

ABSTRACT
The laboratory strain of Pseudomonas aeruginosa, PAO1, activates genes for catabolism of adenosine using quorum sensing (QS). However, this strain is not well-adapted for growth on adenosine, with doubling times greater than 40 h. We previously showed that when PAO1 is grown on adenosine and casein, variants emerge that grow rapidly on adenosine. To understand the mechanism by which this adaptation occurs, we performed whole-genome sequencing of five isolates evolved for rapid growth on adenosine. All five genomes had a gene duplication-amplification (GDA) event covering several genes, including the quorum-regulated nucleoside hydrolase gene, nuh, and PA0148, encoding an adenine deaminase. In addition, two of the growth variants also exhibited a nuh promoter mutation. We recapitulated the rapid growth phenotype with a plasmid containing six genes common to all the GDA events. We also showed that nuh and PA0148, the two genes at either end of the common GDA, were sufficient to confer rapid growth on adenosine. Additionally, we demonstrated that the variant nuh promoter increased basal expression of nuh but maintained its QS regulation. Therefore, GDA in P. aeruginosa confers the ability to grow efficiently on adenosine while maintaining QS regulation of nucleoside catabolism.
IMPORTANCE Pseudomonas aeruginosa thrives in many habitats and is an opportunistic pathogen of humans. In these diverse environments, P. aeruginosa must adapt to use myriad potential carbon sources. P. aeruginosa PAO1 cannot grow efficiently on nucleosides, including adenosine; however, it can acquire this ability through genetic adaptation. We show that the mechanism of adaptation is by amplification of a specific region of the genome and that the amplification preserves the regulation of the adenosine catabolic pathway by quorum sensing. These results demonstrate an underexplored mechanism of adaptation by P. aeruginosa, with implications for phenotypes such as development of antibiotic resistance.
KEYWORDS: nucleoside metabolism, nuh, sociomicrobiology, LasR, quorum sensing
INTRODUCTION
Pseudomonas aeruginosa grows in a wide variety of environments (1). The ecological versatility of P. aeruginosa has been attributed to its large genome, high occurrence of genetic regulatory pathways, and high proportion of genes involved in the metabolism and transport of organic compounds (2). P. aeruginosa is an opportunistic pathogen of humans that can cause chronic lung infections such as those associated with cystic fibrosis patients (3).
To survive in such a broad range of environments, P. aeruginosa must adapt to a myriad of conditions. Such adaptations occur via two general mechanisms. The first is based on regulation affecting gene transcription while the second occurs from genetic alterations (4). These genetic mechanisms include single-nucleotide changes and genetic rearrangements (4). One method of reversible genetic adaptation is gene duplication-amplification (GDA), which results in increased gene copy number (5, 6). GDA at a given locus can occur several orders of magnitude more frequently than spontaneous point mutations (4, 7). GDA has been associated with bacterial survival in extreme or variable conditions, including antimicrobial exposure or growth on suboptimal nutrient sources, and may have a role in rapid coevolution between host and pathogens (8).
Gene amplification allows the genome to expand and contract dynamically, with duplication length changes occurring in approximately 1 out of every 100 dividing cells after the initial duplication has occurred (5, 9, 10). GDA may be transient and therefore difficult to detect because the copy number change can be lost within several generations due to loss of selective conditions or other genetic changes that reduce the benefit from increased gene dosage (4, 6). Modern next-generation sequencing methods allow for more ready detection of GDA, as the generated reads will increase in number in proportion to the magnitude of a GDA (11, 12). In addition to the read depth, “split reads” spanning novel GDA junctions can identify new genomic sequences created by the GDA, compared to the reference genome (12–14).
GDA allows for adaptation to conditions that impose severe demands on regulatory systems (15, 16). Among the genetic regulatory pathways of P. aeruginosa is its quorum sensing (QS) system, which controls the production of a battery of virulence factors, secreted enzymes, and secondary metabolites, based on threshold population densities (17). P. aeruginosa QS is complex and involves multiple transcriptional regulators, including two complete N-acyl-homoserine lactone (AHL) QS systems, LasR-I and RhlR-I (17–20). LasR responds to the signal N-3-oxo-C12-homoserine lactone (3OC12-HSL), which is produced by LasI.
Genes coding for extracellular products are overrepresented in the LasR regulon (21). The list of LasR-induced products includes the secreted protease elastase (22), a “public good” shared among the population (23, 24). When elastase production is required for growth, such as when P. aeruginosa grows on the milk protein casein, LasR-null mutants emerge (23–25). These individual bacteria are “social cheaters” that avail themselves of the elastase produced by QS-intact cooperators but do not engage in QS themselves (23, 24, 26). P. aeruginosa has several mechanisms to deter social cheating in populations, including policing of cheaters by QS-dependent hydrogen cyanide (27), deferring synthesis of QS products until stationary phase (28, 29), and coregulation of intracellular “private goods” with public goods by LasR (25).
Adenosine catabolism (30–32) by P. aeruginosa is dependent on LasR-regulated transcription of nuh (33), which encodes a cellular nucleosidase, a “private good.” Nuh is also required for growth on inosine (25). The nuh gene product hydrolyzes adenosine into adenine and ribose, and inosine into hypoxanthine and ribose (Fig. 1). Adenine is subsequently deaminated to produce hypoxanthine, which is degraded to glyoxylate and urea (30, 31, 33). Ribose can be metabolized through the pentose phosphate pathway (2).
FIG 1.
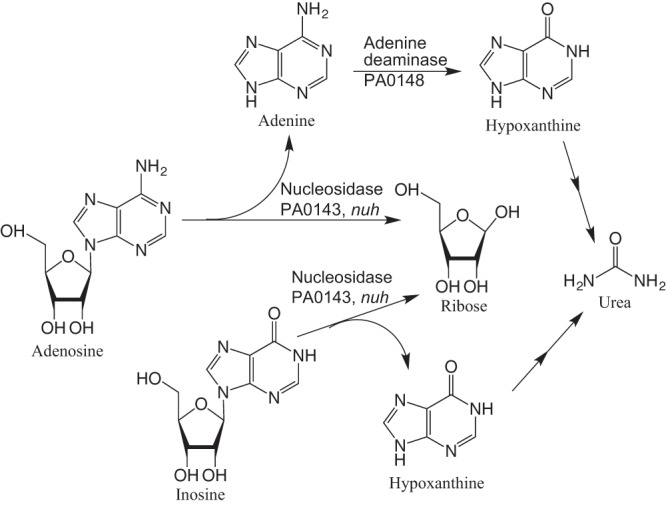
Adenosine catabolism in Pseudomonas aeruginosa. Nuh, the PA0143 gene product, is a nucleosidase that can cleave the base from adenosine or inosine to release ribose (31, 33). PA0148 is involved in the deamination of adenine (35). PA0148 cannot catalyze the deamination of adenosine (35) and there is no gene known to encode an adenosine deaminase in P. aeruginosa. Hypoxanthine is metabolized in multiple reactions to urea (31).
We previously examined the long-term growth of P. aeruginosa strain PAO1 in a minimal medium containing casein and adenosine as carbon and energy sources (25). We showed that the quorum-controlled linkage of elastase to cellular nuh expression suppressed emergence of social cheaters in the population by providing a metabolic incentive to cooperate. We also observed that the parent strain PAO1 grew slowly on adenosine as a carbon and energy source (with doubling times greater than 40 h), but after about 100 doublings in casein plus adenosine, populations transferred to adenosine minimal medium grew relatively rapidly (doubling times of about 5 h).
We investigated the genetic mechanism allowing P. aeruginosa to more efficiently use adenosine as a carbon and energy source, and the impact of adaptation to fast growth on adenosine on regulation by QS. We show that all the adenosine growth variants we examined contained a GDA region that included nuh.
RESULTS
The growth rate of P. aeruginosa PAO1 increases after prolonged growth in the presence of adenosine.
Wild-type P. aeruginosa PAO1 showed a doubling time of about 40 h when grown on photosynthetic medium (PM; see Materials and Methods) containing 1% adenosine (PM-adenosine) (Fig. 2A). We previously reported that daily passage of PAO1 for 25 days on medium containing 0.75% adenosine and 0.25% casein resulted in emergence of adenosine growth variants (25). These variants grew more rapidly on PM-adenosine than the parent strain did (Fig. 2A, variants A and B). We performed a similar experiment with 0.9% adenosine and 0.1% casein medium, and after 14 days, we again identified adenosine growth variants (Fig. 2A, variants C, D, and E). Doubling times in PM-adenosine varied between 5 to 12 h (Fig. 2A and Table 1). Additionally, there was reproducible variation in the lag phase, with 2 of the 5 (variants C and E) isolates demonstrating lag phases of about 50 h, while the other variants demonstrated minimal lag. The phenotypes of variants A to E were stable and growth rates in PM-adenosine were maintained after single isolates were grown for 18 h in LB-morpholinepropanesulfonic acid (LB-MOPS).
FIG 2.

Growth of P. aeruginosa in adenosine broth. (A) Growth of P. aeruginosa PAO1 and five variants (A to E) in PM-adenosine. Cell density was measured as optical density at 600 nm. Isolates A and B arose during passage in 0.75% adenosine, 0.25% casein broth and C to E arose during passage in 0.9% adenosine, 0.1% casein broth. (B) Long-term incubation of PAO1 and nuh, PA0148, lasI, and lasR deletion mutants in PM-adenosine. After approximately 150 h, the growth rates of wild type, LasI− PAO1 (with or without 2 μM 3OC12-HSL), and LasR− PAO1 accelerated. Deletion mutants of nuh and PA0148 mutants were unable to grow on adenosine as a sole carbon and energy source.
TABLE 1.
Doubling times and genetic alterations of adenosine growth variants
| Strain | Promotera | Doubling time (h) | Gene duplication-amplification results |
|||
|---|---|---|---|---|---|---|
| Start nucleotideb | End nucleotideb | Size (kb) | Copy no.c | |||
| Variants | ||||||
| A | Variant | 5 | 132143 | 222860 | 90.7 | 2 |
| B | Variant | 7 | 161949 | 186875 | 24.9 | 10 |
| C | wt | 9 | 146392 | 169812 | 19.7 | 9 |
| D | wt | 5 | 162685 | 175070 | 12.4 | 10 |
| E | wt | 12 | 138531 | 196457 | 57.9 | 3 |
| wt PAO1 | wt | 45 | 1 | |||
Promoter sequence compared to wild-type (wt) PAO1. The variant promoter contains a C-to-T substitution at nucleotide 163308 (based on PAO1 reference on www.pseudomonas.com).
Start and end nucleotides of amplified region from cross-referencing split read and copy number variation analysis using StrandNGS 2.9.
Copy number was estimated using the copy number variant analysis feature of StrandNGS 2.9, divided by 2 to account for the haploid nature of bacteria.
We next asked if adenosine growth variants could emerge from the wild type without casein in the medium. Four colonies of wild-type PAO1 were used to inoculate PM-adenosine. After about 150 h, the growth rate began to accelerate (Fig. 2B). The ability to grow on adenosine was dependent on nuh and the adenine deaminase PA0148, as deletion mutants of the parent strain were unable to grow on adenosine (Fig. 2B). We next asked if the phenotype was dependent on the QS transcription factor LasR. Both a LasR− mutant and a LasI− mutant (with or without the addition of the 3OC12-HSL signal) eventually acquired the adenosine growth variant phenotype (Fig. 2B), suggesting that the adaptation did not involve QS transcription factors.
All adenosine growth variants have a duplicated genomic region encompassing PA0143 to PA0148.
To determine the genetic changes associated with this heritable phenotype, we compared the genomes of the wild type to all five variants. We did not identify a nucleotide substitution, insertion, or deletion common to all adenosine fast-growing variants (see Table S2 in the supplemental material), although two variants (A and B) harbored a mutation in the −35 region of nuh (Tables 1 and S2). All variants showed increased read depth compared to that of the wild type in the region containing nuh (Fig. 3) compared to the average read depth in other parts of the aligned genome.
FIG 3.

Read depth differences in the genomic region containing nuh in adenosine growth variants. Data are displayed as Illumina reads per nucleotide for the genome region PA0008 to PA0305 (www.pseudomonas.com). All variants show increased read depth in a region encompassing nuh (PA0143). The vertical lines delineate a 6-kb minimal region that was amplified in all 5 variants, which included the genes PA0143 to PA0148. The flanking region shows the baseline read depth for each variant. The wild type, PAO1 (slow growth on adenosine), is shown for reference. Note that the y axis scales are not all the same.
The increase in read depth is consistent with the hypothesis that the adenosine growth variant phenotype was directly related to a gene duplication-amplification event (GDA). The length and magnitude of the GDA were variable between the isolates (Table 1 and Fig. 3; see also Table S3 in the supplemental material). In all variants, we confirmed the length of the duplicated region by inference from novel reads identified in our sequencing, i.e., split read analysis (13, 34), and copy number variation analysis (Table S3). We also confirmed the novel junction in variant A by direct sequencing.
A 6-kb genomic region was amplified across all variants (Fig. 3 and Table S3). This region contained several genes coding for enzymes in the nucleoside degradation pathway. The genes included in the GDA were as follows: PA0143 (nuh); PA0144, encoding a hypothetical nucleoside deoxyribosyltransferase; PA0145, encoding another hypothetical nucleoside deoxyribosyltransferase; PA0146, encoding a hypothetical basic membrane protein; PA0147, encoding a hypothetical oxidoreductase; and PA0148, encoding an adenine deaminase (35). Both nuh and PA0144 possess a LasR-RhlR binding motif upstream of the open reading frame (ORF) (36–39). An additional 5-kb region was amplified in all but one variant (variant C); genes included in this 5-kb region of the genome include PA0149, encoding an extracytoplasmic function sigma-70 factor (40); PA0150, encoding an anti-sigma factor; PA0151, encoding a probable TonB-dependent receptor; pcaQ, encoding transcriptional regulator PcaQ (41); pcaH, encoding a protocatechuate 3,4-dioxygenase, beta subunit; and part of pcaG, encoding the alpha subunit of protocatechuate 3,4-dioxygenase, which is important for catabolism of phenolic compounds (42, 43).
Increased gene copy number of the core amplified region confers an adenosine growth variant phenotype.
To determine whether an increase in the gene dosage discussed above was sufficient to confer the adenosine growth variant phenotype, we used the plasmid pBBR1MCS-5 (44) to increase the gene copy number of the 6-kb region common to all variants. Pseudomonas aeruginosa contains approximately 5 copies of this plasmid per cell (45). We cloned the region encompassing PA0143 to PA0148 into pBBR1MCS-5 (see Table S1 in the supplemental material). Both the wild type and the LasR− mutant containing PA0143 to PA0148 mimicked the adenosine-growth variant phenotype (Fig. 4). Furthermore, we reasoned that the key genes were likely nuh (PA0143) and PA0148, as their products can act in concert to hydrolyze and deaminate adenosine (Fig. 1), yielding ribose and hypoxanthine. To determine if nuh and PA0148 were the minimal genes required for the phenotype, we simultaneously overexpressed only nuh and PA0148. The concurrent overexpression of the nucleosidase (Nuh) and the adenine deaminase (the PA0148 gene product) conferred the adenosine growth phenotype and was indistinguishable from growth resulting from the overexpression of the whole region encompassing PA0143 to PA0148 (Fig. 4A).
FIG 4.

Increased copy number of the 6-kb region confers an adenosine-growth variant phenotype. (A) Growth curves of PAO1 expressing the plasmid pJT7, which contains the PA0143 to PA0148 region from PAO1 with either the wild-type (black) or variant (red) nuh promoter. Additionally, we expressed the plasmid pJT10 (blue), which contains only PA0143 and PA0148, in PAO1. (B) Growth curves of PAO1 ΔlasR containing pJT7 with either the wild-type (black) or variant (red) promoter. In both panels, the empty vector pBBR1MCS-5 is shown (orange). Cell density is measured as optical density at 600 nm.
Additionally, we queried whether the nuh promoter mutation in variants A and B accelerated or otherwise changed growth on adenosine, by cloning the PA0143 to PA0148 region containing the variant promoter (Tables 1 and S1) into pBBR1MCS-5. The presence of the variant promoter resulted in accelerated growth compared to the wild type (Fig. 4). Expression of either empty vector (Fig. 4) or pBBR1MCS-5 containing PA0144 or PA0148 alone in PAO1 resulted in no increase in growth rate on 1% adenosine (data not shown). We therefore conclude that duplication of the region encompassing PA0143 through PA0148 is required for fast growth on adenosine.
Overexpression of nuh alone does not result in fast growth on adenosine.
As discussed above, whole-genome sequencing demonstrated that two variants (A and B) harbored a C to T substitution at the second base pair of the −35 box of the nuh promoter (Fig. 5, top). The only other genetic change observed in more than one variant was mutation of psdR. PsdR is a transcriptional repressor of genes involved in the uptake and intracellular degradation of dipeptides in P. aeruginosa (46). Inactivation of PsdR has been shown to improve proteolytic growth on casein (47). Although we identified myriad other mutations, none was common to two or more variants (Table S2).
FIG 5.

Influence of the nuh promoter mutation on nuh expression. (Top) The promoter mutation (var) was found in variants A and B. Shown are the LasR binding site (36), the predicted −10 and −35 sequences, the E. coli consensus (white text) (50, 51), and the ATG start codon. The transcription start site has not been experimentally resolved (66). (Bottom) We used a nuh-gfp fusion to measure transcriptional activation from the wild-type and variant promoters. Data are presented as total fluorescence divided by OD600. In the absence of LasR, GFP was below the limit of detection from the wild-type promoter. Expression of the variant promoter in a LasR− mutant resulted in a 4.5-fold increase in GFP compared to the wild-type promoter in PAO1. In PAO1, the variant promoter was associated with an 18.5-fold increase in GFP compared to the wild-type promoter sequence. All pairwise comparisons are statistically significant using a t test (P < 0.001 in all cases).
In variants A and B, a single allele of nuh was observed that contained the promoter mutation (i.e., every copy of the gene contained the promoter mutation). The C to T promoter substitution results in a −35 sequence closer to the consensus sequence than the wild type, and therefore we predicted that it would result in enhanced transcription of nuh while preserving the LasR binding site (37, 48–51) (Fig. 5, top). We only observed the promoter mutation in the two variants that had grown on adenosine longer than 20 days.
We asked whether overexpression of nuh alone might confer the adenosine-growth variant phenotype. We placed nuh under the control of an arabinose-inducible promoter (pJN105.Nuh) in wild-type PAO1 and found that addition of exogenous l-arabinose (0.1% to 2.0%), which cannot be used by P. aeruginosa as a carbon source (52), did not result in faster growth on adenosine (data not shown).
The nuh promoter mutation retains LasR regulation of nuh.
Next, we assessed the impact of the −35 promoter mutation on nuh transcription by creating a Pnuh-gfp reporter plasmid. Thus, we could relate green fluorescent protein (GFP) expression to nuh transcription associated with either wild-type or variant promoter sequences (Fig. 5, bottom). As expected, expression of the wild-type promoter fusion in PAO1 resulted in a low but detectable level of GFP, and expression in a LasR− background resulted in minimal transcription of gfp. This finding is consistent with previous data (33). The single-nucleotide change found in variants A and B increased transcription (Fig. 5, bottom) in both LasR+ and LasR− backgrounds. The promoter mutation in a LasR+ background resulted in an 18.5-fold increase in GFP fluorescence compared to that in the wild-type promoter. In a LasR− background GFP was only detectable from the variant promoter. The expression of the variant promoter in a LasR− background was 4.5-fold more than the maximum expression of the wild-type promoter in a LasR+ background. Therefore, although transcription of nuh is increased in a LasR− individual harboring the nuh promoter mutation, a LasR+ cell would still have greater levels of gene transcription. This finding is consistent with microarray and proteomics data which show approximately a 5-fold induction of nuh transcription and protein levels by LasR (21, 53).
DISCUSSION
We investigated the mechanism by which P. aeruginosa PAO1 acquires the ability to grow rapidly on adenosine as a sole carbon and energy source, and discovered that this process is mediated in large part by GDA. Amplification of the genomic region of PAO1 from PA0143 to the end of PA0148 significantly reduced the doubling time on adenosine as a sole carbon and energy source. We created a synthetic gene duplication by cloning this 6-kb region into a moderate-copy-number plasmid, and in doing so, we recapitulated the variant phenotype of increased growth rates in 1% adenosine medium (PM-adenosine) (Fig. 4).
We discovered that coexpression of just the two flanking genes of the common GDA, PA0143 and PA0148, was necessary for the phenotype, as plasmids expressing these genes individually did not confer the adenosine growth enhancement phenotype, but a plasmid with both genes did confer rapid growth on adenosine. These findings indicate that P. aeruginosa requires expression of the deaminase encoded by PA0148 in addition to the nuh gene product to effectively use adenosine as a carbon and energy source. This result is consistent with the idea that adenosine catabolism in P. aeruginosa takes place by hydrolysis of adenosine to adenine and ribose, followed by deamination of adenine (Fig. 1), as the PA0148 gene product does not catalyze the deamination of adenosine (35).
Two of the intervening genes included in the GDA, PA0144 and PA0145, have no reported function but are orthologous to those in nucleoside degradation pathways of other pseudomonads (54) and might have a role in catabolism of nucleosides other than adenosine. It is difficult to speculate on the roles of PA0146 and PA0147, as their orthologs are for a hypothetical basic membrane protein and oxidoreductase, respectively, but these genes are not necessary for the phenotype as expression of PA0143 and PA0148 together was sufficient to confer fast growth on adenosine.
In addition to the GDA, we also describe a promoter mutation that was found in two variants. This substitution in the promoter region of nuh results in increased nuh gene transcription (Fig. 5). However, this mutation maintains QS regulation: transcription is 4-fold higher in the presence of active LasR than in a LasR mutant (Fig. 4). This nuh promoter mutation occurred in the two variants (A and B) isolated after 25 days of growth on casein-adenosine broth. Gene amplification followed by subsequent mutation within the duplicated region is well described (4, 6). In the case of our adenosine-growth variant, these two mechanisms appear to be complementary. The GDA is apparently required for the adenosine growth phenotype, as even the isolates with the variant nuh promoter carried multiple copies of the region in question (Table 1).
Upregulation of a designated gene via promoter mutation is estimated to be 5 to 8 orders of magnitude less frequent than GDA of the same gene (55, 56). We do not know the mechanism of acquiring the nuh promoter mutation associated with variants A and B, but it is striking that a single nuh allele is present in all variants. It is possible that the nuh promoter mutation was acquired prior to the GDA; however, we do not know under what circumstances the nuh promoter variant might be beneficial in the absence of GDA. We did not observe a growth benefit in adenosine broth solely with overexpression of nuh, which suggests that any benefit of increased expression of nuh is unresolvable under the conditions of our experiments.
An alternate possibility is that the GDA occurred prior to the mutation. Given that a single nuh allele is seen in all variants, the variant allele would therefore need to replace the wild-type nuh allele. The dynamic nature of GDA may facilitate this by allowing for contraction of gene copies with the wild-type allele, while copies of the variant allele are preserved or further amplified. Previously, it was shown that a fitness-improving mutation reduces selection for amplification and in some instances the copy number may even be reduced to one (9); however, we have shown that amplification of multiple genes is important for the phenotype, and it would be difficult to account for the loss of all duplicated copies of the entire region before reamplification occurred. Another explanation for the selection of the promoter mutation is that selective pressure for improved adenosine growth varies throughout the daily passaging of the evolved cultures. One possibility is that selection occurs at conditions of low cell density such as during the daily passage before growth achieves a threshold population density to activate quorum sensing. The promoter mutation is associated with increased nuh expression (Fig. 5, bottom). There is a noticeable growth advantage associated with the variant promoter compared to the wild-type promoter under conditions where LasR is not present (Fig. 4). This result supports the possibility that the variant nuh allele could arise either before or after an initial GDA due to selection during low population density.
The GDA we observed had an interesting consequence on regulation of adenosine catabolism by the QS transcription factor LasR. We observed that either a LasR− or LasI− mutant can acquire the ability to grow on adenosine as a carbon and energy source (Fig. 2). An essential gene for adenosine catabolism, nuh, is known to have an element of LasR-independent transcription (21, 33). Neither the gene amplification nor the −35 promoter mutation changed this basic feature of nuh regulation. In both cases, the presence of signal-bound LasR resulted in higher transcription of nuh than in the absence of LasR. However, the basal transcription of nuh (in the absence of LasR) was increased by the promoter mutation, decreasing the requirement for LasR. This finding is consistent with the fact that the mutation occurred in the promoter without altering the LasR binding site (Fig. 5, top).
One might predict that the partial decoupling of the intracellular enzyme Nuh from QS and subsequently from elastase production might allow for the invasion of LasR− mutants in media containing both casein and adenosine as carbon and energy sources. However, we did not observe such a phenomenon (25) and our growth experiments with the synthetic duplication demonstrate a growth advantage for QS wild type compared to LasR-null strains (Fig. 4). These data suggest that in PM-adenosine the benefit of lasR mutation is not sufficient to offer a fitness advantage against the wild type, and LasR mutants would be unable to invade a population of LasR wild-type cells.
Our work demonstrates that GDA is a reproducible pathway to enhance nucleoside catabolic abilities in P. aeruginosa PAO1. A GDA encompassing genes PA0143 to PA0148 improves the ability of PAO1 to grow on adenosine as a carbon and energy source. These results offer insights into the social and metabolic environment of P. aeruginosa and have relevance to its adaptation as a pathogen in the myriad settings where this bacterium causes a chronic infection.
MATERIALS AND METHODS
Bacteria, plasmids, and primers.
The bacteria, plasmids, and primers used are listed in Table S1 in the supplemental material. All mutants were created by using allelic exchange (57, 58) with a suicide vector containing in-frame deletions constructed by splice-overlap extension PCR (59) or Escherichia coli DH5α-mediated assembly (60). Gentamicin was used to maintain plasmids in E. coli (10 μg per ml) and P. aeruginosa (100 μg per ml). Where indicated, 2 μM 3OC12-HSL (Cayman Chemical, Ann Arbor, MI) was added to cultures.
To construct plasmids for expression of the entire region encompassing PA0143 to PA0148, we used E. coli DH5α-mediated assembly (60). In this approach, there are separate primers for the vector (designated “V.F” and “V.R” [see Table S1 in the supplemental material]) and for the genomic regions consisting of PA0143 to PA0148 (pJT7 primers). These primers were also employed to create the coexpression plasmid of PA0143 (pJT10_nuh R) and PA0148 (pJT10_PA0148 F) (Table S1).
Plasmids for expression of specific genes in the region of PA0143 to PA0148 were constructed using restriction enzyme-based cloning (61). Individual genes were amplified by PCR and cloned into pBBR1MCS-5 (44). Primers for nuh (pJT4 primers), PA0144 (pJT5 primers), and PA0148 (pJT3 primers) with the associated restriction endonuclease sites are listed in Table S1.
To assess transcription from the nuh promoter, either the wild-type nuh promoter or the variant nuh promoter was fused to gfp in the plasmid pPROBE-GT (62) to form Pnuh-wt or Pnuh-var, respectively. We measured GFP fluorescence after overnight growth of P. aeruginosa electrotransformed with either plasmid. Additionally, nuh was placed under the control of the pbad arabinose-inducible promoter on the plasmid pJN105 (63) using primers Pnuh.HindIII F and Pnuh.BamHI R (Table S1).
Growth conditions.
In all experiments, P. aeruginosa PAO1 (2) was used as the wild-type strain and grown in LB with 50 mM morpholinepropanesulfonic acid (MOPS) at pH 7.0 (LB-MOPS), or in PM minimal medium (64) supplemented with casein, adenosine, or both (total carbon 1%, wt/vol) (25). Cells were grown at 37°C with shaking (225 rpm). To start experiments in PM-casein-adenosine, we prepared overnight cultures in LB-MOPS, inoculated 3 ml of PM-casein-adenosine broth with 20 μl of the overnight culture, and incubated for 36 h. Subsequent daily transfers were 30 μl of the previous day's culture into 3 ml fresh PM-casein-adenosine. Individual variants were selected by plating onto LB agar from passage experiments with 0.75% adenosine and 0.25% casein (variants A and B isolated at 25 days) or 0.9% adenosine and 0.1% casein (variants C, D, and E isolated at 14 days). For arabinose-inducible gene expression, we added 0.01 to 2% l-arabinose to cultures (63, 65). l-Arabinose cannot be used as an energy source by P. aeruginosa (52).
For growth curves and subsequent whole-genome sequencing, isolates were grown overnight, then subcultured in LB-MOPS. Cells at mid-logarithmic phase were used to inoculate 25 ml PM-adenosine in 250-ml flasks at an optical density (OD) at 600 nm (OD600) of 0.02. DNA was isolated from samples taken in early stationary phase (OD = 2.0) for whole-genome sequencing as described below. Growth curves were in triplicate.
Whole-genome sequencing.
Genomic DNA for sequencing was purified from stationary-phase cultures of variants grown in LB-MOPS or PM-adenosine using the Gentra Puregene Yeast/Bact. kit (Qiagen Sciences, Germantown, MD). We used an Illumina MiSeq to complete whole-genome sequencing with paired 150-bp reads. We aligned reads to the published sequence of P. aeruginosa PAO1 (accession NC_002516) using StrandNGS version 2.9 software (Strand Life Sciences, Bangalore, India). This software was used to assess read depth, split read, and calculate copy number differences for the duplications.
Accession number(s).
Raw sequencing reads of adenosine growth variants have been deposited to NCBI SRA BioProject PRJNA342344 under BioSample accessions SAMN06678220, SAMN06678221, SAMN06678222, SAMN06678223, SAMN06678224 (variants A to E), and SAMN06689578 (PAO1).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by U.S. Public Health Service (USPHS) grants K08AI102921 (to A.A.D.) and R01GM59026 (to E.P.G.). A.A.D. is also funded by a Burroughs-Wellcome Fund Career Award for Medical Scientists. We also acknowledge DNA sequencing core support from USPHS grant no. P30DK089507 and the Cystic Fibrosis Foundation (grants SINGH15R0 and R565 CR11).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00261-17.
REFERENCES
- 1.Silby MW, Winstanley C, Godfrey SAC, Levy SB, Jackson RW. 2011. Pseudomonas genomes: diverse and adaptable. FEMS Microbiol Rev 35:652–680. doi: 10.1111/j.1574-6976.2011.00269.x. [DOI] [PubMed] [Google Scholar]
- 2.Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FSL, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GK-S, Wu Z, Paulsen IT, Reizer J, Saier MH, Hancock REW, Lory S, Olson MV. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- 3.Smith EE, Buckley DG, Wu Z, Saenphimmachak C, Hoffman LR, D'Argenio DA, Miller SI, Ramsey BW, Speert DP, Moskowitz SM, Burns JL, Kaul R, Olson MV. 2006. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc Natl Acad Sci U S A 103:8487–8492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andersson DI, Hughes D. 2009. Gene amplification and adaptive evolution in bacteria. Annu Rev Genet 43:167–195. doi: 10.1146/annurev-genet-102108-134805. [DOI] [PubMed] [Google Scholar]
- 5.Elde NC, Child SJ, Eickbush MT, Kitzman JO, Rogers KS, Shendure J, Geballe AP, Malik HS. 2012. Poxviruses deploy genomic accordions to adapt rapidly against host antiviral defenses. Cell 150:831–841. doi: 10.1016/j.cell.2012.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elliott KT, Cuff LE, Neidle EL. 2013. Copy number change: evolving views on gene amplification. Future Microbiol 8:887–899. doi: 10.2217/fmb.13.53. [DOI] [PubMed] [Google Scholar]
- 7.Sun S, Berg OG, Roth JR, Andersson DI. 2009. Contribution of gene amplification to evolution of increased antibiotic resistance in Salmonella typhimurium. Genetics 182:1183–1195. doi: 10.1534/genetics.109.103028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kondrashov FA. 2012. Gene duplication as a mechanism of genomic adaptation to a changing environment. Proc Biol Sci 279:5048–5057. doi: 10.1098/rspb.2012.1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pränting M, Andersson DI. 2011. Escape from growth restriction in small colony variants of Salmonella typhimurium by gene amplification and mutation. Mol Microbiol 79:305–315. doi: 10.1111/j.1365-2958.2010.07458.x. [DOI] [PubMed] [Google Scholar]
- 10.Pettersson ME, Sun S, Andersson DI, Berg OG. 2009. Evolution of new gene functions: simulation and analysis of the amplification model. Genetica 135:309–324. doi: 10.1007/s10709-008-9289-z. [DOI] [PubMed] [Google Scholar]
- 11.Bryant J, Chewapreecha C, Bentley SD. 2012. Developing insights into the mechanisms of evolution of bacterial pathogens from whole-genome sequences. Future Microbiol 7:1283–1296. doi: 10.2217/fmb.12.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tattini L, D'Aurizio R, Magi A. 2015. Detection of genomic structural variants from next generation sequencing data. Front Bioeng Biotechnol 3:92. doi: 10.3389/fbioe.2015.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun S, Ke R, Hughes D, Nilsson M, Andersson DI. 2012. Genome-wide detection of spontaneous chromosomal rearrangements in bacteria. PLoS One 7:e42639. doi: 10.1371/journal.pone.0042639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barrick JE, Colburn G, Deatherage DE, Traverse CC, Strand MD, Borges JJ, Knoester DB, Reba A, Meyer AG. 2014. Identifying structural variation in haploid microbial genomes from short-read resequencing data using breseq. BMC Genomics 15:1039. doi: 10.1186/1471-2164-15-1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson RP, Roth JR. 1977. Tandem genetic duplications in phage and bacteria. Annu Rev Microbiol 31:473–505. doi: 10.1146/annurev.mi.31.100177.002353. [DOI] [PubMed] [Google Scholar]
- 16.Romero D, Palacios R. 1997. Gene amplification and genomic plasticity in prokaryotes. Annu Rev Genet 31:91–111. doi: 10.1146/annurev.genet.31.1.91. [DOI] [PubMed] [Google Scholar]
- 17.Schuster M, Greenberg EP. 2006. A network of networks: quorum-sensing gene regulation in Pseudomonas aeruginosa. Int J Med Microbiol 296:73–81. doi: 10.1016/j.ijmm.2006.01.036. [DOI] [PubMed] [Google Scholar]
- 18.Gambello MJ, Iglewski BH. 1991. Cloning and characterization of the Pseudomonas aeruginosa lasR gene, a transcriptional activator of elastase expression. J Bacteriol 173:3000–3009. doi: 10.1128/jb.173.9.3000-3009.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Passador L, Cook J, Gambello M, Rust L, Iglewski B. 1993. Expression of Pseudomonas aeruginosa virulence genes requires cell-to-cell communication. Science 260:1127–1130. doi: 10.1126/science.8493556. [DOI] [PubMed] [Google Scholar]
- 20.Pearson JP, Gray KM, Passador L, Tucker KD, Eberhard A, Iglewski BH, Greenberg EP. 1994. Structure of the autoinducer required for expression of Pseudomonas aeruginosa virulence genes. Proc Natl Acad Sci U S A 91:197–201. doi: 10.1073/pnas.91.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schuster M, Lostroh CP, Ogi T, Hawkins AC, Harwood CS, Greenberg EP. 2003. Identification, timing, and signal specificity of Pseudomonas aeruginosa quorum-controlled genes: a transcriptome analysis. J Bacteriol 185:2066–2079. doi: 10.1128/JB.185.7.2066-2079.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Toder DS, Ferrell SJ, Nezezon JL, Rust L, Iglewski BH. 1994. lasA and lasB genes of Pseudomonas aeruginosa: analysis of transcription and gene product activity. Infect Immun 62:1320–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sandoz KM, Mitzimberg SM, Schuster M. 2007. Social cheating in Pseudomonas aeruginosa quorum sensing. Proc Natl Acad Sci U S A 104:15876–15881. doi: 10.1073/pnas.0705653104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diggle SP, Griffin AS, Campbell GS, West SA. 2007. Cooperation and conflict in quorum-sensing bacterial populations. Nature 450:411–414. doi: 10.1038/nature06279. [DOI] [PubMed] [Google Scholar]
- 25.Dandekar AA, Chugani S, Greenberg EP. 2012. Bacterial quorum sensing and metabolic incentives to cooperate. Science 338:264–266. doi: 10.1126/science.1227289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilder CN, Diggle SP, Schuster M. 2011. Cooperation and cheating in Pseudomonas aeruginosa: the roles of the las, rhl and pqs quorum-sensing systems. ISME J 5:1332–1343. doi: 10.1038/ismej.2011.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang M, Schaefer AL, Dandekar AA, Greenberg EP. 2015. Quorum sensing and policing of Pseudomonas aeruginosa social cheaters. Proc Natl Acad Sci U S A 112:2187–2191. doi: 10.1073/pnas.1500704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Darch SE, West SA, Winzer K, Diggle SP. 2012. Density-dependent fitness benefits in quorum-sensing bacterial populations. Proc Natl Acad Sci U S A 109:8259–8263. doi: 10.1073/pnas.1118131109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xavier JB, Kim W, Foster KR. 2011. A molecular mechanism that stabilizes cooperative secretions in Pseudomonas aeruginosa. Mol Microbiol 79:166–179. doi: 10.1111/j.1365-2958.2010.07436.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsumoto H, Ohta S, Kobayashi R, Terawaki Y. 1978. Chromosomal location of genes participating in the degradation of purines in Pseudomonas aeruginosa. Mol Gen Genet 167:165–176. doi: 10.1007/BF00266910. [DOI] [PubMed] [Google Scholar]
- 31.Bongaerts GPA, Sin IL, Peters ALJ, Vogels GD. 1977. Purine degradation in Pseudomonas aeruginosa and Pseudomonas testosteroni. Biochim Biophys Acta 499:111–118. doi: 10.1016/0304-4165(77)90233-1. [DOI] [PubMed] [Google Scholar]
- 32.Trijbels F, Vogels GD. 1967. Allantoate and ureidoglycolate degradation by Pseudomonas aeruginosa. Biochim Biophys Acta 132:115–126. doi: 10.1016/0005-2744(67)90197-0. [DOI] [PubMed] [Google Scholar]
- 33.Heurlier K, Haenni M, Guy L, Krishnapillai V, Haas D. 2005. Quorum-sensing-negative (lasR) mutants of Pseudomonas aeruginosa avoid cell lysis and death. J Bacteriol 187:4875–4883. doi: 10.1128/JB.187.14.4875-4883.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chaisson MJ, Pevzner PA. 2008. Short read fragment assembly of bacterial genomes. Genome Res 18:324–330. doi: 10.1101/gr.7088808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goble AM, Zhang Z, Sauder JM, Burley SK, Swaminathan S, Raushel FM. 2011. PA0148 from Pseudomonas aeruginosa catalyzes the deamination of adenine. Biochemistry 50:6589–6597. doi: 10.1021/bi200868u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gilbert KB, Kim TH, Gupta R, Greenberg EP, Schuster M. 2009. Global position analysis of the Pseudomonas aeruginosa quorum-sensing transcription factor LasR. Mol Microbiol 73:1072–1085. doi: 10.1111/j.1365-2958.2009.06832.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whiteley M, Greenberg EP. 2001. Promoter specificity elements in Pseudomonas aeruginosa quorum-sensing-controlled genes. J Bacteriol 183:5529–5534. doi: 10.1128/JB.183.19.5529-5534.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schuster M, Urbanowski ML, Greenberg EP. 2004. Promoter specificity in Pseudomonas aeruginosa quorum sensing revealed by DNA binding of purified LasR. Proc Natl Acad Sci U S A 101:15833–15839. doi: 10.1073/pnas.0407229101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wagner VE, Bushnell D, Passador L, Brooks AI, Iglewski BH. 2003. Microarray analysis of Pseudomonas aeruginosa quorum-sensing regulons: effects of growth phase and environment. J Bacteriol 185:2080–2095. doi: 10.1128/JB.185.7.2080-2095.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Llamas MA, Mooij MJ, Sparrius M, Vandenbroucke-Grauls CMJE, Ratledge C, Bitter W. 2008. Characterization of five novel Pseudomonas aeruginosa cell-surface signalling systems. Mol Microbiol 67:458–472. doi: 10.1111/j.1365-2958.2007.06061.x. [DOI] [PubMed] [Google Scholar]
- 41.Parke D. 1996. Characterization of PcaQ, a LysR-type transcriptional activator required for catabolism of phenolic compounds, from Agrobacterium tumefaciens. J Bacteriol 178:266–272. doi: 10.1128/jb.178.1.266-272.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parke D. 1995. Supraoperonic clustering of pca genes for catabolism of the phenolic compound protocatechuate in Agrobacterium tumefaciens. J Bacteriol 177:3808–3817. doi: 10.1128/jb.177.13.3808-3817.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ohlendorf DH, Lipscomb JD, Weber PC. 1988. Structure and assembly of protocatechuate 3,4-dioxygenase. Nature 336:403–405. doi: 10.1038/336403a0. [DOI] [PubMed] [Google Scholar]
- 44.Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM, Peterson KM. 1995. Four new derivatives of the broad host range cloning vector pBBR1MCS, carrying different antibiotic resistance cassettes. Gene 166:175–176. doi: 10.1016/0378-1119(95)00584-1. [DOI] [PubMed] [Google Scholar]
- 45.Zhao F, Cui Q, Han S, Dong H, Zhang J, Ma F, Zhang Y, Liu W, Zwick M, Wilhelm S, Hausmann R, Syldatk C, Rosenau F, Blank LM. 2015. Enhanced rhamnolipid production of Pseudomonas aeruginosa SG by increasing copy number of rhlAB genes with modified promoter. RSC Adv 5:70546–70552. doi: 10.1039/C5RA13415C. [DOI] [Google Scholar]
- 46.Kiely PD, O'Callaghan J, Abbas A, O'Gara F. 2008. Genetic analysis of genes involved in dipeptide metabolism and cytotoxicity in Pseudomonas aeruginosa PAO1. Microbiology 154:2209–2218. doi: 10.1099/mic.0.2007/015032-0. [DOI] [PubMed] [Google Scholar]
- 47.Asfahl KL, Walsh J, Gilbert K, Schuster M. 2015. Non-social adaptation defers a tragedy of the commons in Pseudomonas aeruginosa quorum sensing. ISME J 9:1734–1746. doi: 10.1038/ismej.2014.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Slock J, VanRiet D, Kolibachuk D, Greenberg EP. 1990. Critical regions of the Vibrio fischeri LuxR protein defined by mutational analysis. J Bacteriol 172:3974–3979. doi: 10.1128/jb.172.7.3974-3979.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Egland KA, Greenberg EP. 1999. Quorum sensing in Vibrio fischeri: Elements of the luxI promoter. Mol Microbiol 31:1197–1204. doi: 10.1046/j.1365-2958.1999.01261.x. [DOI] [PubMed] [Google Scholar]
- 50.Harley CB, Reynolds RP. 1987. Analysis of Escherichia coli promoter sequences. Nucleic Acids Res 15:2343–2361. doi: 10.1093/nar/15.5.2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McLean BW, Wiseman SL, Kropinski AM. 1997. Functional analysis of sigma-70 consensus promoters in Pseudomonas aeruginosa and Escherichia coli. Can J Microbiol 43:981–985. doi: 10.1139/m97-141. [DOI] [PubMed] [Google Scholar]
- 52.Lequette Y, Greenberg EP. 2005. Timing and localization of rhamnolipid synthesis gene expression in Pseudomonas aeruginosa biofilms. J Bacteriol 187:37–44. doi: 10.1128/JB.187.1.37-44.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arevalo-Ferro C, Hentzer M, Reil G, Görg A, Kjelleberg S, Givskov M, Riedel K, Eberl L. 2003. Identification of quorum-sensing regulated proteins in the opportunistic pathogen Pseudomonas aeruginosa by proteomics. Environ Microbiol 5:1350–1369. doi: 10.1046/j.1462-2920.2003.00532.x. [DOI] [PubMed] [Google Scholar]
- 54.Winsor GL, Griffiths EJ, Lo R, Dhillon BK, Shay JA, Brinkman FSL. 2016. Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res 44:D646–D653. doi: 10.1093/nar/gkv1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Andersson DI. 2011. Evolving promiscuously. Proc Natl Acad Sci U S A 108:1199–1200. doi: 10.1073/pnas.1018588108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anderson P, Roth J. 1981. Spontaneous tandem genetic duplications in Salmonella typhimurium arise by unequal recombination between rRNA (rrn) cistrons. Proc Natl Acad Sci U S A 78:3113–3117. doi: 10.1073/pnas.78.5.3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hmelo LR, Borlee BR, Almblad H, Love ME, Randall TE, Tseng BS, Lin C, Irie Y, Storek KM, Yang JJ, Siehnel RJ, Howell PL, Singh PK, Tolker-Nielsen T, Parsek MR, Schweizer HP, Harrison JJ. 2015. Precision-engineering the Pseudomonas aeruginosa genome with two-step allelic exchange. Nat Protoc 10:1820–1841. doi: 10.1038/nprot.2015.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP. 1998. A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212:77–86. doi: 10.1016/S0378-1119(98)00130-9. [DOI] [PubMed] [Google Scholar]
- 59.Horton RM. 1995. PCR-mediated recombination and mutagenesis. Mol Biotechnol 3:93–99. doi: 10.1007/BF02789105. [DOI] [PubMed] [Google Scholar]
- 60.Kostylev M, Otwell AE, Richardson RE, Suzuki Y. 2015. Cloning should be simple: Escherichia coli DH5α-mediated assembly of multiple DNA fragments with short end homologies. PLoS One 10:1–15. doi: 10.1371/journal.pone.0137466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sambrook J, Fritsch E, Maniatis T. 1989. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Plainview, NY. [Google Scholar]
- 62.Miller WG, Leveau JHJ, Lindow SE. 2000. Improved gfp and inaZ broad-host-range promoter-probe vectors. Mol Plant Microbe Interact 13:1243–1250. doi: 10.1094/MPMI.2000.13.11.1243. [DOI] [PubMed] [Google Scholar]
- 63.Newman JR, Fuqua C. 1999. Broad-host-range expression vectors that carry the l-arabinose-inducible Escherichia coli araBAD promoter and the araC regulator. Gene 227:197–203. doi: 10.1016/S0378-1119(98)00601-5. [DOI] [PubMed] [Google Scholar]
- 64.Kim M-K, Harwood CS. 1991. Regulation of benzoate-CoA ligase in Rhodopseudomonas palustris. FEMS Microbiol Lett 83:199–203. doi: 10.1111/j.1574-6968.1991.tb04440.x-i1. [DOI] [Google Scholar]
- 65.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wurtzel O, Yoder-Himes DR, Han K, Dandekar AA, Edelheit S, Greenberg EP, Sorek R, Lory S. 2012. The single-nucleotide resolution transcriptome of Pseudomonas aeruginosa grown in body temperature. PLoS Pathog 8:e1002945. doi: 10.1371/journal.ppat.1002945. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.