

Abstract
Pseudomonas aeruginosa is an opportunistic pathogen that causes severe airway infections in humans. These infections are usually difficult to treat and associated with high mortality rates. While colonizing the human airways, P. aeruginosa could accumulate genetic mutations that often lead to its better adaptability to the host environment. Understanding these evolutionary traits may provide important clues for the development of effective therapies to treat P. aeruginosa infections. In this study, 25 P. aeruginosa isolates were longitudinally sampled from the airways of four ventilator-associated pneumonia (VAP) patients. Pacbio and Illumina sequencing were used to analyse the in vivo evolutionary trajectories of these isolates. Our analysis showed that positive selection dominantly shaped P. aeruginosa genomes during VAP infections and led to three convergent evolution events, including loss-of-function mutations of lasR and mpl, and a pyoverdine-deficient phenotype. Specifically, lasR encodes one of the major transcriptional regulators in quorum sensing, whereas mpl encodes an enzyme responsible for recycling cell wall peptidoglycan. We also found that P. aeruginosa isolated at late stages of VAP infections produce less elastase and are less virulent in vivo than their earlier isolated counterparts, suggesting the short-term in vivo evolution of P. aeruginosa leads to attenuated virulence.
Keywords: Pseudomonas aeruginosa, ventilator-associated pneumonia, in vivo evolution, genomics, adaptation
1. Introduction
The opportunistic pathogen Pseudomonas aeruginosa is one of the leading causes of nosocomial infections worldwide [1]. It was estimated that the average cost for each P. aeruginosa infection case is US $24 700 with a median hospitalization period of 45 days [2]. Among the various types of infections caused by P. aeruginosa, lung infections have become a major concern. Pseudomonas aeruginosa was reported to be the most frequently isolated bacteria from the respiratory tract and one of the major causes of ventilator-associated pneumonia (VAP) [3]. In addition, P. aeruginosa is also the major pathogen causing lung infections and death in patients suffering from cystic fibrosis (CF) and chronic obstructive pulmonary disease [4].
Pseudomonas aeruginosa harbours multiple virulence factors that have been proved to be essential for causing lung infections. For instance, P. aeruginosa quorum-sensing systems and regulated virulence factors such as rhamnolipids and elastase were shown to play important roles in its lung colonization [5,6]. The formation of surface-attached P. aeruginosa biofilm communities was implicated in persistent lung infections of VAP patients and CF patients [7,8], which raises a great challenge for the development of effective antimicrobial therapy.
Previous studies have shown that P. aeruginosa can undergo adaptive evolution during lung infections. Understanding these evolutionary traits may help to predict novel targets for the design of effective treatment strategies [9]. For instance, both lasR and pyoverdine-deficient P. aeruginosa mutants have been identified in the CF lungs [9,10]. LasR is a transcriptional regulator that can activate the expression of various virulence factors in response to N-(3-oxododecanoyl)-l-homoserine lactone signal, whereas pyoverdine is a siderophore by which P. aeruginosa scavenges ferric iron from the environment and within the host [11,12]. The accumulation of these mutants in the CF lungs suggests that remodelling of the quorum-sensing regulatory system and iron metabolism is crucial for the adaptation of P. aeruginosa to the host environment. In addition, lasR mutants were also reported to evolve from the wild-type in mechanically ventilated patients within 10 days of colonization [13]. The evolution of lasR mutants is probably because they could gain fitness advantages by exploiting exoproducts of the wild-type [13]. Although these studies have provided insights into the adaptive evolution of P. aeruginosa during chronic and acute pulmonary infections, there is little evidence showing the evolutionary dynamics of P. aeruginosa during acute pulmonary infections. To address this, we investigated the adaptive evolution of P. aeruginosa during acute VAP infections by sequencing the genomes of 25 P. aeruginosa isolates sequentially sampled from four VAP patients. We show that the short-term in vivo evolution of P. aeruginosa in VAP patients leads to attenuated virulence.
2. Results
2.1. A novel Pseudomonas aeruginosa strain causing nosocomial outbreak of ventilator-associated pneumonia
Four patients developed VAP infections after admission into the intensive care units of a teaching hospital in China from December 2013 to March 2014. Cultivation of the patients' sputum samples and 16s rDNA PCR suggested that P. aeruginosa was responsible for their VAP infections. In the initial screening, we randomly selected three colonies cultured from each sputum sample and compared their colony morphologies, antibiotic resistance and pigment production. We found that the three colonies isolated from the same sputum sample showed no phenotypic variations to each other. Therefore, to investigate the epidemic linkage and adaptive evolution of P. aeruginosa during VAP infections, one single colony from each sputum sample was sequenced to represent the dominant clone. Patient information and the sampling time for each isolate can be found in the electronic supplementary material, table S1.
The genomes of 24 isolates (PA_D1 to PA_D24) were sequenced on an Illumina HiSeq 2500 platform and one isolate (PA_D25) was sequenced on an Illumina MiSeq platform. We further sequenced the earliest and latest isolate from each patient on a Pacific Biosciences RSII sequencer to obtain their concise genome maps. Illumina and Pacific Biosciences sequencing achieved on-average 100× and 50× coverage of the P. aeruginosa genomes, respectively. These sequencing reads were successfully assembled into eight circular genomes and 17 draft genomes. The general characteristics of the eight fully sequenced genomes can be found in the electronic supplementary material, table S2.
To identify the origin of the 25 isolates in our study, we analysed the phylogeny of the 25 genomes against 30 other P. aeruginosa genomes from GenBank. The phylogenetic tree based on core-genome alignment showed that the 25 genomes are closely related to each other and form a monophyletic group (figure 1). In addition, we performed random amplified polymorphic DNA (RAPD) typing on the 25 isolates. The results suggested that the 25 isolates are of the same clone (electronic supplementary material, figure S1).
Figure 1.

Phylogenetic analysis of the 25 sequenced P. aeruginosa genomes and 30 other P. aeruginosa genomes. The 25 P. aeruginosa isolates sequenced in this study cluster together and form a monophyletic group (asterisk indicates isolate sequenced in this study). Phylogenetic inference was then carried out based on 132 511 variant sites using the approximate maximum-likelihood algorithm, with clade confidence estimated with SH-like support values. Accession numbers of the 30 genomes from GenBank are listed in the electronic supplementary material, table S3.
2.2. General characteristics of Pseudomonas aeruginosa PA_D1 genome
As the genomes of the 25 isolates are closely related (figure 1; electronic supplementary material, figure S1), we focused on characterizing PA_D1 genome as a representative strain of this study. The PA_D1 genome consists of a circular chromosome comprising 6 643 823 bp with an average GC content of 66.2%. Annotation on Rapid Annotations using Subsystem Technology (RAST) Server [14] showed that PA_D1 genome is predicted to encode 6210 genes, of which 6135 are protein-encoding genes and 75 are RNA-coding genes (63 tRNAs, 12 rRNAs; electronic supplementary material, table S2). Among the 6135 protein-encoding genes, 4918 can be assigned to putative functions, with the remaining annotated as hypothetical proteins.
Alignment of the PA_D1 genome with five other P. aeruginosa genomes revealed that it harbours several strain-specific genomic regions (figure 2). To identify the cause of these strain-specific genomic regions, we predicted the genomic islands (GIs) present in the PA_D1 genome using IslandViewer 3 server [17]. The 31 predicted GIs in the PA_D1 genome correlate well with its strain-specific regions (figure 2; electronic supplementary material, table S4), suggesting that most of the strain-specific regions can be explained by horizontal gene transfer events. Notably, many of the genes present in the GIs encode products involved in generating transposons, virulence, energy metabolisms, antibiotic resistance and DNA methylation and repair (electronic supplementary material, table S4), suggesting that these GIs are important for PA_D1 to survive under stressful conditions and subsequent spreading in nosocomial settings.
Figure 2.

Sequence conservation between PA_D1 and five other P. aeruginosa genomes. From the innermost to outermost: circle 1, PA_D1, the representative strain in this study; circle 2, NCGM257, a P. aeruginosa strain that causes nosocomial infections in Japan, which is also the closest genome to isolates in this study as shown by the phylogenetic analysis; circle 3, PA14, a highly virulent strain; circle 4, PAO1, the laboratory reference strain; circle 5, DK2, a strain isolated from CF lungs; circle 6, LES431, an endemic strain isolated in UK; circle 7, GIs predicted by IslandViewer 3 [15]; circle 8, antibiotic resistance genes predicted by ResFinder 2.1 server [16].
2.3. Acquisition of antibiotic resistance
To identify the antibiotic resistance profiles of the 25 isolates, we performed antibiotic susceptibility tests. The results showed that all the isolates displayed resistance to various classes of antibiotics, including macrolides (azithromycin), penicillins and penicillin combined with beta-lactamase inhibitor (ampicillin, piperacillin-tazobactam), cephalosporins (cefazolin, cefotetan, ceftazidime, ceftriaxone, cefepime), aminoglycosides (gentamicin, tobramycin), fluoroquinolones (ciprofloxacin, levofloxacin), but remained sensitive to amikacin (table 1). Interestingly, 19 isolates from patient 1, patient 2 and patient 3 remained sensitive to imipenem, whereas the six isolates from patient 4 showed resistance to imipenem, which belongs to carbapenem class of antibiotics. Prediction of resistance gene on ResFinder 2.1 server [16] showed that all the 25 isolates harbour the same set of antibiotic resistance genes. These genes are responsible for resistance to aminoglycosides (aadA2, aacA4, aph(3')-Ic and aph(3')-IIb), beta-lactams (blaOXA−50 and ampC), fluoroquinolones (aac(6')Ib-cr), fosfomycins (fosA), phenicols (cmlA1 and catB7), sulfonamide (sul1), trimethoprim (dfrA1) and tetracycline (tet(C)). Intriguingly, eight out of the 13 predicted antibiotic resistance genes are located in the predicted GIs (figure 2), suggesting that horizontal gene transfer is responsible for the acquisition of antibiotic resistance genes in these isolates.
Table 1.
Antibiotic susceptibility profiles of 25 P. aeruginosa isolates. AMK, amikacin; AMP, ampicillin; ATM, azithromycin; CAZ, ceftazidime; CIP, ciprofloxacin; CRO, ceftriaxone; CTT, cefotetan; CZO, cefazolin; FEP, cefepime; GEM, gentamicin; IMP, imipenem; LVX, levofloxacin; TOB, Tobramycin; TZP, piperacillin-tazobactam. (unit: mg l−1).
| AMK | AMP | ATM | CAZ | CIP | CRO | CTT | CZO | FEP | LVX | GEN | IPM | TOB | TZP | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient 1 | ||||||||||||||
| PA_D1 | 4 | ≥32 | 16 | 4 | ≥4 | ≥64 | ≥64 | ≥64 | 8 | ≥8 | ≥16 | ≤1 | ≥16 | 16 |
| PA_D3 | ≤2 | ≥32 | 16 | 16 | ≥4 | ≥64 | ≥64 | ≥64 | 8 | ≥8 | ≥16 | ≤1 | ≥16 | 64 |
| PA_D8 | ≤2 | ≥32 | 16 | 16 | ≥4 | ≥64 | ≥64 | ≥64 | 8 | ≥8 | ≥16 | ≤1 | ≥16 | 64 |
| PA_D9 | ≤2 | ≥32 | 16 | 16 | ≥4 | ≥64 | ≥64 | ≥64 | 4 | ≥8 | ≥16 | 2 | ≥16 | 32 |
| Patient 2 | ||||||||||||||
| PA_D2 | 4 | ≥32 | 16 | 16 | ≥4 | ≥64 | ≥64 | ≥64 | 8 | ≥8 | ≥16 | ≤1 | ≥16 | 64 |
| PA_D4 | 16 | ≥32 | 16 | 4 | ≥4 | ≥64 | ≥64 | ≥64 | 8 | ≥8 | ≥16 | ≤1 | ≥16 | 64 |
| PA_D6 | ≤2 | ≥32 | 16 | 8 | ≥4 | ≥64 | ≥64 | ≥64 | 8 | ≥8 | ≥16 | ≤1 | ≥16 | 64 |
| PA_D10 | ≤2 | ≥32 | ≥64 | 16 | ≥4 | ≥64 | ≥64 | ≥64 | 8 | ≥8 | ≥16 | ≤1 | ≥16 | 64 |
| PA_D12 | ≤2 | ≥32 | ≥64 | ≥64 | ≥4 | ≥64 | ≥64 | ≥64 | 16 | ≥8 | ≥16 | 2 | ≥16 | ≥128 |
| PA_D19 | ≤2 | ≥32 | 32 | 4 | ≥4 | ≥64 | ≥64 | ≥64 | 8 | ≥8 | ≥16 | 2 | ≥16 | 64 |
| PA_D21 | 4 | ≥32 | 32 | 32 | ≥4 | ≥64 | ≥64 | ≥64 | 8 | ≥8 | ≥16 | ≤1 | ≥16 | 64 |
| Patient 3 | ||||||||||||||
| PA_D5 | ≤2 | ≥32 | 16 | 4 | ≥4 | ≥64 | ≥64 | ≥64 | 4 | ≥8 | ≥16 | ≤1 | ≥16 | 16 |
| PA_D7 | ≤2 | ≥32 | 16 | 16 | ≥4 | ≥64 | ≥64 | ≥64 | 8 | ≥8 | ≥16 | ≤1 | ≥16 | 64 |
| PA_D11 | ≤2 | ≥32 | 16 | 8 | ≥4 | ≥64 | ≥64 | ≥64 | 4 | ≥8 | ≥16 | 2 | ≥16 | 64 |
| PA_D13 | ≤2 | ≥32 | 16 | 16 | ≥4 | ≥64 | ≥64 | ≥64 | 8 | ≥8 | ≥16 | 2 | ≥16 | 64 |
| PA_D14 | ≤2 | ≥32 | 16 | 16 | ≥4 | ≥64 | ≥64 | ≥64 | 8 | ≥8 | ≥16 | 2 | ≥16 | 64 |
| PA_D15 | ≤2 | ≥32 | 16 | 16 | ≥4 | ≥64 | ≥64 | ≥64 | 8 | ≥8 | ≥16 | ≤1 | ≥16 | 64 |
| PA_D18 | ≤2 | ≥32 | 16 | 16 | ≥4 | ≥64 | ≥64 | ≥64 | 8 | ≥8 | ≥16 | ≤1 | ≥16 | 64 |
| PA_D22 | ≤2 | ≥32 | 16 | 8 | ≥4 | ≥64 | ≥64 | ≥64 | 8 | ≥8 | ≥16 | 2 | ≥16 | 64 |
| Patient 4 | ||||||||||||||
| PA_D16 | 4 | ≥32 | ≥64 | ≥64 | ≥4 | ≥64 | ≥64 | ≥64 | ≥64 | ≥8 | ≥16 | ≥16 | ≥16 | ≥128 |
| PA_D17 | 4 | ≥32 | ≥64 | ≥64 | ≥4 | ≥64 | ≥64 | ≥64 | ≥64 | ≥8 | ≥16 | ≥16 | ≥16 | ≥128 |
| PA_D20 | ≤2 | ≥32 | ≥64 | 16 | ≥4 | ≥64 | ≥64 | ≥64 | 32 | ≥8 | ≥16 | ≥16 | ≥16 | 32 |
| PA_D23 | 4 | ≥32 | ≥64 | 16 | ≥4 | ≥64 | ≥64 | ≥64 | 16 | ≥8 | ≥16 | ≥16 | ≥16 | 16 |
| PA_D24 | 4 | ≥32 | ≥64 | 16 | ≥4 | ≥64 | ≥64 | ≥64 | 16 | ≥8 | ≥16 | ≥16 | ≥16 | 16 |
| PA_D25 | ≤2 | ≥32 | ≥64 | 8 | ≥4 | ≥64 | ≥64 | ≥64 | 16 | ≥8 | ≥16 | ≥16 | ≥16 | 16 |
2.4. Evolutionary dynamics of Pseudomonas aeruginosa genome during short-term ventilator-associated pneumonia infection
We further analysed single-nucleotide polymorphisms (SNPs) and short indels between the earliest isolate and late isolates from each patient by mapping the HiSeq or MiSeq reads of late isolates to the genome of earliest isolate. In addition, we noted that some of the isolates displayed an autolytic colony phenotype, which suggests loss of function of lasR [18]. Since our SNP analysis failed to identify lasR mutations in some of these isolates, we further investigated if there are any genome rearrangement events that caused the disruption of lasR. As expected, genome rearrangement was identified in PA_D9 genome, which causes the disruption of both lasR and mpl (electronic supplementary material, figure S2). We then designed primers flanking both genes to screen for gene deletions for all the 25 isolates. The PCR results showed that PA_D8, which was isolated one day before PA_D9 from patient 1, probably underwent the same genome rearrangement that disrupted both lasR and mpl (electronic supplementary material, figure S2). In addition, we noted that PA_D4 and PA_D19 isolated from patient 2 showed deletion of lasR but not mpl (electronic supplementary material, figure S2). Taken together, we summarized all identified SNPs, short indels and gene deletion events in the 25 isolates in table 2.
Table 2.
Mutations identified in 25 isolates. SNPs, short indels and gene deletion events are summarized in this table. In each patient, the earliest isolate was chosen as the ancestor for the identification of mutations of the later isolates. For patient 2, the earliest isolate PA_D2 already harbours a non-synonymous mutation in lasR. Therefore, the detection of mutations in lasR for all the isolates from patient 2 were analysed with PA_D1 as the reference. Bold type indicates convergent evolution events in the P. aeruginosa genomes across the four VAP patients.
| isolate | time | coding region change | amino acid change | type | non-synonymous | remarks on lasR quorum sensing |
|---|---|---|---|---|---|---|
| patient 1 | ||||||
| PA_D1 | Day 1 | — | — | — | — | functional lasR quorum sensing |
| PA_D3 | Day 20 | mpl, 782T > G | Val261Gly | SNV | yes | functional lasR quorum sensing |
| PA_D8 | Day 38 | deletion of lasR | — | — | — | lasR mutant |
| deletion of mpl | — | — | — | |||
| PA_D9 | Day 39 | deletion of lasR | — | — | — | lasR mutant |
| deletion of mpl | — | — | — | |||
| patient 2 | ||||||
| PA_D2 | Day 1 | lasR, 167A > G (compared to PA_D1) | Tyr56Cys | SNV | yes | lasR mutant |
| PA_D4 | Day 14 | deletion of lasR | — | — | — | lasR mutant |
| mpl, 998G > A | Arg333His | SNV | yes | |||
| pelD, 201T > C | — | SNV | no | |||
| PA_D6 | Day 25 | lasR, 167A > G (compared to PA_D1) | Tyr56Cys | SNV | yes | lasR mutant |
| PA_D10 | Day 29 | mtlB, 296G > A | Gly99Asp | SNV | yes | functional lasR quorum sensing |
| pelD, 201T > C | — | SNV | no | |||
| PA_D12 | Day 50 | mpl, 998G > A | Arg333His | SNV | yes | functional lasR quorum sensing |
| ampD, 290G > A | Gly97Asp | SNV | yes | |||
| ampD, 262C > T | Gln88* | SNV | yes | |||
| pelD, 201T > C | — | SNV | no | |||
| PA_D19 | Day 77 | deletion of lasR | — | — | — | lasR mutant |
| mpl, 998G > A | Arg333His | SNV | yes | |||
| pvdS, 139T > C | Phe47Leu | SNV | yes | |||
| hypothetical protein, 286C > T | His96Tyr | SNV | yes | |||
| hypothetical protein, 42delC | Pro16fs | deletion | yes | |||
| BarA sensory histidine kinase, gacS, 1637T > C | Leu546Pro | SNV | yes | |||
| multimodular transpeptidase-transglycosylase, 1558T > C′ | Ser520Pro | SNV | yes | |||
| pelD, 201T > C | — | SNV | no | |||
| PA_D21 | Day 78 | pvdS, 139T > C | Phe47Leu | SNV | yes | functional lasR quorum sensing |
| hypothetical protein, 175A > C | Thr59Pro | SNV | yes | |||
| mtlB, 296G > A | Gly99Asp | SNV | yes | |||
| pelD, 201T > C | — | SNV | no | |||
| patient 3 | ||||||
| PA_D5 | Day 1 | — | — | — | — | functional lasR quorum sensing |
| PA_D7 | Day 8 | mpl, 104_105insC | Met38fs | insertion | yes | |
| lasR, 646C > T | Arg216Trp | SNV | yes | lasR mutant | ||
| PA_D11 | Day 27 | mpl, 104_105insC | Met38fs | insertion | yes | lasR mutant |
| lasR, 646C > T | Arg216Trp | SNV | yes | |||
| putative lipoprotein, 203A > G | Asp68Gly | SNV | yes | |||
| PA_D13 | Day 36 | mpl, 104_105insC | Met38fs | insertion | yes | lasR mutant |
| lasR, 646C > T | Arg216Trp | SNV | yes | |||
| hypothetical protein, 489G > A | Trp163* | SNV | yes | |||
| PA_D14 | Day 43 | mpl, 104_105insC | Met38fs | insertion | yes | lasR mutant |
| lasR, 646C > T | Arg216Trp | SNV | yes | |||
| fliK, 996G > T | — | SNV | no | |||
| PA_D15 | Day 50 | mpl, 104_105insC | Met38fs | insertion | yes | lasR mutant |
| lasR, 646C > T | Arg216Trp | SNV | yes | |||
| PA_D18 | Day 57 | mpl, 104_105insC | Met38fs | insertion | yes | lasR mutant |
| lasR, 646C > T | Arg216Trp | SNV | yes | |||
| PA_D22 | Day 68 | mpl, 104_105insC | Met38fs | insertion | yes | lasR mutant |
| lasR, 646C > T | Arg216Trp | SNV | yes | |||
| pvdS, 77T > C | Val26Ala | SNV | yes | |||
| patient 4 | ||||||
| PA_D16 | Day 1 | — | — | — | — | functional lasR quorum sensing |
| PA_D17 | Day 2 | — | — | — | — | functional lasR quorum sensing |
| PA_D20 | Day 8 | — | — | — | — | functional lasR quorum sensing |
| PA_D23 | Day 21 | lasR, 414delC | Glu139fs | deletion | yes | lasR mutant |
| gltJ, 137G > T | Arg46Leu | SNV | yes | |||
| gltJ, 140G > A | Arg47Gln | SNV | yes | |||
| gltJ, 146T > G | Leu49Arg | SNV | yes | |||
| gltJ, 149T > C | Leu50Pro | SNV | yes | |||
| gltJ, 151T > G | Ser51Ala | SNV | yes | |||
| predicted signal transduction protein, 596C > T | Pro199Leu | SNV | yes | |||
| dipeptide-binding ABC transporter, periplasmic substrate-binding component, 1228_1229insACG | Tyr410_Glu411insAsp | Insertion | yes | |||
| PA_D24 | Day 24 | lasR, 414delC | Glu139fs | deletion | yes | lasR mutant |
| pvdS, 343delC | Leu115fs | deletion | yes | |||
| PA_D25 | Day 25 | lasR, 414delC | Glu139fs | deletion | yes | lasR mutant |
| non-ribosomal peptide synthetase modules, pyoverdine, 5765_5766insG | Ala1925fs | insertion | yes | |||
| dppC, 160_161insC | Tyr56fs | insertion | yes | |||
| DNA primase, phage associated, 2006_2007insG | Ala672fs | insertion | yes | |||
| polymyxin resistance protein ArnT, undecaprenyl phosphate-alpha-L-Ara4N transferase; Melittin resistance protein PqaB, 647T > C | Leu216Pro | SNV | yes | |||
| probable sensor/response regulator hybrid, 649G > A | Gly217Arg | SNV | yes | |||
| NADH:ubiquinone oxidoreductase 49 kD subunit 7, 308C > T | Pro103Leu | SNV | yes | |||
| probable conserved membrane protein, 284T > C | Leu95Pro | SNV | yes | |||
| probable iron–sulfur binding protein YPO1417, 213delC | Gly72fs | deletion | yes | |||
| Kynurenine formamidase, 527T > C | Val176Ala | SNV | yes | |||
| AraC family transcriptional regulator, 88C > T | Arg30Cys | SNV | yes | |||
| potA, 584C > T | Ala195Val | SNV | yes | |||
| Enoyl-CoA hydratase, 466C > T | Pro156Ser | SNV | yes | |||
| 3-deoxy-manno-octulosonate cytidylyltransferase, 8A > G | Gln3Arg | SNV | yes | |||
| gacA, 242T > C | Val81Ala | SNV | yes | |||
| benABC operon transcriptional activator BenR, 866A > G | Asp289Gly | SNV | yes | |||
| hypothetical protein, 176A > G | Asp59Gly | SNV | yes | |||
| Alpha-methylacyl-CoA racemase, 841C > T | Arg281Trp | SNV | yes | |||
| response regulator containing a CheY-like receiver domain and a GGDEF domain, 180G > A | Met60Ile | SNV | yes | |||
| lysine-specific permease, 449G > A | Gly150Asp | SNV | no | |||
| putative translation initiation inhibitor, yjgF family, 184A > G | Ser62Gly | SNV | no | |||
| dppc, 160_161insC | Tyr56fs | insertion | yes | |||
| priA, 241C > T | — | SNV | no | |||
We found that P. aeruginosa evolved rapidly in all four VAP patients (table 2). In patient 1, PA_D3 carrying a non-synonymous mutation in mpl was isolated 20 days after the onset of VAP infection. At the late stage of infection, PA_D8 and PA_D9, both of which harbour disrupted lasR and mpl due to genome rearrangements, have replaced PA_D3 and dominated the airway of patient 1 (table 2; electronic supplementary material, figure S2). In patient 2, the earliest isolate PA_D2 already displayed a quorum-sensing negative phenotype, which is probably caused by a non-synonymous mutation in lasR (table 2), whereas the latest two isolates PA_D19 and PA_D21 carried a non-synonymous mutation in the pvdS gene. In patient 3, non-synonymous mutation in lasR and single-nucleotide insertion in mpl were first identified in PA_D7 isolated on day 8 and were also carried by all subsequent isolates. The latest isolate PA_D22 from patient 3 acquired an additional non-synonymous mutation in pvdS (table 2). In patient 4, the early isolates PA_D16, PA_D17 and PA_D20 were not identified with any mutations in the coding regions of their genomes, whereas a single-nucleotide deletion in lasR was identified in PA_D23, PA_D24 and PA_D25 (table 2). It was noted that both PA_D24 and PA_D25 produce much lower levels of pyoverdine (figure 3), which can be explained by a single-nucleotide deletion in pvdS of PA_D24, and a single-nucleotide insertion in a gene encoding a non-ribosomal peptide synthetase (NRPS) in the pyoverdine synthesis pathway of PA_D25 [19,20] (table 2).
Figure 3.

Decreased pyoverdine production by pvdS and NPRS mutants. The four pvdS mutants (PA_D19, PA_D21, PA_D22 and PA_D24) and PA_D25 produced significantly less pyoverdine compared with their ancestor strains, which do not carry any mutations in genes involved in pyoverdine synthesis pathway. Complementation of PA_D19, PA_D21 and PA_D22 with pUCP18::pvdS restored their pyoverdine production to the similar levels as their ancestors. We could not complement the PA_D24 strain with pUCP18::pvdS due to its high resistance to carbenicillin.
The ratio of non-synonymous substitution rate (dN) to synonymous substitutions rate (dS) could be used to infer the type of selective force that shapes the population during adaptive evolution; a dN/dS ratio of greater than 1 suggests positive selection during evolution. In this study, almost all detected mutations are non-synonymous SNPs, and gene deletions (table 2). This provided the direct evidence that strong positive selective forces have dominated the P. aeruginosa genomes in the VAP lungs.
2.5. Convergent evolution of Pseudomonas aeruginosa genome during in vivo infection
To investigate whether there are any convergent evolution events among the four patients, we compared the mutations carried by the latest isolates from each patient. The evolution of mutations in mpl, which encodes l-alanyl-gamma-d-glutamyl-meso-diaminopimelate ligase [21,22], was identified in the late isolates from patient 1, patient 2 and patient 3. The evolution of mutations in lasR, which encodes a major transcriptional activator of the las quorum-sensing system [23] was found in the late isolates from all the four patients (table 2). We also identified the evolution of pyoverdine-deficient mutants in patient 2, patient 3 and patient 4 (figure 3). This pyoverdine-deficient phenotype is probably due to mutations of pvdS, which encodes a sigma factor that coordinates the expression of multiple proteins involved in pyoverdine synthesis and many virulence factors [19], or a single-nucleotide insertion in a gene predicted to encode an NRPS module required for pyoverdine synthesis pathway [20] (table 2). Specifically, the pyoverdine-deficient mutants were isolated at much later stages of VAP infections than lasR and mpl mutants (table 2).
To confirm whether the pyoverdine-deficient phenotype of the pvdS mutants is due to non-synonymous SNPs in the pvdS gene, we cloned and inserted the wild-type pvdS gene of the ancestor PA_D1 into pUCP18 vector to construct a pUCP18::pvdS plasmid. Complementation of the PA_D19, PA_D21 and PA_D22 with the pUCP18::pvdS completely restored their pyoverdine production to the same level as their ancestors, confirming that the non-synonymous SNPs in pvdS are responsible for the pyoverdine-deficient phenotype of these mutants (figure 3).
2.6. Adaptive evolution of Pseudomonas aeruginosa in the lungs of ventilator-associated pneumonia patient leads to attenuated virulence in vitro and in vivo
Previous studies showed that both las quorum-sensing system and PvdS are required for the expression of various virulence factors, including elastase, rhamnolipids and alkaline proteases [23–27]. The convergent evolution of P. aeruginosa lasR and pvdS mutants during VAP infections has led us to hypothesize that the late isolates are less virulent compared with their earlier isolated counterparts. To verify this, we first assessed the in vitro production of elastase, which is a major virulence factor produced by P. aeruginosa [23], for all the 25 isolates. Interestingly, the earliest isolates from patient 1, patient 3 and patient 4 produced the highest levels of elastase, whereas the late isolates have reduced elastase production (figure 4). In patient 2, however, the earliest two isolates produced low levels of elastase, probably due to mutations of lasR, whereas the three later isolates produced much higher levels of elastase than the earliest isolate, and eventually the latest two isolates produced the lowest levels (figure 4). These results showed that the late isolates from VAP patient are probably less virulent than their early isolated counterparts.
Figure 4.
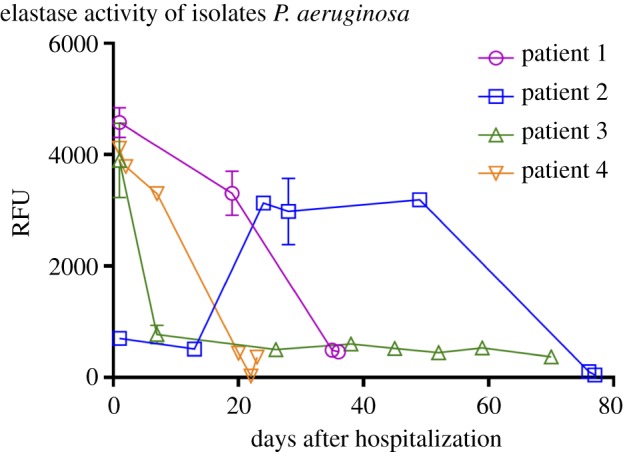
Elastase production of the 25 isolates. Isolates from the same patient were lined and plotted against the time of isolation. In patients 1, 3 and 4, the earliest isolates produced high levels of elastase, whereas the latest isolates produced much lower levels. In patient 2, the earliest two isolates low levels of elastase, whereas the three later isolates produced higher levels and eventually the latest two isolates produced the lowest levels of elastase.
Both quorum sensing and pyoverdine were previously shown to be important for P. aeruginosa to establish infections in vivo [11,28,29]. Therefore, we further employed a murine pulmonary infection model to compare the in vivo virulence of PA_D1 with PA_D21 (pvdS mutant), PA_D5 with PA_D22 (lasR/pvdS mutant), and PA_D16 with PA_D23 (lasR mutant), PA_D24 (lasR/pvdS mutant) and PA_D25 (lasR/NRPS mutant), in order to investigate if the late isolates are less virulent in vivo, and if so, which mutation(s) is the contributing factor. Of note, we cross-compared PA_D1 and PA_D21 because the ancestor strain PA_D2 from patient 2 already harbours a lasR mutation. These strains were administrated intranasally at 106 colony forming units (CFU) per mouse. After 24 h, the total number of bacteria residing in the lungs was numerated to assess the severity of pulmonary infection. As expected, the three ancestor strains PA_D1, PA_D5 and PA_D16 caused severe pulmonary infections in mice (figure 5). Intriguingly, the lasR mutant PA_D23 only showed a mild decrease in CFU compared with its ancestor PA_D16, whereas the pvdS mutant PA_D21 showed a drastic decrease in CFU compared with the wild-type PA_D1 (figure 5). Furthermore, the lasR/pvdS double mutants PA_D22 and PA_D24, and the lasR/NRPS double mutant PA_D25, all displayed the similar phenotype as the pvdS single mutant PA_D21 (figure 5). Taken together, these results confirmed that the late isolates are less virulent in vivo compared with the early isolates, which can largely be explained by mutations in pvdS or NRPS, and/or to a much smaller but significant extent, by mutations in lasR.
Figure 5.

The abilities of early and late isolates to cause acute lung infections in a murine pulmonary infection model. Mice were infected with each strain by intranasal inoculation at 106 CFU per mouse. The total bacterial count recovered from the lungs after 24 h of inoculation are shown in the figure. Comparison between CFU recovered from the lungs of mice infected with (a) PA_D1 (ancestor stain from patient 1) and PA_D21 (pvdS mutant from patient 2), (b) PA_D5 (ancestor stain from patient 3) and PA_D22 (lasR/pvdS mutant from patient 3), (c) PA_D16 (ancestor stain from patient 4) and PA_D23 (lasR mutant from patient 4), PA_D24 (lasR/pvdS mutant from patient 4), PA_D25 (lasR/NRPS mutant from patient 4). Solid lines: the median for each group. Dotted lines: the detection limit of CFU counting (n = 6 for PA_D21 and PA_D16, and n = 7 for the other isolates; *p < 0.05, **p < 0.01, ***p < 0.001, n.s. p > 0.05; Mann–Whitney test).
3. Discussion
In this study, we used both PacBio and Illumina sequencing to obtain the complete and draft genomes of 25 P. aeruginosa clinical isolates belonging to one monophyletic group. Clinical isolates from this group showed multidrug resistance and are responsible for VAP infections in a teaching hospital in China. We showed that these isolates may have acquired various antibiotic resistance genes, many of which are probably acquired through horizontal gene transfer events. This is evidenced by the fact that most of the acquired antibiotic resistance genes are in the GIs (figure 2). Furthermore, we also studied the in vivo evolution of this strain during VAP infections using the high-quality Illumina reads. The time scale for our in vivo evolutionary study is within a period of 78 days, whereas previous studies investigating the genetic adaptation and evolution of P. aeruginosa in the host mainly focused on lung infection in CF patients and the time scale for isolates sampling ranged from six months to several decades [10,30–34]. Our results showed that positive selection has shaped the P. aeruginosa genome and led to three convergent evolution events, which are the evolution of mutations in mpl and lasR and a pyoverdine-deficient phenotype.
In P. aeruginosa, the mpl mutants were shown to overexpress ampC, which leads to a higher resistance to β-lactam antibiotics such as penicillin and cephalosporins [21,22]. The deletion of mpl from P. aeruginosa was reported to cause an approximately 20-fold increase in the β-lactamase activity, which led to a 1.5-fold increase in ceftazidime MIC [21]. The association between mutation of mpl and the increased β-lactam antibiotic resistance was also observed in our results (tables 1 and 2). For example, in patient 1, the two mpl deletion mutants PA_D8 and PA_D9, as well as PA_D3 carrying a non-synonymous mutation in mpl showed a fourfold increase in ceftazidime MIC compared with their ancestor PA_D1. In patient 3, except for the ancestor strain PA_D5, all the other isolates carried a single-nucleotide insertion in mpl. These mutants all showed at least twofold increase of ceftazidime MIC and fourfold increase of piperacillin-tazobactam MIC compared with PA_D5. In patient 2, both PA_D4 and PA_D19 carry the same non-synonymous mutation in mpl, resulting in an amino acid change Arg333His. In these two mutants, we did not observe any increase in β-lactam MICs, probably because this amino acid change does not significantly affect the function of Mpl. Interestingly, the day 50 isolate PA_D12 that harbours a single-nucleotide insertion in the ampD gene showed a fourfold increase of ceftazidime MIC and a more than twofold increase of piperacillin-tazobactam MIC compared with PA_D2. Similar to mpl mutants, the ampD mutant was also reported to overexpress ampC, which leads to increased resistance to β-lactam antibiotics [21,35]. It is possible that the increased resistance of PA_D12 to both ceftazidime and piperacillin-tazobactam is resulted from the inactivation of the ampD gene. Taken together, the evolution of mpl and ampD mutants in VAP patients is probably due to the selective pressure imposed by intensive β-lactam treatments.
Like mpl, gene deletions, single-nucleotide insertions and two types of non-synonymous mutations were identified in the lasR gene of 16 isolates (table 2). PA_D8 and PA_D9 from patient 1, and PA_D4 and PA_D19 from patient 2, have lasR gene deleted from their genomes, whereas all the lasR mutants from patient 4 carry a single-nucleotide deletion in lasR. These seven isolates probably have completely abolished LasR function. The two lasR mutants from patient 2, PA_D2 and PA_D6 synthesize a Try56Cys LasR, which probably has a defective function because the highly conserved Try56 is responsible for LasR to form hydrogen bond with its autoinducer [36]. The seven lasR mutants isolated from patient 3 harbour a Arg216Trp mutation in LasR. Of note, the same type of LasR mutation was reported to emerge during the in vitro growth of P. aeruginosa in rich medium [18]. Arg216 is a conserved residue that lies in α-helix 9 of LasR, which is essential for DNA recognition by LuxR-type transcriptional factors [37]. Mutations at residues Try56 and Arg216 probably affect the ligand binding and target DNA recognition of the LasR protein, respectively. Therefore, we conclude that all the identified lasR mutants in this study have severely impaired, if not completely abolished LasR function.
The five pyoverdine-deficient mutants were isolated from three patients at very late stages of VAP infections (table 2). These mutants harbour either non-synonymous mutations (PA_D19, PA_D21 and PA_D22), a single-nucleotide deletion in pvdS (PA_D24), or a single-nucleotide insertion in a 14 976 bp gene encoding an NRPS module required for pyoverdine synthesis (PA_D25) (table 2). In P. aeruginosa, loss of function of PvdS or NRPS was reported to abolish its pyoverdine biosynthesis, which usually results in a pyoverdine-deficient phenotype [20,38]. As expected, indel mutations in pvdS and the NRPS gene probably have completely abolished the functions of the two genes, resulting in complete loss of pyoverdine production in both PA_D24 and PA_D25. The non-synonymous pvdS 139T > C mutation harboured by PA_D19 and PA_D21 causes a mutation at Phe47, which is a highly conserved residue in the region 2.1 of sigma factors; this mutation was shown to drastically affect the function of PvdS [39]. By contrast, the PvdS Val26Ala mutation carried by PA_D22 does not happen at an essential amino acid residue, but rather adjacent to a conserved residue Leu25 [39]. Therefore, although PA_D22 showed a reduced level of pyoverdine production than its ancestor PA_D5, it still produces a higher amount of pyoverdine than the other three pvdS mutants (figure 3). Nevertheless, complementation of all the three pvdS mutants carrying non-synonymous SNPs with wild-type pvdS have fully restored their pyoverdine production to the same level as their ancestors, confirming that pyoverdine-deficient phenotype is due to mutations in pvdS (figure 3). Previous studies have reported the isolation of pyoverdine-deficient mutants from CF lungs, and the chance of isolating pyoverdine-deficient mutants seemed to increase upon the increment of patient age [9,40,41]. We propose that the convergent evolution of pyoverdine-deficient mutants is possibly due to three reasons. First, the pyoverdine-deficient strains could acquire iron from alternative sources to compensate for the defective ferric iron uptake in the patients’ airways. Second, the reduced pyoverdine synthesis of these mutants may lead to reduced virulence, which may result in less immune attack on pyoverdine-deficient mutants from the host, and hence, enriched pyoverdine-deficient mutants at the infection sites. Third, reduced pyoverdine synthesis has significantly reduced metabolic cost in these mutants, which may associate with better fitness in the host environment.
The pyoverdine-deficient mutants were reported to be deficient in producing exotoxin A and PrpL proteases [42], which are important virulence factors produced by P. aeruginosa. Similarly, loss-of-function mutations of lasR also reduce the production of many virulence factors such as exotoxin A, elastase, rhamnolipids and pyocyanin in P. aeruginosa [23–27]. The convergent evolution of lasR and pyoverdine-deficient mutants in VAP patients suggested that the short-term in vivo evolution selected less virulent P. aeruginosa mutants. In agreement with this, the late isolates did produce lower levels of a major virulence factor elastase in vitro compared with the earlier isolated counterparts (figure 4). We also compared the in vivo virulence of lasR and pyoverdine-deficient mutants using a murine pulmonary infection model. Surprisingly, we found that mutations causing the pyoverdine-deficient phenotype greatly impaired the in vivo virulence of P. aeruginosa, whereas lasR mutation has a much milder effect (figure 5). These findings are in line with previous studies showing that both intact lasR function and pyoverdine synthesis are important for in vivo virulence [11,28,29]. In addition, our results also provide a direct comparison on the in vivo virulence of the two types of mutants isolated from clinical sources.
In conclusion, we identified a novel P. aeruginosa strain responsible for a nosocomial outbreak of VAP infections in China. We showed that this strain could undergo rapid adaptive evolution during VAP infections, which led to increased resistance to β-lactams, a defective quorum-sensing system, reduced production of pyoverdine, and attenuated virulence in vitro and in vivo. Our findings provide novel insights into the short-term evolution of P. aeruginosa in the human airways during acute VAP infections.
4. Methods
4.1. Isolate collection and characterization
All P. aeruginosa isolates were collected from clinical specimens. Sputum samples from VAP patients were streaked onto blood agar plate and incubated at 35°C for 18 h. Three colonies from each cultivation were randomly selected and sub-cultured in LB broth and stored in 25% glycerol under −80°C. The 16S rRNA gene of each isolate was amplified using primers 27F (5′-GAGTTTGATCCTGGCTCAG-3′) and 1492R (5′-GGTTACCTTGTTACGACTT-3′) [43]. PCR products were purified and sequenced to identify the species of each isolate. RAPD typing was performed using primer 272 as previously described [44]. Susceptibilities to various antibiotics were determined by the VITEK 2 Compact system (bioMérieux).
4.2. Sequencing, assembly and annotation
Genomic DNA from a single colony of each isolate was purified using Blood and Cell Culture DNA Midi Kit (Qiagen) and sequenced on PacBio RS II platform, Illumina HiSeq 2500 platform or Illumina MiSeq platform. The sequencing data were used to assemble the full-length genomes and identify SNPs in this study. All sequencing data used in this study are available on NCBI registered under BioProject no. PRJNA294254. The full-length genome of eight isolates was assembled from long reads obtained from PacBio RS II system using HGAP3 pipeline. The assembled genomes were then corrected with high-quality Illumina HiSeq reads using CLC Genomic Workbench 8.5 (CLC Bio, Qiagen) assisted with manual curation to further minimize sequencing errors and close gaps in the assembled genomes. The corrected full-genome files were uploaded to the Rapid Annotations using Subsystem Technology (RAST) server for bacterial genome annotation [14].
4.3. Comparative genomics and phylogenetic analysis
Core-genome alignment was carried out using Parsnp v. 1.1.2 [45], yielding an alignment with 4 561 475 putatively homologous sites. Of these, 136 126 sites were found to contain potential single-nucleotide variants. Recombinant sites in the alignment were then identified with PhiPack [46] (version integrated with the Parsnp). In total, 132 511 variant sites remained after removing putative recombinant and low-quality variant sites. Phylogenetic inference was then carried out on the variant sites using the approximate maximum-likelihood algorithm implemented in FastTree2 (version integrated with Parsnp), with clade confidence estimated with SH-like support values [47]. Finally, branch lengths in the inferred tree were rescaled from substitutions per variant site to represent substitutions per core-genome site. Genomic islands of PA_D1 genome were predicted by IslandViewer 3 server [15]. Antibiotic resistance genes were predicted from the 25 assembled genomes were done using the ResFinder 2.1 server [16]. Comparison of PA_D1 genome against other five full P. aeruginosa genomes was done by BLAST search using BLAST Ring Image Generator 0.95 [48], with locations of genomic islands and antibiotic resistance genes marked in the output image as instructed by the manual.
4.4. Detection of nucleotide differences
Nucleotide differences were detected and evaluated using CLC Genomic Workbench 8.5. Paired-end reads in FASTQ formats were mapped to the annotated PA_D1 reference genome and further compared with each other to generate lists of SNPs and short indels. A frequency cut-off of more than 80% was set to minimize false SNPs due to sequencing error. PCR was used to verify detected gene deletions. Genome rearrangements were detected by ProgressiveMauve v. 2.3.1 [49]. Primers lasR_F (5′-GGAATTCCTTCTCGGACTGCCGTACAAC-3′) and lasR_R (5′-GCTCTAGAGCAAATTACCGATCGCCAG-3′) were used to amplify the entire coding region of lasR, whereas mpl_F (5′-CAACACCCTGTATCGCAAGC-3′) and mpl_R (5′-ATCGCCAGGGTAATGCGTTC-3′) were used to amplify mpl. PCR products were resolved in 1% agarose gel. The sizes of the amplified products are 865 bp and 1608 bp, respectively.
4.5. Quantification of elastase and pyoverdine
Bacterial strains were grown in ABT minimal medium [50] supplemented with 5 g l–1 glucose and 2 g l–1 casamino acids. The filtered supernatants of overnight culture were used to quantify the productions of elastase and pyoverdine in vitro. The quantification of elastase production was performed in a 96-well plate using EnzCheck Elastase Assay Kit (Thermo Fisher Scientific) as instructed by the manual. Elastase activity was measured by Infinite M200 PRO system (Tecan) and the results were shown as emission at 530 nm. For the quantification of pyoverdine, filtered supernatant was measured for fluorescence signal emitted at 460 nm upon excitation at 398 nm using Infinite M200 PRO system (Tecan). Both experiments were performed in at least triplicates and the results are shown as mean ± standard deviation in the figures.
4.6. Pvds complementation
The wild-type pvdS gene was amplified from PA_D1 genomic DNA using primers pvdS_EcoRI_F (5′-CCGAATTCCGCAGCAAGGTGATTTCCAT-3′) and pvdS_KpnI_R (5′-CCGGTACCCTGCGAGAAGGAGTTCGACT-3′), and inserted into EcoRI and KpnI sites in pUCP18 vector to construct pvdS complementation plasmid pUCP18::pvdS. The pUCP18::pvdS plasmid was transformed into PA_D19, PA_D21 and PA_D22 by electroporation.
4.7. Animal model
A murine pulmonary infection model was used to evaluate the virulence of P. aeruginosa strains. Briefly, bacterial cells of overnight culture were washed three times and re-suspended in phosphate-buffered saline. Bacterial suspensions were administrated intranasally to each mouse at 20 µl per mouse under anaesthesia (approx. 1 × 106 CFU per mouse). After 24 h, all mice were sacrificed and the lungs were harvested and kept in ice-cold 0.9% NaCl saline. The bacterial cells residing in the lungs were suspended into the 0.9% NaCl saline by homogenization using a Bio-Gen PRO200 Homogenizer (Pro Scientific). CFU was quantified by serial dilution and plating on LB agar.
4.8. Nucleotide sequence accession numbers
The whole-genome Illumina shotgun sequencing for P. aeruginosa PA_D1 to PA_D25 has been deposited at GenBank under the accession numbers SRX2066370, SRX2066389, SRX2066394, SRX2066397, SRX2066399, SRX2066406, SRX2066408, SRX2066409, SRX2066415, SRX2066423, SRX2066444, SRX2066449, SRX2066453, SRX2066456, SRX2066475, SRX2066480, SRX2066484, SRX2066486, SRX2066488, SRX2066489, SRX2066521, SRX2066560, SRX2066602, SRX2066605 and SRX2066607. The whole-genome shotgun sequencing on Pacbio RS II platform for the 8 isolates are deposited under accession numbers SRX2066384, SRX2066390, SRX2066402, SRX2066416, SRX2066482, SRX2066542, SRX2066581 and SRX2066616.
Supplementary Material
Ethics
The use of clinical specimen samples was approved by the First Affiliated Hospital of Guangxi Medical University, registered under the reference no. 2014-KY-028. All patients have given informed consent. The study was performed in accordance with approved guidelines, and all experimental protocols were approved by the First Affiliated Hospital of Guangxi Medical University. The animal model protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of Nanyang Technological University, under the permit number A-0228 AZ. Animal experiments were performed in accordance to the NACLAR Guidelines of Animal and Birds (Care and Use of Animals for Scientific Purposes) Rules by Agri-Food & Authority (AVA) of Singapore.
Authors' contributions
L.Y., S.C.S. and Y.D. designed the experiments; K.W. and Y.C. designed the clinical sample collection scheme; K.W., J.K., H.L., Z.Y., Y.X., H.W. and S.C. collected clinical specimens, and isolated and screened bacterial strains; D.I.D.-M. performed whole-genome sequencing; Y.D. analysed sequencing data, carried out laboratory experiments and interpreted results; A.E.D. and G.S.K. conducted phylogenetic analysis; M.M.S. and Y.D. did animal work; Y.D. wrote the manuscript; L.Y., S.C.S., G.S.K., K.W. and Y.C. commented on the manuscript.
Competing interests
The authors declare no conflict of interest.
Funding
The genomic analysis part of the research is supported by the National Research Foundation and Ministry of Education Singapore under its Research Centre of Excellence Programme AcRF TIER 2 (MOE2014-T2-2-172) and TIER3 (MOE2013-T3-1-003) from Ministry of Education, Singapore. The clinical part of the research was supported by the National Natural Sciences Foundation of China (grant nos. 81260663 and 81460003).
References
- 1.Vincent JL, Bihari DJ, Suter PM, Bruining HA, White J, Nicolas-Chanoin MH, Wolff M, Spencer RC, Hemmer M. 1995. The prevalence of nosocomial infection in intensive care units in Europe: results of the European Prevalence of Infection in Intensive Care (EPIC) study. JAMA 274, 639–644. (doi:10.1001/jama.1995.03530080055041) [PubMed] [Google Scholar]
- 2.Bou R, Lorente L, Aguilar A, Perpinan J, Ramos P, Peris M, Gonzalez D. 2009. Hospital economic impact of an outbreak of Pseudomonas aeruginosa infections. J. Hosp. Infect. 71, 138–142. (doi:10.1016/j.jhin.2008.07.018) [DOI] [PubMed] [Google Scholar]
- 3.Fujitani S, Sun HY, Yu VL, Weingarten JA. 2011. Pneumonia due to Pseudomonas aeruginosa: part I: epidemiology, clinical diagnosis, and source. Chest 139, 909–919. (doi:10.1378/chest.10-0166) [DOI] [PubMed] [Google Scholar]
- 4.Govan JR, Brown PH, Maddison J, Doherty CJ, Nelson JW, Dodd M, Greening AP, Webb AK. 1993. Evidence for transmission of Pseudomonas cepacia by social contact in cystic fibrosis. Lancet 342, 15–19. (doi:10.1016/0140-6736(93)91881-L) [DOI] [PubMed] [Google Scholar]
- 5.Kohler T, Guanella R, Carlet J, van Delden C. 2010. Quorum sensing-dependent virulence during Pseudomonas aeruginosa colonisation and pneumonia in mechanically ventilated patients. Thorax 65, 703–710. (doi:10.1136/thx.2009.133082) [DOI] [PubMed] [Google Scholar]
- 6.Hentzer M, et al. 2009. Attenuation of Pseudomonas aeruginosa virulence by quorum sensing inhibitors. EMBO J. 22, 3803–3815. (doi:10.1093/emboj/cdg366) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gil-Perotin S, Ramirez P, Marti V, Sahuquillo JM, Gonzalez E, Calleja I, Menendez R, Bonastre J. 2012. Implications of endotracheal tube biofilm in ventilator-associated pneumonia response: a state of concept. Crit. Care 16, R93 (doi:10.1186/cc11357) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bjarnsholt T, Jensen PO, Fiandaca MJ, Pedersen J, Hansen CR, Andersen CB, Pressler T, Givskov M, Høiby N. 2009. Pseudomonas aeruginosa biofilms in the respiratory tract of cystic fibrosis patients. Pediatr. Pulmonol. 44, 547–558. (doi:10.1002/ppul.21011) [DOI] [PubMed] [Google Scholar]
- 9.Marvig RL, Sommer LM, Molin S, Johansen HK. 2015. Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat. Genet. 47, 57–64. (doi:10.1038/ng.3148) [DOI] [PubMed] [Google Scholar]
- 10.Smith EE, et al. 2006. Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc. Natl Acad. Sci. USA 103, 8487–8492. (doi:10.1073/pnas.0602138103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lehoux DE, Sanschagrin F, Levesque RC. 2000. Genomics of the 35-kb pvd locus and analysis of novel pvdIJK genes implicated in pyoverdine biosynthesis in Pseudomonas aeruginosa. FEMS. Microbiol. Lett. 190, 141–146. (doi:10.1111/j.1574-6968.2000.tb09276.x) [DOI] [PubMed] [Google Scholar]
- 12.Latifi A, Foglino M, Tanaka K, Williams P, Lazdunski A. 1996. A hierarchical quorum-sensing cascade in Pseudomonas aeruginosa links the transcriptional activators LasR and RhIR (VsmR) to expression of the stationary-phase sigma factor RpoS. Mol. Microbiol. 21, 1137–1146. (doi:10.1046/j.1365-2958.1996.00063.x) [DOI] [PubMed] [Google Scholar]
- 13.Kohler T, Buckling A, van Delden C. 2009. Cooperation and virulence of clinical Pseudomonas aeruginosa populations. Proc. Natl Acad. Sci. USA 106, 6339–6344. (doi:10.1073/pnas.0811741106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aziz RK, et al. 2008. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9, 75 (doi:10.1186/1471-2164-9-75) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Langille MG, Brinkman FS. 2009. IslandViewer: an integrated interface for computational identification and visualization of genomic islands. Bioinformatics 25, 664–665. (doi:10.1093/bioinformatics/btp030) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, Aarestrup FM, Larsen MV. 2012. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. (doi:10.1093/jac/dks261) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dhillon BK, et al. 2015. IslandViewer 3: more flexible, interactive genomic island discovery, visualization and analysis. Nucleic Acids Res. 43(W1), W104–W108. (doi:10.1093/nar/gkv401) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.D'Argenio DA, et al. 2007. Growth phenotypes of Pseudomonas aeruginosa lasR mutants adapted to the airways of cystic fibrosis patients. Mol. Microbiol. 64, 512–533. (doi:10.1111/j.1365-2958.2007.05678.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cunliffe HE, Merriman TR, Lamont IL. 1995. Cloning and characterization of pvdS, a gene required for pyoverdine synthesis in Pseudomonas aeruginosa: PvdS is probably an alternative sigma factor. J. Bacteriol. 177, 2744–2750. (doi:10.1128/jb.177.10.2744-2750.1995) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mossialos D, et al. 2002. Identification of new, conserved, non-ribosomal peptide synthetases from fluorescent pseudomonads involved in the biosynthesis of the siderophore pyoverdine. Mol. Microbiol. 45, 1673–1685. (doi:10.1046/j.1365-2958.2002.03120.x) [DOI] [PubMed] [Google Scholar]
- 21.Alvarez-Ortega C, Wiegand I, Olivares J, Hancock RE, Martínez JL. 2010. Genetic determinants involved in the susceptibility of Pseudomonas aeruginosa to β-lactam antibiotics. Antimicrob. Agents Chemother. 54, 4159–4167. (doi:10.1128/AAC.00257-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsutsumi Y, Tomita H, Tanimoto K. 2013. Identification of novel genes responsible for overexpression of ampC in Pseudomonas aeruginosa PAO1. Antimicrob. Agents Chemother. 57, 5987–5993. (doi:10.1128/AAC.01291-13) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pearson JP, Pesci EC, Iglewski BH. 1997. Roles of Pseudomonas aeruginosa las and rhl quorum-sensing systems in control of elastase and rhamnolipid biosynthesis genes. J. Bacteriol. 179, 5756–5767. (doi:10.1128/jb.179.18.5756-5767.1997) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gambello MJ, Kaye S, Iglewski BH. 1993. LasR of Pseudomonas aeruginosa is a transcriptional activator of the alkaline protease gene (apr) and an enhancer of exotoxin A expression. Infect. Immun. 61, 1180–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Latifi A, Winson MK, Foglino M, Bycroft BW, Stewart GS, Lazdunski A, Williams P. 1995. Multiple homologues of LuxR and LuxI control expression of virulence determinants and secondary metabolites through quorum sensing in Pseudomonas aeruginosa PAO1. Mol. Microbiol. 17, 333–343. (doi:10.1111/j.1365-2958.1995.mmi_17020333.x) [DOI] [PubMed] [Google Scholar]
- 26.Ochsner UA, Reiser J. 1995. Autoinducer-mediated regulation of rhamnolipid biosurfactant synthesis in Pseudomonas aeruginosa. Proc. Natl Acad. Sci. USA 92, 6424–6428. (doi:10.1073/pnas.92.14.6424) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Toder D, Ferrell S, Nezezon J, Rust L, Iglewski B. 1994. lasA and lasB genes of Pseudomonas aeruginosa: analysis of transcription and gene product activity. Infect. Immun. 62, 1320–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pearson JP, Feldman M, Iglewski BH, Prince A. 2000. Pseudomonas aeruginosa cell-to-cell signaling is required for virulence in a model of acute pulmonary infection. Infect. Immun. 68, 4331–4334. (doi:10.1128/IAI.68.7.4331-4334.2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Minandri F, Imperi F, Frangipani E, Bonchi C, Visaggio D, Facchini M, Pasquali P, Bragonzi A, Visca P. 2016. Role of iron uptake systems in Pseudomonas aeruginosa virulence and airway infection. Infect. Immun. 84, 2324–2335. (doi:10.1128/IAI.00098-16) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang L, et al. 2011. Evolutionary dynamics of bacteria in a human host environment. Proc. Natl Acad. Sci. USA 108, 7481–7486. (doi:10.1073/pnas.1018249108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mena A, et al. 2008. Genetic adaptation of Pseudomonas aeruginosa to the airways of cystic fibrosis patients is catalyzed by hypermutation. J. Bacteriol. 190, 7910–7917. (doi:10.1128/JB.01147-08) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huse HK, Kwon T, Zlosnik JE, Speert DP, Marcotte EM, Whiteley M. 2010. Parallel evolution in Pseudomonas aeruginosa over 39,000 generations in vivo. MBio 1, e00199-10 (doi:10.1128/mBio.00199-10) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marvig RL, Johansen HK, Molin S, Jelsbak L.. 2013. Genome analysis of a transmissible lineage of pseudomonas aeruginosa reveals pathoadaptive mutations and distinct evolutionary paths of hypermutators. PLoS Genet. 9, e1003741 (doi:10.1371/journal.pgen.1003741) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feliziani S, Marvig RL, Lujan AM, Moyano AJ, Di Rienzo JA, Krogh Johansen H, Molin S, Smania AM. 2014. Coexistence and within-host evolution of diversified lineages of hypermutable Pseudomonas aeruginosa in long-term cystic fibrosis infections. PLoS Genet. 10, e1004651 (doi:10.1371/journal.pgen.1004651) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Langaee TY, Gagnon L, Huletsky A. 2000. Inactivation of the ampD gene in Pseudomonas aeruginosa leads to moderate-basal-level and Hyperinducible AmpC β-lactamase expression. Antimicrob. Agents Chemother. 44, 583–589. (doi:10.1128/AAC.44.3.583-589.2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bottomley MJ, Muraglia E, Bazzo R, Carfì A. 2007. Molecular insights into quorum sensing in the human pathogen Pseudomonas aeruginosa from the structure of the virulence regulator LasR bound to its autoinducer. J. Biol. Chem. 282, 13 592–13 600. (doi:10.1074/jbc.M700556200) [DOI] [PubMed] [Google Scholar]
- 37.Vannini A, Volpari C, Gargioli C, Muraglia E, Cortese R, De Francesco R, Neddermann P, Di Marco S. 2002. The crystal structure of the quorum sensing protein TraR bound to its autoinducer and target DNA. EMBO J. 21, 4393–4401. (doi:10.1093/emboj/cdf459) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ochsner UA, Wilderman PJ, Vasil AI, Vasil ML. 2002. GeneChip expression analysis of the iron starvation response in Pseudomonas aeruginosa: identification of novel pyoverdine biosynthesis genes. Mol. Microbiol. 45, 1277–1287. (doi:10.1046/j.1365-2958.2002.03084.x) [DOI] [PubMed] [Google Scholar]
- 39.Wilson MJ, Lamont IL. 2006. Mutational analysis of an extracytoplasmic-function sigma factor to investigate its interactions with RNA polymerase and DNA. J. Bacteriol. 188, 1935–1942. (doi:10.1128/JB.188.5.1935-1942.2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Vos D, De Chial M, Cochez C, Jansen S, Tümmler B, Meyer J-M, Cornelis P. 2001. Study of pyoverdine type and production by Pseudomonas aeruginosa isolated from cystic fibrosis patients: prevalence of type II pyoverdine isolates and accumulation of pyoverdine-negative mutations. Arch. Microbiol. 175, 384–388. (doi:10.1007/s002030100278) [DOI] [PubMed] [Google Scholar]
- 41.Martin LW, Reid DW, Sharples KJ, Lamont IL. 2011. Pseudomonas siderophores in the sputum of patients with cystic fibrosis. Biometals 24, 1059–1067. (doi:10.1007/s10534-011-9464-z) [DOI] [PubMed] [Google Scholar]
- 42.Lamont IL, Beare PA, Ochsner U, Vasil AI, Vasil ML. 2002. Siderophore-mediated signaling regulates virulence factor production in Pseudomonas aeruginosa. Proc. Natl Acad. Sci. USA 99, 7072–7077. (doi:10.1073/pnas.092016999) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Flanagan J, et al. 2007. Loss of bacterial diversity during antibiotic treatment of intubated patients colonized with Pseudomonas aeruginosa. J. Clin. Microbiol. 45, 1954–1962. (doi:10.1128/JCM.02187-06) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mahenthiralingam E, Campbell ME, Foster J, Lam JS, Speert DP. 1996. Random amplified polymorphic DNA typing of Pseudomonas aeruginosa isolates recovered from patients with cystic fibrosis. J. Clin. Microbiol. 34, 1129–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Treangen TJ, Ondov BD, Koren S, Phillippy AM.. 2014. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 15, 1 (doi:10.1186/s13059-014-0524-x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bruen TC, Philippe H, Bryant D. 2006. A simple and robust statistical test for detecting the presence of recombination. Genetics 172, 2665–2681. (doi:10.1534/genetics.105.048975) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Price MN, Dehal PS, Arkin AP. 2010. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490 (doi:10.1371/journal.pone.0009490) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alikhan N-F, Petty NK, Zakour NLB, Beatson SA.. 2011. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12, 402 (doi:10.1186/1471-2164-12-402) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Darling AE, Mau B, Perna NT.. 2010. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5, e11147 (doi:10.1371/journal.pone.0011147) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.JosephClark D, Maaløe O. 1967. DNA replication and the division cycle in Escherichia coli. J. Mol. Biol. 23, 99–112. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.