

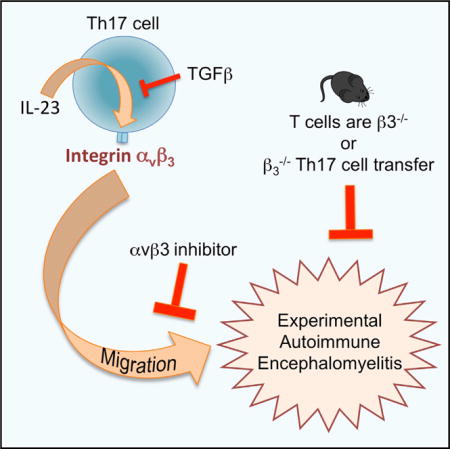
SUMMARY
Inerleukin-23 (IL-23) is required for inflammatory Th17 cell function in experimental autoimmune encephalomyelitis (EAE), and IL-23 blockade reduces the number of effector Th17 cells in the CNS. We report that pro-inflammatory Th17 cells express high integrin β3 that is IL-23 dependent. Integrin β3 was not upregulated on all activated T cells; rather, integrin β3 was upregulated along with its functional partner integrin αv on effector Th17 cells and “ex-Th17” cells, and αvβ3hi RORγt+ cells expanded during EAE. Integrin αvβ3 inhibitors ameliorated clinical signs of EAE, and integrin β3 deficiency on CD4+ T cells alone was sufficient to block EAE induction. Furthermore, integrin-β3-deficient Th17 cells, but not Th1 cells, were impaired in their ability to induce EAE. Integrin β3−/− T cells induced smaller demyelinated lesions and showed reduced spread and accumulation within the CNS, corresponding with impaired extracellular-matrix-mediated migration. Hence, integrin β3 is required for Th17 cell-mediated autoimmune CNS inflammation.
Graphical abstract
INTRODUCTION
Th17-mediated inflammation is highly dependent on signals from interleukin-23 (IL-23), an IL-6 family member cytokine composed of the common IL-12/IL-23 p40 subunit paired with the unique p19 subunit (Aggarwal et al., 2003; Cua et al., 2003; Oppmann et al., 2000; Reboldi et al., 2009). The IL-23 receptor (IL-23R) is not highly expressed on naive CD4+ T cells, and accordingly, IL-23 is not required for the early upregulation of the putative Th17 transcription factor RORγt or for expression of IL-17 (Zúñiga et al., 2013; Ivanov et al., 2006). Rather, IL-23 is required for Th17 cell proliferation and the switch to effector phenotype after the initial signals for differentiation have been provided by transforming growth factor β (TGF-β), IL-6, and IL-1 (Mangan et al., 2006; Veldhoen et al., 2006; Bettelli et al., 2006; Chung et al., 2009). The latter two cytokines induce upregulation of the IL-23 receptor (IL-23R), thus allowing IL-23 signals to come into play as Th17 cell differentiation progresses (Zhou et al., 2007). Hence, it is possible to induce early Th17 cells in the absence of IL-23 signals in vivo. However, beginning 1 week post-immunization, IL-23R-deficient Th17 cells show reduced proliferation, lose IL-17 production, and generate few IL-2−IL7RhiCD27lo effector phenotype cells (McGeachy et al., 2009). IL-23 is also required for granulocyte-monocyte colony stimulating factor (GM-CSF) production by Th17 cells, which is critical for EAE induction (Codarri et al., 2011; El-Behi et al., 2011). Mice deficient in IL-23 or IL-23R are therefore highly resistant to Th17-mediated autoimmune inflammation, and monoclonal antibodies targeting IL-23 or IL-17 are proving highly efficacious in clinical treatment of psoriasis and are currently being trialed in multiple sclerosis (MS) and other autoimmune diseases.
In the experimental autoimmune encephalomyelitis (EAE) model of MS, IL-23R-deficient Th17 cells show defective accumulation in the CNS (McGeachy et al., 2009). Fewer cells in the blood could partially explain this defect. Alternatively, IL-23R signaling may confer a migratory advantage on Th17 effector cells. CCR6 is the key Th17-expressed chemokine receptor thought to allow initial entry of Th17 cells into the CNS by promoting migration through the choroid plexus (Reboldi et al., 2009). However, IL-23 is not required for expression of CCR6 (McGeachy et al., 2009).
Integrins are cell-surface receptors that promote migration of cells into inflamed tissue sites through interactions with inflamed endothelium and stromal extracellular matrix (ECM) components. Integrin blockade is used therapeutically in MS and Crohn’s disease; natalizumab is a monoclonal antibody targeting integrin α4-mediated migration of inflammatory T cells into the brain and gut. While highly effective in some patients, natalizumab therapy carries the risk of progressive multifocal leukoencephalopathy, caused by a rare but frequently fatal uncontrolled John Cunningham (JC) virus infection in the brain that occurs due to the inability of virus-specific T cells, including Th1 cells, to migrate to the CNS after α4 blockade (Hellwig and Gold, 2011; Aly et al., 2011). Furthermore, recent data indicate that integrin α4 is not absolutely required for Th17 cell entry to the CNS (Glatigny et al., 2011; Rothhammer et al., 2011). Identification of integrins that are specifically expressed on Th17 cells, and particularly in response to IL-23, therefore has great therapeutic potential.
Integrin β3 (Itgb3) is a member of the RGD family of integrins with two described heterodimeric partners: αIIb is expressed on platelets, while αv is expressed on a wide variety of cells and pairs with β1, β5, β6, and β8 as well as β3 (Hynes, 2002). Integrin β3 expression is increased in Th17-associated diseases such as psoriasis (Goedkoop et al., 2004), psoriatic arthritis (Cañete et al., 2004), rheumatoid arthritis (Kurohori et al., 1995), and MS (Murugaiyan et al., 2008). However, the functions of integrin β3 have not been closely studied on immune cells. Integrin αvβ3 is known to bind ECM proteins, including vitronectin and fibronectin, which show increased expression in the CNS in both EAE and MS (Han et al., 2008; Teesalu et al., 2001). Integrin αvβ3 also binds osteopontin, which is strongly associated with autoimmune diseases, including MS (Steinman, 2009). Given these intriguing connections with the IL-23/Th17 axis and integrin αvβ3, we therefore directly tested the expression and function of this integrin heterodimer on Th17 cells in the context of autoimmune disease. We focused our studies largely on the β3 integrin, since it partners only with αv to form a functional receptor on T cells, while αv can form receptors with multiple α integrins.
RESULTS
IL-23R-Dependent Th17 Cells Express Integrin β3
To investigate expression of integrin β3 on Th17 cells in vivo, we used an OTII adoptive transfer system known to drive robust antigen-specific Th17 cell development (McGeachy et al., 2009; Chen et al., 2011; Chung et al., 2009). OTII cells activated by immunization with OVA(323–339) in complete Freund’s adjuvant (CFA) contained a clear population of IL-17-expressing cells that had uniformly high expression of integrin β3 (Figure 1A). Similarly, IL-23R+ cells were mostly integrin β3hi (Figure 1B), while the majority of activated but IL-17− or IL-23R− cells were integrin β3lo. Th17 cells are dependent on expression of IL-23R for development of effector functions in vivo (McGeachy et al., 2009). Integrin β3 expression peaked on wild-type (WT) OTII Th17 cells on day 10 and was strongly dependent on IL-23 signaling, since IL-23R−/− OTII cells showed a significant defect in their ability to upregulate integrin β3 on Th17 cells (Figures 1C and 1D). IL-17 production is decreased in the absence of IL-23R by day 10 post-immunization, and so we confirmed that the defect in upregulation was consistent in IL-23R−/− OTII cells when gating only on remaining IL-17+ cells (Figure 1E). Integrin β3 expression was not dependent on IL-12 in vivo, as transfer of WT OTII cells into IL-12-deficient hosts did not reduce expression on resulting Th17 cells (data not shown).
Figure 1. IL-23R-Dependent Inflammatory Th17 Cells Are Integrin β3hi.
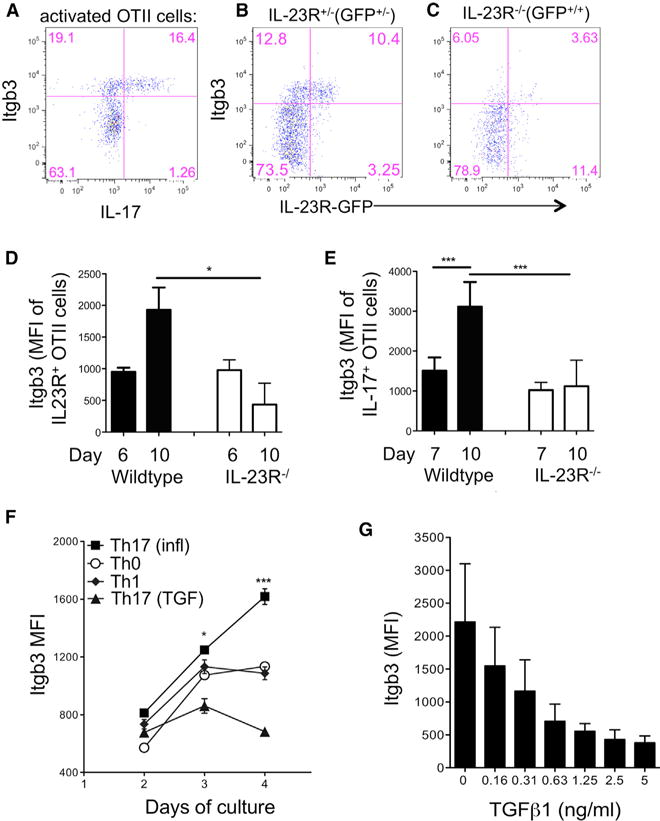
CD4+ T cells from CD45.1+ IL-23R−/− GFP, IL-23R+/− GFP or WT OTII mice were adoptively transferred into WT B6 recipients that were immunized the following day with OVA(323–339) in CFA.
(A) dLN cells were stimulated with PMA/ionomycin on day 10 and OTII cells analyzed by flow cytometry for co-expression of IL-17 and integrin β3, gating on CD4+CD45.1+ cells.
(B) Integrin β3 expression on unstimulated IL-23R+/− GFP+/− CD45.1+ OTII cells taken from dLNs on day 10.
(C) Integrin β3 expression on IL-23R-deficient (IL-23R−/− GFP+/+) CD45.1+ OTII cells taken from dLNs on day 10.
(D and E) Pooled data for integrin β3 expression on wild-type and IL-23R−/− cells as described above, gating on unstimulated IL-23R(GFP)+ OTII cells (D) and IL-17+OTII cells following PMA/ionomycin stimulation (E).
(F) CD4+ T cells from B6 mice were activated with plate-bound anti-CD3 under the following conditions: IL-23, IL-6, IL-1 (Th17(i)), TGF-β + IL-23, IL-6, IL-1 (Th17(TGF)), IL-12 (Th1), or no cytokines (Th0), and expression of integrin β3 was assessed by flow cytometry on indicated days of culture.
(G) Expression of integrin β3 on CD4+ cells cultured in the presence of IL-23, IL-6, and IL-1 with indicated concentrations of TGF-β, analyzed by flow cytometry on day 4.
Data are representative of three separate experiments with four mice per group (A–E) or performed in triplicate (F and G). Statistical significance was assessed by one-way ANOVA. Error bars indicate mean ± SD.
TGF-β strongly promotes Th17 development when measured by expression of RORγt and IL-17, and T cells lacking TGF-β signaling show a defect in Th17 development and maintenance in vivo (Zúñiga et al., 2013). However, high levels of TGF-β signaling result in non-inflammatory Th17 cells that produce IL-10 and are unable to transfer disease in vivo (McGeachy et al., 2007; Ghoreschi et al., 2010). In contrast, stimulation of T cells in the presence of IL-23, IL-6, and IL-1 induces highly inflammatory Th17 cells (Th17(i) cells) (Ghoreschi et al., 2010). In vitro, Th17(i) showed a time-dependent upregulation of integrin β3 that was significantly greater than Th0 and Th1 cells (Figure 1F). Furthermore, TGF-β suppressed integrin β3 expression by Th17(i) cells in a dose-dependent manner (Figures 1F and 1G). Together, these data support the conclusion that inflammatory Th17 cells express high integrin β3 that is promoted by IL-23 signaling but suppressed by high levels of TGF-β.
Integrin β3hi RORγt+ Th17 Cells Increase in EAE
We next investigated integrin β3 expression by Th17 cells during inflammation in the EAE model. Corresponding with the previous results, IL-17-expressing cells in lymph nodes (LNs) and CNS of mice with EAE were integrin β3hi (Figure 2A). Since phorbol 12-myristate 13-acetate (PMA)/ionomycin stimulation likely overestimates the frequency of MOG-specific Th17 cells, we also confirmed that MOG-specific IL-17 producers are high expressors of integrin β3 in EAE (Figure 2B), and furthermore, integrin β3 was not altered by PMA/ionomycin stimulation (Figure 2B).
Figure 2. Integrin β3 Expression on Th17 Cells in EAE.
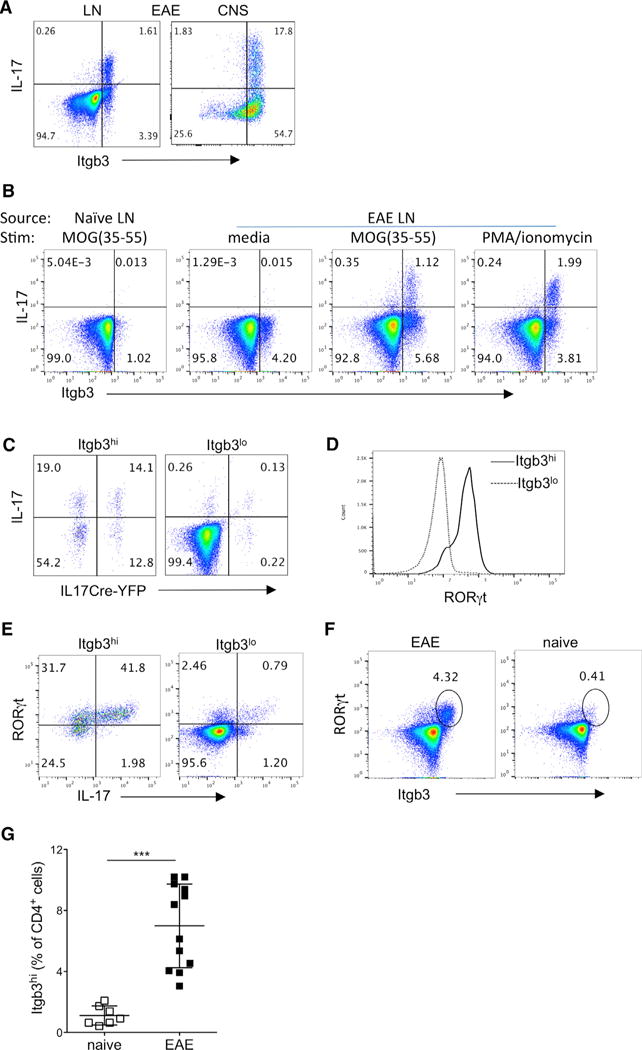
CD4+ T cells from dLNs and CNS were analyzed by flow cytometry during active EAE (day 12–16) induced in IL-17Cre-YFP fate-tracking mice.
(A) Representative flow cytometry plots of integrin β3 and intracellular IL-17 staining following PMA/ionomycin stimulation in live CD4+ cells from indicated sites.
(B) IL-17 and integrin β3 co-expression in live CD4+ T cells from naive and EAE LNs, stimulated as indicated (all EAE plots are from same donor for comparison).
(C) EAE LN cells were stimulated with PMA/ionomycin, and live CD4+ cells were then gated according to high or low integrin β3 expression to determine expression of intracellular IL-17 protein and IL17Cre-mediated YFP.
(D) Histogram comparing RORγt expression in integrin β3hi and integrin β3lo CD4+ EAE LN cells.
(E) EAE LN cells stimulated with PMA/ionomycin and analyzed for expression of IL-17 and RORγt in integrin β3hi and integrin β3lo CD4+ cells.
(F) Representative plots of RORγt+integrin β3hi population within live CD4+ cells from EAE LNs and naive LNs.
(G) Percentage of integrin β3hi cells gated from live CD4+ cells, with data pooled from three separate experiments.
Data are representative of at least three separate experiments, with two to four mice per group. Statistical significance was analyzed by one-way ANOVA. Error bars indicate mean ± SD.
In both the OTII model and EAE, it was apparent that a fairly large proportion of integrin β3hi cells did not express IL-17 in response to stimulation. Since conversion of Th17 cells with loss of IL-17 production is quite prevalent in EAE, we used IL-17CreYFP fate-tracking mice and demonstrated that current and prior Th17 lineage cells accounted for approximately half of integrin β3hi cells in draining LNs (dLNs) (Figures 2C and S1A). In contrast, very few Th17 cells were found in the integrin β3lo population (Figure 2C). It is known that the IL17CreYFP cells under-report IL-17 expression due to the threshold required for Cre expression (Hirota et al., 2011). In addition, IL-17 production is not the only factor defining Th17 cells. We therefore analyzed expression of the putative Th17 cell transcription factor RORγt and found that most integrin β3hi cells also expressed RORγt (Figure 2D), and some of the integrin β3hi RORγt+ cells co-expressed IL-17 (Figure 2E). Interestingly, RORγt+ integrin β3hi cells were almost absent in naive animals (Figure 2F), and integrin β3hi T cells were significantly increased in EAE compared to naive LNs (Figure 2G).
Integrin β3 and Th1 Cells in EAE
Compared to Th17 cells, interferon-γ-positive (IFN-γ+) cells had less consistent integrin β3 expression, and only approximately half of IFN-γ+ cells were integrin β3hi in EAE (Figure 3A). This was particularly apparent in dLN, where IL-17+ cells had significantly higher integrin β3 expression overall than IFN-γ+ cells (Figure 3B). Fate-tracking studies in EAE previously demonstrated that a high proportion of IFNγ+ T cells are in fact derived from Th17 precursors, termed “ex-Th17” cells (Hirota et al., 2011). We therefore employed IL-17CreYFP fate-tracking mice to further interrogate the relationship between integrin β3 expression and the Th17 cell lineage in IFN-γ-producing cells. YFP expression marks cells that have produced high amounts of IL-17 at some time in their differentiation history. In both LN and CNS, YFP-expressing cells (regardless of IFN-γ production) showed significantly higher integrin β3 expression than YFP “true Th1” cells, which had similar levels to cells that were double negative for both IFN-γ and YFP (Figure 3C). Hence, integrin β3 expression is retained on converted ex-Th17 cells, similarly to IL-1R1 (Hirota et al., 2011).
Figure 3. Integrin β3 and Th1 Cells in EAE.
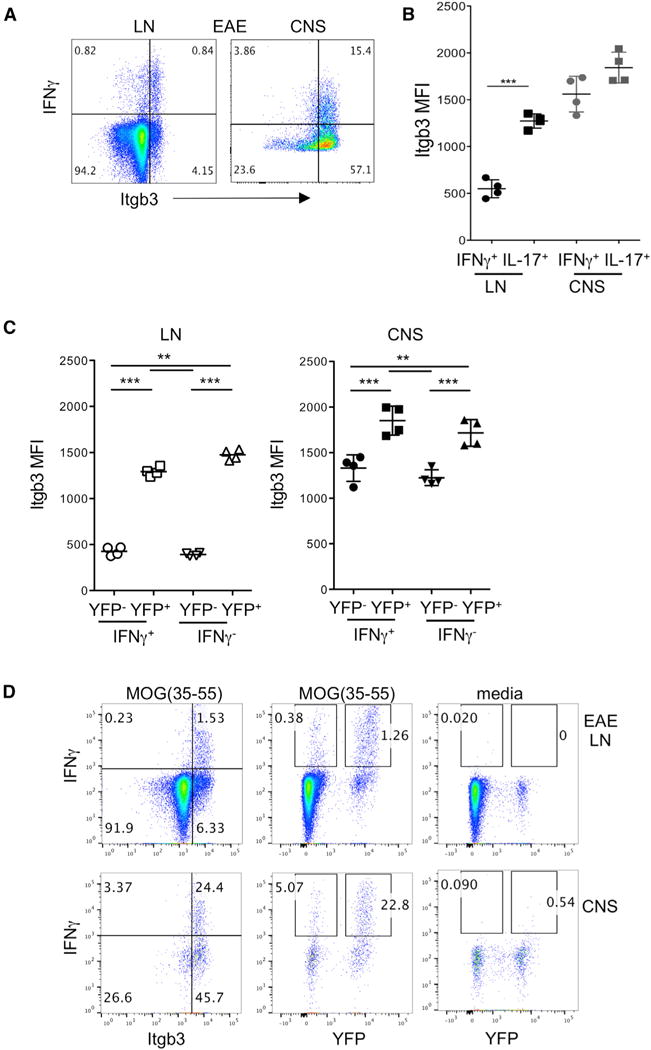
CD4+ T cells from dLNs and CNS were analyzed by flow cytometry during active EAE (day 12–16) induced in IL-17Cre-YFP fate-tracking mice.
(A) Representative flow cytometry plots of integrin β3 and intracellular IFN-γ staining following PMA/ionomycin stimulation in live CD4+ cells from indicated sites.
(B) Geometric mean fluorescence intensity (MFI) of integrin β3 expression on cells positive by intracellular cytokine staining for IFN-γ and IL-17 at indicated sites.
(C) Geometric MFI of integrin β3 expression on LNs and CNS CD4+ T cells gated according to co-expression of IFN-γ and YFP as indicated.
(D) IFN-γ and integrin β3 co-expression by live CD4+ T cells from EAE LNs and CNS, stimulated with MOG(35–55) overnight, or with no stimulation in media-only control.
Data are representative of at least three experiments with three to five mice per group. Statistical significance was analyzed by one-way ANOVA. Error bars indicate mean ± SD. See also Figure S3.
We observed that some IFN-γ-producing cells were also present in T cells from naive mice (data not shown), suggesting that PMA/ionomycin stimulation can induce non-specific Th1 responses. We therefore stimulated EAE dLN and CNS cells with MOG(35–55). In contrast to PMA/ionomycin induced responses, EAE T cells that produced IFN-γ in response to MOG were mostly integrin β3hi (Figure 3D). Furthermore, the majority of MOG-responsive IFN-γ-producing cells were also YFP+, and the proportion of YFP+ cells was greatly increased in CNS. Hence, these data support the Th17-driven nature of the EAE model as previously reported by Hirota et al. (2011), with few true Th1 cells present, particularly if one analyzes MOG-driven responses.
Co-expression of Integrins on Th17 and T Regulatory Cells in EAE versus Naive LN
Integrins are expressed as heterodimers to form functional receptors, pairing an α and β chain. On leukocytes, integrin β3 is only known to have one partner: integrin αv. As predicted, integrin β3hi cells were also integrin αvhi, and this αvβ3hi population was largely absent in naive animals (Figure 4A). The staining pattern of these two integrins gave a consistently linear pattern, an effect that can occur when autofluorescence or compensation problems occur. However, the absence of this specific population in naive LN, along with fluorescence-minus-one staining controls (Figure S2A), instead suggests that integrin β3 and integrin αv are co-expressed, as would be expected for a heterodimeric receptor. To confirm the association between αv and β3 on Th17 cells, we performed co-immunoprecipitation experiments, and as predicted by the literature, β3 was detected upon αv immunoprecipitation, but not the IgG control, and vice versa (Figures 4B and S2B).
Figure 4. Integrin β3 Expression by Activated and Effector T Cells.
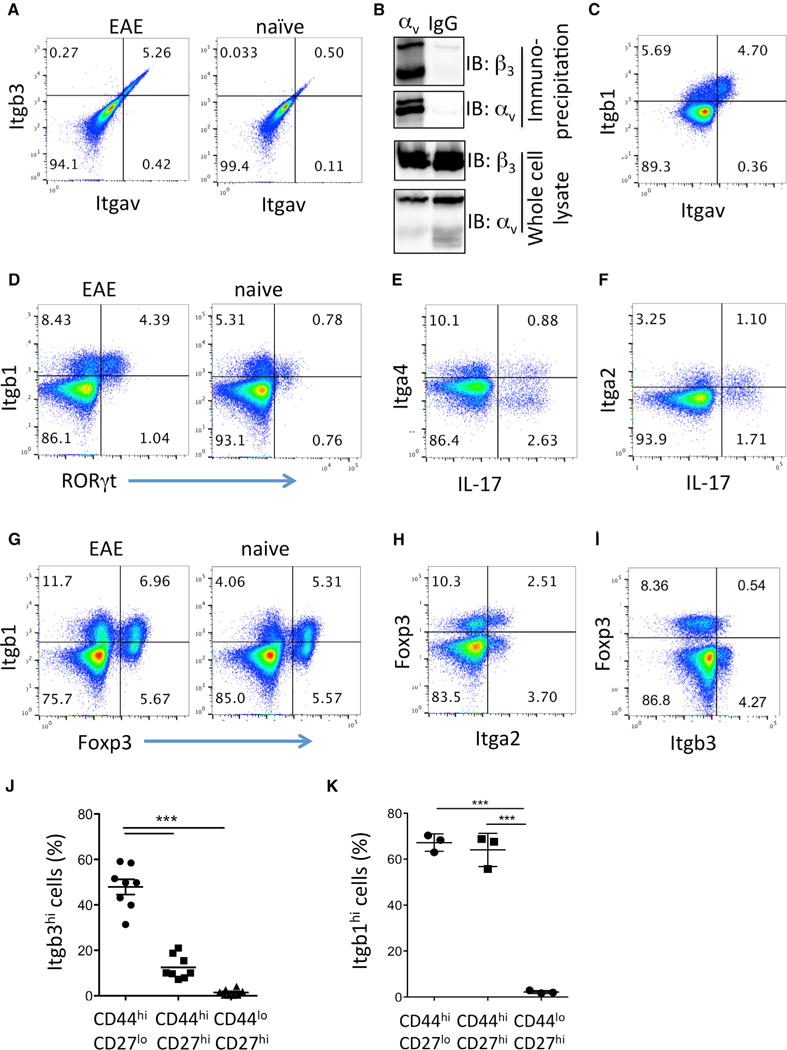
Live CD4+ T cells from dLNs were analyzed by flow cytometry during active EAE (day 12–16) and from naive controls as indicated.
(A) Co-expression of integrin β3 and integrin αv.
(B) Co-immunoprecipitation of αv and β3 from in vitro-differentiated Th17 cells as described in Figure 1F. Lysates were immunoprecipitated with anti-αv or IgG control, and immunoblot was performed for β3 and αv as indicated.
(C) Co-expression of integrin β1 and integrin αv in EAE.
(D) Co-expression of RORγt and integrin β1.
(E) Co-expression of IL-17 and integrin α4.
(F) Co-expression of IL-17 and integrin α2.
(G) Co-expression of integrin β1 and Foxp3.
(H) Co-expression of integrin α2 and Foxp3.
(I) Co-expression of integrin β3 and Foxp3.
(J) Proportion of effector (CD44hiCD27lo), activated/memory (CD44hiCD27hi), and naive (CD44loCD27hi) CD4+ cells that were integrin β3hi in EAE LNs.
(K) Proportion of effector (CD44hiCD27lo), activated/memory (CD44hiCD27hi), and naive (CD44loCD27hi) CD4+ cells that were integrin β1hi.
Data are representative of at least three independent experiments with two to four mice per group; data in (I) and (J) were pooled from two separate experiments. Statistical significance was analyzed by one-way ANOVA. Error bars indicate mean ± SD.
Integrin αv also partners with integrin β1, and αvβ1 was recently shown to be important for Th1 cell migration in tissues (Overstreet et al., 2013). In EAE, the expanded integrin-αv-positive T cells also expressed integrin β1 (Figure 4C). However, the pattern of staining was different to αv with β3, and approximately half of integrin β1hi cells did not express integrin αv (Figure 4C). This suggested that integrin β1hi cells were not exclusively Th17 cells, and indeed, this was confirmed by analyzing co-expression of integrin β1 and RORγt (Figure 4D). Furthermore, while RORγt+ integrin β1hi cells expanded in EAE compared to naive LN, a population of integrin β1hi cells was consistently present in naive animals (Figure 4D). Unlike integrin β3, there are multiple potential partners for β1 heterodimers. Integrin α4 was expressed on few Th17 cells (Figure 4E), in accordance with previous findings that Th17 cells are less dependent on integrin α4 than Th1 cells (Glatigny et al., 2011; Rothhammer et al., 2011). Integrin α6 was only present on a few T cells in EAE and not on Th17 cells (Figure S3A). Integrin α2 has been reported to be expressed with β1 on Th17 cells (Gagliani et al., 2015; El Azreq et al., 2013). In EAE, less than half of Th17 cells expressed integrin α2 (Figure 4F). Integrin α2β1 has also been reported on T regulatory cells (Gagliani et al., 2015). Indeed, integrin β1 was expressed on approximately half of Foxp3+ regulatory T cells in LN from both naive and EAE mice (Figure 4G), although a smaller proportion of regulatory cells also expressed integrin α2 (Figure 4H). In contrast, most Foxp3+ T regulatory cells were integrin β3lo (Figure 4I). As expected for expanded helper T cells, integrin β3hi T cells were exclusively found in the CD44hi-activated cell population during EAE and were especially enriched in CD44hiCD27lo effector T cells (Figure 4J). However, it is important to note that integrin β3hi T cells constituted <20% of the CD44hiCD27hi-activated T cell population and only half of the CD44hiCD27lo effector T cell population (Figure 4J). As suggested by our finding that integrin β1hi cells were present in naive animals and expressed by other T helper populations including regulatory T cells, integrin β1 was expressed on ~75% of both activated and effector T populations (Figure 4K). The levels of integrin β1 were high on both IL-17+ and IFN-γ+ T cell populations in the LN and CNS, and did not vary between ex-Th17 IL-17Cre-YFP+ and YFP− cells (Figures S3B and S3C). Although not an extensive analysis of integrin expression by helper T cell subsets, taken together, these data point to integrin β3 forming a heterodimer with αv specifically on Th17 cells that expand in response to EAE induction. Th17 cells also express other integrins, particularly integrin β1, and some Th17 cells expressed α2 or α4. However, none of these integrins showed Th17-constrained expression to the degree of integrin αvβ3. Therefore, we focused our functional analyses on the requirements for integrin αvβ3 by specifically blocking β3 in Th17 cells.
Integrin αvβ3 Is Functionally Important for Th17-Mediated CNS Inflammation
To test the requirement for integrin β3 in Th17-mediated EAE, MOG-reactive cells were expanded from immunized Itgb3−/− or from control Itgb3+/− donor LN in the presence of IL-23 before transfer to naive recipients. As expected, integrin β3 deficiency significantly reduced the ability of IL-23-driven cells to transfer EAE (Figure 5A). In contrast, Itgb3−/− Th1 cells expanded in presence of IL-12 were capable of transferring EAE (Figure 5B). The incidence of EAE in WT Th17 cell recipients was 12/15 (80%) compared to 5/15 (30%) in recipients of Itgb3−/− Th17 cells, and correspondingly few CNS lesions and infiltrating CD4+ cells were found in these resistant mice (Figure 5C).
Figure 5. Integrin β3 Is Important for Th17-Mediated EAE Induction.
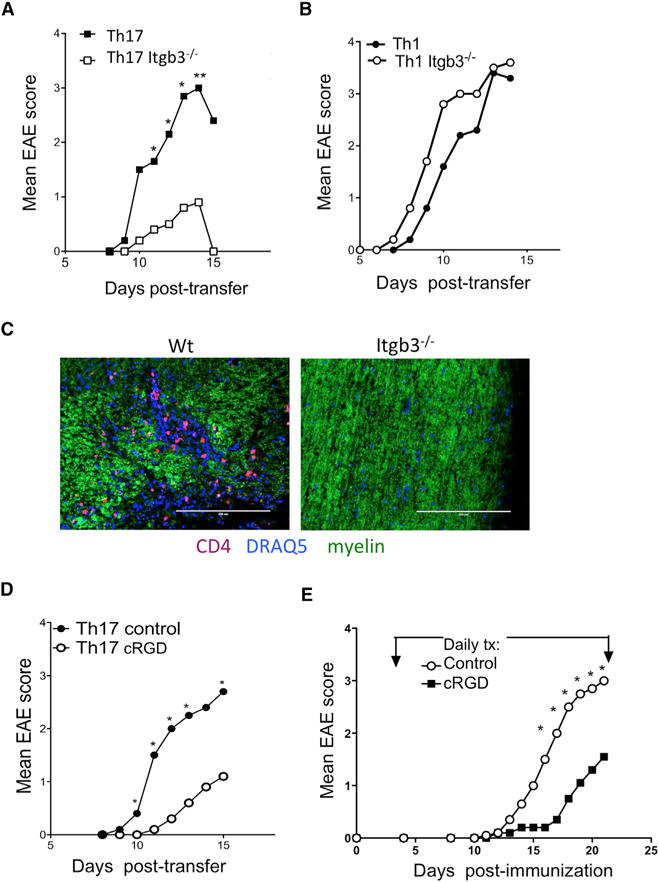
(A) EAE was passively induced by stimulating T cells from MOG(35–55)-immunized Itgb3−/− and Itgb3+/+ donors with MOG(35–55) in presence of IL-23 to expand Th17 cells before transfer into wild-type recipients.
(B) Passive EAE induction by T cells taken from immunized Itgb3−/− and Itgb3+/+ donors and stimulated with MOG(35–55) in presence of IL-12 to expand Th1 cells before transfer into wild-type recipients.
(C) Representative immunofluorescence staining of spinal cord sections from recipients of MOG-stimulated WT or Itgb3−/− Th17 cells, analyzed at peak of disease, showing CD4+ T cells, nuclear stain (DRAQ5), and myelin (scale bars indicate 200 μm).
(D) C57B/6 mice received passive transfer of WT MOG(35–55)-stimulated Th17 cells, with cRGD (cilengitide) inhibitor treatment administered daily from day of transfer.
(E) C57Bl/6 mice were immunized to induce EAE and treated daily from day 4 with 100 μg cRGDfv to inhibit integrin β3 or PBS control. Clinical signs were monitored.
Data are pooled from four (A), two (B and D), and three (E) experiments with four to six mice per group, and statistical significance of EAE scores was assessed by a Mann-Whitney test separately for each time point.
We wanted to confirm that defects in ability of Itgb3−/− Th17 cells to transfer EAE were not due to developmental effects or priming in the knockout donors. We therefore transferred WT Th17 cells and administered cilengitide, a modified compound of cRGDfv, developed to have increased sensitivity for integrin αvβ3 (Mas-Moruno et al., 2010; Reardon et al., 2008). Integrin αvβ3 inhibition also significantly reduced severity of Th17-mediated EAE (Figure 5D), and these data support the role of integrin αvβ3 in the effector phase rather than early activation of Th17 cells. We also confirmed that αvβ3 inhibition by cilengitide administration from day 4 after active immunization with MOG(35–55) resulted in significantly reduced EAE severity compared to control mice (Figure 5E). Together, these data support a critical targetable role for integrin αvβ3 expression by Th17 cells for their pro-inflammatory functions in autoimmune disease.
To further confirm the requirement for integrin αvβ3 in Th17 inflammatory cell function, we generated Itgb3−/− 2D2 mice, in which CD4+ T cells bear a transgenic T cell receptor (TCR) reactive to MOG(35–55). Th17 cells were differentiated from these mice in vitro, resulting in equivalent expression of IL-17 (Figure 6A). Following resimulation according to the protocol established by Jäger et al. (2009), equal numbers of effector Th17 cells were transferred to naive recipients. Itgb3−/− 2D2 Th17 cells were again found to be significantly impaired in their capacity to induce EAE (Figure 6B). Spinal cords from recipients of WT 2D2 Th17 cells showed large demyelinated lesions, with extensive infiltration of CD4+ T cells consistently apparent even beyond the main inflammatory cellular focus (Figures 6C and 6D). In contrast, spinal cords from recipients of Itgb3−/− 2D2 Th17 cells had smaller demyelinated lesions and fewer inflammatory infiltrates, and the CD4+ T cells appeared to be more contained within the inflammatory cell infiltrate, in contrast to the rather diffuse distribution of WT 2D2 cells (Figures 6C and 6D).
Figure 6. Role of Integrin β3 in 2D2 Th17 Cell Transfer.
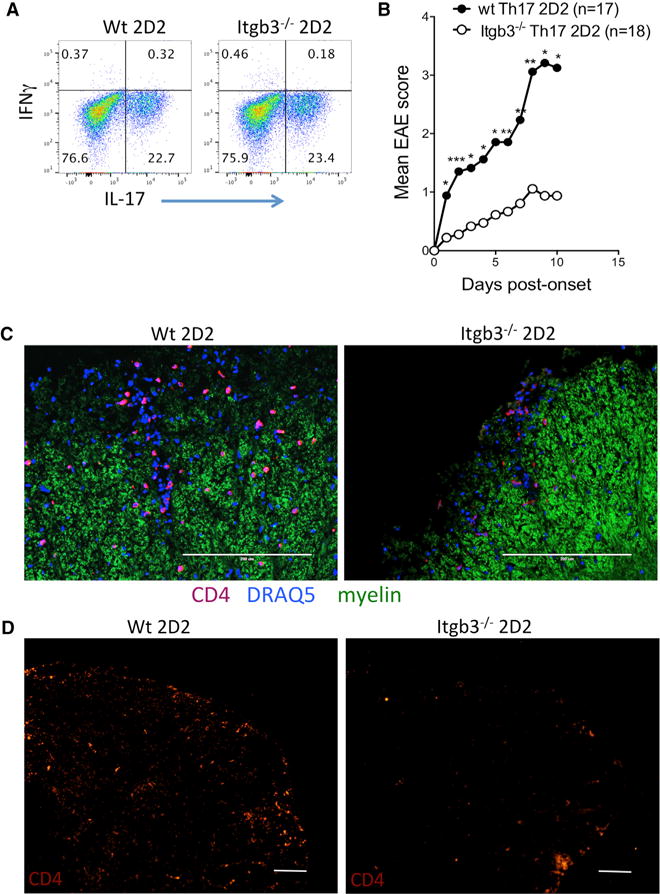
(A) Itgb3−/− 2D2 and Itgb3+/+2D2 donor cells were activated under Th17-inducing conditions and IL-17 production confirmed by flow cytometry.
(B) Passive EAE induction by Itgb3−/− 2D2 and Itgb3+/+ 2D2 donor cells Th17 differentiated in vitro. Day of onset varied between experiments, so day 0 was designated as first day clinical signs were observed in the control group in order to determine statistical significance of pooled experiments.
(C) Representative immunofluorescence staining of spinal cord sections from recipients of MOG-stimulated WT or Itgb3−/− 2D2 Th17 cells, analyzed at peak of disease, showing CD4+ T cells, nuclear stain (DRAQ5), and myelin (scale bars indicate 200 μm).
(D) Representative immunofluorescence staining of spinal cord sections from recipients of MOG-stimulated WT or Itgb3−/− 2D2 Th17 cells, showing CD4 distribution across upper right quadrant of spinal cord cross-section.
Data are pooled from four experiments with four to five mice per group. Statistical significance of EAE scores was assessed by Mann-Whitney separately for each time point.
Integrin β3-Deficient Th17 Cells Are Expanded but Fail to Accumulate in the CNS during EAE
To determine the T cell-specific impact of integrin β3 deficiency during active EAE induction, we isolated CD4+ T cells from Itgb3−/− mice and transferred them into RAG1−/− mice, which lack T and B cells but have normal expression of integrin β3 in vasculature and innate immune cells. Recipients of Itgb3−/− T cells were resistant to EAE induction by immunization with MOG(35–55), with only 20% of mice showing mild clinical signs, compared to 100% incidence and greater severity in controls that received CD4+ T cells isolated from Itgb3+/− littermates (Figure 7A). There was no difference in the number of IL-17-producing cells in dLNs prior to onset of EAE (Figure 7B), indicating that activation and expansion of Th17 cells does not require integrin β3 in vivo. Likewise, the number of IFN-γ-producing cells was not affected by integrin β3 deficiency (Figure 7C).
Figure 7. Integrin β3 Is Required for Accumulation of Th17 Cells in CNS.
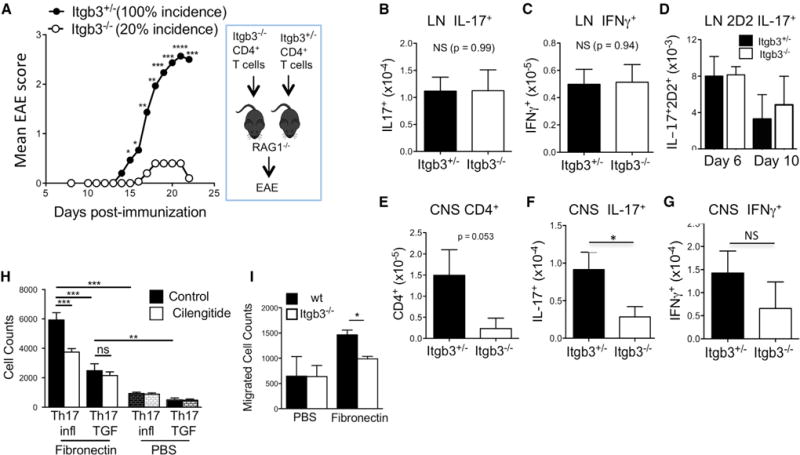
Itgb3−/− and Itgb3+/− CD4+ T cells were transferred into separate RAG−/− recipients that were immunized for EAE.
(A) Mean EAE clinical scores monitored.
(B) Absolute number of IL-17+ T cells in LNs on day 12 post-immunization.
(C) Absolute number of IFN-γ+ T cells in LNs day 12.
(D) CD4+ T cells from naive wt or Itgb3−/− 2D2 mice were transferred into naive CD45.1 recipients that were immunized for EAE. Numbers of IL-17+ 2D2 cells in dLNs were analyzed on days 6 and 10 post-immunization.
(E) Mice were transferred and immunized as in A, and absolute number of CD4+ cells in CNS analyzed on day 22.
(F) Absolute number of IL-17+ T cells in CNS on day 22.
(G) Absolute number of IFN-γ+ T cells in CNS on day 22.
(H) Transwell migration assay of Th17 cells cultured under Th17(i) integrin β3-promoting conditions or under Th17(TGF) integrin β3-suppressing conditions, in presence or absence of cRGD inhibitor, wells were coated with fibronectin or PBS as indicated.
(I) Transwell migration assay of WT and Itgb3−/− T cells cultured under Th17(i) conditions.
Data shown are pooled from three experiments with three to five mice per group, except (D), which was repeated twice, and (H) and (I), which are representative of three separate experiments. Statistical significance was assessed by Student’s t test, except for (H), which was assessed was by one-way ANOVA, and (A), which was assessed by Mann-Whitney test separately for each time point of EAE. Error bars indicate mean ± SD.
To further confirm that integrin β3 is not required for Th17 differentiation in vivo, we transferred WT or Itgb3−/− 2D2 T cells into WT recipients and immunized with MOG in CFA. In this model, we could specifically analyze MOG-specific T cell expansion and differentiation. As reported previously for 2D2 adoptive transfers, we were unable to track 2D2 cells in vivo after day 10 post-immunization (Reboldi et al., 2009). However, the number of IL-17-producing 2D2 T cells generated in dLNs was not affected by integrin β3 deficiency (Figure 7D), supporting our conclusion that integrin β3 is not required for Th17 cell differentiation.
We then examined the CNS infiltration of Itgb3−/− T cells during the clinically active phase of EAE. Similarly to the passive transfer results, few Itgb3−/− CD4+ T cells were found in the CNS (Figure 7E), corresponding with the failure to induce EAE. Likewise, the absolute number of cytokine producing cells, particularly IL-17+ cells, in the CNS was greatly reduced in Itgb3−/− recipients (Figures 7F and 7G). These data are consistent with a model in which integrin β3 is not required for early Th17 differentiation, but is required for effector Th17 cells to initiate and sustain inflammation. A similar observation was made for integrin β1 in Th1-mediated dermal inflammation (Overstreet et al., 2013), in which ECM-mediated migration of Th1 cells within interstitial tissue resulted in impaired effector function. We therefore tested the capacity of integrin β3 to mediate migration of Th17 cells to ECM using a transwell migration assay. Inflammatory Th17 cells (Itgb3hi) showed strong fibronectin-induced migration, which was blocked by the αvβ3 inhibitor cilengitide (Figure 7H). As a negative control, we tested migration of Th17 cells generated in the presence of TGF-β (Itgb3lo but Itgb1hi). These Th17 (TGF) cells (with low integrin β3 expression) also showed fibronectin-induced migration. However, Th17(TGF) cell migration was significantly reduced compared to Th17(i) cells, and was also not inhibited by cilengitide. This suggests that integrin β3 plays a dominant role in inflammatory Th17 migration that is only partially redundant with other migratory mechanisms such as integrin β1. Finally, we confirmed that Itgb3−/− T cells cultured under Th17(i) conditions have impaired fibronectin-induced migratory capacity (Figure 7I). Hence, integrin αvβ3 plays a role in ECM-directed migration that correlates with impaired accumulation of integrin-β3-deficient cells in the CNS after EAE induction by active immunization or passive transfer.
DISCUSSION
IL-23 is known to be important for Th17 effector functions, but not for early differentiation. Our data describe a role for integrin β3 expression by IL-23R-dependent inflammatory Th17 cells. Integrin β3 was almost exclusively expressed on RORγt+ Th17 cells during EAE, expression directly correlated with expression of integrin αv, and co-immunoprecipitation experiments confirmed that Th17 cells express αvβ3. In LNs from naive animals, very few integrin β3hi cells were present, corresponding with the paucity of Th17 cells in peripheral LNs in absence of inflammation. Th17 cells also expressed integrin β1, although this integrin appears to be much more broadly expressed, and was found to be present in naive animals and on regulatory T cells. Th17 cells have been reported to express α2β1, as have regulatory T cells (Gagliani et al., 2015; El Azreq et al., 2013). Integrin α4β1 is also expressed by inflammatory T cells and is targeted by natalizumab in MS. However, we found that α4 expression was low on Th17 cells, fitting with reports that Th17 cells are less dependent on α4β1 compared to Th1 cells (Rothhammer et al., 2011; Glatigny et al., 2011). It is possible that integrin α2β1 or α4β1 (or indeed other integrins) expression partially compensates in Itgb3−/− Th17 cells, to allow partial EAE induction. We have not observed any increase in integrin β1 expression on Itgb3−/− T cells, while αv expression is typically reduced (A.L.-H., unpublished data). Since chemokine receptors, such as CCR6, also mediate entry into the CNS, it is likely that the small lesions established by Itgb3−/− Th17 cells reflect this redundancy in migratory mechanisms and in fact highlight the strength of Th17 cell dependence on this particular integrin for EAE induction.
High integrin β3 expression was maintained on IFN-γ-producing ex-Th17 cells identified through fate-tracking studies. In contrast to the commonly proposed role of IL-23 in stabilizing the Th17 phenotype, three independent labs have shown that IL-23 signaling is in fact required for conversion of Th17 cells to Th1 (Duhen et al., 2013; Haines et al., 2013; Hirota et al., 2011). IL-23-mediated conversion corresponds with a predominance of Th1/Th17 double producers typically found in sites of IL-23-associated tissue inflammation. Hirota et al. similarly demonstrated that IL1R1 was maintained on ex-Th17 cells in EAE (Hirota et al., 2011). Therefore, the high expression of integrin β3 on converted ex-Th17 cells supports the role of IL-23 signaling in this process. The finding that integrin β3hi cells are enriched in CD44hiCD27lo effector populations also corresponds with previous data showing that IL-23 is required for the transition of activated Th17 cells to CD44hiCD27lo effector Th17 cells (McGeachy et al., 2009). It was also interesting to note that MOG-induced IFN-γ production particularly strongly associated with ex-Th17 cells, and with high integrin β3 expression, in comparison to PMA/ionomycin stimulation, further supporting that active induction of EAE by immunization with MOG(35–55) induces a predominantly Th17 response. It would therefore be interesting to compare expression of integrins in a strongly Th1-dependent context, such as infection with known Th1-dependent pathogens.
Integrin αvβ3 has many ligands. Several are ECM proteins such as fibronectin and vitronectin, and these are strongly upregulated in the CNS during EAE as well as in brain samples from patients with MS (Teesalu et al., 2001; Han et al., 2008). Many of these ECM proteins have been proposed to contribute to CNS inflammation in their capacity as coagulation and pro-angiogenic factors (Steinman, 2008). Our data suggest an additional function in promoting migration of inflammatory T cells into the CNS parenchyma to establish inflammatory foci. Early migration into the CNS is thought to bring T cells to the subarachnoid space via the blood-cerebrospinal barrier (Sallusto et al., 2012; Goverman, 2009; Ransohoff et al., 2003) and can be mediated by chemokine receptors, particularly CCR6 (Reboldi et al., 2009). A second stage of T cell activation and migration within the CNS is then required to establish the inflammatory signals that lead to demyelination (Goverman, 2009), leading to fulminant neurological damage. This stage is not well defined, and our data support a role for integrin αvβ3 in this process. Similar defects in interstitial migration have been reported for Th1 cells in an integrin-αvβ1-dependent manner (Overstreet et al., 2013).
Integrin αvβ3 signaling has been reported to mediate additional functions apart from migration. In a Th2 transfer model, integrin αvβ3 was found to promote contact-dependent activation of microglia by myelin-reactive T cells (Roy et al., 2007). That study focused on the protective effect of Th2-induced neurotrophins in vitro. However, it is feasible that reduced microglia activation by Itgb3−/− Th17 cells arriving in the CNS could also contribute to their inability to establish inflammation and recruitment of further inflammatory immune cells. Integrin αvβ3 signaling may also promote CD8+ T cell cytotoxic functions (Lacy-Hulbert et al., 2007; Doucey et al., 2003; Ma et al., 1997). Osteopontin, an integrin αvβ3 ligand, has been shown to enhance CD4+ IL-17 production in an integrin-β3-dependent manner in EAE (Murugaiyan et al., 2008) and in concanavalin A (con-A)-induced hepatitis (Diao et al., 2012), although we could not find a role for osteopontin in IL-17 production in our systems (data not shown). Osteopontin and vitronectin interactions with integrin αvβ3 are important in endothelial cell survival as well as migration during angiogenesis (Scatena et al., 1998; Courter et al., 2005). Osteopontin also promotes survival of encephalitogenic T cells in EAE (Hur et al., 2007), although specific receptors have not been identified. While we cannot formally rule out a contribution of direct osteopontin signaling through integrin αvβ3 on Th17 cells, in our study, we did not find consistent evidence for poor survival or reduced IL-17 production in Itgb3−/− T cells. Osteopontin binds several receptors that are present in the absence of integrin αvβ3, which could account for some of the differences between Itgb3−/− and osteopontin-deficient mice. However, it does remain feasible that the pro-inflammatory functions of integrin αvβ3 extend beyond tissue migration.
The finding that cilengitide, a compound already being evaluated in humans for cancer therapy, was able to reduce EAE severity supports further investigation into the potential of targeting integrin αvβ3 therapeutically. Blockade of αv using a similar inhibitor, cRGDfV, has previously been shown to block EAE development when administered from time of immunization (Acharya et al., 2010), an effect that was attributed to the role of integrin αv role in activating TGF-β on dendritic cells during initial Th17 cell activation. We therefore initiated cilengitide treatment on day 4 after immunization, at which time the initial presentation of antigen with TGF-β signaling is expected to have occurred. The efficacy of cilengitide for ameliorating EAE induced by previously activated Th17 cells further supports the use of this therapy to block relapses in diseases such as MS.
In summary, we report that integrin αvβ3 is highly expressed on effector Th17 cells in an IL-23-dependent manner. Although integrin-αvβ3-expressing cells showed an activated phenotype, integrin αvβ3 expression was not a marker of all activated T cells or Th1 cells, but it was enriched for cells of the Th17 lineage that expanded during EAE. This was in contrast to integrin β1 which showed broader expression on activated T cells. Furthermore, Th17 cells required integrin β3 expression for efficient induction of EAE by either passive transfer of activated cells or active immunization with MOG(35–55). This role for integrin αvβ3 expression corresponds with previous reports suggesting a role for IL-23 in promoting LN egress and migration into the CNS (McGeachy et al., 2009). Hence, this study identifies integrin αvβ3 as an IL-23-dependent Th17-expressed molecule with important functions that could be targeted by therapeutic intervention.
EXPERIMENTAL PROCEDURES
Mice
C57Bl/6, CD45.1+, OTII, IL-17Cre, ROSAfl/flYFP, and RAG1−/− and Itgb3 (integrinβ3) knockout mice were purchased from Jackson Laboratory. IL-23 RGFP reporter mice were a kind gift from Dr. Vijay Kuchroo (Brigham and Woman’s Hospital, Harvard Medical School).
Study Approval
Animals were housed under specific-pathogen-free conditions in an Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC)-approved facility and all animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh.
OTII Adoptive Cell Transfers
Recipient mice (B6, CD45.2+) received 105 CD45.1+ Il23ra−/− or Il23ra+/− OTII CD4+ T cells intravenously 1 day before immunization with 100 μg OVA(323–339) (Bio synthesis) in 10 mg/ml CFA (Difco Laboratories) subcutaneously in the flank. On day 6/7 and day 10 post-immunization, mice were sacrificed and dLNs were harvested; cells were stimulated with PMA and ionomycin for 4 hr in the presence of golgiplug before flow cytometry analysis.
In Vitro CD4+ T Cell Differentiation
CD4+ T cells from spleens and lymph nodes of naive mice were purified by magnetic separation (Miltenyi Biotec). T cells were activated by plate-bound anti-CD3 (clone 145-TC11, 5 μg ml−1; BioXcell) in IMDM medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 50 μM 2-β-mercaptoethanol, HEPES, and Na pyruvate for 3–4 days in the presence of recombinant mouse IL-1β, IL-23 (each 20 ng/ml), and IL-6 (50 ng/ml) for Th17(i), with IL-6 plus TGF-β1 (5 ng ml−1) for Th17(TGF); all cytokines were obtained from R&D Systems. In all Th17 cell cultures, anti-IFN-γ neutralizing antibodies (5 μg ml−1, BioXcell) were added. For Th1 cultures, IL-12 (PeproTech) was added at 20 ng/ml.
2D2 Adoptive Cell Transfers
Recipient mice (B6, CD45.1+) received 105 CD45.1− Itgb3−/− or Itgb3+/− 2D2 CD4+ T cells 1 day before immunization according to EAE immunization protocol. On day 6 and day 10 post-immunization, mice were sacrificed and dLNs were harvested; cells were stimulated with PMA and ionomycin for 4 hr in the presence of golgiplug before flow cytometry analysis.
EAE Induction
Active immunization: mice were immunized subcutaneously with 100 μg MOG(35–55) (Bio synthesis) emulsified in 200 μl CFA (Difco Laboratories) containing 100 μg heat-killed Mycobacterium tuberculosis H37Ra (Difco Laboratories) distributed in four sites on the flank. 200 ng pertussis toxin (List Biological Laboratories) was given intraperitoneally on days 0 and 2. For RAG−/− transfer experiments, dLNs and spleen were harvested from donor C57Bl/6 mice, Itgb3−/− mice, or Itgb3+/− littermates, and CD4+ cells were isolated by magnetic separation using CD4 microbeads (Miltenyi Biotec). 3–10 million CD4+ cells were transferred intraperitoneally to naive RAG1−/− recipients, which were immunized the following day as described above.
For passive transfer of EAE, donor Itgb3−/− mice or Itgb3+/− littermates were immunized with MOG(35–55) in CFA. On day 8, LNs and spleen were harvested and cells were cultured with 20 μg/ml MOG(35–55) in the presence of 20 ng/ml IL-23 (Th17) or IL-12 plus IL-2 (Th1). 3 days later, cell cultures were harvested and 2.5 × 107 cells were transferred into naive B6 recipients, with injection of pertussis toxin on day 0 and day 2. Alternatively, LNs and spleen were harvested from WT or Itgb3−/− 2D2 mice and stimulated in vitro according to protocol described by Jäger et al. (2009).
For the RGD blockade of integrin β3, 100 μg cRGD peptide (Peptide Institute) in PBS (HyClone) was injected subcutaneously daily from day 4 until day 8 and then from day 15 to day 21, twice daily from day 9 to day 14. The control group received equal volume of PBS.
EAE was assessed according to the following clinical grades: 1, flaccid tail; 2, impaired righting reflex and hindlimb weakness; 3, partial hindlimb paralysis; 4, complete hindlimb paralysis; 5, hindlimb paralysis with partial forelimb paralysis; and 6, moribund/dead.
Flow Cytometry
The following fluorescent-activated cell sorting (FACS) antibodies were purchased from BD Biosciences: CD4 (RM4-5), CD44 (IM7), CD27 (LG.3A10), integrin αv (RMV-7), IFN-γ (XMG1.2), and IL-17 (TC11-18H10). The following were purchased from eBioscience: integrin β1 (eBioHMb1-1), integrin β3 (2C9.G3), integrin α2 (DX5), integrin α4 (R1–2), integrin α6 (GoH3), RORγt (AKFJS9), Foxp3 (FJK-16 s). For cytokine analysis, cells were cultured in complete medium (RPMI media containing 10% fetal calf serum (FCS), supplemented with Pen-Strep, L-glutamine, HEPES, sodium pyruvate, and 2-ME) with 50 ng/ml PMA and 500 ng/ml ionomycin (both Sigma-Aldrich) in the presence of Golgiplug (BD Biosciences) for 3 to 4 hr followed by FACS staining and analysis. For intracellular cytokines, staining was performed using Cytofixcytoperm kit from BD; RORγt and Foxp3 intracellular stains were performed using an eBioscience Foxp3 staining kit according to the manufacturer’s instructions.
Immunoprecipitations and Western Blotting
Western blotting and immunoprecipitations were performed as described previously (Garg et al., 2015, McCarty et al., 2005). Anti-αV (EMD Millipore) and anti-β3 (Santa Cruz Biotechnology) antibodies were used for immunoprecipitation and anti-αV and anti-β3 (Cell Signaling Technology) antibodies were used for western blotting. Blots were developed using a FluorChem E imager (Protein Simple).
Immunofluorescence Staining
Mice were sacrificed and perfused with PBS followed by 2% PFA. Spinal cords were removed and post-fixed in 2% PFA followed by immersion in 30% sucrose overnight; tissue was then embedded and frozen in Optimal Cutting Temperature (OCT; Tissue-Tek). 6-μm sections were stained with CD4-BV421 (BD Biosciences), FluoroMyelin Green (Invitrogen), and nuclear stain DRAQ5 (BD Biosciences). Slides were mounted with Gelvatol mounting media and images acquired on Evos FL Auto microscope.
Migration Assays
CD4+ T cells were isolated from spleen of C57Bl/6 or Itgb3−/− mice by negative selection and magnetic sorting (Miltenyi Biotech). To generate Th17 cells, T cells were cultured in RPMI 1640 medium supplemented with 10% FCS, IL-6, IL-23, IL-1β, and anti-IFN-γ antibody, with or without TGF-β1, in plates coated with anti-CD3 antibody. Cells were harvested between days 5–7 of culture and resuspended in migration medium (RPMI with 2% BSA). Migration was assayed in 96-well cell-permeable chambers (5-μm pore size polycarbonate membranes) which had been pre-coated by incubation with rat fibronectin (Sigma; 10 μg/ml), vitronectin (Sigma; 1 μg/ml) or no matrix at 37°C for 2 hr, and remaining protein binding sites blocked with RPMI/BSA for 1 hr. 4 × 104 T cells were added to the upper chambers, and RPMI containing 1% FCS was added to the lower chambers. In some cases, GRGDNP peptide (Enzo Life Sciences) was added to upper chambers at 2 μg/ml. After 2 hr, the upper chambers were removed, and migrated cells in the lower chamber were stained with Calcein AM (1 μM; Invitrogen) and visualized and counted by imaging plate reader (Cytation 3; BioTek).
Statistics
One-way ANOVA (for multiple groups) or Student’s t tests were performed for experiments with parametric values (such as FACS %); Mann-Whitney U test was performed for EAE experiments, analyzing scores for each day separately. p values are shown as *p < 0.05, **p < 0.01, and ***p < 0.001, where statistical significance was found, and all data are presented as means + SD.
Supplementary Material
Highlights.
Inflammatory Th17 cells express high levels of integrin αvβ3
Integrin β3 expression is IL-23 dependent and suppressed by TGF-β
Chemical or genetic αvβ3 inhibition reduced Th17-mediated disease in EAE
β3 blockade reduces Th17 migration in the CNS during EAE and in vitro
Acknowledgments
We thank Dr. Sarah Gaffen, Dr. Partha Biswas, and Dr. Amanda Poholek for helpful discussion and comments. This study was funded by National Institutes of Health (NIH) grants AI110822-01 (to M.J.M.) and DK093695 (to A.H.-L.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes three figures and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.06.065.
AUTHOR CONTRIBUTIONS
F.D., A.V.G., K.K., S.M., D.G.K., G.H., M.M., M.H., A.L.-H., and M.J.M. performed experiments; F.D., A.V.G., A.L.-H., and M.M. analyzed data; and M.M. designed the experiments and wrote the paper.
References
- Acharya M, Mukhopadhyay S, Païdassi H, Jamil T, Chow C, Kissler S, Stuart LM, Hynes RO, Lacy-Hulbert A. αv Integrin expression by DCs is required for Th17 cell differentiation and development of experimental autoimmune encephalomyelitis in mice. J Clin Invest. 2010;120:4445–4452. doi: 10.1172/JCI43796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- Aly L, Yousef S, Schippling S, Jelcic I, Breiden P, Matschke J, Schulz R, Bofill-Mas S, Jones L, Demina V, et al. Central role of JC virus-specific CD4+ lymphocytes in progressive multi-focal leucoencephalopathy-immune reconstitution inflammatory syndrome. Brain. 2011;134:2687–2702. doi: 10.1093/brain/awr206. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Cañete JD, Pablos JL, Sanmartí R, Mallofré C, Marsal S, Maymó J, Gratacós J, Mezquita J, Mezquita C, Cid MC. Antiangiogenic effects of anti-tumor necrosis factor alpha therapy with infliximab in psoriatic arthritis. Arthritis Rheum. 2004;50:1636–1641. doi: 10.1002/art.20181. [DOI] [PubMed] [Google Scholar]
- Chen Y, Haines CJ, Gutcher I, Hochweller K, Blumenschein WM, McClanahan T, Hämmerling G, Li MO, Cua DJ, McGeachy MJ. Foxp3(+) regulatory T cells promote T helper 17 cell development in vivo through regulation of interleukin-2. Immunity. 2011;34:409–421. doi: 10.1016/j.immuni.2011.02.011. [DOI] [PubMed] [Google Scholar]
- Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, Dong C. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30:576–587. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codarri L, Gyülvészi G, Tosevski V, Hesske L, Fontana A, Magnenat L, Suter T, Becher B. RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12:560–567. doi: 10.1038/ni.2027. [DOI] [PubMed] [Google Scholar]
- Courter DL, Lomas L, Scatena M, Giachelli CM. Src kinase activity is required for integrin alphaVbeta3-mediated activation of nuclear factor-kappaB. J Biol Chem. 2005;280:12145–12151. doi: 10.1074/jbc.M412555200. [DOI] [PubMed] [Google Scholar]
- Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 2003;421:744–748. doi: 10.1038/nature01355. [DOI] [PubMed] [Google Scholar]
- Diao H, Liu X, Wu Z, Kang L, Cui G, Morimoto J, Denhardt DT, Rittling S, Iwakura Y, Uede T, Li L. Osteopontin regulates interleukin-17 production in hepatitis. Cytokine. 2012;60:129–137. doi: 10.1016/j.cyto.2012.06.287. [DOI] [PubMed] [Google Scholar]
- Doucey MA, Legler DF, Faroudi M, Boucheron N, Baumgaertner P, Naeher D, Cebecauer M, Hudrisier D, Rüegg C, Palmer E, et al. The beta1 and beta3 integrins promote T cell receptor-mediated cytotoxic T lymphocyte activation. J Biol Chem. 2003;278:26983–26991. doi: 10.1074/jbc.M302709200. [DOI] [PubMed] [Google Scholar]
- Duhen R, Glatigny S, Arbelaez CA, Blair TC, Oukka M, Bettelli E. Cutting edge: the pathogenicity of IFN-γ-producing Th17 cells is independent of T-bet. J Immunol. 2013;190:4478–4482. doi: 10.4049/jimmunol.1203172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Azreq MA, Boisvert M, Cesaro A, Pagé N, Loubaki L, Allaeys I, Chakir J, Poubelle PE, Tessier PA, Aoudjit F. α2β1 integrin regulates Th17 cell activity and its neutralization decreases the severity of collagen-induced arthritis. J Immunol. 2013;191:5941–5950. doi: 10.4049/jimmunol.1301940. [DOI] [PubMed] [Google Scholar]
- El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, Zhang GX, Dittel BN, Rostami A. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12:568–575. doi: 10.1038/ni.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagliani N, Amezcua Vesely MC, Iseppon A, Brockmann L, Xu H, Palm NW, de Zoete MR, Licona-Limón P, Paiva RS, Ching T, et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature. 2015;523:221–225. doi: 10.1038/nature14452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg AV, Amatya N, Chen K, Cruz JA, Grover P, Whibley N, Conti HR, Hernandez Mir G, Sirakova T, Childs EC, et al. MCPIP1 endoribonuclease activity negatively regulates interleukin-17-mediated signaling and inflammation. Immunity. 2015;43:475–487. doi: 10.1016/j.immuni.2015.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-β signalling. Nature. 2010;467:967–971. doi: 10.1038/nature09447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatigny S, Duhen R, Oukka M, Bettelli E. Cutting edge: loss of α4 integrin expression differentially affects the homing of Th1 and Th17 cells. J Immunol. 2011;187:6176–6179. doi: 10.4049/jimmunol.1102515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedkoop AY, Kraan MC, Picavet DI, de Rie MA, Teunissen MB, Bos JD, Tak PP. Deactivation of endothelium and reduction in angiogenesis in psoriatic skin and synovium by low dose infliximab therapy in combination with stable methotrexate therapy: a prospective single-centre study. Arthritis Res Ther. 2004;6:R326–R334. doi: 10.1186/ar1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goverman J. Autoimmune T cell responses in the central nervous system. Nat Rev Immunol. 2009;9:393–407. doi: 10.1038/nri2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haines CJ, Chen Y, Blumenschein WM, Jain R, Chang C, Joyce-Shaikh B, Porth K, Boniface K, Mattson J, Basham B, et al. Autoimmune memory T helper 17 cell function and expansion are dependent on interleukin-23. Cell Rep. 2013;3:1378–1388. doi: 10.1016/j.celrep.2013.03.035. [DOI] [PubMed] [Google Scholar]
- Han MH, Hwang SI, Roy DB, Lundgren DH, Price JV, Ousman SS, Fernald GH, Gerlitz B, Robinson WH, Baranzini SE, et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature. 2008;451:1076–1081. doi: 10.1038/nature06559. [DOI] [PubMed] [Google Scholar]
- Hellwig K, Gold R. Progressive multifocal leukoencephalopathy and natalizumab. J Neurol. 2011;258:1920–1928. doi: 10.1007/s00415-011-6116-8. [DOI] [PubMed] [Google Scholar]
- Hirota K, Duarte JH, Veldhoen M, Hornsby E, Li Y, Cua DJ, Ahlfors H, Wilhelm C, Tolaini M, Menzel U, et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol. 2011;12:255–263. doi: 10.1038/ni.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur EM, Youssef S, Haws ME, Zhang SY, Sobel RA, Steinman L. Osteopontin-induced relapse and progression of autoimmune brain disease through enhanced survival of activated T cells. Nat Immunol. 2007;8:74–83. doi: 10.1038/ni1415. [DOI] [PubMed] [Google Scholar]
- Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- Jäger A, Dardalhon V, Sobel RA, Bettelli E, Kuchroo VK. Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J Immunol. 2009;183:7169–7177. doi: 10.4049/jimmunol.0901906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurohori Y, Sato K, Suzuki S, Kashiwazaki S. Adhesion molecule expression on peripheral blood mononuclear cells in rheumatoid arthritis: positive correlation between the proportion of L-selectin and disease activity. Clin Rheumatol. 1995;14:335–341. doi: 10.1007/BF02208350. [DOI] [PubMed] [Google Scholar]
- Lacy-Hulbert A, Ueno T, Ito T, Jurewicz M, Izawa A, Smith RN, Chase CM, Tanaka K, Fiorina P, Russell PS, et al. Beta 3 integrins regulate lymphocyte migration and cytokine responses in heart transplant rejection. Am J Transplant. 2007;7:1080–1090. doi: 10.1111/j.1600-6143.2007.01757.x. [DOI] [PubMed] [Google Scholar]
- Ma EA, Lou O, Berg NN, Ostergaard HL. Cytotoxic T lymphocytes express a beta3 integrin which can induce the phosphorylation of focal adhesion kinase and the related PYK-2. Eur J Immunol. 1997;27:329–335. doi: 10.1002/eji.1830270147. [DOI] [PubMed] [Google Scholar]
- Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- Mas-Moruno C, Rechenmacher F, Kessler H. Cilengitide: the first anti-angiogenic small molecule drug candidate design, synthesis and clinical evaluation. Anticancer Agents Med Chem. 2010;10:753–768. doi: 10.2174/187152010794728639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty JH, Cook AA, Hynes RO. An interaction between alphavbeta8 integrin and Band 4.1B via a highly conserved region of the Band 4.1 C-terminal domain. Proc Natl Acad Sci USA. 2005;102:13479–13483. doi: 10.1073/pnas.0506068102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- McGeachy MJ, Chen Y, Tato CM, Laurence A, Joyce-Shaikh B, Blumenschein WM, McClanahan TK, O’Shea JJ, Cua DJ. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol. 2009;10:314–324. doi: 10.1038/ni.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murugaiyan G, Mittal A, Weiner HL. Increased osteopontin expression in dendritic cells amplifies IL-17 production by CD4+ T cells in experimental autoimmune encephalomyelitis and in multiple sclerosis. J Immunol. 2008;181:7480–7488. doi: 10.4049/jimmunol.181.11.7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity. 2000;13:715–725. doi: 10.1016/s1074-7613(00)00070-4. [DOI] [PubMed] [Google Scholar]
- Overstreet MG, Gaylo A, Angermann BR, Hughson A, Hyun YM, Lambert K, Acharya M, Billroth-Maclurg AC, Rosenberg AF, Topham DJ, et al. Inflammation-induced interstitial migration of effector CD4+ T cells is dependent on integrin αV. Nat Immunol. 2013;14:949–958. doi: 10.1038/ni.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM, Kivisäkk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol. 2003;3:569–581. doi: 10.1038/nri1130. [DOI] [PubMed] [Google Scholar]
- Reardon DA, Nabors LB, Stupp R, Mikkelsen T. Cilengitide: an integrin-targeting arginine-glycine-aspartic acid peptide with promising activity for glioblastoma multiforme. Expert Opin Investig Drugs. 2008;17:1225–1235. doi: 10.1517/13543784.17.8.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, Uccelli A, Lanzavecchia A, Engelhardt B, Sallusto F. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol. 2009;10:514–523. doi: 10.1038/ni.1716. [DOI] [PubMed] [Google Scholar]
- Rothhammer V, Heink S, Petermann F, Srivastava R, Claussen MC, Hemmer B, Korn T. Th17 lymphocytes traffic to the central nervous system independently of α4 integrin expression during EAE. J Exp Med. 2011;208:2465–2476. doi: 10.1084/jem.20110434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Liu X, Pahan K. Myelin basic protein-primed T cells induce neurotrophins in glial cells via alphavbeta3 [corrected] integrin. J Biol Chem. 2007;282:32222–32232. doi: 10.1074/jbc.M702899200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F, Impellizzieri D, Basso C, Laroni A, Uccelli A, Lanzavecchia A, Engelhardt B. T-cell trafficking in the central nervous system. Immunol Rev. 2012;248:216–227. doi: 10.1111/j.1600-065X.2012.01140.x. [DOI] [PubMed] [Google Scholar]
- Scatena M, Almeida M, Chaisson ML, Fausto N, Nicosia RF, Giachelli CM. NF-kappaB mediates alphavbeta3 integrin-induced endothelial cell survival. J Cell Biol. 1998;141:1083–1093. doi: 10.1083/jcb.141.4.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman L. New targets for treatment of multiple sclerosis. J Neurol Sci. 2008;274:1–4. doi: 10.1016/j.jns.2008.06.040. [DOI] [PubMed] [Google Scholar]
- Steinman L. Shifting therapeutic attention in MS to osteopontin, type 1 and type 2 IFN. Eur J Immunol. 2009;39:2358–2360. doi: 10.1002/eji.200939814. [DOI] [PubMed] [Google Scholar]
- Teesalu T, Hinkkanen AE, Vaheri A. Coordinated induction of extracellular proteolysis systems during experimental autoimmune encephalomyelitis in mice. Am J Pathol. 2001;159:2227–2237. doi: 10.1016/S0002-9440(10)63073-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldhoen M, Hocking RJ, Flavell RA, Stockinger B. Signals mediated by transforming growth factor-beta initiate autoimmune encephalomyelitis, but chronic inflammation is needed to sustain disease. Nat Immunol. 2006;7:1151–1156. doi: 10.1038/ni1391. [DOI] [PubMed] [Google Scholar]
- Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- Zúñiga LA, Jain R, Haines C, Cua DJ. Th17 cell development: from the cradle to the grave. Immunol Rev. 2013;252:78–88. doi: 10.1111/imr.12036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.