

Abstract
Genome size varies considerably across taxa, and extensive research effort has gone into understanding whether variation can be explained by differences in key ecological and life-history traits among species. The extreme environmental conditions that characterize the deep sea have been hypothesized to promote large genome sizes in eukaryotes. Here we test this supposition by examining genome sizes among 13 species of deep-sea amphipods from the Mariana, Kermadec and New Hebrides trenches. Genome sizes were estimated using flow cytometry and found to vary nine-fold, ranging from 4.06 pg (4.04 Gb) in Paralicella caperesca to 34.79 pg (34.02 Gb) in Alicella gigantea. Phylogenetic independent contrast analysis identified a relationship between genome size and maximum body size, though this was largely driven by those species that display size gigantism. There was a distinct shift in the genome size trait diversification rate in the supergiant amphipod A. gigantea relative to the rest of the group. The variation in genome size observed is striking and argues against genome size being driven by a common evolutionary history, ecological niche and life-history strategy in deep-sea amphipods.
Keywords: genome size, deep sea, amphipods, gigantism, adaptation, evolution
1. Introduction
Understanding the causes and consequences of the extraordinary variation in genome sizes found among eukaryotes is an enduring issue in ecology and evolution. Genome sizes range from less than 2.3 Mb in the protist Encephalitozoon intestinalis [1] to over 149 Gb in the canopy plant Paris japonica [2]. Genomes can increase or decrease in size through a variety of mechanisms, including whole-genome duplication [3], the loss or gain of individual genes or gene families [4], recombination events [5,6] or the gradual accumulation of ‘junk’ genetic material such as pseudogenes [7], transposable elements (TEs) [8] or repeat arrays [9].
Despite a growing body of data on genome sizes from across a broad taxonomy, plus a recognition that genome size variation is at least partly determined by natural selection, there is still little consensus on the underlying ecological [10] or environmental [11] drivers of genome size. Indeed, the lack of any overarching phylogenetic signature for genome size, and the high variation that can occur within taxonomic lineages, suggests that genome size diversity is a consequence of multiple factors with varying influence in different taxa and in different habitats. Notwithstanding, multiple hypotheses have been proposed that attempt to provide a general explanation or single driver for genome size in particular taxonomic groups. The majority of focus has been on the interrelationships between genome size, nuclear envelope size, cell size and body size, and the ecological or physiological traits that can influence one or more of these individual components [12].
There are well-established positive relationships between genome size and nucleus size, and between nucleus size and cell size in all major taxonomic groups. It has been suggested that these are a consequence of large cells requiring large genomes for structural reasons, and larger cells necessitating larger nuclei to maintain efficient transport of mRNA into the cytoplasm [13]. Similarly, there are established links between key ecological, physiological and life-history traits and cell size that could have downstream effects on genome size evolution and diversity. For example, fast growth rate and high metabolic rates of species with r-selected life-history traits can select for small cell size through mechanical constraints associated with faster cell replication and metabolic activity, and/or a requirement to allocate phosphorous from DNA to RNA because of the demand for ribosomes to allow protein synthesis during rapid growth [14,15]. Both scenarios would then predict small genome sizes, which is indeed a characteristic feature of r-selected species [12,16].
These logical links between life history, cell size and genome size are readily observed in amphipod crustaceans. There is a 460-fold variation in crustacean genome sizes [17] and 100-fold variation within the amphipods [18], which reflects the wide range of marine, semi-terrestrial and freshwater habitats the taxa occupy. This has facilitated attempts to establish links between ecological or evolutionary constraint with genome size. For example, in several arctic species of amphipods, the reduced temperature lowers metabolic activity and growth rate relative to sub-polar and temperate regions. This would predict larger than average genome size and this has been largely shown to be the case [18]. Indeed, the largest crustacean genome is found in the arctic amphipod Ampelisca macrocephola [18].
Here we expand our understanding of the extent to which genome size can be considered a determinant of life-history, or vice versa, in Crustacea by providing, (to our knowledge), the first reports of genome sizes for deep-sea lyssianassoid amphipods collected from bathyal (1000–3000 m), abyssal (3000–6000 m) and hadal (6000 to approx. 11 000 m) depths. All aspects of the ecology and life history of the deep-sea amphipods would predict that they possess large genome sizes. In the classical r-K-A life-history continuum [19,20] the deep-sea amphipods are categorized as adversity or ‘A-selected’ species as a result of their low fecundity, slow development rate and by occupying ecological niches in poorly productive but predictable environments [21]. As with the arctic species, this would predict low metabolic rates, large cells and concomitantly large genomes.
Moreover, it has also been suggested that with the reduced temperature and increased hydrostatic pressure associated with greater ocean depth there should be an increase in cell size and life span in exactly the same way that Bergmann's principle generates the eco-geographical pattern of larger body size at higher latitude [22]. This would, in turn, also predict larger genome sizes in the amphipods relative to their shallow water equivalents. Indeed, this deep-sea extension of Bergmann's rule has been considered to generate the size gigantism characteristic of some deep-sea amphipods, notably in the genus Eurythenes [23] and more obviously the ‘supergiant’ amphipod Alicella gigantea [24] that can grow to lengths of over 25 cm.
The high hydrostatic pressures that define the deep-sea environment are also posited to drive large genome sizes [25,26]. It is well recognized that the deep sea has been colonized multiple times from shallower waters following climate-induced dysoxic mass extinction events [27,28]. The physiological effects of high hydrostatic pressure will limit the extent of range expansion of shallow water species, and those pioneer species that do reach the deep sea will have experienced an environmental stress that, among other things, disrupts the epigenetic control of TEs leading to TE proliferation and increased genome size [29–31].
Here we directly test the hypothesis that deep-sea amphipods have large genomes. We use flow cytometry to estimate genome sizes across 13 species of the Lysianassoidea from the Kermadec, Mariana and New Hebrides trenches and examine results in a phylogenetically controlled way to identify ecological and life-history correlates of genome size. We compare genome sizes with a broader range of amphipods, with particular recourse to key studies that have characterized genome sizes in arctic species [18] and across the depths of Lake Baikal [32].
2. Material and methods
2.1. Sample collection
Amphipods were collected over the course of three research cruises: in 2013 to the Kermadec Trench (approx. 26°43′ S 175°11′ W), New Hebrides Trench (approx. 21°13′ S 168°14′ E) and South Fiji Basin (approx. 24°58′ S 171°3′ E); and in 2014 to the Mariana Trench (approx. 18°49′ N 149°50′ E). In all cases an autonomous, full ocean depth rated lander vehicle was deployed to the seafloor at various depths (for details see table 1) which incorporated small baited funnel traps for sample collection [33]. Upon recovery of the lander vehicle, samples were frozen in liquid nitrogen and stored at −80°C.
Table 1.
Table of 13 deep-sea amphipod species with calculated C-values, associated GenBank Accession Numbers and ecological data. (Ecological data were compiled from data in this study and wider literature.)
| GenBank accession numbers |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| species | C-value ± s.e. | genome size (Gb) | 16S | COI | 18S | depth range (m) | max. depth (m) | median depth (m) | max. body length (mm) |
| Lanceola sp. | — | — | KP456062 | KP713953 | KT372894 | — | — | — | — |
| Abyssorchomene sp. | 9.81 | 9.59 | KX034333 | KX365238 | KX365242 | 1010 | 2500 | 1995 | 9 |
| Abyssorchomene chevreuxi | 16.46 | 16.10 | KX034329 | KP713882 | KP347454 | 3300 | 5400 | 3750 | 14 |
| Abyssorchomene distinctusa | 15.30 ± 0.04 | 14.96 | KX034327 | KP713886 | KT372892 | 3800 | 6007 | 3004 | 14 |
| Unidentified amphipod | 9.09 | 8.89 | KX034299 | KX365239 | KX365243 | 4984 | 7484 | 4992 | — |
| Alicella giganteaa | 34.79 ± 1.43 | 34.02 | KX034290 | KP713894 | KP347467 | 5280 | 7000 | 4360 | 340 |
| Cyclocaris sp. | 4.73 | 4.62 | KX034301 | KP713899 | KT372890 | 1907 | 6007 | 5034 | 15 |
| Eurythenes magellanicusa | 18.35 ± 0.74 | 17.95 | KX034311 | KP713957 | KP347469 | 1229 | 5329 | 3486 | 85 |
| Eurythenes maldorora | 18.86 ± 3.56 | 18.45 | KX034310 | KX365240 | KX365244 | 3160 | 6230 | 4650 | 100 |
| Hirondellea dubiaa | 4.74 ± 0.56 | 4.64 | KX034251 | KP713906 | KP347459 | 6218 | 11, 000 | 7891 | 12 |
| Paracallisoma sp. | 19.54 | 19.11 | KX034319 | KX365241 | KX365245 | 1726 | 5100 | 4237 | 28 |
| Paralicella caperescaa | 4.06 ± 0.54 | 3.97 | KX034272 | KP713921 | KP347463 | 5925 | 7415 | 4453 | 18 |
| Paralicella tenuipesa | 4.13 ± 0.59 | 4.04 | KX034284 | KP713931 | KP347464 | 4915 | 7415 | 4958 | 14 |
| Valettietta anacantha | 7.80 | 7.63 | KX034322 | KP713950 | KT372893 | 6007 | 6007 | 3004 | 15 |
aSamples with replicates.
2.2. Phylogenetic reconstruction
Species were identified and phylogenetic relationships were ascertained based upon DNA sequence variation at two mitochondrial (COI and 16S rDNA) and one nuclear (18S rDNA) loci, according to Ritchie et al. [33]. In total, amphipod samples were sorted into 13 species and eight genera belonging to six families, all within the Lysianassoidea superfamily.
DNA sequence electropherograms were examined in MEGA v. 6.0.5 [34] and nucleotide alignments were made using webPRANK [35]. Individuals were identified to species or genus level using default parameters on NCBI BLASTn [36]. All species returned a 99–100% identity match to a BLAST hit with the exception of the unknown amphipod which returned positive matches to Lysianassoidea amphipods but without a high enough identity match to confidently assign it to either species or genus level.
For phylogenetic reconstruction the optimal evolutionary model for the dataset was identified using JModelTest 2.1.6 [37] using both the Akaike information criterion (AIC) and the Bayesian information criterion (BIC). Both AIC and BIC identified the general time-reversal substitution model (GTR + I + G) for COI and 18S rDNA, and the Hasegawa, Kishino and Yano model (HYK + G) as the best-fit model for the 16S rDNA dataset. Phylogenetic reconstruction was conducted using a Bayesian approach in *BEAST [38] where the analysis was given two runs each for 50 000 000 generations sampling 500 000 trees (every 100 generations) using the models of sequence evolution estimated by JModelTest but with the parameters estimated by *BEAST and Lanceola sp. was included as the outgroup. The first 150 000 trees were discarded as burn-in where the partition frequencies among the remaining trees gives the posterior probabilities to provide an estimate of clade credibility. Convergence of both runs was evaluated using Tracer v. 1.4.1 [39]. Trees were visualized using FigTree v. 1.4.2 [40].
2.3. Genome size estimation
Nuclear genome sizes were estimated using a flow cytometry approach where individual cell suspensions were prepared using a standard protocol [41] using whole amphipods in 1 ml of ice-cold Galbraith buffer [42]. Replicate estimates for individual species were conducted where possible (table 1). Owing to the high lipid content of deep-sea amphipods preparations were centrifuged at ×800g for 10 m to pellet cells and allow for the removal of the buffer suspension containing the unwanted lipids. Pelleted cells were re-suspended in 1 ml of phosphate-buffered saline (PBS) and stored at 4°C.
Chicken erythrocyte nuclei (CEN) from Gallus gallus domesticus were added to cell suspensions as an internal size standard and co-stained using propidium iodide at a final concentration of 50 ppm before incubation in the dark for 20 m at 4°C. Relative fluorescence of co-stained nuclei of samples were quantified using a FACSCalibur flow cytometer (BD Biosciences, USA) with an argon-ion laser emitting 15 mW of light at 488 nm. A minimum of 10 000 nuclei per sample were measured using Cell ProQuest software (BD Biosciences).
The relative fluorescence of nuclei peaks of interest were isolated using BD FACSDiva v. 7.0 software (BD Biosciences, NJ, USA). Haploid nuclear DNA content (C-value) of the samples were estimated from the fluorescence intensity of the sample and internal size standard using the haploid genome size of CENs which is 1.25 pg. C-values were subsequently converted into genome sizes using the standard conversion of 1 pg = 978 Mb as described in [43].
2.4. Statistical analysis
2.4.1. Diversification rate analysis
To examine the patterns of diversification rate variation in genome size we used Bayesian analysis of macroevolutionary mixtures (BAMM) [44] and the R package BAMMtools [45]. Tree appropriate rate prior parameters were determined using the setBAMMpriors function in BAMMtools before two separate rjMCMC runs were conducted in BAMM. Each BAMM analysis was run for 10 000 000 generations where parameters were sampled every 50 000 generations and the first 100 000 generations were discarded as burn-in. MCMC runs were checked for convergence. The credible shifts and net diversification rates across the tree were computed using BAMMtools. It was not possible to account for incomplete lineage sampling as there are no diversity estimates available for Lysianassoidea amphipods although the samples investigated cover a good spread of the known diversity.
2.4.2. Independent contrast analysis
Independent contrast analysis is used to transform phylogenetic information into independent values that can be used to detect co-variance between traits or variables of interest. Here Felsenstein's independent contrasts method [46] was used to examine correlations between genome size (pg), depth range (m), maximum depth (m), median depth (m) and maximum body length (mm) while controlling for the influence of phylogenetic signal. Phylogenetically independent contrasts were conducted using PDAP v. 1.07 (Phenotypic Diversity Analysis Package) [47] implemented within Mesquite v. 3.04 [48].
2.4.3. Regression analysis
Linear regressions were also implemented to examine relationships between genome size (pg), maximum depth (m) and maximum body length (mm) across a range of amphipod species including those from the deep sea, arctic and Lake Baikal. Genome sizes, maximum depths and maximum body lengths for each species were collated from data collected in this study and taken from the wider literature (electronic supplementary material, table S1).
3. Results
3.1. Genome size estimation
Haploid genome size estimates for the 13 Lysianassoidea amphipod species examined in this study are presented in table 1. In total, genome sizes varied nine-fold from 4.06 ± 0.54 pg in Paralicella caperesca to 34.79 ± 1.43 pg in A. gigantea. The mean genome size across the species was 11.28 pg (11.03 Gb).
3.1.1. Diversification rate analysis
All 13 species were successfully sequenced at 250 bp of the mitochondrial 16S rRNA gene, 627 bp of the COI gene and 599 bp of the 18S gene for a combined amplicon length of 1476 bp. GenBank accession numbers are provided in table 1 and a coalescent Bayesian tree is given in figure 1.
Figure 1.

Mean phylorate plot for genome size evolution in Lysianssoidea amphipods using the BAMM MCC phylogeny is overlaid on the *BEAST phylogeny where branch colours indicate instantaneous rates of phenotypic evolution with rates divided into bins using the Jenks natural breaks method. The distinct shift configuration is indicated by a star where the shift was shown in the majority of the shift configurations sampled during simulation of the posterior (f = 0.59). Estimated C-values (pg) for the amphipods are also plotted.
The BAMM diversification analysis reached a stationary state before 500 000 generations in both independent runs and using a Poisson prior (PP) of 1.0 it identified the most probable number of genome size diversification rate shifts was estimated as 1 (PP = 0.39) followed by 2 (PP = 0.21) and 0 (PP = 20). The mean phylorate plot shows an increase in mean diversification rate for genome size at the branch of the ‘supergiant’ A. gigantea (depicted as a star in figure 1). Individual rate-shift configurations sampled by BAMM were also investigated. The most probable scenario sampled showed a significant rate increase at the A. gigantea branch (PP = 0.59) and the remainder of the scenarios samples showed no significant rate changes across the phylogeny (PP = 0.41).
A macroevolutionary cohort matrix of BAMM analyses for genome size evolution shows the pairwise probabilities that species share a common macroevolutionary rate regime (figure 2). There is a relatively high probability of shared rate regimes across the whole Lysianassoidea superfamily with the exception of A. gigantea which has a distinct macroevolutionary rate regime. Within the Lysianassoidea there are also groups that have higher than average pairwise probabilities of shared rate regimes. All species within the Abyssorchomene group have a high probability of shared rate regimes with each other and the unknown amphipod species which is likely to be another Abyssorchomene species given its phylogenetic placement. Both Paralicella species have a high probability of shared rate regimes with Valettietta anacantha. Both Eurythenes species also have a high probability of shared rate regimes between themselves and with Paracallisoma sp.
Figure 2.
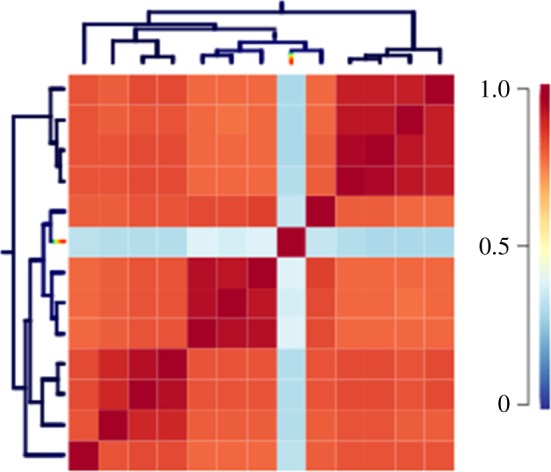
Macroevolutionary cohort matrix for genome size evolution in Lysianassoidea amphipods where each cell shows the pairwise probability that two species shares a common macroevolutionary rate regime. Pairwise probabilities are shown using a temperature scale from blue (p = 0) to red (p = 1). The BAMM MCC phylogeny is shown on the left and top of the cohort matrix for reference but the pairwise probabilities are calculated from a sample of trees from the posterior distribution of topologies and branch lengths.
3.1.2. Independent contrast analysis
Within the deep-sea amphipods a phylogenetically independent contrast analysis showed no strong or significant interactions between genome size and depth range (r = 0.063, p > 0.05), maximum depth (r = 0.019, p > 0.05) or median depth (r = 0.075, p > 0.05). A significant correlation was shown between genome size and maximum body length (r = 0.890, p < 0.001) however when the ‘supergiant’ amphipod A. gigantea was removed from the analysis the strength of this correlation was reduced (r = 0.504, p = 0.09).
3.1.3. Regression analysis
The deep-sea amphipods do not have a larger genome size relative to other amphipods groups that have been studied (electronic supplementary material, figure S1). The average genome size of deep-sea amphipods was smaller than the arctic species (t9 = −1.18, p = 0.27) but greater than the freshwater Lake Baikal species (t12 = 2.80, p = 0.01). Arctic species also exhibited greater genome sizes than Lake Baikal species (t7 = 2.17, p = 0.07).
Overall the positive relationship between genome size and body size shown for the deep-sea amphipods is retained across all the amphipod species (R2 = 0.3067) (electronic supplementary material, figure S2). However, a positive relationship between genome size and maximum depth is also shown (R2 = 0.3409), though this is primarily driven by the amphipods from Lake Baikal (electronic supplementary material, figure S3).
4. Discussion
The salient finding of this study is that there is considerable genome size variation within the deep-sea Lysianassoidea amphipods. Among the 13 species examined there is a nine-fold change in genome size ranging from 4.06 pg (3.97 Gb) in P. caperesca to 34.79 pg (34.02 Gb) in A. gigantea. The mean genome size across the species was 11.28 pg (11.03 Gb). Given that the majority of all animal genomes recorded are less than 5 pg (4.89 Gb) and genomes over 10 pg (9.78 Gb) are considered to be large [49], then among these deep-sea amphipods analysed here there are six large genomes, five small genomes and two of intermediate size. Genome sizes for previously recorded amphipod species range from 0.94 pg (0.92 Gb) to 64.62 pg (63.33 Gb) with a mean genome size of 9.08 pg (8.89 Gb). This places the deep-sea Lysianassoidea amphipods at the larger end of genome size spectrum for amphipods.
As such, while there are clearly some large genomes among the deep-sea amphipods, large genome sizes are not a characteristic of the group. While their average genome size is larger than the amphipods in the freshwater Lake Baikal this is not true for the average genome size observed in arctic environments. This is sufficient to reject the hypothesis that genome size is determined by their common position in the r-K-A life-history continuum or an equivalent ecological niche in the extreme deep-sea environment.
It was predicted that the low temperature [22], high hydrostatic pressure [25,26] and A-selected life-history would select for large genome sizes across the group, but this is clearly not the case. Even among sympatric pairs of deep-sea species there is a large range of genome sizes suggesting an absence of a dominant environment variable influencing genome size. Moreover, independent contrast analysis found no relationship with depth which might have been expected if hydrostatic pressure was the primary selection pressure driving large genome sizes. Indeed, the patterns observed are somewhat counterintuitive given that the deepest living amphipod Hirondellea dubia has a small genome size of 4.74 pg at approximately 11 000 m whereas Eurythenes magellanicus has a genome size of 18.35 pg with a maximum known depth of 5329 m.
There was clear genome size rate diversification in genome size evolution across the group, with A. gigantea showing an enhanced rate of phenotypic evolution at a relatively derived position in the overall phylogeny. This is consistent with the conjecture that larger genomes are secondarily derived from smaller genomes [26], though this does not appear to be a consistent feature across the phylogeny, nor does it shed light on the processes that underpin genome size increase.
One clear pattern that was apparent both in deep-sea species and in the broader amphipod group was the positive relationship between genome size and body size [22,50]. The two genera that exhibit size gigantism had considerably larger genomes, with the giant amphipods Eurythenes spp. that reach maximum body sizes of 85 and 100 mm [51] having intermediate-large genome sizes of 18.35 and 18.86 pg, respectively, and the ‘supergiant’ amphipod A. gigantea with a maximum body size of 340 mm ([52] in [53]) having a large genome size of 34.79 pg. The majority of the Lysianassoidea are considerably smaller than Eurythenes and Alicella with average body sizes of approximately 9–14 mm and the majority of these having smaller genomes below 10 pg. If the largest species, A. gigantea, is removed from the analysis the relationship becomes marginal and certainly for the smaller bodied amphipods there is no clear relationship between body size and genome size. This is in accordance with the assertion that A. gigantea has an accelerated genome size diversification rate which is significantly distinct from the remainder of the Lysianassoidea.
Flow cytometry provides a rapid, economical and accessible approach for investigating genome size variation across taxa, but current data do not encompass a wide enough range of species and habitats, and it provides limited information on the changes in genome content and structure that drives the observed diversity [54]. Understanding the patterns and drivers of genome size evolution in deep-sea amphipods would benefit from the addition of phylogenetically similar shallow water counterparts to allow more extensive comparisons to be made. Another challenge moving forward is to establish whether the variation observed for the Lysianassoidea amphipods reflects changes in gene content, is a consequence of gene duplication or is influenced by the occurrence of TEs. Generally, the latter might be expected to be a major driver given the extreme and stressful environmental conditions associated with the deep sea. This might result in the increase of TEs associated with the disruption of epigenetic control [55–57]. Indeed, a growing body of genomic data available for Paralicella tenuipes [58] has shown both the presence of copia retrotransposons and evidence for several duplication events for two important heat-shock proteins [59]. Notwithstanding, the vastly accelerated genome size expansion shown in A. gigantea may also identify a whole-genome duplication event rather than solely being attributed to an accumulation of TEs, but this would require further investigation.
Overall, the occurrence of high genome size variation within a relatively small taxonomic group of deep-sea amphipods occupying an equivalent habitat and ecological niche emphasizes how problematic it can be to identify simple drivers of diversity, especially from correlative assessment. In all likelihood, the large variation in genome size will either be attributable to multiple factors acting in concert or, with different drivers operating in different taxonomic groups, habitats and in different times.
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Acknowledgements
We thank the chief scientists, crew and company of the New Zealand RV Kaharoa (KAH1301 and KAH1310) and the United States RV Falkor (Cruise FK141109). From NIWA, we thank Malcolm Clark, Ashley Rowden, Kareen Schnabel, and Sadie Mills for logistical support at the NIWA Invertebrate Collection. We thank NOAA Marine National Monuments, Richard Hall and Eric Breuer for their support and collaboration. We also thank Attila Bebes and the Iain Fraser Cytometry Centre (IFCC) for technical assistance.
Ethics
No permission of research and animal ethics was necessary. No endangered species were collected for the study and specimens were fixed with minimum harm. Permission to collect samples was granted by the New Zealand Ministry for Primary Industries (NIWA Special Permit 421—Kermadec Trench), the French Ministry for Foreign Affairs (Permit 1081—New Hebrides Trench) and NOAA Marine National Monuments (no permit required—Mariana Trench).
Data accessibility
All data necessary to reproduce the results in this paper can be found in the electronic supplementary material.
Authors' contributions
Fieldwork and sample collection was carried out by H.R., A.J.J. and S.B.P. H.R. processed samples, undertook all laboratory work and carried out all analysis. H.R., A.J.J. and S.B.P equally contributed to writing the manuscript and gave final approval for publication.
Competing interests
We have no competing interests.
Funding
This work was supported by the HADEEP projects, funded by the Nippon Foundation, Japan (2009765188); the Natural Environment Research Council (NERC), UK (NE/E007171/1); Total Foundation, France; National Institute of Water and Atmospheric Research (NIWA), New Zealand (CO1_0906); Schmidt Ocean Institute, USA (FK141109) (A.J.J. and S.B.P); Marine Alliance for Science and Technology for Scotland (MASTS) (HR09011 and DSSG15) (H.R., A.J.J., S.B.P); and the Leverhulme Trust (S.B.P.).
References
- 1.Méténier G, Vivarès CP. 2001. Molecular characteristics and physiology of microsporidia. Microbes Infect. 3, 407–415. (doi:10.1016/S1286-4579(01)01398-3) [DOI] [PubMed] [Google Scholar]
- 2.Pellicer J, Fay MF, Leitch IJ. 2010. The largest eukaryotic genome of them all? Bot. J. Linn. Soc. 164, 10–15. (doi:10.1111/j.1095-8339.2010.01072.x) [Google Scholar]
- 3.Lien S, et al. 2016. The Atlantic salmon genome provides insights into rediploidization. Nature 533, 200–205. (doi:10.1038/nature17164) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Zee JP, Schlueter JA, Schlueter S, Dixon P, Brito Sierra CA, Hill CA. 2016. Paralog analyses reveal gene duplication events and genes under positive selection in Ixodes scapularis and other ixodid ticks. BMC Genomics 17, 241 (doi:10.1186/s12864-015-2350-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Devos KM, Brown JKM, Bennetzen JL. 2002. Genome size reduction through illegitimate recombination counteracts genome expansion in Arabidopsis. Genome Res. 12, 1075–1079. (doi:10.1101/gr.132102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vitte C, Panaud O. 2005. LTR retrotransposons and flowering plant genome size: emergence of the increase/decrease model. Cytogenet. Genome Res. 110, 91–107. (doi:10.1159/000084941) [DOI] [PubMed] [Google Scholar]
- 7.Sisu C, et al. 2014. Comparative analysis of pseudogenes across three phyla. Proc. Natl Acad. Sci. USA 111, 13 361–13 366. (doi:10.1073/pnas.1407293111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kidwell MG. 2002. Transposable elements and the evolution of genome size in eukaryotes. Genetica 115, 49–63. (doi:10.1023/A:1016072014259) [DOI] [PubMed] [Google Scholar]
- 9.Hancock JM. 2002. Genome size and the accumulation of simple sequence repeats: implications of new data from genome sequencing projects. Genetica 115, 93–103. (doi:10.1023/A:1016028332006) [DOI] [PubMed] [Google Scholar]
- 10.Konstantinidis KT, Tiedje JM. 2004. Trends between gene content and genome size in prokaryotic species with larger genomes. Proc. Natl Acad. Sci. USA 101, 3160–3165. (doi:10.1073/pnas.0308653100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nevo E. 2001. Evolution of genome-phenome diversity under environmental stress. Proc. Natl Acad. Sci. USA 98, 6233–6240. (doi:10.1073/pnas.101109298) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gregory TR. 2002. Genome size and developmental complexity. Genetica 115, 131–146. (doi:10.1023/A:1016032400147) [DOI] [PubMed] [Google Scholar]
- 13.Cavalier-Smith T. 1985. Cell volume and the evolution of eukaryotic genome size. In The evolution of genome size (ed. T Cavalier-Smith), pp. 104–184. Chichester, UK: Wiley. [Google Scholar]
- 14.Hessen DO, Jensen TC, Kyle M, Elser JJ. 2007. RNA responses to N- and P-limitation; reciprocal regulation of stoichiometry and growth rate in Brachionus. Funct. Ecol. 21, 956–962. (doi:10.1111/j.1365-2435.2007.01306.x) [Google Scholar]
- 15.Hessen DO, Persson J. 2009. Genome size as a determinant of growth and life-history traits in crustaceans. Biol. J. Linn. Soc. 98, 393–399. (doi:10.1111/j.1095-8312.2009.01285.x) [Google Scholar]
- 16.Gregory TR. 2005. Genome size evolution in animals. The evolution of the genome. San Diego, CA: Elsevier. [Google Scholar]
- 17.Gregory TR. 2015. Animal genome size database. See http://www.genomesize.com .
- 18.Rees DJ, Dufresne F, Glémet H, Belzile C. 2007. Amphipod genome sizes: first estimates for Arctic species reveal genomic giants. Genome 50, 151–158. (doi:10.1139/G06-155) [DOI] [PubMed] [Google Scholar]
- 19.Southwood TRE. 1977. Habitat, the templet for ecological strategies? J. Anim. Ecol. 46, 336 (doi:10.2307/3817) [Google Scholar]
- 20.Greenslade PJM. 1983. Adversity selection and the habitat templet. Am. Nat. 122, 352–365. (doi:10.1086/284140) [Google Scholar]
- 21.Sainte-Marie B. 1991. A review of the reproductive bionomics of aquatic gammaridean amphipods: variation of life history traits with latitude, depth, salinity and superfamily. Hydrobiologia 223, 189–227. (doi:10.1007/BF00047641) [Google Scholar]
- 22.Timofeev SF. 2001. Bergmann's principle and deep-water gigantism in marine crustaceans. Biol. Bull. Russ. Acad. Sci. 28, 646–650. (doi:10.1023/A:1012336823275) [PubMed] [Google Scholar]
- 23.Eustace RM, Ritchie H, Kilgallen NM, Piertney SB, Jamieson AJ. 2016. Morphological and ontogenetic stratification of abyssal and hadal Eurythenes gryllus sensu lato (Amphipoda: Lysianassoidea) from the Peru-Chile trench. Deep Sea Res. Part I Oceanogr. Res. Pap. 109, 91–98. (doi:10.1016/j.dsr.2015.11.005) [Google Scholar]
- 24.Jamieson AJ, Lacey NC, Lörz A, Rowden AA, Piertney SB. 2013. The supergiant amphipod Alicella gigantea (Crustacea: Alicellidae) from hadal depths in the Kermadec Trench, SW Pacific Ocean. Deep Sea Res. Part II Top. Stud. Oceanogr. 92, 107–113. (doi:10.1016/j.dsr2.2012.12.002) [Google Scholar]
- 25.Chénais B, Caruso A, Hiard S, Casse N. 2012. The impact of transposable elements on eukaryotic genomes: from genome size increase to genetic adaptation to stressful environments. Gene 509, 7–15. (doi:10.1016/j.gene.2012.07.042) [DOI] [PubMed] [Google Scholar]
- 26.Libertini A, Vitturi R, Gregorini A, Colomba M. 2009. Karyotypes, banding patterns and nuclear DNA content in Crepidula unguiformis Lamarck, 1822, and Naticarius stercusmuscarum (Gmelin, 1791) (Mollusca, Caenogastropoda). Malacologia 51, 111–118. (doi:10.4002/040.051.0107) [Google Scholar]
- 27.Rogers AD. 2007. Evolution and biodiversity of Antarctic organisms: a molecular perspective. Phil. Trans. R. Soc. B 362, 2191–2214. (doi:10.1098/rstb.2006.1948) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wignall PB, Twitchett RJ. 1996. Oceanic anoxia and the end Permian mass extinction. Science 272, 1155–1158. (doi:10.1126/science.272.5265.1155) [DOI] [PubMed] [Google Scholar]
- 29.Brown A, Thatje S. 2014. Explaining bathymetric diversity patterns in marine benthic invertebrates and demersal fishes: physiological contributions to adaptation of life at depth. Biol. Rev. 89, 406–426. (doi:10.1111/brv.12061) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Long LK, Lin XY, Zhai JZ, Kou HP, Yang W, Liu B. 2006. Heritable alternation in DNA methylation pattern occurred specifically at mobile elements in rice plants following hydrostatic pressurization. Biochem. Biophys. Res. Commun. 340, 369–376. (doi:10.1016/j.bbrc.2005.12.015) [DOI] [PubMed] [Google Scholar]
- 31.Rebollo R, Horard B, Hubert B, Vieira C. 2010. Jumping genes and epigenetics: towards new species. Gene 454, 1–7. (doi:10.1016/j.gene.2010.01.003) [DOI] [PubMed] [Google Scholar]
- 32.Jeffrey NW, Yampolsky L, Gregory TR. 2016. Nuclear DNA content correlates with depth, body size, and diversification rate in amphipod crustaceans from ancient Lake Baikal, Russia. Genome 60, 303–309. (doi:10.1139/gen-2016-0128) [DOI] [PubMed] [Google Scholar]
- 33.Ritchie H, Jamieson AJ, Piertney SB.. 2015. Phylogenetic relationships among hadal amphipods of the superfamily Lysianassoidea: implications for taxonomy and biogeography. Deep Sea Res. Part I Oceanogr. Res. Pap. 105, 119–131. (doi:10.1016/j.dsr.2015.08.014) [Google Scholar]
- 34.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. 2013. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. (doi:10.1093/molbev/mst197) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Löytynoja A, Goldman N.. 2010. webPRANK: a phylogeny-aware multiple sequence aligner with interactive alignment browser. BMC Bioinformatics 11, 579 (doi:10.1186/1471-2105-11-579) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215, 403–410. (doi:10.1016/S0022-2836(05)80360-2) [DOI] [PubMed] [Google Scholar]
- 37.Darriba D, Taboada GL, Doallo R, Posada D. 2012. jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772 (doi:10.1038/nmeth.2109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Drummond AJ, Suchard MA, Xie D, Rambaut A. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973. (doi:10.1093/molbev/mss075) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Drummond AJ, Rambaut A.. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214 (doi:10.1186/1471-2148-7-214) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rambaut A. 2002. FigTree 1.4. Inst. Evol. Biol. Univ. Edinburgh. See: http://tree.bio.ed.ac.uk/software/figtree.
- 41.Bennett MD, Leitch IJ, Price HJ, Johnston JS. 2003. Comparisons with Caenorhabditis (∼100 Mb) and Drosophila (∼175 Mb) using flow cytometry show genome size in Arabidopsis to be ∼157 Mb and thus ∼25% larger than the Arabidopsis genome initiative estimate of ∼125 Mb. Ann. Bot. 91, 547–557. (doi:10.1093/aob/mcg057) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Galbraith DW, Harkins KR, Maddox JM, Ayres NM, Sharma DP, Firoozabady E. 1983. Rapid flow cytometric analysis of the cell cycle in intact plant tissues. Science 220, 1049–1051. (doi:10.1126/science.220.4601.1049) [DOI] [PubMed] [Google Scholar]
- 43.Dolezel J, Bartos J, Voglmayr H, Greilhuber J. 2003. Nuclear DNA content and genome size of trout and human. Cytometry. A 51, 127–128. (doi:10.1002/cyto.a.10013) [DOI] [PubMed] [Google Scholar]
- 44.Rabosky DL. 2014. Automatic detection of key innovations, rate shifts, and diversity-dependence on phylogenetic trees. PLoS ONE 9, e89543 (doi:10.1371/journal.pone.0089543) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rabosky DL, Grundler M, Anderson C, Title P, Shi JJ, Brown JW, Huang H, Larson JG. 2014. BAMMtools: an R package for the analysis of evolutionary dynamics on phylogenetic trees. Methods Ecol. Evol. 5, 701–707. (doi:10.1111/2041-210X.12199) [Google Scholar]
- 46.Felsenstein J. 1985. Phylogenies and the comparative method. Am. Nat. 125, 1–15. (doi:10.1086/284325) [Google Scholar]
- 47.Midford PE, Garland T, Jr, Maddison WP. 2005. PDAP package of Mesquite. See www.mesquiteproject.org/pdap_mesquite .
- 48.Maddison WP, Maddison DR. 2001. Mesquite: a modular system for evolutionary analysis. See http://mesquiteproject.org.
- 49.Dufresne F, Jeffrey N. 2011. A guided tour of large genome size in animals: what we know and where we are heading. Chromosome Res. 19, 925–938. (doi:10.1007/s10577-011-9248-x) [DOI] [PubMed] [Google Scholar]
- 50.Gregory TR. 2000. Nucleotypic effects without nuclei: genome size and erythrocyte size in mammals. Genome 43, 895–901. (doi:10.1139/g00-069) [DOI] [PubMed] [Google Scholar]
- 51.d'Udekem d'Acoz C, Havermans C. 2015. Contribution to the systematics of the genus Eurythenes SI Smith in Scudder, 1882 (Crustacea: Amphipoda: Lysianassoidea: Eurytheneidae). Zootaxa 3971, 1–80. (doi:10.11646/zootaxa.3971.1.1) [Google Scholar]
- 52.Harrison CS, Hida TS, Seki MP. 1983. Hawaiian seabird feeding ecology. Wildl. Monogr. 85, 3–71. [Google Scholar]
- 53.Barnard JL, Ingram CL. 1986. The supergiant amphipod Alicella gigantea Chevreux from the north Pacific gyre. J. Crustac. Biol. 6, 825–839. (doi:10.2307/1548395) [Google Scholar]
- 54.Elliott TA, Gregory TR.. 2015. What's in a genome? the C-value enigma and the evolution of eukaryotic genome content. Phil. Trans. R. Soc. B 370, 20140331 (doi:10.1098/rstb.2014.0331) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kazazian HH. 2004. Mobile elements: drivers of genome evolution. Science 303, 1626–1632. (doi:10.1126/science.1089670) [DOI] [PubMed] [Google Scholar]
- 56.Bennetzen JL. 2005. Transposable elements, gene creation and genome rearrangement in flowering plants. Curr. Opin. Genet. Dev. 15, 621–627. (doi:10.1016/j.gde.2005.09.010) [DOI] [PubMed] [Google Scholar]
- 57.Sessegolo C, Burlet N, Haudry A. 2016. Strong phylogenetic inertia on genome size and transposable element content among 26 species of flies. Biol. Lett. 12, 521–524. (doi:10.1098/rsbl.2016.0407) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ritchie H, Jamieson AJ, Piertney SB. 2016. Isolation and characterization of microsatellite DNA markers in the deep-sea amphipod Paralicella tenuipes by Illumina MiSeq Sequencing. J. Hered. 107, 367–371. (doi:10.1093/jhered/esw019) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ritchie H, Jamieson AJ, Piertney SB.. In press Heat-shock protein adaptation in abyssal and hadal amphipods. Deep Sea Res. Part II Top. Stud. Oceanogr. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data necessary to reproduce the results in this paper can be found in the electronic supplementary material.