

Abstract
Oligomers equipped with a sequence of phenol and pyridine N-oxide groups form duplexes via H-bonding interactions between these recognition units. Reductive amination chemistry was used to synthesize all possible 3-mer sequences: AAA, AAD, ADA, DAA, ADD, DAD, DDA, and DDD. Pairwise interactions between the oligomers were investigated using NMR titration and dilution experiments in toluene. The measured association constants vary by 3 orders of magnitude (102 to 105 M–1). Antiparallel sequence-complementary oligomers generally form more stable complexes than mismatched duplexes. Mismatched duplexes that have an excess of H-bond donors are stabilized by the interaction of two phenol donors with one pyridine N-oxide acceptor. Oligomers that have a H-bond donor and acceptor on the ends of the chain can fold to form intramolecular H-bonds in the free state. The 1,3-folding equilibrium competes with duplex formation and lowers the stability of duplexes involving these sequences. As a result, some of the mismatch duplexes are more stable than some of the sequence-complementary duplexes. However, the most stable mismatch duplexes contain DDD and compete with the most stable sequence-complementary duplex, AAA·DDD, so in mixtures that contain all eight sequences, sequence-complementary duplexes dominate. Even higher fidelity sequence selectivity can be achieved if alternating donor–acceptor sequences are avoided.
Introduction
The encoded recognition properties of nucleic acids are currently unrivaled in any other material. High-fidelity sequence-selective duplex formation is the molecular basis for replication of the genetic information encoded by DNA and is finding widespread applications in the programmed assembly of complex nucleic acid nanostructures.1 There is no fundamental reason that these properties should be restricted to biological polymers, and a range of synthetic nucleic acid analogues have been demonstrated to form duplexes.2−5 In principle, any synthetic polymer equipped with complementary recognition units has the potential to show sequence-selective duplex formation and the associated properties found in nucleic acids. Figure 1a shows a minimalist blueprint for such polymers. A two-letter recognition alphabet would be sufficient to encode sequence information in binary form. Then all that is required is reliable chemistry for the synthesis of oligomers and a compatible backbone to link the components together.
Figure 1.
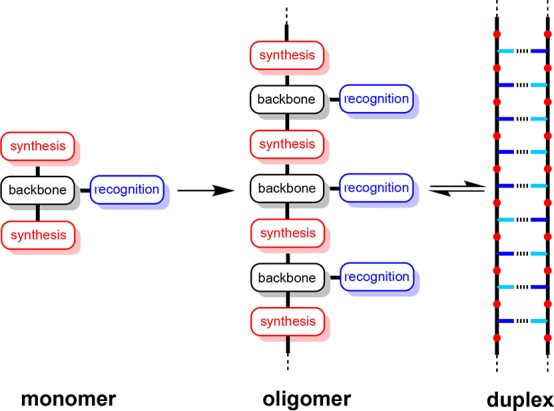
Blueprint for assembly of a polymer that forms a duplex with sequence selectivity based on a two-letter recognition alphabet. The key design components are the covalent chemistry used for synthesis (red), the noncovalent chemistry used for recognition (blue), and the backbone linker that determines the geometric complementarity of the two chains (black).
A number of synthetic duplex-forming oligomers have been reported,6−10 and in some of these systems, it was possible to investigate the effect of changing the sequence of the building blocks. Lehn et al. showed that oligomeric bipyridine and terpyridine ligands form duplexes with complementary metal ions, demonstrating both length and sequence selectivity.6 Gong et al. have described oligomers that form duplexes due to H-bonding interactions between amide groups located in the backbone. It was possible to control the recognition properties of these systems by changing both the sequence and the geometrical spacing of H-bond donors and acceptors along the chain.7 Yashima et al. have demonstrated sequence-selective duplex formation between oligomers equipped with carboxylate and amidinium recognition units that form salt bridges.8
We have been investigating a range of different duplex-forming oligomer systems based on the blueprint in Figure 1.11 The most promising system that we have characterized to date is shown in Figure 2. Strong H-bonding interactions between the phenol and pyridine N-oxide recognition units give rise to stable duplexes in toluene solution. For duplexes formed between homo-oligomers, the stability increases by an order of magnitude for every additional recognition unit in the chain, which is indicative of cooperative H-bond formation along the duplex. The X-ray crystal structure of the duplex formed by the self-complementary AD 2-mer is shown in Figure 2b.11e The recognition units are too far apart in the duplex for the long-range secondary electrostatic interactions that are observed in other H-bonded arrays to be important in this system.12 The solution phase self-assembly properties of the AD 2-mer also show that there is no intramolecular 1,2-folding between adjacent H-bond donors and acceptors in the monomeric free state. This system is therefore ideally suited for a more detailed investigation of the selectivity of duplex formation for longer mixed sequence oligomers.
Figure 2.
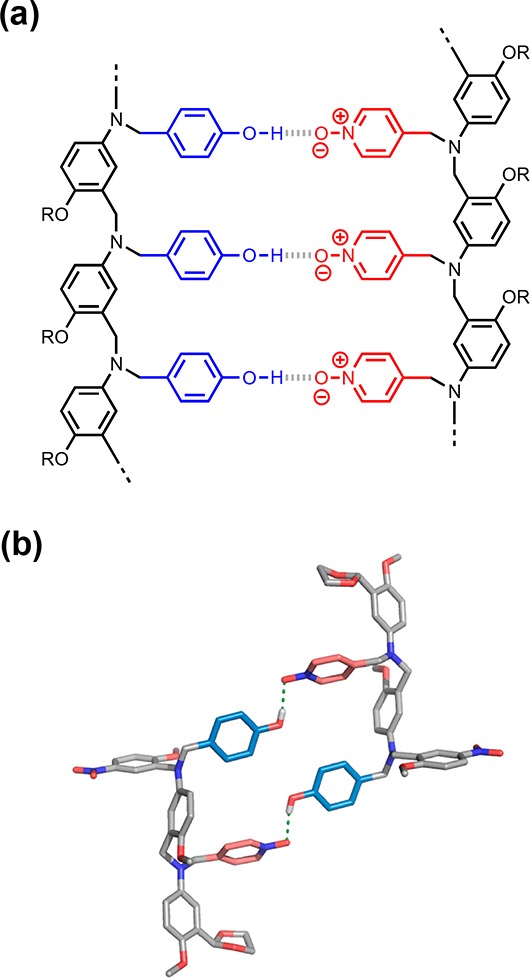
(a) H-bonded duplex formed by a phenol homo-oligomer and a pyridine N-oxide homo-oligomer. (b) X-ray crystal structure of the duplex formed by the corresponding mixed phenol–pyridine N-oxide 2-mer.11e
The simplest systems for which the sequence selectivity of duplex formation can be studied are the mixed sequence 3-mers. Figure 3 shows the structures of all possible 3-mer sequences of the system shown in Figure 2. In this paper, we describe the synthesis of these eight compounds and measurement of the pairwise binding affinities in toluene. The results allow quantification of the fidelity of the single H-bond recognition system and provide insights into competing processes that could be targeted to improve the sequence selectivity of duplex formation.
Figure 3.
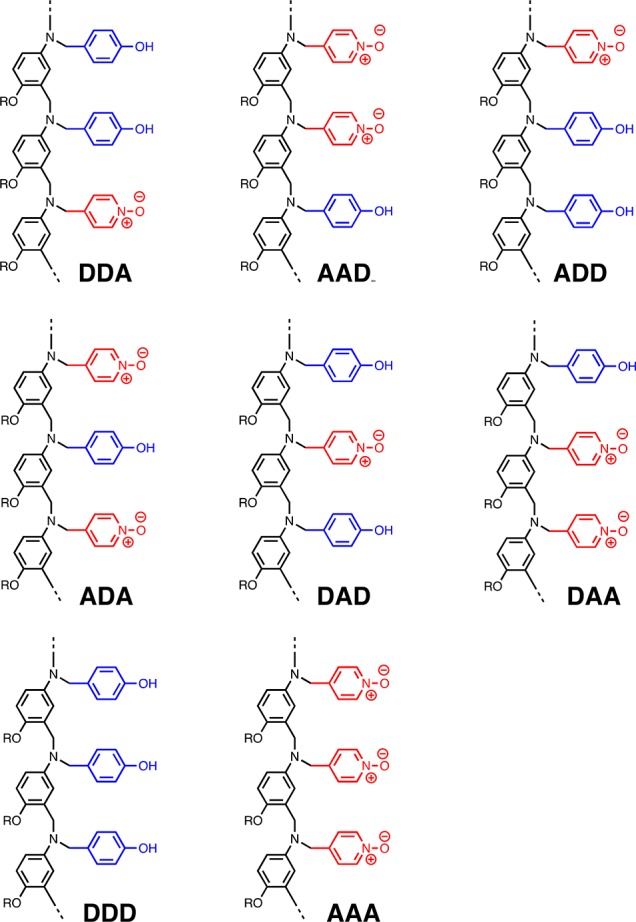
Eight different 3-mer sequences of H-bond donors (D) and acceptors (A).
Results and Discussion
Synthesis
The 3-mers were synthesized from monomers 1 and 2 by sequential acetal deprotection and reductive amination reactions (Scheme 1). Synthesis of the monomer building blocks, 1 and 2, was described previously.11a,11c The phenol group of the H-bond donor monomer, 1, was protected as the triisopropylsilyl ether, and the protecting groups were removed using tetra-n-butylammonium fluoride (TBAF) in the final step of the synthesis. In some cases, the acetal protecting group on the end of the 3-mer was removed during workup, so these compounds were isolated as the aldehyde as indicated in Scheme 1. The H-bond acceptor properties of aldehydes and acetals are both poor compared with pyridine N-oxide, so the presence of different terminal groups should not significantly affect the assembly properties of the oligomers.
Scheme 1. (i) 1 or 2, NaBH(OAc)3; (ii) Aqueous HCl; (iii) TBAF.

The use of amino–aldehyde monomer units confers directionality on the oligomer backbone, so we will describe the sequence of recognition units in the direction of synthesis, starting from the amino-terminal end (the nitrobenzyl group) to the aldehyde-terminal end (acetal or aldehyde group). For example, the oligomers described as ADD and DDA in Scheme 1 differ in the orientation of the backbone with respect to the sequence of the recognition units.
NMR Titrations
Interactions between all pairwise combinations of the 3-mer sequences were investigated by 1H NMR titration and dilution experiments in toluene-d8. The titration data all fit well to 1:1 binding isotherms, and the dilution data fit well to dimerization isotherms. The resulting association constants are reported in Table 1. The stabilities of the complexes span 3 orders of magnitude. The most stable complex is the sequence-complementary AAA·DDD duplex, which has an association constant of 105 M–1, but some of the other sequence-complementary complexes are significantly less stable.
Table 1. Association Constants (log K/M–1) for Formation of 1:1 Complexes Measured by 1H NMR Titrations and Dilutions in Toluene-d8 at 298 Ka.
| DDD | ADD | DAD | AAD | DDA | ADA | DAA | AAA | |
|---|---|---|---|---|---|---|---|---|
| DDD | 0.6 | 2.3 | 3.0 | 4.2 | n.d.b | 4.2 | 4.0 | 5.0 |
| ADD | 2.3 | 3.3 | 3.9 | 3.1 | 3.4 | 3.0 | 3.2 | |
| DAD | n.d. | 3.8 | 3.6 | 4.4 | 3.9 | 3.9 | ||
| AAD | 2.1 | 3.3 | 3.0 | 2.9 | n.d. | |||
| DDA | n.d. | 3.7 | 3.6 | 3.2 | ||||
| ADA | 2.9 | 3.4 | n.d. | |||||
| DAA | n.d. | 2.7 | ||||||
| AAA | 1.6 |
Errors in log K are less than ±0.2.
n.d. For some complexes with low association constants, reliable determination of the association constant was not possible.
For each sequence-complementary combination of recognition units, up to four different duplexes are possible due to the directionality of the backbone. For example, Figure 4a shows the structures of four different duplexes that have the same arrangement of H-bonded recognition units but different backbone directions. Using the N-to-C terminal description of sequence, these four duplexes are designated DDA·AAD, DDA·DAA, ADD·AAD, and ADD·DAA. In these systems, the structure of the duplex is dictated by the sequence of the recognition units, so it should be possible to distinguish parallel (DDA·AAD and ADD·DAA) and antiparallel (DDA·DAA and ADD·AAD) arrangements of the backbone. If the sequence of recognition units is symmetric, then it is possible for parallel and antiparallel duplexes to coexist in equilibrium. For example, for the AAA·DDD duplex both parallel and antiparallel directions of the backbone are compatible with the arrangement of the recognition units (Figure 4b). To simplify the discussion, we will start by considering only the arrangement of the recognition units, but we will return to the directionality of the backbone later.
Figure 4.
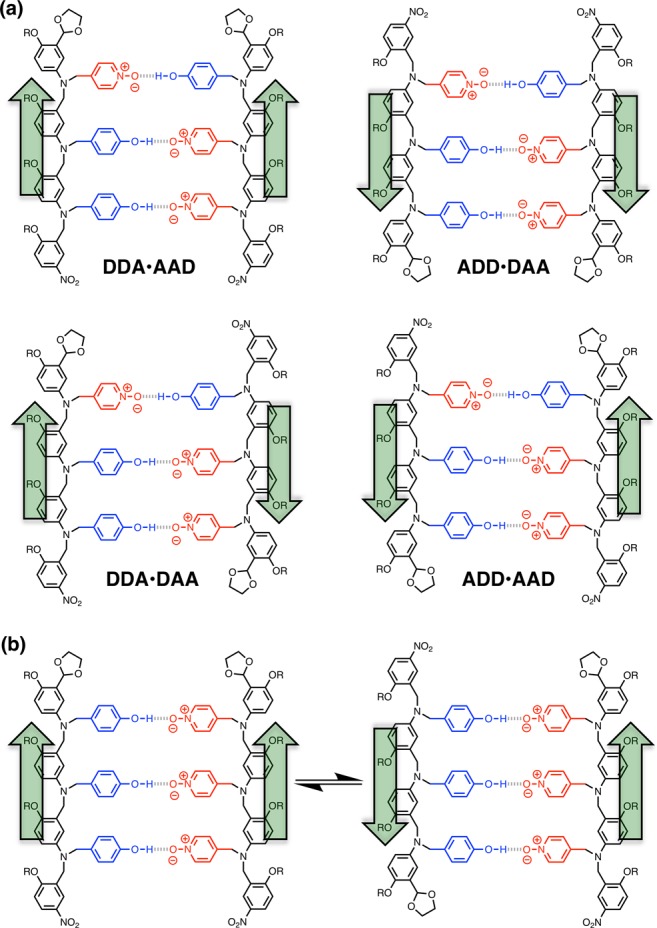
Structures of duplexes with parallel and antiparallel backbones. (a) Duplexes where the sequence of recognition units dictates the backbone arrangement: parallel for DDA·AAD and ADD·DAA and antiparallel for DDA·DAA and ADD·AAD. (b) Duplex where the sequence of recognition units allows parallel and antiparallel backbone arrangements to coexist in equilibrium: AAA·DDD.
Single-Site Mismatch Analysis
Figure 5 shows an analysis of the data in Table 1 comparing the stabilities of duplexes formed by complementary sequences of recognition units with the stabilities of the corresponding duplexes containing a single mismatch. Where different arrangements of the backbone are possible, the results for all backbone arrangements are plotted side by side in the same bar of the chart. For example, the association constants for the four duplexes illustrated in Figure 4a are shown as four different values that make up the first sequence-complementary entry in Figure 5c. The data in Table 1 can therefore be analyzed in terms of three sequence-complementary trimer duplexes: the homo-oligomer duplex, AAA·DDD, the alternating oligomer duplex, ADA·DAD, and the four duplexes shown in Figure 4a. If a single recognition unit is modified in a sequence-complementary 3-mer duplex, then a total of three A → D and three D → A mutations are possible. For symmetric sequences of recognition units, some of the mutations are equivalent, and in these cases, the data appear twice in Figure 5.
Figure 5.
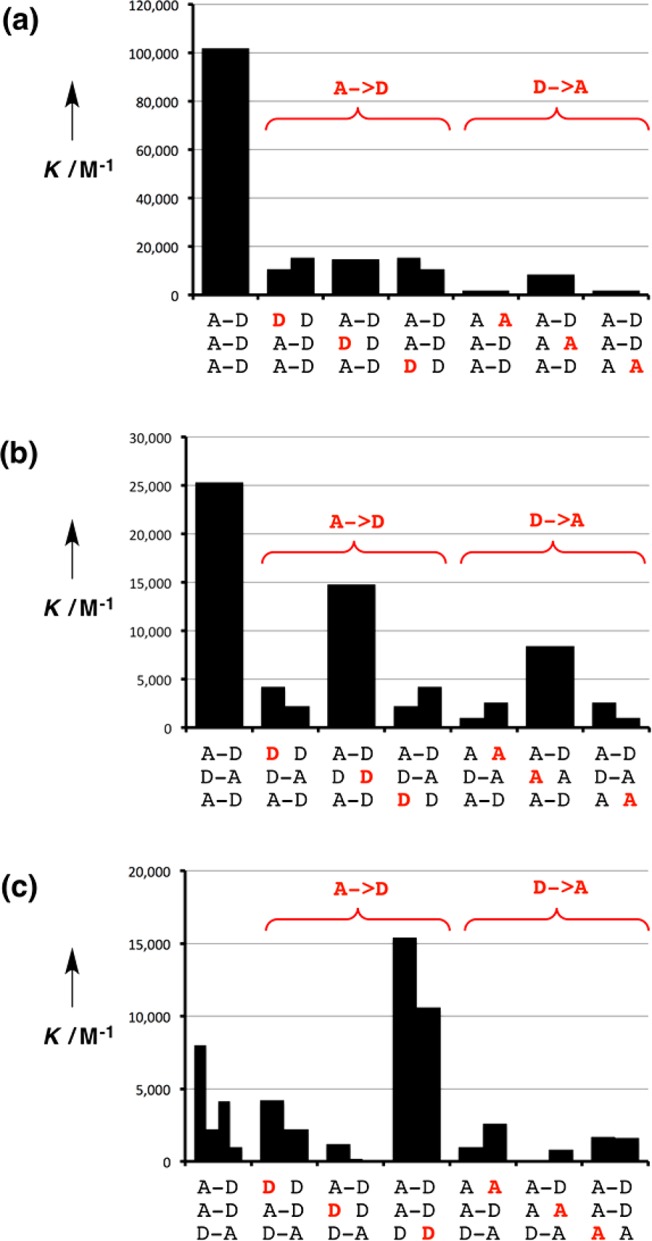
Effects of single A → D and D → A mutations (red) on the stabilities of sequence-complementary duplexes. (a) AAA·DDD. (b) ADA·DAD. (c) DDA·AAD, DDA·DAA, ADD·AAD, and ADD·DAA. Duplexes with different arrangements of the backbone are plotted on the same bar of the chart.
Figure 5a shows that AAA·DDD is the most stable duplex and that mutation of any of the recognition units leads to a decrease in stability of an order of magnitude. The stability of the ADA·DAD duplex is 4 times lower than that of AAA·DDD (Figure 5b). Again the sequence-complementary duplex is the most stable complex for this system, but some of the mismatch sequences are surprisingly stable. For example, the mismatched duplex involving DDD is only 2-fold lower in stability than the sequence-matched duplex. Figure 5c shows that for the third type of duplex the stabilities of the sequence-complementary complexes span an order of magnitude, and they are 10–100 times less stable than AAA·DDD. Moreover, the sequence-matched duplexes are not the most stable complexes in Figure 5c, and the mismatched duplexes involving DDD are more stable.
Stabilization of D-Rich Complexes
Closer examination of Figure 5 reveals some interesting patterns. In general, the A → D mutations give complexes that are more stable than the D → A mutations. For example in Figure 5a, the D → A mutants have stabilities of (2–8) × 103 M–1, whereas the A → D mutants have stabilities of (1–2) × 104 M–1. These values can be compared with the stabilities of the corresponding 2-mer duplexes, AA·DD and AD·AD, where only two H-bonds are made ((2–5) × 103 M–1). The association constants for formation of the 2-mer duplexes are comparable to the values for the D → A mismatch 3-mer complexes and significantly lower than the values for the A → D mismatch 3-mer complexes, suggesting that A → D mutations introduce additional stabilizing interactions. There is a fundamental difference between the D → A and A → D mutations: phenol has one H-bond donor site and so can only interact with one H-bond acceptor; in contrast, pyridine N-oxide can accept more than one H-bond from multiple donors. Thus, a D → A mutation removes all possibility of forming a H-bonding interaction, because there are no additional H-bond donor sites in the oligomers that could interact with the new mismatch pyridine N-oxide acceptor. However, when an A → D mutation is made, the two unpaired phenols that do not have complementary pyridine N-oxide partners to interact with can form additional interactions with pyridine N-oxides that are already H-bonded to complementary phenols.
Molecular mechanics calculations on the structures of the duplexes support this hypothesis. Figure 6 shows the lowest energy conformations of three different duplexes: AAD·DDA, ADA·DDD, and AAA·DAD. The sequence-complementary 3-mers form a duplex with three H-bonds as expected (Figure 6a). In the mismatch duplex that has an excess of H-bond donor recognition units (Figure 6b), one of the pyridine N-oxide acceptors is H-bonded to one phenol donor, but the other pyridine N-oxide is H-bonded to two phenol donors (this structure also shows an additional phenol–phenol interaction). In the mismatch duplex that has an excess of H-bond acceptor recognition units (Figure 6c), two intermolecular H-bonds are formed as expected, and the unsatisfied acceptor units dangle freely from the side of the duplex.
Figure 6.
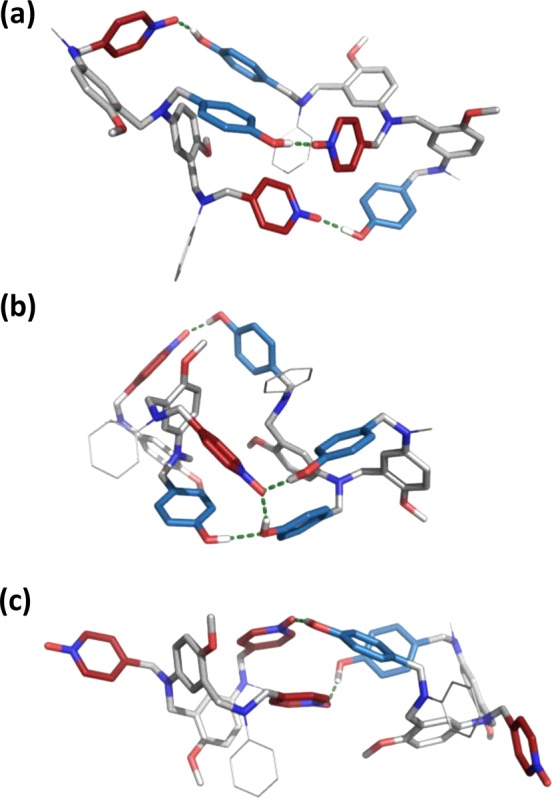
Lowest energy conformations of duplexes calculated using molecular mechanics conformational searches for (a) ADA·DAD, (b) ADA·DDD, and (c) AAA·DAD.13 The backbone is shown in gray, the H-bond donor recognition units are in blue, and the H-bond acceptor units are in red. The terminal groups were simplified to methyl and phenyl and are shown as lines for clarity.
The thermodynamic consequences of doubly H-bonded acceptor units can be tested directly by measuring the interaction of a simple pyridine N-oxide monomer with the DD 2-mer. 1H NMR titrations of p-cresol (D) or DD into 4-methylpyridine N-oxide (A) were carried out in toluene-d8. Performing the titrations in this way ensures that the concentration of A is too low for the 2:1 A2·DD complex to be formed. The titration data fit well to 1:1 binding isotherms in both cases, and the resulting association constants were 3.3 ± 0.8 × 102 M–1 for the A·D complex and 1.7 ± 0.2 × 103 M–1 for the A·DD complex. The larger association constant observed for the compound with two H-bond donor sites suggests that interactions of the type illustrated in Figure 6b stabilize 3-mer duplexes with A → D mutations.
The observed equilibrium constant for the formation of the 1:1 A·DD complex is given by eq 1.
| 1 |
where K1 and K2 are the stepwise equilibrium constants illustrated in Figure 7.
Figure 7.
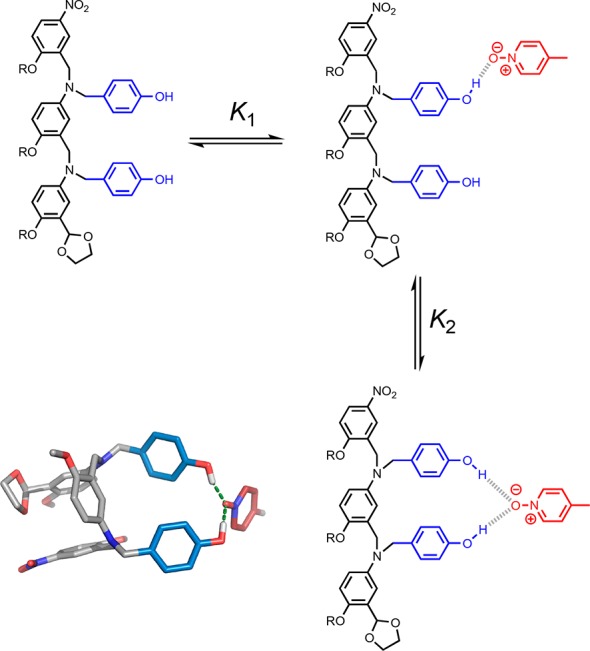
Pyridine N-oxide can accept two H-bonds, leading to enhanced stability in complexes with an excess of H-bond donors. The stepwise equilibrium constants for formation of the doubly H-bonded complex between DD and A are K1 and K2. The global minimum conformation of the 1:1 complex obtained from a molecular mechanics conformational search is shown (right).13 The backbone is shown in gray, the H-bond donor recognition units are in blue, and the H-bond acceptor unit is in red.
Rearranging eq 1 gives eq 2, which allows estimation of the value of K2, the equilibrium constant for formation of a second intramolecular H-bond to a H-bonded pyridine N-oxide, assuming that the value of K1 is 2KA·D. The statistical factor of 2 accounts for the degeneracy of the singly H-bonded complex.
| 2 |
The value of K2 for the system shown in Figure 7 is 3, which means that the doubly H-bonded complex represents 75% of the bound state. The presence of the second H-bond donor in the DD·A complex increases the observed association constant by a factor of 5 compared with the D·A complex, where only one H-bond can be formed. This value represents an upper limit on the increase in association constant that is expected due to formation of a second intramolecular H-bond to a bound pyridine N-oxide in the 3-mer mismatch duplexes, because the overall geometry of the duplex is likely to restrict the possible arrangements of the recognition units. However, stabilization by a factor of 5 is consistent with the higher association constants observed for D-rich mismatch complexes in Figure 5.
Intramolecular Folding
The analysis above indicates the complexes with D → A mutations are not perturbed by additional interactions involving unsatisfied recognition units. However, there is some variation in the stabilities of the complexes with D → A mutations. In both Figure 5a and b, making D → A mutations at the chain ends leads to complexes that are significantly less stable than mutating the recognition unit in the center. The common feature of the less stable complexes is that they contain oligomers that have a H-bond donor at one end of the chain and a H-bond acceptor at the other. Such sequences could fold via intramolecular H-bonding interactions between the terminal recognition units, and folding would compete with duplex formation. This observation would also account for the exceptionally low stability of the sequence-complementary duplexes in Figure 5c, because for these systems, both oligomers can fold in the unbound state (Figure 8).
Figure 8.
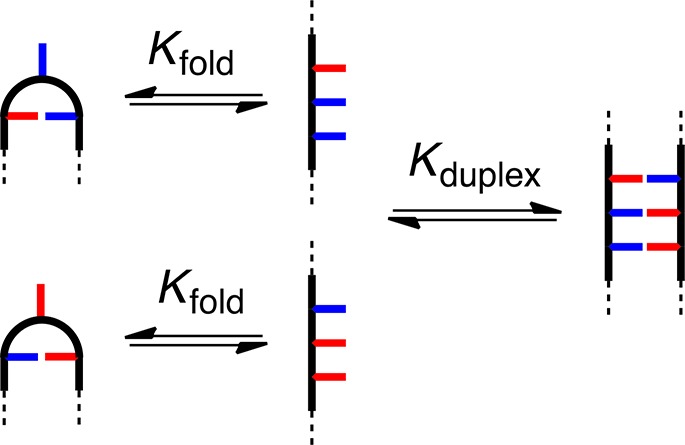
1,3-Folding competes with duplex formation.
The potential of the oligomers to fold was investigated using molecular mechanics calculations. Figure 9 shows an overlay of the lowest energy conformation found for each of the oligomers AAD, ADD, DAA, and DDA. In all four cases, there is an intramolecular H-bond between the terminal recognition units, and the backbones adopt very similar conformations in order to achieve this interaction. Thus, there appears to be a well-defined conformation of the backbone that places the recognition units in an arrangement that allows intramolecular H-bonding in a 1,3-folded state.
Figure 9.
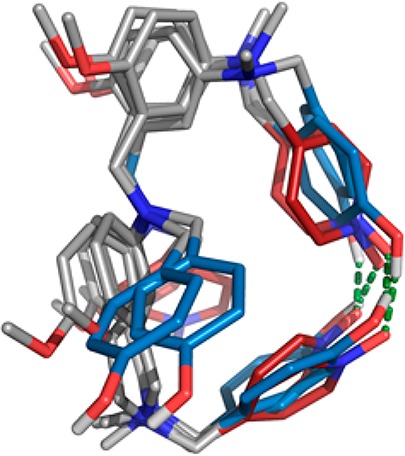
Overlay of the lowest energy conformation calculated for AAD, ADD, DAA, and DDA using molecular mechanics conformational searches.13 Intramolecular H-bonds (green) are observed between the terminal recognition units in all cases. The backbone is shown in gray, H-bond donor recognition units are in blue, and H-bond acceptor recognition units are in red. The terminal aromatic groups and solubilizing groups are omitted for clarity.
It is possible to estimate the extent of folding experimentally by comparing the stabilities of complexes involving oligomers that can and cannot fold. We have shown previously that 1,2-folding between neighboring recognition units does not compete with duplex formation in AD 2-mers, and so we assume that none of the 3-mers discussed here suffer from 1,2-folding. For the AAA, DDD, ADA, and DAD sequences, intramolecular 1,3-folding is not possible, so folding equilibria can only compete for duplex formation for complexes involving AAD, ADD, DDA, and DAA. Complexes with A → D mutations are complicated by additional H-bonding interactions as discussed above, so we will consider only complexes with D → A mutations. A direct comparison can be made between AAA·DAD, where 1,3-folding is not possible, and AAA·DDA, AAA·ADD, ADA·DAA, and ADA·AAD, where one of the two binding partners can form an intramolecular H-bond between the terminal recognition units. The four duplexes that compete with 1,3-folding equilibria have very similar association constants (1.6 × 103, 1.7 × 103, 2.6 × 103, and 1.0 × 103 M–1), and these values are on average 5 times lower than the association constant for AAA·DAD (8.4 × 103 M–1), where there are no competing folding equilibria.
For duplexes where one of the two binding partners folds, the observed association constant, Kobs, is given by eq 3.
| 3 |
where Kfold is the equilibrium constant for 1,3-folding, and Kduplex is the association constant for formation of the duplex from the unfolded state (Figure 8).
Equation 3 can be rearranged to estimate the value of Kfold using the association constants measured for complexes that compete with one intramolecular folding equilibrium (Kobs) and complexes that do not (Kduplex) (eq 4).
| 4 |
The analysis of the D → A mismatch complexes described above therefore indicates that Kfold for 1,3-folding is approximately 4 for oligomers with complementary terminal recognition units. The folded state therefore represents 80% of the monomeric unbound state for these oligomers, with 20% in the unfolded state. For complexes where both binding partners fold, the observed association constant for duplex formation is given by eq 5.
| 5 |
Using Kfold = 4 in eq 5 suggests that for sequence-complementary duplexes where both binding partners can form intramolecular H-bonds between the terminal recognition units, the stability of the duplex will be reduced by a factor of 25 compared with sequences where 1,3-folding is not possible. This estimate accounts rather well for the results shown in Figure 5c: the association constants for formation of the four sequence-complementary duplexes are between 10 and 100 times lower than the association constant for formation of the AAA·DDD duplex. It should be noted that although 1,3-folding competes with duplex formation in these systems, folding does not abolish duplex formation. For example, in a 10 mM sample of a 1:1 mixture of AAD and ADD, the population of duplex is 10 times greater than the population of the 1,3-folded state.
Backbone Arrangement in Duplexes
Although the duplexes illustrated in Figure 4a have the same arrangement of recognition units, the measured association constants span an order of magnitude (see the first sequence-complementary bar in Figure 5c). The major difference between the structures of these duplexes is the parallel and antiparallel arrangement of the backbone. The association constants for formation of the two antiparallel duplexes (4.2 × 103 and 8.0 × 103 M–1) are higher than for formation of the two parallel duplexes (1.0 × 103 and 2.2 × 103 M–1). These results suggest that on average the antiparallel arrangement of the backbone is preferred by a factor of 4. For systems where the arrangement of the backbone is not dictated by the sequence of the recognition units, a similar preference is expected, i.e., a 20% population with the backbone in a parallel arrangement in equilibrium with 80% in an antiparallel arrangement.
Sequence Selectivity in Mixtures
In order to assess the sequence selectivity of duplex formation in this system, it is important to define what is meant by selectivity. The selectivity of a recognition event depends on what the competition is. For example, consider formation of the ADD·AAD duplex. If ADD competes with DDD for duplex formation with AAD, then DDD will win because, as illustrated in Figure 5c, the AAD·DDD mismatch complex is more stable than the sequence-complementary duplex. However, if this competition is repeated in the presence of AAA, then the two sequence-complementary duplexes AAA·DDD and ADD·AAD will be formed, because the AAA·DDD duplex is much more stable than any other complex in this system.
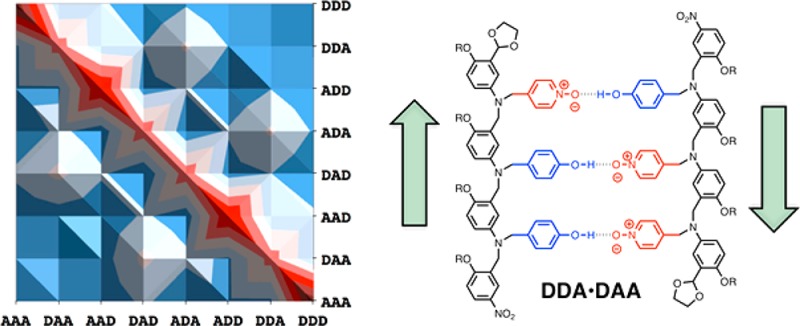
The association constants in Table 1 can be used to estimate the speciation of complexes in mixtures of the 3-mers. Figure 10a illustrates the populations of all possible duplexes calculated for an equimolar mixture of all eight 3-mers at a concentration at which all of the compounds are fully bound (>95% bound at 100 mM).14 There are six complexes for which association constants are not reported in Table 1. However, the titration experiments suggest that these are all weak binding systems, and assigning association constants in the range 102–103 M–1 to these complexes has no significant effect on the speciation of the other complexes or the appearance of Figure 10a. The sequences in Figure 10a are organized so that all of the antiparallel sequence-complementary duplexes lie on the diagonal of this plot, and it is clear that these duplexes are the most populated complexes (blue regions). At first sight, this result is counterintuitive, because, as illustrated in Figure 5, some of the mismatch duplexes are more stable than the corresponding sequence-complementary duplexes. However, in a system where all sequences compete for optimal binding partners, the effects that are apparent in the mismatch analysis are damped, and the result is that sequence-complementary duplexes dominate.
Figure 10.
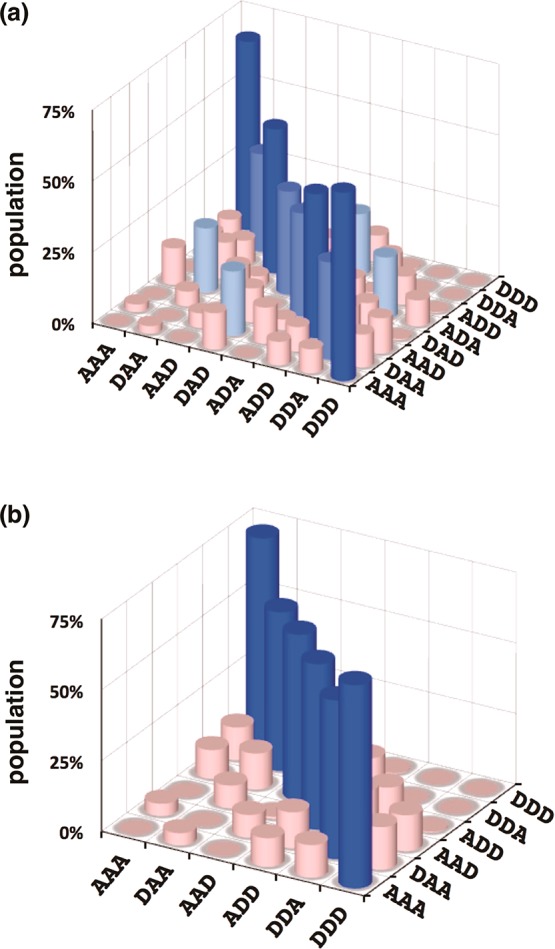
Calculated populations of duplexes formed in a 100 mM equimolar mixture of 3-mers in toluene (>50% dark blue, >30% royal blue, >20% pale blue, <20% pink). (a) Mixture of all eight trimer sequences. (b) Mixture that does not contain the alternating ADA and DAD sequences. Each duplex appears twice, and antiparallel sequence-complementary duplexes lie along the diagonal.
The noncomplementary duplexes that compete most effectively with sequence-complementary duplex formation are ADA·DDA and DAD·DAA. These two duplexes correspond to the four off-diagonal peaks in Figure 10a (each duplex appears twice due to symmetry). The populations of the corresponding sequence-complementary duplexes (ADA·DAD and DDA·DAA, which each appear twice on the diagonal in Figure 10) are somewhat reduced by population of the two mismatch duplexes. The two mismatch complexes are both less stable than the sequence-complementary ADA·DAD duplex, but they are both slightly more stable than the sequence-complementary DDA·DAA duplex. The appearance of mismatch duplexes is therefore the result of competition between all of the different possible complexes in the system. The off-diagonal mismatch peaks in Figure 10a suggest that longer mixed sequence oligomers are unlikely to exhibit high-fidelity sequence recognition. However, the identity of the mismatch duplexes provides a clue as to how this fidelity might be improved. Although the ADA·DAD duplex is relatively stable, these two 3-mer sequences participate in the most significant mismatch duplexes. The speciation in a mixture of the other six 3-mers that does not contain ADA or DAD is illustrated in Figure 10b. In this case, the sequence selectivity is excellent, with a high degree of discrimination between matched and mismatched duplexes. This result provides an important strategy for enhancing the fidelity of sequence recognition in longer oligomers. If alternating donor–acceptor sequences are avoided, then mismatch duplexes will be suppressed.
Conclusions
Selective recognition between oligomers programmed with information encoded in the form of a sequence of recognition sites is the basis for the unique chemical properties of nucleic acids. We describe a synthetic oligomer system that recapitulates some of these properties. Oligomers equipped with a sequence of phenols (H-bond donors, D) and pyridine N-oxides (H-bond acceptors, A) show sequence-selective duplex formation due to H-bonding interactions between complementary recognition sites. This paper describes the synthesis of all eight 3-mer sequences and measurement of the pairwise binding affinities of the oligomers in toluene. The stabilities of the complexes vary by 3 orders of magnitude depending on sequence complementarity. There are three factors that govern the overall stabilities of the complexes in addition to the number of complementary H-bonding interactions.
1. Backbone Orientation
For the oligomer sequences AAD, DAA, ADD, and DDA, it is possible to characterize the relative stabilities of duplexes that have parallel and antiparallel backbones, because the orientation of the backbones is dictated by the sequence of recognition units. These systems show that the antiparallel arrangement of the backbones is more stable than the parallel arrangement by a factor of 4. The other duplexes presumably exist as a 80:20 mixture of the two backbone arrangements.
2. Doubly H-Bonded Acceptors
A single site mismatch analysis reveals that an A → D mutation leads to unexpectedly stable complexes, because the pyridine N-oxide recognition units can accept a second H-bond from an unpaired phenol recognition unit. These additional H-bonding interactions can stabilize D-rich mismatch complexes by up to a factor of 5.
3. 1,3-Folding
We have previously shown that 1,2-folding of adjacent complementary recognition sites does not take place in this system. However, for 3-mers that have a H-bond donor and acceptor at each end of the oligomer, 1,3-folding is significant in the monomeric free state. Folding equilibria compete with duplex formation and reduce the stability of the corresponding duplex by a factor of 5.
The latter two factors conspire to make the stabilities of some of the mismatch complexes greater than the stabilities of some of the sequence-complementary duplexes. However, the measured association constants show that in a mixture of all eight 3-mer sequences the sequence-complementary duplexes are the predominant species present in solution. The most problematic sequence from the point of view of mismatched duplex formation is DDD, which competes effectively with the fully matched sequence in a number of cases. However, DDD has a much higher affinity for AAA than for any other sequence, and so, in the presence of one equivalent of AAA, DDD will not form a mismatched duplex with other sequences. Thus, the fidelity of the recognition system in a complex mixture is higher than might be expected by comparing the stabilities of individual duplexes. Moreover, if alternating donor–acceptor sequences are avoided, it is possible to show that competition from mismatched duplexes can be almost completely eliminated. It should therefore be possible to extend these studies to longer oligomers to obtain high-fidelity sequence recognition.
This issue of doubly H-bonded acceptors can be addressed in a straightforward manner by replacing the pyridine N-oxide recognition units with pyridines, which can only form a single H-bond with phenol. We have shown previously that although the pyridine–phenol H-bond is weaker than the pyridine N-oxide–phenol interaction, the increased conformational restriction imposed by the oriented pyridine nitrogen lone pair compensates to yield stable duplexes.11c The issue of folding equilibria is more difficult. Folding will always compete with duplex formation in synthetic information molecules of this type, because the oligomers carry mutually complementary recognition units. However, the properties of nucleic acids show that, for long oligomers, sequence complementarity can be used to ensure that duplex formation predominates or that the absence of a complementary partner can be used to ensure that intramolecular folding predominates. The same should be true of the systems described here, and this duality of behavior offers interesting avenues for future research. There are some differences between the synthetic H-bonded duplexes and nucleic acid duplexes that may lead to differences in behavior. In nucleic acids, formation of the first base pair is thermodynamically unfavorable, which leads to a nucleation and growth mechanism of duplex assembly, whereas formation of the first base pair in the synthetic duplexes is thermodynamically favorable. Nucleic acid duplexes form compact structures that promote cooperativity and selectivity, whereas the synthetic duplexes are less organized. However, the well-defined assembly properties of nucleic acids are not apparent for short oligomers and only emerge for sequences several bases long. Work on longer synthetic oligomers will reveal whether more organized structures emerge for larger molecules, how the fidelity of sequence recognition is affected, and the impact on the kinetics of strand exchange.
Acknowledgments
We thank the Engineering and Physical Sciences Research Council (EP/J008044/2) and European Research Council (ERC-2012-AdG 320539-duplex) for funding.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b06619.
Detailed experimental procedures and 1H and 13C of all compounds, NMR titration and dilution spectra, molecular modeling methods (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Crick F.; Watson J. Nature 1953, 171, 964. 10.1038/171964b0. [DOI] [PubMed] [Google Scholar]; b Ellington A. D.; Szostak J. W. Nature 1990, 346, 818. 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]; c Rothemund P. W. K. Nature 2006, 440, 297. 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]; d Aldaye F. A.; Palmer A. L.; Sleiman H. F. Science 2008, 321, 1795. 10.1126/science.1154533. [DOI] [PubMed] [Google Scholar]; e Seeman N. C. Nano Lett. 2010, 10, 1971. 10.1021/nl101262u. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Ke Y.; Ong L. L.; Shih W. M.; Yin P. Science 2012, 338, 1177. 10.1126/science.1227268. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Pinheiro A. V.; Han D.; Shih W. M.; Yan H. Nat. Nanotechnol. 2011, 6, 763. 10.1038/nnano.2011.187. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Zhang F.; Nangreave J.; Liu Y.; Yan H. J. Am. Chem. Soc. 2014, 136, 11198. 10.1021/ja505101a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Eschenmoser A. Science 1999, 284, 2118. 10.1126/science.284.5423.2118. [DOI] [PubMed] [Google Scholar]; b Benner S. A. Acc. Chem. Res. 2004, 37, 784. 10.1021/ar040004z. [DOI] [PubMed] [Google Scholar]; c Benner S. A.; Chen F.; Yang Z. In Chemical Synthetic Biology; Luisi P. L.; Chiarabelli C., Eds.; John Wiley & Sons, Ltd: Chichester, UK, 2011; p 69. [Google Scholar]; d Benner S. A. Biol. Theory 2013, 8, 357. 10.1007/s13752-013-0142-y. [DOI] [Google Scholar]; e Wilson C.; Keef A. D. Curr. Opin. Chem. Biol. 2006, 10, 607. 10.1016/j.cbpa.2006.10.001. [DOI] [PubMed] [Google Scholar]; f Appella D. H. Curr. Opin. Chem. Biol. 2009, 13, 687. 10.1016/j.cbpa.2009.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Kool E. T. Curr. Opin. Chem. Biol. 2000, 4, 602. 10.1016/S1367-5931(00)00141-1. [DOI] [PubMed] [Google Scholar]
- Sugar modifications of nucleic acids:; a Schöning K.; Scholz P.; Guntha S.; Wu X.; Krishnamurthy R.; Eschenmoser A. Science 2000, 290, 1347. 10.1126/science.290.5495.1347. [DOI] [PubMed] [Google Scholar]; b van Aerschot A.; Verheggen I.; Hendrix C.; Herdewijn P. Angew. Chem., Int. Ed. Engl. 1995, 34, 1338. 10.1002/anie.199513381. [DOI] [Google Scholar]; c Renneberg D.; Leumann C. J. J. Am. Chem. Soc. 2002, 124, 5993. 10.1021/ja025569+. [DOI] [PubMed] [Google Scholar]; d Braasch D. A.; Corey D. R. Chem. Biol. 2001, 8, 1. 10.1016/S1074-5521(00)00058-2. [DOI] [PubMed] [Google Scholar]; e Singh S. K.; Koshkin A. A.; Wengel J. Chem. Commun. 1998, 455. 10.1039/a708608c. [DOI] [Google Scholar]; f Nguyen H. V.; Zhao Z. Y.; Sallustrau A.; Horswell S. L.; Male L.; Mulas A.; Tucker J. H. R. Chem. Commun. 2012, 48, 12165. 10.1039/c2cc36428j. [DOI] [PubMed] [Google Scholar]; g Zhang L.; Peritz A.; Meggers E. J. Am. Chem. Soc. 2005, 127, 4174. 10.1021/ja042564z. [DOI] [PubMed] [Google Scholar]; h Schlegel M. K.; Peritz A. E.; Kittigowittana K.; Zhang L.; Meggers E. ChemBioChem 2007, 8, 927. 10.1002/cbic.200600435. [DOI] [PubMed] [Google Scholar]; i Karri P.; Punna V.; Kim K.; Krishnamurthy R. Angew. Chem., Int. Ed. 2013, 52, 5840. 10.1002/anie.201300795. [DOI] [PubMed] [Google Scholar]
- Phosphate modifications of nucleic acids:; a Benner S. A.; Hutter D. Bioorg. Chem. 2002, 30, 62. 10.1006/bioo.2001.1232. [DOI] [PubMed] [Google Scholar]; b Huang Z.; Schneider K. C.; Benner S. A. J. Org. Chem. 1991, 56, 3869. 10.1021/jo00012a018. [DOI] [Google Scholar]; c Huang Z.; Benner S. A. J. Org. Chem. 2002, 67, 3996. 10.1021/jo0003910. [DOI] [PubMed] [Google Scholar]; d Richert C.; Roughton A. L.; Benner S. A. J. Am. Chem. Soc. 1996, 118, 4518. 10.1021/ja952322m. [DOI] [Google Scholar]; e Li P.; Sergueeva Z. A.; Dobrikov M.; Shaw B. R. Chem. Rev. 2007, 107, 4746. 10.1021/cr050009p. [DOI] [PubMed] [Google Scholar]; f Isobe H.; Fujino T.; Yamazaki N.; Guillot-Nieckowski M.; Nakamura E. Org. Lett. 2008, 10, 3729. 10.1021/ol801230k. [DOI] [PubMed] [Google Scholar]; g Eriksson M.; Nielsen P. E. Q. Q. Rev. Biophys. 1996, 29, 369. 10.1017/S0033583500005886. [DOI] [PubMed] [Google Scholar]; h Nielsen P. E. Chem. Biodiversity 2010, 7, 786. 10.1002/cbdv.201000005. [DOI] [PubMed] [Google Scholar]; i Nielsen P. E.; Egholm M. Curr. Issues Mol. Biol. 1999, 1, 89. [PubMed] [Google Scholar]; j Nielsen P. E.; Haaima G. Chem. Soc. Rev. 1997, 26, 73. 10.1039/cs9972600073. [DOI] [Google Scholar]; k Ura Y.; Beierle J. M.; Leman L. J.; Orgel L. E.; Ghadiri M. R. Science 2009, 325, 73. 10.1126/science.1174577. [DOI] [PubMed] [Google Scholar]
- Base modification of nucleic acids:; a Piccirilli J. A.; Krauch T.; Moroney S. E.; Benner S. A. Nature 1990, 343, 33. 10.1038/343033a0. [DOI] [PubMed] [Google Scholar]; b Yang Z.; Hutter D.; Sheng P.; Sismour A. M.; Benner S. A. Nucleic Acids Res. 2006, 34, 6095. 10.1093/nar/gkl633. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Wojciechowski F.; Leumann C. J. Chem. Soc. Rev. 2011, 40, 5669. 10.1039/c1cs15027h. [DOI] [PubMed] [Google Scholar]; d Benner S. A. Curr. Opin. Chem. Biol. 2012, 16, 581. 10.1016/j.cbpa.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Liu H.; Gao J.; Lynch S. R.; Saito Y. D.; Maynard L.; Kool E. T. Science 2003, 302, 868. 10.1126/science.1088334. [DOI] [PubMed] [Google Scholar]; f Kool E. T. Acc. Chem. Res. 2002, 35, 936. 10.1021/ar000183u. [DOI] [PubMed] [Google Scholar]; g Kool E. T.; Lu H.; Kim S. J.; Tan S.; Wilson J. N.; Gao J.; Liu H. Nucleic Acids Symp. Ser. 2006, 50, 15. 10.1093/nass/nrl008. [DOI] [PubMed] [Google Scholar]; h Wilson J. N.; Kool E. T. Org. Biomol. Chem. 2006, 4, 4265. 10.1039/b612284c. [DOI] [PubMed] [Google Scholar]
- a Kramer R.; Lehn J.-M.; Marquis-Rigault A. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 5394. 10.1073/pnas.90.12.5394. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Marquis A.; Smith V.; Harrowfield J.; Lehn J.-M.; Herschbach H.; Sanvito R.; Leize-Wagner E.; van Dorsselaer A. Chem. - Eur. J. 2006, 12, 5632. 10.1002/chem.200600143. [DOI] [PubMed] [Google Scholar]
- a Gong B.; Yan Y.; Zeng H.; Skrzypczak-Jankunn E.; Kim Y. W.; Zhu J.; Ickes H. J. Am. Chem. Soc. 1999, 121, 5607. 10.1021/ja990904o. [DOI] [Google Scholar]; b Gong B. Synlett 2001, 2001, 582. 10.1055/s-2001-13350. [DOI] [Google Scholar]; c Zeng H.; Miller R. S.; Flowers R. A.; Gong B. J. Am. Chem. Soc. 2000, 122, 2635. 10.1021/ja9942742. [DOI] [Google Scholar]; d Zeng H.; Ickes H.; Flowers R. A.; Gong B. J. Org. Chem. 2001, 66, 3574. 10.1021/jo010250d. [DOI] [PubMed] [Google Scholar]; e Gong B. Polym. Int. 2007, 56, 436. 10.1002/pi.2175. [DOI] [Google Scholar]; f Gong B. Acc. Chem. Res. 2012, 45, 2077. 10.1021/ar300007k. [DOI] [PubMed] [Google Scholar]; g Yang Y.; Yang Z. Y.; Yi Y. P.; Xiang J. F.; Chen C. F.; Wan L. J.; Shuai Z. G. J. Org. Chem. 2007, 72, 4936. 10.1021/jo070525a. [DOI] [PubMed] [Google Scholar]
- a Tanaka Y.; Katagiri H.; Furusho Y.; Yashima E. Angew. Chem. 2005, 117, 3935. 10.1002/ange.200501028. [DOI] [PubMed] [Google Scholar]; b Ito H.; Furusho Y.; Hasegawa T.; Yashima E. J. Am. Chem. Soc. 2008, 130, 14008. 10.1021/ja806194e. [DOI] [PubMed] [Google Scholar]; c Yamada H.; Furusho Y.; Ito H.; Yashima E. Chem. Commun. 2010, 46, 3487. 10.1039/c002170a. [DOI] [PubMed] [Google Scholar]; d Yashima E.; Ousaka N.; Taura D.; Shimomura K.; Ikai T.; Maeda K. Chem. Rev. 2016, 116, 13752. 10.1021/acs.chemrev.6b00354. [DOI] [PubMed] [Google Scholar]
- a Anderson H. L. Inorg. Chem. 1994, 33, 972. 10.1021/ic00083a022. [DOI] [Google Scholar]; b Taylor P. N.; Anderson H. L. J. Am. Chem. Soc. 1999, 121, 11538. 10.1021/ja992821d. [DOI] [Google Scholar]; c Berl V.; Huc I.; Khoury R. G.; Krische M. J.; Lehn J.-M. Nature 2000, 407, 720. 10.1038/35037545. [DOI] [PubMed] [Google Scholar]; d Berl V.; Huc I.; Khoury R. G.; Lehn J.-M. Chem. - Eur. J. 2001, 7, 2810.. [DOI] [PubMed] [Google Scholar]; e Sánchez-Quesada J.; Seel C.; Prados P.; de Mendoza J.; Dalcol I.; Giralt E. J. Am. Chem. Soc. 1996, 118, 277. 10.1021/ja953243d. [DOI] [Google Scholar]; f Bisson A. P.; Carver F. J.; Eggleston D. S.; Haltiwanger R. C.; Hunter C. A.; Livingstone D. L.; McCabe J. F.; Rotger C.; Rowan A. E. J. Am. Chem. Soc. 2000, 122, 8856. 10.1021/ja0012671. [DOI] [Google Scholar]; g Bisson A. P.; Hunter C. A. Chem. Commun. 1996, 1723. 10.1039/cc9960001723. [DOI] [Google Scholar]; h Chu W.-J.; Yang Y.; Chen C.-F. Org. Lett. 2010, 12, 3156. 10.1021/ol101068n. [DOI] [PubMed] [Google Scholar]; i Chu W.-J.; Chen J.; Chen C.-F.; Yang Y.; Shuai Z. J. Org. Chem. 2012, 77, 7815. 10.1021/jo301434a. [DOI] [PubMed] [Google Scholar]; j Archer E. A.; Krische M. J. J. Am. Chem. Soc. 2002, 124, 5074. 10.1021/ja012696h. [DOI] [PubMed] [Google Scholar]; k Gong H.; Krische M. J. J. Am. Chem. Soc. 2005, 127, 1719. 10.1021/ja044566p. [DOI] [PubMed] [Google Scholar]
- a Dervan P. B.; Burli R. W. Curr. Opin. Chem. Biol. 1999, 3, 688. 10.1016/S1367-5931(99)00027-7. [DOI] [PubMed] [Google Scholar]; b Renneberg D.; Dervan P. B. J. Am. Chem. Soc. 2003, 125, 5707. 10.1021/ja0300158. [DOI] [PubMed] [Google Scholar]; c Doss R. M.; Marques M. A.; Foister S.; Chenoweth D. M.; Dervan P. B. J. Am. Chem. Soc. 2006, 128, 9074. 10.1021/ja0621795. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Meier J. L.; Yu A. S.; Korf I.; Segal D. J.; Dervan P. B. J. Am. Chem. Soc. 2012, 134, 17814. 10.1021/ja308888c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Stross A. E.; Iadevaia G.; Hunter C. A. Chem. Sci. 2016, 7, 94. 10.1039/C5SC03414K. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Iadevaia G.; Stross A. E.; Neumann A.; Hunter C. A. Chem. Sci. 2016, 7, 1760. 10.1039/C5SC04467G. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Stross A. E.; Iadevaia G.; Hunter C. A. Chem. Sci. 2016, 7, 5686. 10.1039/C6SC01884J. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Núñez-Villanueva D.; Hunter C. A. Chem. Sci. 2017, 8, 206. 10.1039/C6SC02995G. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Núñez-Villanueva D.; Iadevaia G.; Stross A. E.; Jinks M. A.; Swain J. A.; Hunter C. A. J. Am. Chem. Soc. 2017, 139, 6654. 10.1021/jacs.7b01357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Jorgensen W. L.; Pranata J. J. Am. Chem. Soc. 1990, 112, 2008. 10.1021/ja00161a061. [DOI] [Google Scholar]; b Jeong K. S.; Tjivikua T.; Muehldorf A.; Deslongchamps G.; Famulok M.; Rebek J. J. Am. Chem. Soc. 1991, 113, 201. 10.1021/ja00001a029. [DOI] [Google Scholar]; c Murray T. J.; Zimmerman S. C. J. Am. Chem. Soc. 1992, 114, 4010. 10.1021/ja00036a079. [DOI] [Google Scholar]
- MacroModel, version 9.8; Schrodinger, LLC: New York, 2014.
- For analyses of fidelity in other systems, see:; a Todd E. M.; Quinn J. R.; Park T.; Zimmerman S. C. Isr. J. Chem. 2005, 45, 381. 10.1560/DQCJ-1K9J-1TBT-DK5M. [DOI] [Google Scholar]; b Park T.; Todd E. M.; Nakashima S.; Zimmerman S. C. J. Am. Chem. Soc. 2005, 127, 18133. 10.1021/ja0545517. [DOI] [PubMed] [Google Scholar]; c Mukhopadhyay P.; Zavalij P. Y.; Isaacs L. J. Am. Chem. Soc. 2006, 128, 14093. 10.1021/ja063390j. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Pellizzaro M. L.; Houton K. A.; Wilson A. J. Chem. Sci. 2013, 4, 1825. 10.1039/c3sc22194f. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.