

We describe a mapping of plasticity expression, mediated by different mechanisms, among three distinct GABA afferents to ventral tegmental area (VTA) dopamine neurons: the rostromedial tegmental nucleus, the nucleus accumbens, and the local GABA neurons within the VTA known to synapse on VTA dopamine neurons. This work is the first demonstration that discrete plasticity mechanisms recruit overlapping but different subsets of GABA inputs to VTA dopamine neurons.
Keywords: ventral tegmental area, dopamine neuron, GABA inhibition, long-term potentiation, rostromedial tegmental nucleus, nucleus accumbens
Abstract
The in vivo firing pattern of ventral tegmental area (VTA) dopamine neurons is controlled by GABA afferents originating primarily from the nucleus accumbens (NAc), rostromedial tegmental nucleus (RMTg), and local GABA neurons within the VTA. Although different forms of plasticity have been observed from GABA inputs to VTA dopamine neurons, one dependent on cyclic GMP synthesis and the other on adenylyl cyclase activation, it is unknown whether plasticity is differentially expressed in each. Using an optogenetic strategy, we show that identified inhibitory postsynaptic currents (IPSCs) from local VTA GABA neurons and NAc afferents exhibit a cyclic GMP-dependent long-term potentiation (LTP) that is capable of inhibiting the firing activity of dopamine neurons. However, this form of LTP was not induced from RMTg afferents. Only an adenylyl cyclase-mediated increase in IPSCs was exhibited by all three inputs. Thus discrete plasticity mechanisms recruit overlapping but different subsets of GABA inputs to VTA dopamine neurons.
NEW & NOTEWORTHY We describe a mapping of plasticity expression, mediated by different mechanisms, among three distinct GABA afferents to ventral tegmental area (VTA) dopamine neurons: the rostromedial tegmental nucleus, the nucleus accumbens, and the local GABA neurons within the VTA known to synapse on VTA dopamine neurons. This work is the first demonstration that discrete plasticity mechanisms recruit overlapping but different subsets of GABA inputs to VTA dopamine neurons.
ventral tegmental area (VTA) dopamine neurons in vivo fire irregularly, shifting from tonic firing to short-latency bursts and pauses of action potentials during reward and sequence learning (Jin and Costa 2010; Schultz 2002). Burst firing and pauses in activity are readily induced by phasic disruptions of excitatory and/or inhibitory synaptic balance (Lobb et al. 2011). Activation of GABAA receptors alone is sufficient to suppress burst firing in dopamine neurons, whereas blockade of GABAA receptors facilitates burst firing (Paladini et al. 1999b; Paladini and Tepper 1999; Lobb et al. 2011). Synaptic plasticity of the inputs contributing to this GABA tone could readily shift the output of a dopamine neuron.
GABA afferents to VTA dopamine neurons originate from multiple sources (Kaufling et al. 2010a; Watabe-Uchida et al. 2012), and different forms of synaptic plasticity have been demonstrated. One form of plasticity demonstrated at GABA inputs to VTA dopamine neurons involves a heterosynaptic mechanism: activation of postsynaptic NMDA receptors by glutamate afferents results in retrograde release of nitric oxide to activate soluble guanylyl cyclase, located at presynaptic GABA terminals, and synthesize cyclic GMP (Nugent et al. 2007). Another form of plasticity at GABA afferents to VTA dopamine neurons involves signaling via adenylyl cyclase activation and has been shown to be influenced by exposure to drugs of abuse (Bonci and Williams 1997; Melis et al. 2002). The potential exists, therefore, for heterogeneous expression of synaptic plasticity among the different populations of GABA afferents to dopamine neurons. Three of the densest GABA projections to VTA dopamine neurons (Kaufling et al. 2010a; Watabe-Uchida et al. 2012) include local VTA GABA neurons (Johnson and North 1992), the rostromedial tegmental nucleus (RMTg; Jhou et al. 2009b), and the nucleus accumbens (NAc; Nauta et al. 1978). The RMTg inhibits dopamine neurons in response to aversive stimuli (Jhou et al. 2009a). The NAc has been reported to feedback upon and inhibit dopamine neuron activity (Einhorn et al. 1988; Rahman and McBride 2000). Activation of VTA GABA neurons disrupts reward consumption and reduces the excitability of dopamine neurons (Tan et al. 2012; van Zessen et al. 2012). We hypothesized that input-specific inhibitory postsynaptic currents (IPSCs) from NAc, VTA GABA neurons, and/or the RMTg could be induced to express long-term potentiation (LTP) if they contain the molecular components necessary for induction (e.g., adenylyl cyclase, cyclic GMP). In a series of separate experiments, we infected GABA neurons from each of the three afferents with a virus encoding channelrhodopsin (ChR2) and used either an adenylyl cyclase- or cyclic GMP-mediated synaptic plasticity protocol for each input (Melis et al. 2002; Nugent et al. 2007). We showed that all three afferents exhibited potentiated responses following activation of adenylyl cyclase. However, only the projections from local VTA GABA neurons and NAc exhibited cyclic GMP-dependent potentiation. We found that the RMTg, a major controller of dopamine neuron activity, did not undergo heterosynaptic LTP due to a lack in the downstream targets of cyclic GMP, activation of which is necessary for the nitric oxide-dependent induction of LTP in GABA afferents to the VTA (Nugent et al. 2007).
MATERIALS AND METHODS
General surgical procedures.
Mice from both Balb/c (7 male, 11 female) and the 016962 Stock Slc32a1<tm2-(cre) Lowl>/J strains (Vgat-ires-Cre; The Jackson Laboratory; 7 male, 6 female) had specific nuclei infected with ChR2 by stereotaxic injection of an adeno-associated virus. GABA neurons in the RMTg and the NAc were targeted in Balb/c mice using injections of the AAV1-CamKII-ChR2-EYFP viral construct. Local VTA GABA neurons were targeted in Vgat-ires-Cre mice using injections of the rAAV5/EF1α-DIO-hChR2-(H134R)-mCherry viral construct. A range of 50–300 nl of virus was bilaterally injected using appropriate stereotaxic coordinates relative to bregma. Injections were made into the RMTg [−4.0 mm anteroposterior (AP), 0.3 mm mediolateral (ML), −4.6 mm dorsoventral (DV)], VTA [−3.6 mm AP, 0.3 mm ML, −4.6 mm DV], and NAc [+1.5 mm AP, 1.0 mm ML, −4.6 mm DV]. RMTg injections (50 nl) were aimed caudal to the decussation of the superior cerebellar peduncle to avoid rostral spread into the VTA. VTA injections (150 nl) were aimed rostral to medial lemniscus to avoid infection of the RMTg. NAc injections (300 nl) were aimed dorsal to the anterior commissure to avoid leakage into the lateral ventricles. All mice were housed for a minimum of 4 wk before experiments to allow expression of the virus at synaptic terminals. The mice were grouped according to the nucleus targeted for injection. All procedures were performed in accordance with the University of Texas at San Antonio policy, and the Institutional Animal Care and Use Committee approved all of the experimental procedures.
Slice preparation.
Horizontal brain slices were cut from Balb/c and Vgat-ires-Cre mice. Mice were anesthetized by isoflurane inhalation and decapitated. The brain was rapidly removed and cooled. Slices (250 μm) were cut using a vibratome (Microm HM 650V) in ice-cold cutting artificial cerebrospinal fluid (ACSF) containing (in mM) 110 choline chloride, 2.5 KCl, 1.25 NaH2PO4, 7 MgCl2, 0.5 CaCl2, 10 dextrose, 25 NaHCO3, 1.3 ascorbic acid, and 2.4 sodium pyruvate, and continuously bubbled with 95% O2–5% CO2. Cutting solution was bubbled for 15 min before cooling. Slices were then incubated for 15 min in warm (35°C) recording ACSF (in mM: 126 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1.2 MgCl2, 2 CaCl2, 10 dextrose, 25 NaHCO3, 1.3 ascorbic acid, 2.4 sodium pyruvate) before recordings and then kept at room temperature for the rest of the day.
Electrophysiology.
Slices were transferred to a recording chamber filled with recording ACSF and continually fed at a rate of 2 ml/min by a gravity feed system. An inline heater maintained a bath temperature between 32 and 34°C. The VTA was identified as medial to the medial terminal nucleus of the accessory optic tract. Whole cell recordings were made on the somata of dopamine neurons by use of an Axopatch 200B amplifier and were visualized with an Olympus microscope (BX51W1) using a Dodt imaging system. Glass electrodes were made using borosilicate glass and pulled using a Flaming/Brown pipette puller to a tip resistance of 4–6 MΩ. The electrodes were filled with internal solution containing (in mM) 128 K-gluconate, 10 HEPES, 2 MgCl2, 0.2 EGTA, 0.0001 CaCl2, 8.05 biocytin, 4 Na-ATP, and 0.4 Na-GTP. After a gigaohm seal was formed, dopamine neurons were tentatively identified by a cell-attached spike width >1.8 ms, depolarization block during a 500-ms, 200-pA current injection, and a spontaneous firing frequency of 1–4 Hz (Chieng et al. 2011; Ford et al. 2006; Grace and Bunney 1984). Dopamine neuron identity was confirmed after experiments by labeling biocytin-filled neurons with streptavidin and costaining for tyrosine hydroxylase expression (Fig. 1). In all experiments, 10 µM NBQX was added to block AMPA receptor-mediated synaptic currents. VTA dopamine neurons were voltage-clamped at −60 mV except during high-frequency stimulation, when neurons were in current-clamp mode. Electrical IPSCs were stimulated with a 0.05-ms pulse using a bipolar stimulating electrode inserted into the VTA. Optically stimulated IPSCs were elicited by transmitting 473-nm light (2-ms pulse) through an optical fiber directed at the VTA in the slice at an acute angle to the recording chamber. IPSC stimulus intensities were adjusted to, and sampled at, a level that produced 50–75% of the maximum measurable evoked current to avoid postsynaptic receptor saturation. Electrically and optically stimulated IPSCs were evoked in every recording at a rate of 0.1 Hz at all times except during high-frequency stimulation. IPSC amplitudes were calculated by comparing the peak amplitude of the stimulus response with a baseline measurement just before stimulation. The peak amplitude was measured by sampling between 3 and 20 ms following stimulation. After a 10-min stable baseline was established, cyclic GMP-mediated LTP was induced by electrically stimulating afferents at 100 Hz for 1 s (high-frequency stimulation); this train was repeated twice with a 20-s interval. For adenylyl cyclase-mediated synaptic plasticity, forskolin was bath applied for 20 min at 20 µM after a 10-min stable baseline was established. IPSCs were continually evoked for up to 30 min postinduction. Series resistance was monitored online by measuring the peak of the capacitance transient in response to a 5-mV hyperpolarizing voltage step applied at the end of each sweep. Traces were discarded whenever the series resistance varied by more than 20%.
Fig. 1.

ChR2 expressed at GABA inputs to dopamine neurons. A: image of a coronal brain section expressing ChR2 in the nucleus accumbens (NAc; green). aca, Anterior commissure; cc, corpus callosum; CPu, caudate/putamen; D, dorsal; V, ventral; L, lateral; M, medial. B: image of a horizontal brain section expressing ChR2 in the ventral tegmental area (VTA; green). cp, Cerebral peduncle; IPF, interpeduncular fossa; ml, medial lemniscus; ns, nigrostriatal bundle; SNR, substantia nigra pars reticulata; R, rostral; C, caudal. Local VTA GABA neurons were targeted in Vgat-ires-Cre mice using injections of the rAAV5/EF1α-DIO-hChR2-(H134R)-mCherry viral construct. C: image of a horizontal brain section expressing ChR2 in the rostromedial tegmental nucleus (RMTg). RRF, retrorubral field; PPN, pedunculopontine nucleus; xscp, decussation of the superior cerebellar peduncle. D–G: filled neuron costained with tyrosine hydroxylase (arrows). D: horizontal midbrain section stained for tyrosine hydroxylase (TH; blue) with a recorded neuron (biocytin fill; red), with dashed inset reference for filled neuron in E–G. E: VTA neuron filled with biocytin during recording, labeled with streptavidin coupled to Alexa594. F: TH staining of slice with rabbit anti-TH primary and goat anti-rabbit secondary. G: merged image showing biocytin-filled neuron is costained with TH.
For measuring optically stimulated inhibition, cells with a minimum firing rate of 1.5 Hz were tested for innervation by ChR2-infected inputs by application of an optical stimulation train. If the stimulation train successfully inhibited the tonic activity of the cell, then the recording proceeded. The activity was recorded in a 10-s period. The first 5 s were a baseline recording of tonic firing. From 5 to 7 s, an optical stimulation train was applied to the cell. The cell remained undisturbed for the remaining 3 s, and then the protocol was repeated four more times. Trains that inhibited the dopamine neuron firing rate more than 50% were not further analyzed. Once baseline sampling of inhibition was complete, LTP induction was attempted by bath application of 100 µM 8-(4-chlorophenylthio) guanosine-39,59 monophosphate (8-pCPT-cGMP). Following induction, the inhibition from the optical stimulation trains were resampled and compared with baseline.
Immunofluorescence.
After recordings, slices with biocytin-filled cells were immersed in 4% paraformaldehyde for 1 h. The slices were then washed with PBS, blocked with 5% normal goat serum in PBS-TX-100 for 2 h at room temperature, and incubated in primary antibody for tyrosine hydroxylase (1:500; catalog no. AB152, Millipore; RRID:AB_390204) overnight at 4°C. The floating sections were then washed and incubated in secondary antibody and streptavidin (1:500, for visualizing biocytin) for 2 h at room temperature, washed, stained with DAPI, mounted to glass microscope slides with ProLong Gold Antifade, and covered with a coverslip. The mounting medium was allowed to cure for 24 h before imaging. Imaging was performed using a Zeiss 710 LSM confocal microscope with Zen 2011 software (Carl Zeiss, Oberkochen, Germany) or a Deltavision Personal DV system equipped with high numerical aperture objectives and a CoolSnap HQ2 cooled charge-coupled device camera (Applied Precision, Issaquah, WA). Images were prepared for publication using SoftWorx (Applied Precision), ImageJ (RRID:SCR_003070; Schneider et al. 2012), and Illustrator software (Adobe Systems; RRID:SCR_010279).
GABA/soluble guanylyl cyclase.
Mice were anaesthetized and perfused intracardially with ice-cold PBS followed by 4% paraformaldehyde. Brains were removed and immersed in ice-cold paraformaldehyde. After 2 h, the brains were incubated in 30% sucrose at 4°C until they sank (~48 h). The brains were then washed with PBS and embedded in O.C.T. The brains were cryosectioned to yield 50-μm parasagittal slices. The slices were then incubated as floating sections in a blocking solution containing 10% normal horse serum in 1× PBS, 0.5% Triton X-100, and 0.2% BSA for 1 h at room temperature. Slices were then incubated in primary antibody for soluble guanylyl cyclase (1:1,000; catalog no. G4280, Sigma; RRID:AB_259901), and primary antibody for GABA (1:750; catalog no. A0310, Sigma; RRID:AB_476667) overnight at 4°C in carrier solution containing 1% normal horse serum, 0.2% BSA, and 0.05% Triton X-100 in 1× PBS. The floating sections were then washed and incubated in secondary antibody (Alexa594, RRID:AB_2534095; and Alexa488, RRID:AB_138404) for 2 h at room temperature, washed, mounted to glass microscope slides with ProLong Gold Antifade, and covered with a coverslip. The mounting medium was allowed to cure for 24 h before imaging.
RMTg/soluble guanylyl cyclase.
For soluble guanylyl cyclase staining of RMTg neurons, a solution of fluorescent microspheres (100 nl, excitation/emission = 460/505 nm; Lumafluor) was injected into the VTA. After 3 days were allowed for retrograde trafficking, the mice were anesthetized and brains were prepared for sectioning as described above. The brains were cryosectioned to yield 100-µm parasagittal slices and were incubated in primary antibody for soluble guanylyl cyclase (1:1,000; catalog no. G4280, Sigma; RRID:AB_259901) overnight at 4°C, followed by incubation in secondary antibody (Alexa594, RRID:AB_2534095) for 2 h at room temperature. Slices were then washed and mounted as described above.
Chemicals.
All chemicals were obtained from Tocris Bioscience or Sigma-Aldrich unless otherwise stated.
Analysis and statistics.
Voltage- and current-clamp traces were analyzed offline using AxographX. Ratio paired t-tests and one-way ANOVA were performed using the software package GraphPad Prism (GraphPad Software, La Jolla, CA; RRID:SCR_002798). A P value of <0.05 was used as our significance level for all tests. All numerical data are expressed as the mean value ± the sample-corrected SE. Levels of LTP are reported as averaged IPSC amplitudes for 5 min before LTP induction compared with averaged IPSC amplitudes during the 5-min period from 15 to 20 min after induction. For agonist induction, the 5-min period from 15 to 20 min is compared with a 5-min predrug baseline.
RESULTS
NAc and VTA GABA neuron projections to VTA dopamine neurons express cyclic GMP-mediated LTP, but RMTg does not.
VTA dopamine neurons receive GABA input from a variety of nuclei, including local VTA GABA neurons, the RMTg, and the NAc. Intracranial infusion of adeno-associated virus containing ChR2 into each of these nuclei resulted in expression of ChR2 in cell bodies at their respective injection sites (Fig. 1). For VTA injections, GABA neurons were selectively targeted in Vgat-ires-Cre mice using injections of the rAAV5/EF1α-DIO-hChR2-(H134R)-mCherry viral construct. If fluorescent cell bodies were observed outside of the intended injection site, cells from that injected animal were not analyzed. The Cre-driven mCherry construct used in the VTA was never observed to be expressed in cell bodies or axons outside of the injection site; it was therefore deemed unsuitable for infection of long-range projections while also minimizing risk of contamination from spillage into RMTg.
For every recorded dopamine neuron, IPSCs were induced by a single 2-ms optical stimulation to elicit responses from identified inhibitory afferents. The decay time constants of the IPSCs elicited for all three input specific currents were comparable to the decay kinetics for GABAA receptors, with no significant differences detected among the inputs (F = 1.481, df = 4, 32, P = 0.231; one-way ANOVA, Tukey’s test). Cyclic GMP-dependent LTP was first induced using a 100-Hz electrical stimulation protocol (Nugent et al. 2007). A significant normalized fold change from initial baseline (final current amplitude/baseline current amplitude) was successfully induced by high-frequency stimulation (HFS) from only two of the three afferents studied. NAc-specific IPSCs were successfully potentiated (1.481 ± 0.05-fold change from baseline, P = 0.013, n = 6). IPSCs from VTA GABA neurons onto VTA dopamine neurons also expressed LTP (1.178 ± 0.01-fold change from baseline, P < 0.001, n = 10). However, RMTg-specific IPSCs showed no potentiation following induction by high-frequency electrical stimulation induction (0.999 ± 0.01-fold change from baseline, P = 0.977, n = 10) (Fig. 2). Although there appears to be a difference in the electrical HFS-induced mean fold change from baseline between NAc and VTA GABA, no significant difference was observed for NAc inputs vs. VTA GABA inputs (P = 0.361, Kruskal-Wallis test). However, the mean fold change from baseline observed for both NAc and VTA inputs was significantly larger than the lack of fold change observed from RMTg inputs (P < 0.0001 and P = 0.005, respectively; Kruskal-Wallis test, Dunn’s test). No correlation was observed between baseline photo-evoked current amplitude and post-plasticity induction fold change for electrical HFS (r = −0.334, P = 0.24, n = 16). The raw change in current (final current amplitude – baseline current amplitude) resulting from HFS plasticity induction was compared between NAc and VTA GABA-mediated currents. No significant differences were observed in the amount of raw current amplitude added by plasticity induction between these two inputs (P = 0.591, unpaired, Welch’s correction). To test whether plasticity expression was pre- or postsynaptic for electrical HFS, the coefficient of variation (CV) of peak current amplitude was measured for electrically evoked IPSCs before and after plasticity induction. The CV2 of synaptic current amplitude is negatively correlated with release probability (Yang and Calakos 2013). The 1/CV2 measurement for nonspecific electrical IPSCs was increased following electrical HFS (baseline: 60.22 ± 12.06, after HFS: 68.90 ± 11.55; P = 0.03, n = 18), suggesting that this plasticity mechanism is indeed mediated by a presynaptic increase in neurotransmitter release probability.
Fig. 2.

High-frequency electrical stimulation induces cyclic GMP-mediated LTP at a subset of optically stimulated, identified inputs. A: potentiation of NAc-specific IPSCs following high-frequency electrical stimulation (HFS; n = 6 cells, P = 0.013). B: potentiation of VTA GABA-specific IPSCs following HFS (n = 10 cells, P < 0.001). C: no potentiation available from RMTg-specific IPSCs following HFS (n = 10 cells, P = 0.977). Insets in A–C are representative input-specific traces before (Pre) and after (Post) induction of LTP. Data are means ± SE. D: no correlation was observed between baseline photo-evoked current amplitude and the current fold change (final current amplitude/baseline current amplitude) following LTP induction by electrical HFS (r = −0.334, P = 0.24, n = 16). E: no significant differences were observed in the raw current amplitude increase (final current amplitude − baseline current amplitude) induced by LTP between NAc and VTA GABA inputs (P = 0.591, unpaired, Welch’s correction). F: the mean 1/CV2 measurement for electrical IPSC amplitudes was increased following electrical HFS (baseline mean: 60.22 ± 12.06; mean after HFS: 68.90 ± 11.55; P = 0.03, n = 18), suggesting a presynaptic increase in neurotransmitter release probability.
Electrically stimulated IPSCs express cyclic GMP-mediated LTP in the same recordings, whereas optically stimulated IPSCs of RMTg afferents do not.
To investigate the lack of LTP from RMTg inputs, electrical IPSCs were additionally recorded during the same recordings from a subset of neurons with RMTg-specific IPSCs. For these dopamine neurons, electrical IPSCs were induced by a single 0.05-ms electrical pulse 1 s after the 2-ms optical stimulation to elicit responses from nonspecific and identified RMTg afferents, respectively. Again, RMTg-specific IPSCs showed no potentiation following LTP induction (0.982 ± 0.02-fold change from baseline, P = 0.375, n = 5), even though electrical IPSCs recorded from the same neurons successfully exhibited LTP (1.285 ± 0.02-fold change from baseline, P = 0.002, n = 5; Fig. 3).
Fig. 3.
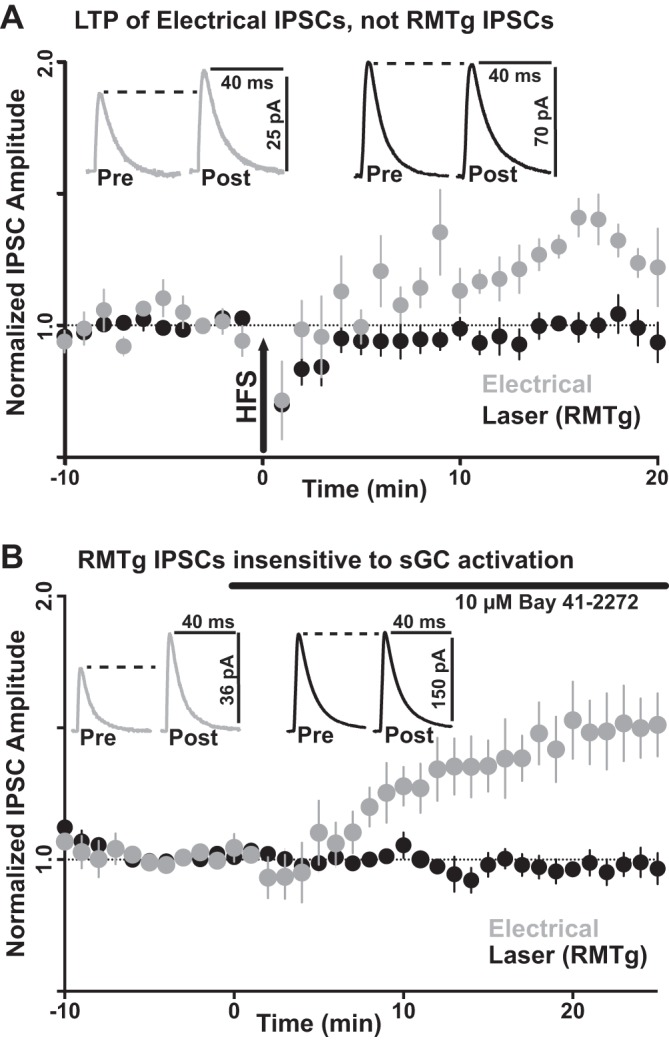
LTP Induction of nonspecific electrical and identified RMTg-specific optical IPSCs. A: electrical induction. RMTg-specific IPSCs showed no potentiation following induction by high-frequency stimulation (HFS; n = 5 cells, P = 0.375), even though paired electrical IPSCs during the same recordings successfully expressed LTP (n = 5 cells, P = 0.002). B: agonist induction. RMTg-specific IPSCs showed no potentiation following bath application of an activator (BAY 41-2272; 10 µM) for the enzyme in the synthesis of GTP to cyclic GMP, soluble guanylyl cyclase (sGC; n = 11 cells, P = 0.227). However, unpaired electrically induced IPSCs successfully expressed LTP (n = 10 cells, P = 0.001).
One possibility for the lack of LTP at RMTg-specific currents would be insufficient retrograde activation of soluble guanylyl cyclase at presynaptic terminals during high-frequency electrical stimulation. To test for the availability of soluble guanylyl cyclase, the specific activator BAY 41-2272 (10 µM) was bath applied to both nonspecific electrical and RMTg-specific optical IPSCs. As with the 100-Hz electrical induction, RMTg-specific IPSCs showed no potentiation following bath application of BAY 41-2272 (0.927 ± 0.03-fold change from baseline, P = 0.227, n = 11). However, as with HFS, electrical IPSCs successfully exhibited LTP following bath application of BAY 41-2272 (1.457 ± 0.04-fold change from baseline, P = 0.001, n = 10).
RMTg neurons express soluble guanylyl cyclase.
Some critical deficit renders RMTg inputs insensitive to potentiation by HFS or bath application of soluble guanylyl cyclase activator BAY 41-2272. We hypothesized that the cell bodies of inputs lacking cGMP-mediated LTP would stain negative for the soluble guanylyl cyclase enzyme required for induction. Because soluble guanylyl cyclase is a cytosolic protein, a neuron’s cell body should positively label if that neuron expresses soluble guanylyl cyclase at all. We stained parasagittal brain slices with antibodies for GABA and soluble guanylyl cyclase. Counter to our predictions, we observed that the proportion of GABA-positive cells that also costained with soluble guanylyl cyclase in the RMTg was not significantly different from that in the VTA (RMTg: 50.99 ± 5.47%; VTA: 51.46 ± 2.42%; n = 3 animals, P = 0.942). To isolate and stain neurons specifically projecting from the RMTg to VTA, green fluorescent microspheres were injected into the VTA to retrogradely label the RMTg. Three days after injection, brain sections were stained for soluble guanylyl cyclase. Within the RMTg, 24.1 ± 2.52% of the retrogradely labeled neurons were observed to express soluble guanylyl cyclase (n = 5 animals; Fig. 4).
Fig. 4.

Soluble guanylyl cyclase expression levels in RMTg neurons projecting to VTA. A: mouse brain slices (50 µm) were costained with antibodies for GABA and soluble guanylyl cyclase. RMTg inset refers to images in B–D. VTA inset refers to images in E–G. The proportion of GABA-positive cells that also costained with soluble guanylyl cyclase in the RMTg was not significantly different from that in the VTA (RMTg: 50.99 ± 5.47%; VTA: 51.46 ± 2.42%; n = 3 animals, P = 0.942). B and E: images (×20 magnification) of GABA staining in the RMTg and VTA, respectively. C and F: images (×20 magnification) of soluble guanylyl cyclase staining in the RMTg and VTA, respectively. D and G: merged images of GABA and soluble guanylyl cyclase staining for the RMTg and VTA, respectively. H: mice were injected with fluorescent microspheres (100 nl) into the VTA. RMTg is defined as the retrogradely labeled region dorsal and caudal to the VTA. RMTg inset refers to images in I–K. Within the RMTg, only 24.1 ± 2.52% of the retrogradely labeled neurons were observed to express soluble guanylyl cyclase (n = 5 animals). I: images (×40 magnification) of retrogradely labeled neurons in the RMTg. J: images (×40 magnification) of soluble guanylyl cyclase staining in the RMTg. K: merged images of retrogradely labeled neurons with soluble guanylyl cyclase staining for the RMTg.
Direct addition of cyclic GMP analog induces LTP capable of inducing a pause dopamine neuron firing.
Even if an undetected decrease in soluble guanylyl cyclase levels was preventing the expression of LTP in RMTg afferents, circumventing that step by adding the product of soluble guanylyl cyclase activity should elicit LTP. We attempted to induce LTP in all three afferents (RMTg, NAc, and local VTA GABA neurons) by bath application of an analog of the subsequent byproduct of soluble guanylyl cyclase activation, cyclic GMP. Additionally, there remained to be seen whether an input would be more effective at inhibiting tonic firing following LTP. Therefore, cells were also recorded in current clamp. Those cells with a minimum firing rate of 1.5 Hz were tested for innervation by ChR2-infected inputs by optical stimulation delivered at half-maximal laser intensity. If the optical stimulation successfully slowed the tonic activity of the cell, we then proceeded with the recording. We recorded firing activity during a 10-s period. From 5 to 7 s, a 20-Hz optical stimulation train was applied to the cell. Trains that inhibited the dopamine neuron firing rate more than 50% were not further analyzed. Once baseline sampling of inhibition was complete, we attempted LTP induction by bath application of 100 µM 8-pCPT-cGMP (15–20 min). After induction, the inhibition from the optical stimulation trains were recorded and compared with baseline (Fig. 5).
Fig. 5.

Cyclic GMP-mediated LTP enhancement of inhibition. A: representative traces of tonic firing inhibition (20-Hz optical stimulation train for 2 s) before (top; pre LTP) and after (bottom; post LTP) LTP induction at NAc afferents by bath application of the cyclic GMP analog 8-pCPT-cGMP (100 µM). B: representative traces of tonic firing inhibition (20-Hz optical stimulation train for 2 s) before (top; pre LTP) and after (bottom; post LTP) LTP induction at GABA neurons within the VTA that project to VTA dopamine neurons by bath application of the cyclic GMP analog 8-pCPT-cGMP (100 µM). C: representative traces of tonic firing inhibition (20-Hz optical stimulation train for 2 s) before (top; pre LTP) and after (bottom; post LTP) LTP induction at RMTg afferents by bath application of the cyclic GMP analog 8-pCPT-cGMP (100 µM). D: summary of input-specific inhibition. After LTP induction at NAc inputs, optically induced inhibition of firing was potentiated (n = 6 cells, *P = 0.014). From VTA GABA inputs, inhibition of firing rate was also facilitated (n = 7 cells, **P = 0.004). However, no change in inhibition was observed from RMTg-specific inputs following bath application of the cyclic GMP analog 8-pCPT-cGMP (n = 5 cells, P = 0.845). Data are medians with each data point plotted.
After LTP induction at NAc inputs, inhibition of firing rate increased from 22.84 ± 7.50% to 69.53 ± 19.30% (P = 0.014, n = 6) of background firing. From VTA GABA inputs, inhibition of firing rate increased from 27.71 ± 6.42% to 64.22 ± 13.18% (P = 0.004, n = 7). Finally, as with the other cyclic GMP-dependent induction protocols, no change in inhibition was observed from identified RMTg inputs following attempted LTP induction with 8-pCPT-cGMP (from 21.70 ± 3.00% to 20.87 ± 2.01%, P = 0.845, n = 5), indicating that the lack of LTP expression at RMTg afferents is not due to reduced expression of soluble guanylyl cyclase.
VTA GABA neuron, NAc, and RMTg afferents express adenylyl cyclase-mediated potentiation of IPSCs.
We also measured normalized fold changes in IPSC baselines induced by activation of the adenylyl cyclase-mediated pathway to determine whether RMTg afferents were capable of expressing any form of plasticity. We used the adenylyl cyclase agonist forskolin to induce facilitation of IPSCs from identified local VTA GABA neurons and from NAc- and RMTg-specific inputs (Melis et al. 2002). Forskolin was bath applied for 20 min at 20 μM after a 10-min stable baseline was established. Unlike activation of cyclic GMP synthesis, activation of adenylyl cyclase successfully induced enhancement of RMTg-specific IPSCs (1.824 ± 0.04-fold change from baseline, P = 0.001, n = 5; Fig. 6). Similar to the effects on RMTg-specific IPSCs, activation of adenylyl cyclase with forskolin also increased the amplitude of IPSCs elicited from optical stimulation of local GABA neurons within the VTA (1.457 ± 0.036 of baseline, P = 0.005, n = 5) and afferents originating from NAc (1.798 ± 0.053 of baseline, P = 0.004, n = 5). No correlation was observed between baseline photo-evoked current amplitude and post-plasticity induction fold change for forskolin bath application (r = −0.472, P = 0.10, n = 15). The raw change in current resulting from forskolin bath application was compared between all three input-specific photo-evoked currents. No significant differences were observed in the amount of raw current amplitude added by plasticity induction between these three inputs (F = 1.849, df = 2, 12, P = 0.257; one-way ANOVA, Tukey’s test). To test for the presynaptic or postsynaptic location of plasticity expression following forskolin bath application, the CV of peak current amplitudes were measured for electrically evoked IPSCs before and after plasticity induction. The mean 1/CV2 measurement for nonspecific electrical IPSC amplitudes was increased following forskolin bath application (baseline: 87.59 ± 24.46; after forskolin: 140.4 ± 33.99; P = 0.02, n = 14), suggesting that this plasticity mechanism is indeed mediated by a presynaptic increase in neurotransmitter release probability.
Fig. 6.

Bath-applied forskolin-mediated potentiation is expressed by all 3 identified inputs. A: optically stimulated, identified IPSCs were evoked from NAc before and after bath application of an activator of adenylyl cyclase (forskolin; 20 µM). Forskolin induced potentiation of NAc-specific IPSCs (n = 5 cells, P = 0.004). B: optically stimulated, identified IPSCs were evoked from GABA neurons within the VTA that project to VTA dopamine neurons before and after bath application of an activator (forskolin; 20 µM) of the enzyme for the conversion of ATP to cyclic AMP, adenylyl cyclase. Forskolin bath application induced potentiation of VTA GABA-specific IPSCs (n = 5 cells, P = 0.005). C: unlike the cyclic GMP-dependent pathway, activation of the cyclic AMP-dependent pathway with forskolin bath application induced an increase of identified RMTg-specific IPSC amplitudes (n = 5 cells, P = 0.001). Data in A–C are means ± SE. D: no correlation was observed between baseline photo-evoked current amplitude and post-plasticity induction fold change (final current amplitude/baseline current amplitude) for forskolin bath application (r = −0.472, P = 0.10, n = 15). E: no significant differences were observed in the raw current amplitude increase (final current amplitude − baseline current amplitude) added by plasticity induction between these 3 inputs (F = 1.849, df = 2, 12, P = 0.257; one-way ANOVA, Tukey’s test). F: the mean 1/CV2 measurement for nonspecific electrical IPSC amplitudes was increased following forskolin bath application (baseline: 87.59 ± 24.46; after forskolin: 140.4 ± 33.99; P = 0.02, n = 14), suggesting that this plasticity mechanism is mediated by a presynaptic increase in neurotransmitter release probability.
DISCUSSION
The ability to learn how to secure biologically rewarding resources and avoid aversive threats is critical to the survival of any organism. Plasticity of inputs to dopamine neurons is needed for the ability to both store learned patterns of reward presentation and revise that stored information as necessary (Schultz et al. 1997). Although there have been many breakthroughs in understanding reward learning by studying changes in excitatory inputs to dopamine neurons (Kauer and Malenka 2007), more still remains to be learned from the relatively understudied plasticity of GABA inputs.
GABA afferents control the activity of VTA dopamine neurons and behavior.
In behaving animals, a pause in dopamine neuron firing signals the absence of an expected reward (Schultz 2002). A candidate mechanism for pauses is IPSCs generated by GABA synaptic afferents (Lobb et al. 2010; Paladini et al. 1999a). In addition to opposing tonic firing, activation of GABAA receptors can also suppress burst firing in dopamine neurons (Lobb et al. 2011; Paladini et al. 1999b). This suggests that plasticity of GABA synapses on dopamine neurons may be important for gating phasic bursts and inducing pauses of spontaneous firing. All GABA inputs to dopamine neurons are not necessarily equivalent in function. It has been demonstrated that cGMP- and forskolin/cAMP-mediated plasticity are distinct parallel pathways. Blocking either forskolin/cAMP- or cGMP-mediated plasticity induction upon nonspecific electrical IPSCs does not occlude induction of the other. However, expression of one does occlude expression of the other, suggesting convergence on a common downstream effector (Nugent et al. 2009). In this report we show evidence that three inhibitory inputs to VTA dopamine neurons releasing the same neurotransmitter are differentially modified by these two distinct plasticity mechanisms.
The NAc provides inhibition of dopamine neurons that is reciprocal to its innervation by the VTA reward circuit (Rahman and McBride 2000); our findings suggest that the gain of this feedback is modulated by both adenylyl cyclase- and guanylyl cyclase-mediated physiological signals. Optogenetic stimulation of VTA GABA neurons is known to disrupt reward seeking and consumption and to decrease the excitability of VTA dopamine neurons (Tan et al. 2012; van Zessen et al. 2012), suggesting that potentiation of these neurons’ GABA signaling could have profound consequences for the initiation and/or termination of reward-related behaviors. Therefore, it is perhaps not surprising to observe plasticity at VTA GABA inputs through the adenylyl cyclase- and cyclic GMP-mediated pathways. Acute stress disables nitric oxide-mediated (cyclic GMP dependent) LTP, which in turn facilitates reinstatement of drug seeking behavior in rodents (Graziane et al. 2013; Niehaus et al. 2010). In vivo exposure to ethanol induces adenylyl cyclase-mediated (cyclic AMP dependent) LTP and is associated with increased alcohol consumption (Camarini and Hodge 2004; Melis et al. 2002; Spanagel and Weiss 1999). The RMTg supplies potent inhibition of dopamine neurons (Jhou et al. 2009b). It provides aversive reward salience information to the circuit and directly regulates phasic firing activity (Jhou et al. 2009a). Stimulating the RMTg in vivo is already sufficient to induce pauses in dopamine neurons without synaptic plasticity (Jhou et al. 2013). However, RMTg inputs do not exhibit cyclic GMP-dependent LTP, suggesting that plasticity of this input would be insensitive to acute in vivo stress. Only adenylyl cyclase-mediated synaptic facilitation was readily available at RMTg inputs.
Exposure to drugs of abuse has been shown to induce diverse cyclic AMP / PKA-mediated changes in cellular function and gene expression in brain structures such as the NAc, where these adaptations are suggested to underlie psychological aspects of drug addiction (Dohrman et al. 1996; Nestler 2016; Terwilliger et al. 1991). In vivo cocaine exposure, in addition to disabling potentiation of direct inhibition of VTA dopamine neurons, was also previously shown to potentiate inhibitory projections from the NAc to local VTA GABA neurons (Bocklisch et al. 2013). This PKA-mediated indirect disinhibition of dopamine, paired with PKG-mediated disabling of direct inhibition of dopamine, likely acts in concert to promote maladaptive behaviors following cocaine exposure. Local infusion of PKA activators and inhibitors into the NAc revealed a cyclic AMP-dependent pathway whose activation blunts rewarding effects of opiates and cocaine (Barrot et al. 2002; Carlezon et al. 1998; Miserendino and Nestler 1995), drives increased self-administration (Larson et al. 2011), and increases the excitability of NAc spiny neurons (Dong et al. 2006). Activation of this cyclic AMP signaling pathway within the NAc is thought to play a role in mediating drug tolerance and dependence (Nestler 2016). Within the VTA (before the distinction of the “tail of the VTA” as a discrete nucleus), exposure to drugs of abuse was shown to have profound influences on GABA signaling to dopamine neurons. Cocaine and morphine exposure was found to cause D1 receptor activation to diminish GABAB receptor-mediated IPSP amplitudes, instead of normally augmenting them. Disruption of cyclic AMP metabolism blocked this inhibition (Bonci and Williams 1996). Morphine exposure was also found to enhance GABAA IPSCs recorded from dopamine neurons within the VTA, and additionally sensitized them to both facilitation by adenylyl cyclase agonists and inhibition by PKA antagonists (Bonci and Williams 1997). The finding that the RMTg is recruited by ethanol exposure/adenylyl cyclase-mediated enhancement of GABA IPSCs coincides with previous evidence that this structure is also significantly influenced by exposure to drugs of abuse (Bourdy and Barrot 2012; Kaufling et al. 2010b). That the sign of the GABA signaling modulation varies across different drugs studied (Lecca et al. 2011), however, suggests a complicated interplay between the circuits processing reward and aversion signals. Our current finding that the diversity of GABAergic pathways into the VTA are heterogeneous in their expression of synaptic plasticity reveals a need for an input specific strategy for disentangling this complexity.
Lack of GMP-mediated LTP from RMTg is not due to decreased levels of soluble guanylyl cyclase.
Because expression of soluble guanylyl cyclase, the target of nitric oxide signaling, is required of any nucleus expressing cyclic GMP-mediated LTP, it follows that expression of soluble guanylyl cyclase in the cell body of an input might correlate with plasticity from that input’s synapses. In situ hybridization and RT-PCR studies have shown that the NAc, an input we have shown to be positive for cyclic GMP-mediated LTP, is also positive for soluble guanylyl cyclase (Gibb and Garthwaite 2001; Matsuoka et al. 1992). Therefore, the RMTg should be negative for expression of soluble guanylyl cyclase, because no plasticity is observed at these inputs by either HFS or bath application of a soluble guanylyl cyclase activator (BAY 41-2272). However, our findings show that GABA-positive neurons within the RMTg show equivalent colabeling for soluble guanylyl cyclase to GABA-positive neurons within the VTA. This result would suggest that there is no correlation between soluble guanylyl cyclase expression and cGMP-mediated plasticity.
In an additional assay, VTA neurons were targeted with fluorescent retrograde microspheres, and retrogradely labeled RMTg neurons were stained for soluble guanylyl cyclase. RMTg neurons retrogradely labeled from VTA were found to have low levels in coexpression of soluble guanylyl cyclase, but still not near zero as we predicted. Finally, we went one step beyond soluble guanylyl cyclase activation by bath applying the cyclic GMP analog 8-pCPT-cGMP (100 µM) on all three identified inputs to discern what effect cyclic GMP-dependent potentiation could have on input-specific GABA inhibition of dopamine neuron firing. Because bypassing the expression of soluble guanylyl cyclase did not rescue facilitation of RMTG inputs, as evidenced by failure to enhance their inhibition of VTA dopamine neurons, our findings suggest that the critical deficiency in this signaling pathway must instead lie downstream of cyclic GMP within the RMTg. These results highlight the possibility that GABA neurons within the VTA and those within RMTg constitute different populations, at least with respect to LTP induction dependent on the conversion of GTP to cyclic GMP.
Heterogeneous LTP expression among GABA inputs to VTA dopamine neurons.
We have shown that GABA inputs from the NAc and the VTA can be potentiated from a physiological signal that does not recruit the RMTg. Cyclic GMP-mediated LTP, induced by either high-frequency electrical stimulation or agonist bath application, was found at inputs from the NAc and VTA GABA neurons, but not from the RMTg. However, the RMTg, together with the NAc and VTA GABA neurons, was found to be plastic under an alternative, adenylyl cyclase-mediated synaptic enhancement induction protocol, suggesting a potential modulation by in vivo ethanol exposure (Camarini and Hodge 2004; Melis et al. 2002; Spanagel and Weiss 1999). Taking these findings together, we conclude that discrete plasticity mechanisms for modulating GABA-mediated inhibition of VTA dopamine neurons act on diverse subsets of GABA inputs. The convergence of GABA inputs on dopamine neurons from a diversity of sources likely represents a multitude of circuits modulating dopamine neuron activity, with varying degrees of overlap in their pathways. There are other structures that send GABA inputs to VTA DA neurons. The bed nucleus of stria terminalis (BNST), for example, sends both glutamatergic and GABAergic projections to VTA neurons, in vivo stimulation of which produces aversive/anxiogenic and rewarding/anxiolytic behavioral phenotypes, respectively (Jennings et al. 2013). These functionally opposed BNST inputs could likely express cGMP-mediated LTP to serve as a heterosynaptic gain equilibration of salient reward signal integration, when not occluded by stress or drugs of abuse. Input-specific investigation, however, will be required to confirm this hypothesis until future studies can reveal a predictive marker for LTP expression. Differences in projection target may also impact expression of inhibitory plasticity. Mild physical and psychological stressors have been shown to elevate dopamine release in the medial prefrontal cortex while not significantly elevating dopamine release in the NAc or dorsal striatum (Herman et al. 1982; Roth et al. 1988).These data suggest that medial prefrontal cortex projecting dopamine neurons are likely more susceptible and sensitive to stress-induced modulation of inhibitory plasticity. Unraveling the heterogeneous expression of synaptic plasticity among GABA inputs to VTA dopamine neurons is an important step forward in understanding the role of GABA inhibition in shaping reward prediction error calculations that organisms make in adapting to their experience and environment.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
GRANTS
This work was supported by National Institutes of Health (NIH) Grants MH107229, DA030530, and DA038453 (to C. A. Paladini), and GM060655 (to D. V. Simmons), and by NIH Research Centers at Minority Institutions Grant G12MD007591 (University of Texas at San Antonio.
AUTHOR CONTRIBUTIONS
D.V.S. and C.A.P. conceived and designed research; D.V.S. and A.K.P. performed experiments; D.V.S. analyzed data; D.V.S. and C.A.P. interpreted results of experiments; D.V.S. prepared figures; D.V.S. and A.K.P. drafted manuscript; D.V.S., A.K.P., and C.A.P. edited and revised manuscript; C.A.P. approved final version of manuscript.
REFERENCES
- Barrot M, Olivier JD, Perrotti LI, DiLeone RJ, Berton O, Eisch AJ, Impey S, Storm DR, Neve RL, Yin JC, Zachariou V, Nestler EJ. CREB activity in the nucleus accumbens shell controls gating of behavioral responses to emotional stimuli. Proc Natl Acad Sci USA 99: 11435–11440, 2002. doi: 10.1073/pnas.172091899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocklisch C, Pascoli V, Wong JC, House DR, Yvon C, de Roo M, Tan KR, Lüscher C. Cocaine disinhibits dopamine neurons by potentiation of GABA transmission in the ventral tegmental area. Science 341: 1521–1525, 2013. doi: 10.1126/science.1237059. [DOI] [PubMed] [Google Scholar]
- Bonci A, Williams JT. A common mechanism mediates long-term changes in synaptic transmission after chronic cocaine and morphine. Neuron 16: 631–639, 1996. doi: 10.1016/S0896-6273(00)80082-3. [DOI] [PubMed] [Google Scholar]
- Bonci A, Williams JT. Increased probability of GABA release during withdrawal from morphine. J Neurosci 17: 796–803, 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdy R, Barrot M. A new control center for dopaminergic systems: pulling the VTA by the tail. Trends Neurosci 35: 681–690, 2012. doi: 10.1016/j.tins.2012.06.007. [DOI] [PubMed] [Google Scholar]
- Camarini R, Hodge CW. Ethanol preexposure increases ethanol self-administration in C57BL/6J and DBA/2J mice. Pharmacol Biochem Behav 79: 623–632, 2004. doi: 10.1016/j.pbb.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Carlezon WA Jr, Thome J, Olson VG, Lane-Ladd SB, Brodkin ES, Hiroi N, Duman RS, Neve RL, Nestler EJ. Regulation of cocaine reward by CREB. Science 282: 2272–2275, 1998. doi: 10.1126/science.282.5397.2272. [DOI] [PubMed] [Google Scholar]
- Chieng B, Azriel Y, Mohammadi S, Christie MJ. Distinct cellular properties of identified dopaminergic and GABAergic neurons in the mouse ventral tegmental area. J Physiol 589: 3775–3787, 2011. doi: 10.1113/jphysiol.2011.210807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohrman DP, Diamond I, Gordon AS. Ethanol causes translocation of cAMP-dependent protein kinase catalytic subunit to the nucleus. Proc Natl Acad Sci USA 93: 10217–10221, 1996. doi: 10.1073/pnas.93.19.10217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Green T, Saal D, Marie H, Neve R, Nestler EJ, Malenka RC. CREB modulates excitability of nucleus accumbens neurons. Nat Neurosci 9: 475–477, 2006. doi: 10.1038/nn1661. [DOI] [PubMed] [Google Scholar]
- Einhorn LC, Johansen PA, White FJ. Electrophysiological effects of cocaine in the mesoaccumbens dopamine system: studies in the ventral tegmental area. J Neurosci 8: 100–112, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CP, Mark GP, Williams JT. Properties and opioid inhibition of mesolimbic dopamine neurons vary according to target location. J Neurosci 26: 2788–2797, 2006. doi: 10.1523/JNEUROSCI.4331-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibb BJ, Garthwaite J. Subunits of the nitric oxide receptor, soluble guanylyl cyclase, expressed in rat brain. Eur J Neurosci 13: 539–544, 2001. doi: 10.1046/j.1460-9568.2001.01421.x. [DOI] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: single spike firing. J Neurosci 4: 2866–2876, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graziane NM, Polter AM, Briand LA, Pierce RC, Kauer JA. Kappa opioid receptors regulate stress-induced cocaine seeking and synaptic plasticity. Neuron 77: 942–954, 2013. doi: 10.1016/j.neuron.2012.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman JP, Guillonneau D, Dantzer R, Scatton B, Semerdjian-Rouquier L, Le Moal M. Differential effects of inescapable footshocks and of stimuli previously paired with inescapable footshocks on dopamine turnover in cortical and limbic areas of the rat. Life Sci 30: 2207–2214, 1982. doi: 10.1016/0024-3205(82)90295-8. [DOI] [PubMed] [Google Scholar]
- Jennings JH, Sparta DR, Stamatakis AM, Ung RL, Pleil KE, Kash TL, Stuber GD. Distinct extended amygdala circuits for divergent motivational states. Nature 496: 224–228, 2013. doi: 10.1038/nature12041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhou TC, Fields HL, Baxter MG, Saper CB, Holland PC. The rostromedial tegmental nucleus (RMTg), a GABAergic afferent to midbrain dopamine neurons, encodes aversive stimuli and inhibits motor responses. Neuron 61: 786–800, 2009a. doi: 10.1016/j.neuron.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhou TC, Geisler S, Marinelli M, Degarmo BA, Zahm DS. The mesopontine rostromedial tegmental nucleus: a structure targeted by the lateral habenula that projects to the ventral tegmental area of Tsai and substantia nigra compacta. J Comp Neurol 513: 566–596, 2009b. doi: 10.1002/cne.21891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhou TC, Good CH, Rowley CS, Xu SP, Wang H, Burnham NW, Hoffman AF, Lupica CR, Ikemoto S. Cocaine drives aversive conditioning via delayed activation of dopamine-responsive habenular and midbrain pathways. J Neurosci 33: 7501–7512, 2013. doi: 10.1523/JNEUROSCI.3634-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X, Costa RM. Start/stop signals emerge in nigrostriatal circuits during sequence learning. Nature 466: 457–462, 2010. doi: 10.1038/nature09263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SW, North RA. Two types of neurone in the rat ventral tegmental area and their synaptic inputs. J Physiol 450: 455–468, 1992. doi: 10.1113/jphysiol.1992.sp019136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nat Rev Neurosci 8: 844–858, 2007. doi: 10.1038/nrn2234. [DOI] [PubMed] [Google Scholar]
- Kaufling J, Veinante P, Pawlowski SA, Freund-Mercier MJ, Barrot M. γ-Aminobutyric acid cells with cocaine-induced ΔFosB in the ventral tegmental area innervate mesolimbic neurons. Biol Psychiatry 67: 88–92, 2010a. doi: 10.1016/j.biopsych.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Kaufling J, Waltisperger E, Bourdy R, Valera A, Veinante P, Freund-Mercier MJ, Barrot M. Pharmacological recruitment of the GABAergic tail of the ventral tegmental area by acute drug exposure. Br J Pharmacol 161: 1677–1691, 2010b. doi: 10.1111/j.1476-5381.2010.00984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson EB, Graham DL, Arzaga RR, Buzin N, Webb J, Green TA, Bass CE, Neve RL, Terwilliger EF, Nestler EJ, Self DW. Overexpression of CREB in the nucleus accumbens shell increases cocaine reinforcement in self-administering rats. J Neurosci 31: 16447–16457, 2011. doi: 10.1523/JNEUROSCI.3070-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecca S, Melis M, Luchicchi A, Ennas MG, Castelli MP, Muntoni AL, Pistis M. Effects of drugs of abuse on putative rostromedial tegmental neurons, inhibitory afferents to midbrain dopamine cells. Neuropsychopharmacology 36: 589–602, 2011. doi: 10.1038/npp.2010.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobb CJ, Wilson CJ, Paladini CA. A dynamic role for GABA receptors on the firing pattern of midbrain dopaminergic neurons. J Neurophysiol 104: 403–413, 2010. doi: 10.1152/jn.00204.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobb CJ, Wilson CJ, Paladini CA. High-frequency, short-latency disinhibition bursting of midbrain dopaminergic neurons. J Neurophysiol 105: 2501–2511, 2011. doi: 10.1152/jn.01076.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka I, Giuili G, Poyard M, Stengel D, Parma J, Guellaen G, Hanoune J. Localization of adenylyl and guanylyl cyclase in rat brain by in situ hybridization: comparison with calmodulin mRNA distribution. J Neurosci 12: 3350–3360, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melis M, Camarini R, Ungless MA, Bonci A. Long-lasting potentiation of GABAergic synapses in dopamine neurons after a single in vivo ethanol exposure. J Neurosci 22: 2074–2082, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miserendino MJ, Nestler EJ. Behavioral sensitization to cocaine: modulation by the cyclic AMP system in the nucleus accumbens. Brain Res 674: 299–306, 1995. doi: 10.1016/0006-8993(95)00030-T. [DOI] [PubMed] [Google Scholar]
- Nauta WJ, Smith GP, Faull RL, Domesick VB. Efferent connections and nigral afferents of the nucleus accumbens septi in the rat. Neuroscience 3: 385–401, 1978. doi: 10.1016/0306-4522(78)90041-6. [DOI] [PubMed] [Google Scholar]
- Nestler EJ. Reflections on: “A general role for adaptations in G-Proteins and the cyclic AMP system in mediating the chronic actions of morphine and cocaine on neuronal function”. Brain Res 1645: 71–74, 2016. doi: 10.1016/j.brainres.2015.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehaus JL, Murali M, Kauer JA. Drugs of abuse and stress impair LTP at inhibitory synapses in the ventral tegmental area. Eur J Neurosci 32: 108–117, 2010. doi: 10.1111/j.1460-9568.2010.07256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent FS, Niehaus JL, Kauer JA. PKG and PKA signaling in LTP at GABAergic synapses. Neuropsychopharmacology 34: 1829–1842, 2009. doi: 10.1038/npp.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nugent FS, Penick EC, Kauer JA. Opioids block long-term potentiation of inhibitory synapses. Nature 446: 1086–1090, 2007. doi: 10.1038/nature05726. [DOI] [PubMed] [Google Scholar]
- Paladini CA, Celada P, Tepper JM. Striatal, pallidal, and pars reticulata evoked inhibition of nigrostriatal dopaminergic neurons is mediated by GABAA receptors in vivo. Neuroscience 89: 799–812, 1999a. doi: 10.1016/S0306-4522(98)00355-8. [DOI] [PubMed] [Google Scholar]
- Paladini CA, Iribe Y, Tepper JM. GABAA receptor stimulation blocks NMDA-induced bursting of dopaminergic neurons in vitro by decreasing input resistance. Brain Res 832: 145–151, 1999b. doi: 10.1016/S0006-8993(99)01484-5. [DOI] [PubMed] [Google Scholar]
- Paladini CA, Tepper JM. GABA(A) and GABA(B) antagonists differentially affect the firing pattern of substantia nigra dopaminergic neurons in vivo. Synapse 32: 165–176, 1999. doi:. [DOI] [PubMed] [Google Scholar]
- Rahman S, McBride WJ. Feedback control of mesolimbic somatodendritic dopamine release in rat brain. J Neurochem 74: 684–692, 2000. doi: 10.1046/j.1471-4159.2000.740684.x. [DOI] [PubMed] [Google Scholar]
- Roth RH, Tam SY, Ida Y, Yang JX, Deutch AY. Stress and the mesocorticolimbic dopamine systems. Ann N Y Acad Sci 537: 138–147, 1988. doi: 10.1111/j.1749-6632.1988.tb42102.x. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9: 671–675, 2012. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz W. Getting formal with dopamine and reward. Neuron 36: 241–263, 2002. doi: 10.1016/S0896-6273(02)00967-4. [DOI] [PubMed] [Google Scholar]
- Schultz W, Dayan P, Montague PR. A neural substrate of prediction and reward. Science 275: 1593–1599, 1997. doi: 10.1126/science.275.5306.1593. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Weiss F. The dopamine hypothesis of reward: past and current status. Trends Neurosci 22: 521–527, 1999. doi: 10.1016/S0166-2236(99)01447-2. [DOI] [PubMed] [Google Scholar]
- Tan KR, Yvon C, Turiault M, Mirzabekov JJ, Doehner J, Labouèbe G, Deisseroth K, Tye KM, Lüscher C. GABA neurons of the VTA drive conditioned place aversion. Neuron 73: 1173–1183, 2012. doi: 10.1016/j.neuron.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger RZ, Beitner-Johnson D, Sevarino KA, Crain SM, Nestler EJ. A general role for adaptations in G-proteins and the cyclic AMP system in mediating the chronic actions of morphine and cocaine on neuronal function. Brain Res 548: 100–110, 1991. doi: 10.1016/0006-8993(91)91111-D. [DOI] [PubMed] [Google Scholar]
- van Zessen R, Phillips JL, Budygin EA, Stuber GD. Activation of VTA GABA neurons disrupts reward consumption. Neuron 73: 1184–1194, 2012. doi: 10.1016/j.neuron.2012.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watabe-Uchida M, Zhu L, Ogawa SK, Vamanrao A, Uchida N. Whole-brain mapping of direct inputs to midbrain dopamine neurons. Neuron 74: 858–873, 2012. doi: 10.1016/j.neuron.2012.03.017. [DOI] [PubMed] [Google Scholar]
- Yang Y, Calakos N. Presynaptic long-term plasticity. Front Synaptic Neurosci 5: 8, 2013. doi: 10.3389/fnsyn.2013.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]