomains (3rd generation CAR) have superior effector function than 2nd generation CAR T cells remains controversial and depends on the used tumor model (37–39).
Besides incorporating costimulatory signaling domains into CARs, investigators have also explored the expression of costimulatory molecules on the cell surface of CAR T cells (40). A study by Zhao and colleagues suggests that T cells expressing CAR.CD28.ζ and 41BBL have superior effector function in comparison to T cells expressing CAR.ζ, CD80 and 41BBL, or CAR.41BB.CD28.ζ on their cell surface (41). Here, we explored if provision of costimulatory molecules improved the effector function of CD19-ENG T cells, which secrete BiTEs that only activate CD3ζ. We successfully generated CD19-ENG T cells expressing CD80 and/or 41BBL. Expression of both molecules did not change the phenotype of CD19-ENG T cells and did not result in upregulation of exhaustion markers (PD-1, LAG3, TIM3), which has been observed in T cells that express CARs that are constitutively active (tonic signaling) (42). However, expressing CD19-ENG in T cells led to a decrease of naïve (CD45RO− CCR7+) T cells in comparison to NT T cells, which might reflect the presence of residual B-cells in our culture system at the time of transduction, resulting in T-cell activation through CD19-ENGs. Although CD19-ENG T cells did not produce significant amounts of IFNγ in the absence of CD19+ target cells, baseline tonic signaling by CD19-ENG bound to the T-cell surface cannot be excluded as another mechanism for a decrease of naïve T cells. Further studies are needed to investigate the mechanism of these phenotypic changes including molecular and epigenetic studies (43,44).
Expression of both CD80 and 41BBL was necessary for a significant increase in antigen-dependent IL2 production in comparison to CD19-ENG T cells. This differs from CARs in which a single costimulatory endodomain is sufficient (45,46). However, it is consistent with findings by others that expression of CD80 and 41BBL is required on the cell surface of CAR.ζ T cells for significant antigen-dependent IL2 production (40,41). Expression of both CD80 and 41BBL was also required for optimal cytolytic activity and expansion of CD19-ENG T cells in the sequential killing assays we performed. In these assays, CD19-ENG T cells expressing either CD80, 41BBL or no costimulatory molecule were re-exposed to tumor cells every 3 to 4 days to mimic the in vivo situation in which tumor cells are exposed to tumor cells constantly when they first arrive at tumor sites. All CD19-ENG T-cell populations expanded better in the presence of BV173 than Nalm 6, most likely due to the upregulation of PD-L1 on Nalm 6 cells in the presence of IFNγ. Here we explored only one combination of costimulatory molecules. In the setting of CAR.ζ T cells, investigators have compared the benefits of expressing CD80 with several members of the tumor necrosis factor ligand family, including 41BBL, CD70, OX40L, and CD30L (40). Their results indicate that combining CD80 and 41BBL costimulation is most effective in enhancing the effector function of CAR.ζ T cells (40).
In vivo, expression of CD80 and 41BBL on the cell surface of CD19-ENG T cells resulted in a significant increase in their antitumor activity, confirming the in vitro sequential killing assay result. This result was not due to nonspecific tumor killing because control EphA2-ENG.41BBL/CD80 T cells had no antitumor activity. CD19-ENG.41BBL/CD80 T cells did not induce significant xenogenic graft versus host disease (GvHD), as judged by weight and/or fur loss, during the 80-day observation period post T-cell injection nor was it observed in the T-cell persistence experiment with a follow-up of 93 days post T-cell injection. Future studies are planned to confirm our findings by performing a detailed histological analysis of organs of euthanized mice.
Besides expressing costimulatory molecules on the cell surface of ENG T cells, other strategies could be explored to provide costimulation. For example, others have generated fusion proteins that consist of a tumor-associated antigen (TAA)-specific scFv and the ectodomain (ECD) of CD80 or 41BBL (32,33). T cells incubated with BiTEs, TAA.scFv-CD80.ECD, and TAA.scFv-41BBL.ECD recombinant proteins result in improved IFNγ production and T-cell proliferation in comparison to T cells that were incubated with BiTEs and TAA.scFv-CD80.ECD (33,47). These results mirror our findings that provision of two costimulatory signals results in enhanced effector function after BiTE-mediated T-cell activation.
In summary, our study demonstrates that provision of CD80 and 41BBL enhances the effector function of CD19-ENG T cells. Our results are informative not only for the future clinical development of ENG T cells for hematological and solid malignancies, but also for immunotherapeutic approaches that rely on the infusion of recombinant BiTE proteins or oncolytic viruses that are genetically engineered to produce BiTEs.
Supplementary Material
Acknowledgments
This work was supported by NIH grants P50 CA126752 and P30 CA125123, Leukemia and Lymphoma Society SCOR grant 7001-14, the Alex Lemonade Stand Foundation and the St. Baldrick’s Foundation.
Footnotes
Conflict-of-Interest Statement: MPV and SG have patent applications in the field of immune- and/or gene therapy for cancer.
Authorship: MPV, SG designed the study. MPV, AS, AV, PN, AT conducted experiments. All authors contributed to data analysis and writing of the manuscript.
References
- 1.Raetz EA, Bhatla T. Where do we stand in the treatment of relapsed acute lymphoblastic leukemia? Hematology American Society of Hematology Education Program. 2012;2012:129–36. doi: 10.1182/asheducation-2012.1.129. [DOI] [PubMed] [Google Scholar]
- 2.Forman SJ, Rowe JM. The myth of the second remission of acute leukemia in the adult. Blood. 2013;121:1077–82. doi: 10.1182/blood-2012-08-234492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bhojwani D, Pui CH. Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol. 2013;14:e205–17. doi: 10.1016/S1470-2045(12)70580-6. [DOI] [PubMed] [Google Scholar]
- 4.Gokbuget N, Stanze D, Beck J, Diedrich H, Horst HA, Huttmann A, et al. Outcome of relapsed adult lymphoblastic leukemia depends on response to salvage chemotherapy, prognostic factors, and performance of stem cell transplantation. Blood. 2012;120:2032–41. doi: 10.1182/blood-2011-12-399287. [DOI] [PubMed] [Google Scholar]
- 5.Bargou R, Leo E, Zugmaier G, Klinger M, Goebeler M, Knop S, et al. Tumor regression in cancer patients by very low doses of a T cell-engaging antibody. Science. 2008;321:974–7. doi: 10.1126/science.1158545. [DOI] [PubMed] [Google Scholar]
- 6.Baeuerle PA, Kufer P, Bargou R. BiTE: Teaching antibodies to engage T-cells for cancer therapy. CurrOpinMolTher. 2009;11:22–30. [PubMed] [Google Scholar]
- 7.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. NEnglJMed. 2011;365:725–33. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Topp MS, Kufer P, Gokbuget N, Goebeler M, Klinger M, Neumann S, et al. Targeted therapy with the T-cell-engaging antibody blinatumomab of chemotherapy-refractory minimal residual disease in B-lineage acute lymphoblastic leukemia patients results in high response rate and prolonged leukemia-free survival. JClinOncol. 2011;29:2493–8. doi: 10.1200/JCO.2010.32.7270. [DOI] [PubMed] [Google Scholar]
- 9.Klinger M, Brandl C, Zugmaier G, Hijazi Y, Bargou RC, Topp MS, et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood. 2012;119:6226–33. doi: 10.1182/blood-2012-01-400515. [DOI] [PubMed] [Google Scholar]
- 10.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. NEnglJMed. 2013;368:1509–18. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. SciTranslMed. 2014;6:224ra25. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Topp MS, Gokbuget N, Stein AS, Zugmaier G, O’Brien S, Bargou RC, et al. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. 2015;16:57–66. doi: 10.1016/S1470-2045(14)71170-2. [DOI] [PubMed] [Google Scholar]
- 13.Stieglmaier J, Benjamin J, Nagorsen D. Utilizing the BiTE (bispecific T-cell engager) platform for immunotherapy of cancer. Expert opinion on biological therapy. 2015;15:1093–9. doi: 10.1517/14712598.2015.1041373. [DOI] [PubMed] [Google Scholar]
- 14.Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–28. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.von Stackelberg A, Locatelli F, Zugmaier G, Handgretinger R, Trippett TM, Rizzari C, et al. Phase I/Phase II Study of Blinatumomab in Pediatric Patients With Relapsed/Refractory Acute Lymphoblastic Leukemia. J Clin Oncol. 2016;34:4381–9. doi: 10.1200/JCO.2016.67.3301. [DOI] [PubMed] [Google Scholar]
- 16.Aldoss I, Bargou RC, Nagorsen D, Friberg GR, Baeuerle PA, Forman SJ. Redirecting T cells to eradicate B-cell acute lymphoblastic leukemia: bispecific T-cell engagers and chimeric antigen receptors. Leukemia. 2017 doi: 10.1038/leu.2016.391. [DOI] [PubMed] [Google Scholar]
- 17.Teachey DT, Rheingold SR, Maude SL, Zugmaier G, Barrett DM, Seif AE, et al. Cytokine release syndrome after blinatumomab treatment related to abnormal macrophage activation and ameliorated with cytokine-directed therapy. Blood. 2013;121:5154–7. doi: 10.1182/blood-2013-02-485623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teachey DT, Lacey SF, Shaw PA, Melenhorst JJ, Maude SL, Frey N, et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer discovery. 2016;6:664–79. doi: 10.1158/2159-8290.CD-16-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Velasquez MP, Torres D, Iwahori K, Kakarla S, Arber C, Rodriguez-Cruz T, et al. T cells expressing CD19-specific Engager Molecules for the Immunotherapy of CD19-positive Malignancies. Scientific reports. 2016;6:27130. doi: 10.1038/srep27130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu X, Barrett DM, Jiang S, Fang C, Kalos M, Grupp SA, et al. Improved anti-leukemia activities of adoptively transferred T cells expressing bispecific T-cell engager in mice. Blood cancer journal. 2016;6:e430. doi: 10.1038/bcj.2016.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shaffer DR, Savoldo B, Yi Z, Chow KK, Kakarla S, Spencer DM, et al. T cells redirected against CD70 for the immunotherapy of CD70-positive malignancies. Blood. 2011;117:4304–14. doi: 10.1182/blood-2010-04-278218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arber C, Feng X, Abhyankar H, Romero E, Wu MF, Heslop HE, et al. Survivin-specific T cell receptor targets tumor but not T cells. J Clin Invest. 2015;125:157–68. doi: 10.1172/JCI75876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iwahori K, Kakarla S, Velasquez MP, Yu F, Yi Z, Gerken C, et al. Engager T cells: a new class of antigen-specific T cells that redirect bystander T cells. Molecular therapy : the journal of the American Society of Gene Therapy. 2015;23:171–8. doi: 10.1038/mt.2014.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chow KK, Naik S, Kakarla S, Brawley VS, Shaffer DR, Yi Z, et al. T Cells Redirected to EphA2 for the Immunotherapy of Glioblastoma. MolTher. 2013;21:629–37. doi: 10.1038/mt.2012.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O, et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR. CD19-T cells and are preserved by IL-7 and IL-15. Blood. 2014;123:3750–9. doi: 10.1182/blood-2014-01-552174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krenciute GPB, Yi Z, Wu MF, Liu H, Dotti G, Balyasnikova IV, Gottschalk S. Transgenic expression of IL15 improves antiglioma activity of IL13Rα2-specific CAR T cells, but highlights the need to target multiple antigens. 2017 doi: 10.1158/2326-6066.CIR-16-0376. submitted; under revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alvarez-Fernandez C, Escriba-Garcia L, Vidal S, Sierra J, Briones J. A short CD3/CD28 costimulation combined with IL-21 enhance the generation of human memory stem T cells for adoptive immunotherapy. Journal of translational medicine. 2016;14:214. doi: 10.1186/s12967-016-0973-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sabatino M, Hu J, Sommariva M, Gautam S, Fellowes V, Hocker JD, et al. Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human B-cell malignancies. Blood. 2016;128:519–28. doi: 10.1182/blood-2015-11-683847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang S, Ji Y, Gattinoni L, Zhang L, Yu Z, Restifo NP, et al. Modulating the differentiation status of ex vivo-cultured anti-tumor T cells using cytokine cocktails. Cancer immunology, immunotherapy : CII. 2013;62:727–36. doi: 10.1007/s00262-012-1378-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. NatRevImmunol. 2013;13:227–42. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beyranvand Nejad E, van der Sluis TC, van Duikeren S, Yagita H, Janssen GM, van Veelen PA, et al. Tumor Eradication by Cisplatin Is Sustained by CD80/86-Mediated Costimulation of CD8+ T Cells. Cancer Res. 2016;76:6017–29. doi: 10.1158/0008-5472.CAN-16-0881. [DOI] [PubMed] [Google Scholar]
- 32.Notter M, Willinger T, Erben U, Thiel E. Targeting of a B7-1 (CD80) immunoglobulin G fusion protein to acute myeloid leukemia blasts increases their costimulatory activity for autologous remission T cells. Blood. 2001;97:3138–45. doi: 10.1182/blood.v97.10.3138. [DOI] [PubMed] [Google Scholar]
- 33.Muller D, Frey K, Kontermann RE. A novel antibody-4-1BBL fusion protein for targeted costimulation in cancer immunotherapy. JImmunother. 2008;31:714–22. doi: 10.1097/CJI.0b013e31818353e9. [DOI] [PubMed] [Google Scholar]
- 34.Brocker T, Karjalainen K. Signals through T cell receptor-zeta chain alone are insufficient to prime resting T lymphocytes. JExpMed. 1995;181:1653–9. doi: 10.1084/jem.181.5.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRzeta/CD28 receptor. NatBiotechnol. 2002;20:70–5. doi: 10.1038/nbt0102-70. [DOI] [PubMed] [Google Scholar]
- 36.Imai C, Mihara K, Andreansky M, Nicholson IC, Pui CH, Geiger TL, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18:676–84. doi: 10.1038/sj.leu.2403302. [DOI] [PubMed] [Google Scholar]
- 37.Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. MolTher. 2009;17:1453–64. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. MolTher. 2010;18:413–20. doi: 10.1038/mt.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. ProcNatlAcadSciUSA. 2009;106:3360–5. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stephan MT, Ponomarev V, Brentjens RJ, Chang AH, Dobrenkov KV, Heller G, et al. T cell-encoded CD80 and 4-1BBL induce auto- and transcostimulation, resulting in potent tumor rejection. NatMed. 2007;13:1440–9. doi: 10.1038/nm1676. [DOI] [PubMed] [Google Scholar]
- 41.Zhao Z, Condomines M, van der Stegen SJ, Perna F, Kloss CC, Gunset G, et al. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell. 2015;28:415–28. doi: 10.1016/j.ccell.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nature medicine. 2015;21:581–90. doi: 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. NatMed. 2011;17:1290–7. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abdelsamed HA, Moustaki A, Fan Y, Dogra P, Ghoneim HE, Zebley CC, et al. Human memory CD8 T cell effector potential is epigenetically preserved during in vivo homeostasis. J Exp Med. 2017 doi: 10.1084/jem.20161760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–28. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. NEnglJMed. 2014;371:1507–17. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hornig N, Kermer V, Frey K, Diebolder P, Kontermann RE, Muller D. Combination of a bispecific antibody and costimulatory antibody-ligand fusion proteins for targeted cancer immunotherapy. JImmunother. 2012;35:418–29. doi: 10.1097/CJI.0b013e3182594387. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.